Introduction
Sepsis is a severe inflammatory disease in response
to bacterial infection (1). It is
characterized by systemic inflammatory response syndrome (SIRS) and
is frequently caused by hemorrhage, trauma and abdominal surgery
(1). In the intensive care unit,
severe sepsis is considered as a leading cause of death due to the
occurrence of SIRS and multiple organ dysfunctions (2,3).
Thus, it is imperative to understand the underlying pathogenesis
and search for effective intervention and therapy for sepsis.
Previous study has demonstrated that cardiac
dysfunction is induced in ~50% of critically ill patients with
sepsis, and the recovery of cardiac function is considered as a key
predictor for survival (4). It is
reported that cardiac microvascular dysfunction is associated with
cardiac dysfunction in patients with sepsis (5). Importantly, cardiac microvascular
endothelial cells (CMVECs), accounting for 1/3 of all heart cells,
exert an important influence on the normal condition of coronary
microvessels and adjacent cardiomyocytes (6). In addition, CMVEC dysfunction, such
as hyperpermeability and apoptosis caused by inflammatory
responses, is found in myocardial ischemia/reperfusion injury
(7,8). Although cardiac microvascular
dysfunction is confirmed in sepsis, few studies have investigated
the function of CMVECs in sepsis.
Ulinastatin, a protease inhibitor, exhibits
anti-inflammatory activity by inhibiting the expression of
pro-inflammatory factors and blocking the signaling pathways
related to inflammation (9).
Several animal experiments have demonstrated a protective role of
ulinastatin against sepsis by suppressing the inflammatory response
(10–12). Randomized controlled clinical
studies have further revealed that ulinastatin can significantly
improve organ failure and reduce mortality in patients with sepsis
(13,14). However, it is still unclear
whether the role of ulinastatin in sepsis is associated with the
function of CMVECs.
Currently, increasing evidence has demonstrated that
long non-coding RNAs (lncRNAs) are involved in various diseases
(15), including
neurodegenerative diseases (16),
cardiovascular diseases (17) and
cancers (18) by regulating
target gene transcription. Understanding the underlying mechanisms
of lncRNAs in sepsis may contribute to an effective treatment for
sepsis. Metastasis-associated lung adenocarcinoma transcript 1
(MALAT1) was originally known as an lncRNA in patients with
non-small cell lung cancer (NSCLC) who have a high risk of
metastasis. A recent study reported that MALAT1 is associated with
cell proliferation and migration in cancer cells (19). Nevertheless, its role in CMVEC
dysfunction of sepsis is still poorly investigated. In the present
study, a sepsis model in the rat was induced by cecal ligation and
puncture (CLP), and rat CMVECs were isolated and treated by
lipopolysaccharide (LPS) or/and ulinastatin. We aimed to evaluate
the effects of ulinastatin on the permeability and apoptosis of
LPS-induced CMVECs and investigate its molecular mechanisms.
Materials and methods
Ethics statement
Healthy male Sprague-Dawley (SD) rats (~100 g of
body weight) were purchased from Beijing Vital River Laboratory
Animal Technology Co., Ltd. (Beijing, China). Approval from the
Institutional Animal Care and Use Committee of the First Affiliated
Hospital of Xinjiang Medical University was obtained prior to all
animal experiments.
Animal model of sepsis
A total of 50 SD rats were randomly assigned into
two groups: a sham group (n=10) and a sepsis group (n=40). All rats
were acclimatized for 3–5 days before the experiments. The rat
sepsis model was established using CLP according to a previously
described (20). CLP induced
intra-abdominal infection and the release of LPS, resulting in SIRS
and sepsis. Briefly, the rats were anesthetized by an
intraperitoneal injection of 10% chloral hydrate (3 ml/kg; Sigma,
St. Louis, MO, USA) and fixed in a supine position. A 1.5-cm
longitudinal incision was made along the abdominal midline, then
the caecum was exposed and separated. For rats in the sepsis group,
the caecum below the ileocaecal valve was ligated using 3.0 suture.
Then, a 18-gauge needle was used to puncture through the caecum 3
times, and the feces was extruded by gently squeezing the caecum.
Lastly, the separated caecum was returned to the abdominal cavity
and the incision was sutured layer by layer. Rats in the sham group
were treated with similar procedures but without ligation or
puncture. The entire operation was carried out under aseptic
conditions. The rats were sacrificed 24 h post-operatively, and the
heart tissues were collected for the following experiments.
Isolation of CMVECs
CMVECs were isolated from SD rats following a
previously reported method (21).
In brief, two SD rats were anesthetized and then intraperitoneally
injected with heparin (300 IU/g; Sigma) for 20 min. Then, the rats
were sacrificed by cervical dislocation. After sterilization with
75% alcohol, the thoracic cavity was opened. Then, the heart was
removed and rinsed with sterile D-Hanks' containing heparin. Next,
the connective tissue, atria, right ventricle and interventricular
septum were removed, and epicardial and endocardial cells from the
left ventricle were devitalized by immersing in 75% ethanol for
20–30 sec. The remaining tissue was minced and rinsed with
D-Hanks', and then incubated in 0.02% collagenase type II
(dissolved in D-Hanks') for 30 min and 0.02% trypsin (both from
Sigma) for another 10 min at 37°C. The dissociated cells were
centrifuged at 1,000 rpm for 10 min, and then resuspended in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Cambridge,
MA, USA) containing 15% fetal bovine serum (FBS). Lastly, the cells
were cultured in laminin-coated dishes (Corning, Inc., Corning, NY,
USA).
Cell treatment
LPS was used to induce the sepsis cell model. To
determine the optimum inducement time and concentration, the
primary CMVECs were treated with various concentrations (0, 10, 50,
100 ng/ml, 1 and 2 µg/ml) for 0, 3, 6, 9 and 12 h,
respectively. To investigate the effect of ulinastatin on the
LPS-induced CMVECs, the CMVECs were treated with LPS and LPS +
ulinastatin (1000, 3000 or 5000 U/l), respectively. In addition,
the treated cells were also transfected with the lncRNA MALAT1
overexpression vector (named as pc-MALAT1; RiboBio, Guangzhou,
China) using Lipofectamine 2000 (Invitrogen).
Assessment of cell permeability
To evaluate cell permeability, transendothelial
electrical resistance (TER) was measured using STX2 electrode and
EVOM2 meter (World Precision Instruments, Sarasota, FL, USA).
CMVECs cells were seeded on fibronectin-coated Transwell membrane
inserts (0.4-µm pore size; Corning, Inc.). Then, confluent
CMVECs were treated with LPS, LPS + ulinastatin or LPS +
ulinastatin + lncRNA MALAT1 overexpression vector, respectively.
The value of TER was calculated as the resistance value multiplied
by the membrane area.
Cell apoptosis assay
The Annexin V-FITC apoptosis detection kit
(Invitrogen) was used to quantify cell apoptosis. Briefly, the
treated cells were collected by digestion with Trypsin and
centrifugation at 1,500 rpm for 6 min. After being washed with
phosphate-buffered saline (PBS), the cells were mixed with
FITC-Annexin V and propidium iodide (PI) for 15 min at 25°C in the
dark. Cells were then added to 400 µl 1X binding buffer and
detected using a flow cytometer. Annexin V-positive and PI-negative
cells were considered as apoptotic cells.
Reactive oxygen species (ROS) assay
The ROS level was assessed by
2,7-dichlorofluorescein diacetate (DCFH-DA; Sigma). After being
washed with PBS, the treated cells were incubated with serum-free
DMEM containing 10 µM DCFH-DA for 20 min at 37°C in the
dark. Next, the cells were collected and analyzed using a Spectra
Max M5 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
The fluorescent intensities were measured at a 535 nm
absorbance.
Real-time-quantitative polymerase chain
reaction (RT-qPCR) analysis
Total RNA extraction was carried out using TRIzol
reagent (Invitrogen), and complementary DNA was obtained by the
reverse transcription of total RNA using the MultiScribe RT kit
(Applied Biosystems, Foster City, CA, USA). Then, the levels of
target genes, including lncRNA MALAT1, B-cell lymphoma-2 (Bcl-2),
Bax, caspase-3, EZH2, DAB2IP and Brachyury were detected using
SYBR® Premix Ex Taq™ (Takara Biotechnology Co., Ltd.,
Dalian, China). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH)
served as an internal reference. Primers for these genes are listed
in Table I. The relative
quantification of these genes was carried out using the comparative
threshold (Ct) cycle method (2−ΔΔCt).
 | Table IPrimer sequences for specific
genes. |
Table I
Primer sequences for specific
genes.
| Gene | Primer
sequences |
|---|
| RT-qPCR |
| MALAT1 | F:
5′-AAAGCAAGGTCTCCCCACAAG-3′ |
| R:
5′-GGTCTGTGCTAGATCAAAAGGCA-3′ |
| Bcl-2 | F:
5′-CATCAGGAAGGCTAGAGTTACC-3′ |
| R:
5′-CAGACATTCGGAGACCACAC-3′ |
| Bax | F:
5′-GATGCGTCCACCAAGAAG-3′ |
| R:
5′-AGTTGAAGTTGCCGTCAG-3′ |
| Caspase-3 | F:
5′-TGTCATCTCGCTCTGGTACG-3′ |
| R:
5′-AAATGACCCCTTCATCACCA-3′ |
| ZEH2 | F:
5′-TTGTTGGCGGAAGCGTGTAAAATC-3′ |
| R:
5′-TCCCTAGTCCCGCGCAATGAGC-3′ |
| DAB2IP | F:
5′-ACACGCCATGGAGCCCGACT-3′ |
| R:
5′-GAAGCCCGTGACCCGGAACG-3′ |
| Brachyury | F:
5′-AGGTGGGGAAGTTTCCTTCT-3′ |
| R:
5′-GCAAATGAGGTCCTTTTGGT-3′ |
| GAPDH | F:
5′-AGCTTCGGCACATATTTCATCTG-3′ |
| R:
5′-CGTTCACTCCCATGACAAACA-3′ |
| ChIP-qPCR |
| DAB2IP | F:
5′-CCTGCTTTCTGTTTCCTTCTCCTG-3′ |
| R:
5′-TTGAACCACCTCCTCCTCCCTCTC-3′ |
| Brachyury | F:
5′-AGGTGGGGAAGTTTCCTTCT-3′ |
| R:
5′-GCAAATGAGGTCCTTTTGGT-3′ |
Western blotting
RIPA buffer (Sangon Biotech, Shanghai, China) was
used to extract protein from the heart tissues and cells. The
measurement of protein concentration was conducted using BCA
Protein Quantitative assay (Pierce, Appleton, WI, USA). Protein
samples were subjected to sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE), and then blotted onto nitrocellulose
membranes (Millipore, Billerica, MA, USA). After being blocked with
5% nonfat milk in Tris-buffered saline Tween-20 (TBST), the
membranes were incubated with rat anti-Bcl-2, Bax, caspase-3, EZH2,
DAB2IP, Brachyury and GAPDH monoclonal antibodies (1:500–1,000;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) overnight at
4°C, respectively. After being washed with TBST, the membranes were
incubated with goat anti-rat secondary antibody (1:5,000; Santa
Cruz Biotechnology, Inc.) for 2 h at room temperature. Ultimately,
the proteins were detected with Enhanced chemiluminescence
(Millipore, Bedford, MA, USA).
RNA immunoprecipitation (RIP) assay
Magna RIP RNA-Binding Protein Immunoprecipitation
kit (Millipore) was used to perform RIP assay following the
manufacturer's instructions. In brief, CMVECs were treated with LPS
for 6 h. Then, the cells were collected and treated with RIPA
buffer. After centrifugation, the supernatant was incubated with
protein-A/G-Sepharose beads, anti-EZH2 monoclonal antibody and IgG
overnight at 4°C. Subsequently, co-precipitated RNA was extracted
and then detected by RT-qPCR.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assay was carried out using QuikChIP kit
(Imgenex, San Diego, CA, USA). In brief, CMVECs were transfected
with two siRNAs targeting MALAT1 (si-MALAT1-1 and si-MALAT1-2;
Dharmacon, Mickleton, NJ, USA) using Lipofectamine 2000 for 48 h.
Then, protein-chromatin cross-linking of cells was induced using 1%
formaldehyde for 10 min at 37°C and quenched by glycine. Next, the
cells were lysed in lysis buffer and chromatin DNA from cell
lysates was sonicated into fragments. Next, chromatin DNA fragments
were subjected to immunoprecipitation with EZH2 or
methyltransferase catalyzing histone H3 lysine 27 trimethylation
(H3K27me3) antibody. After reverse cross-linking, DNA fragments
were purified and then detected by RT-qPCR.
Statistical analysis
Statistical analyses were carried out using SPSS
19.0 (IBM Corp., Armonk, NY, USA). The results of all experiments
are expressed as the mean ± SD and analyzed by t-test. A
statistically significant difference was defined as a p-value
<0.05.
Results
Ulinastatin reduces LPS-induced CMVEC
hyperpermeability
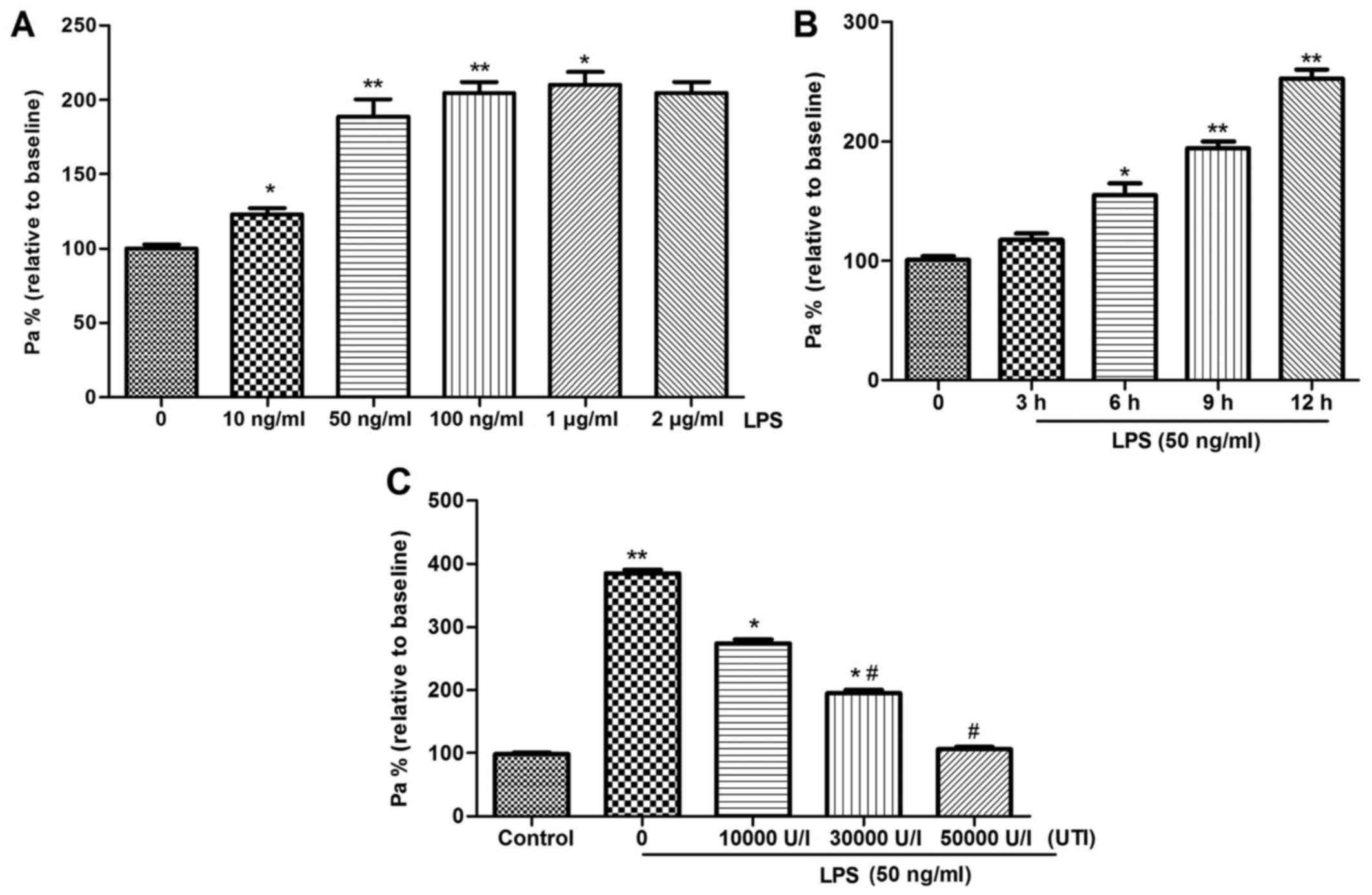
The results of TER showed that compared with the
untreated cells, cell permeability was significantly increased
(p<0.05) in the LPS-induced cells in a dose- and time-dependent
manner (Fig. 1A and B). Notably,
cell permeability was elevated when the dose of LPS exceeded 50
ng/ml and the treatment time was >6 h (Fig. 1A and B). Thus, the optimum
inducement time and concentration was determined as 6 h and 50
ng/ml. In addition, the presence of ulinastatin markedly inhibited
(p<0.05) cell permeability in LPS-induced cells, and 5,000 U/l
ulinastatin led to the more obvious improvement (Fig. 1C).
Ulinastatin reduces LPS-induced CMVEC
apoptosis
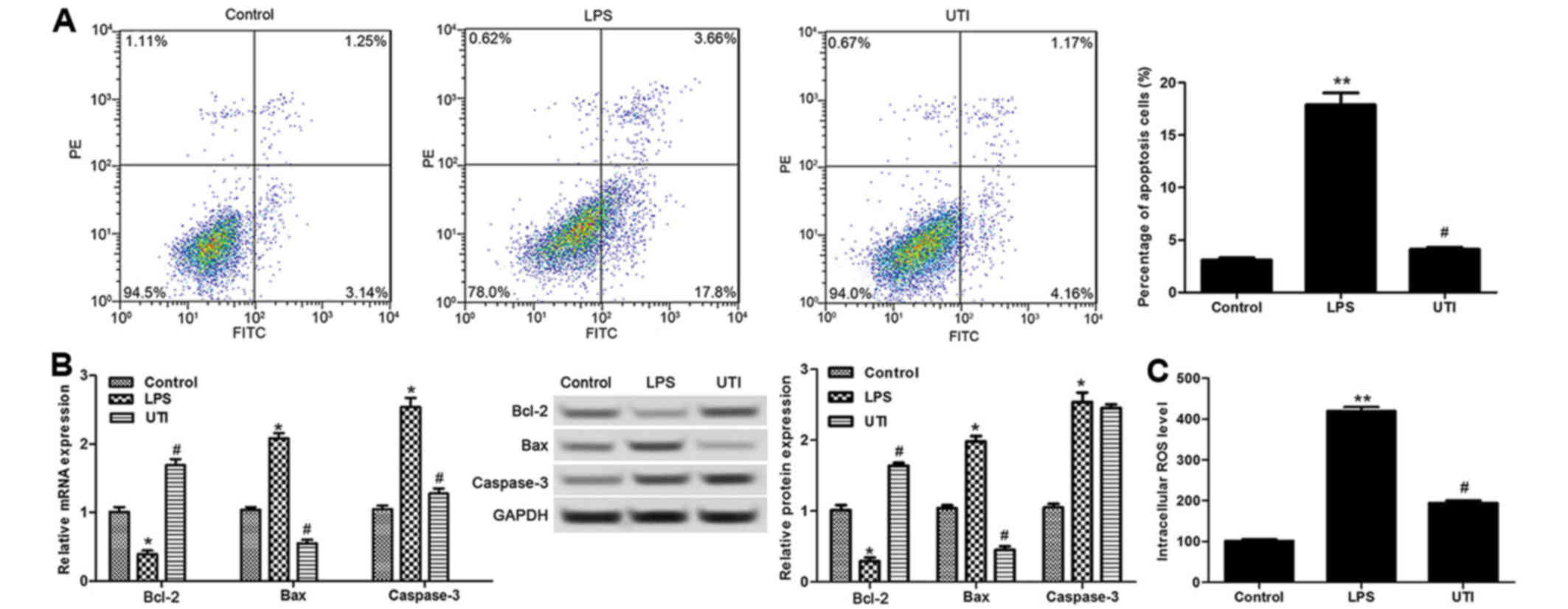
Next, we analyzed the effect of ulinastatin on CMVEC
apoptosis. The results revealed that compared with the untreated
cells, the percentage of apoptotic cells was markedly increased in
the LPS-induced cells (p<0.01), while it was obviously decreased
after treatment with ulinastatin (p<0.05) (Fig. 2A). Meanwhile, the expression
levels of cell apoptosis-related proteins, including Bcl-2,
caspase-3 and Bax, were detected by RT-qPCR and western blotting,
respectively. The results showed that LPS-induced cells showed a
lower Bcl-2 level as well as higher Bax and caspase-3 levels than
the untreated cells (p<0.05) (Fig.
2B). After treatment with ulinastatin, the mRNA and protein
levels of Bcl-2 were significantly increased, and the levels of
caspase-3 and Bax were markedly decreased in the LPS-induced cells
(p<0.05) (Fig. 2B). Also,
compared with the untreated cells, LPS induced a higher ROS level
(p<0.01) (Fig. 2C), while
ulinastatin markedly inhibited the increase in ROS level induced by
LPS (p<0.05) (Fig. 2C).
Ulinastatin inhibits the expression of
lncRNA MALAT1 and EZH2 in LPS-induced CMVECs
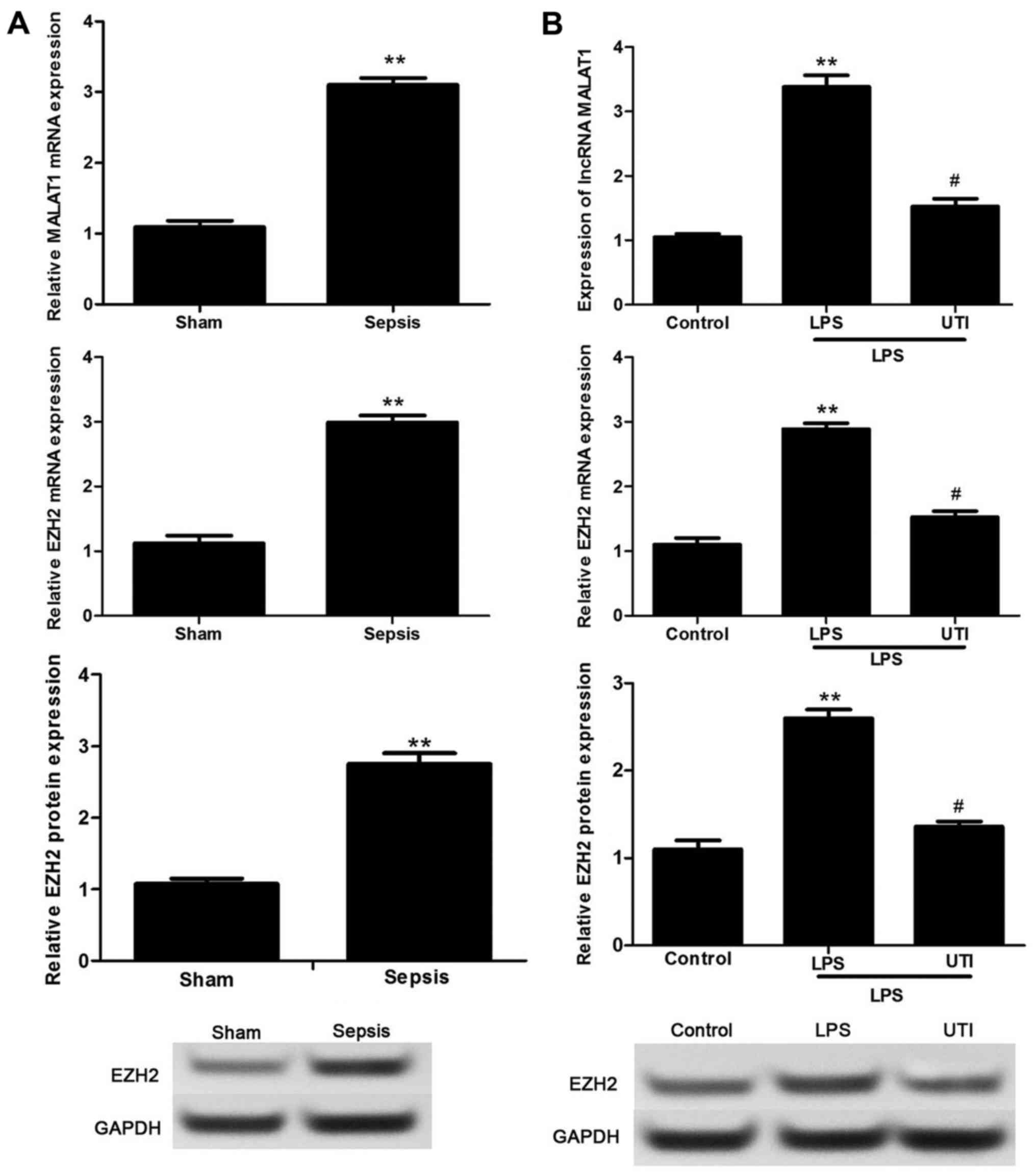
To investigate the mechanism of ulinastatin in
LPS-induced CMVECs, the expression levels of lncRNA MALAT1 and EZH2
were detected. The results revealed that the levels of lncRNA
MALAT1 and EZH2 were significantly upregulated in the hearts of rat
from the sepsis group in comparison with these levels in the sham
group (p<0.01) (Fig. 3A).
Similarly, in vitro experiments showed that LPS markedly
elevated the expression levels of lncRNA MALAT1 and EZH2 compared
with the levels in the untreated cells (p<0.01) (Fig. 3B), while ulinastatin inhibited the
upregulation of lncRNA MALAT1 and EZH2 (p<0.05) (Fig. 3B).
lncRNA MALAT1 interacts with EZH2 in
CMVECs
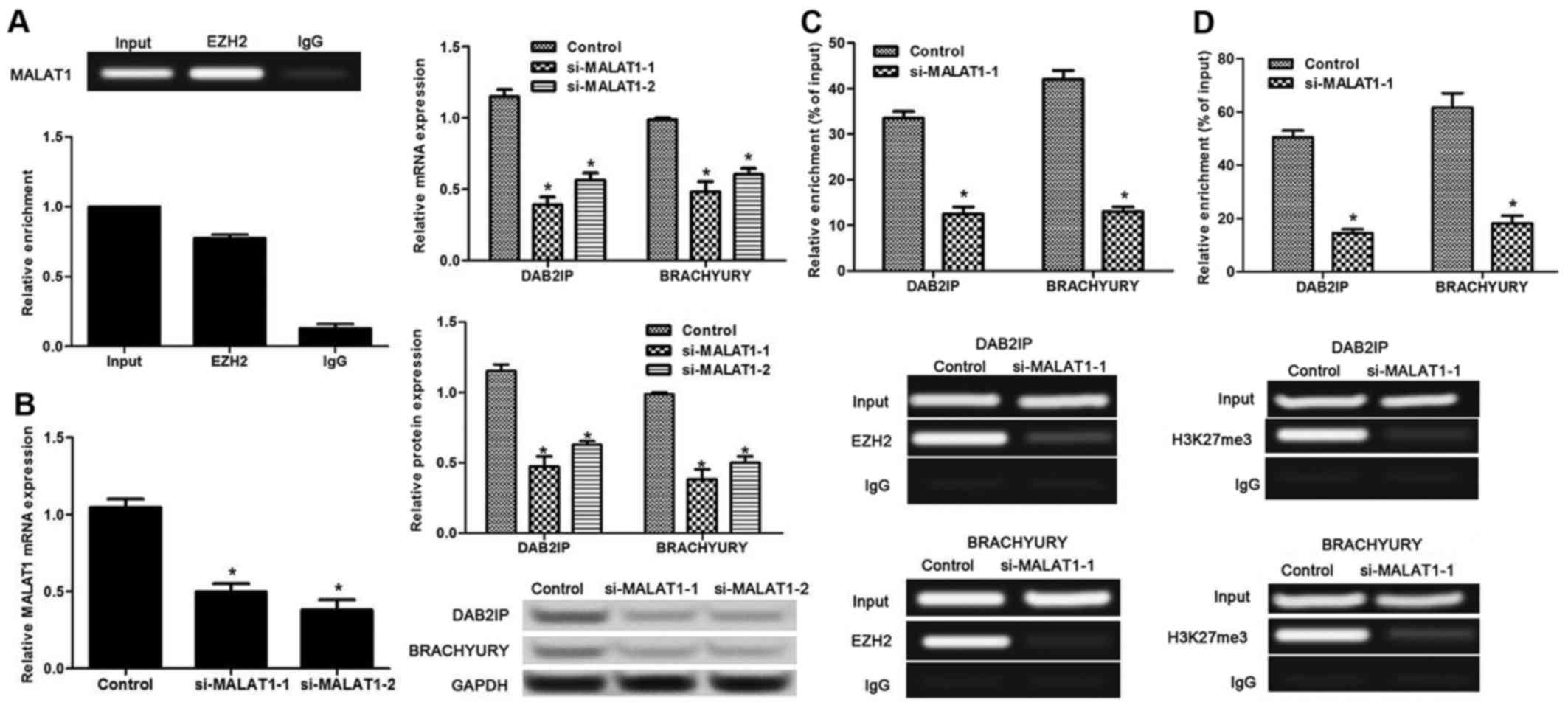
RTP assay was performed to evaluate the relationship
between lncRNA MALAT1 and EZH2. The results revealed that lncRNA
MALAT1 could bind to EZH2 (Fig.
4A), indicating the interaction of lncRNA MALAT1 and EZH2.
Then, the lncRNA MALAT1 level was successfully inhibited by the
transfection of si-MALAT1-1 and si-MALAT1-2 (p<0.05) (Fig. 4B). Silencing of lncRNA MALAT1
markedly inhibited the expression of EZH2 target genes, DAB2IP and
Brachyury (p<0.05) (Fig. 4B)
compared with that in the untreated cells. Furthermore, the effect
of lncRNA MALAT1 on EZH2 was evaluated by ChIP assay. The results
revealed that when lncRNA MALAT1-knockdown cells were precipitated
with EZH2 or H3K27me3 (the composition of EZH2) antibody, the
expression levels of DAB2IP and Brachyury were both inhibited
compared with levels in the untreated cells (p<0.05) (Fig. 4C and D).
Ulinastatin protects against LPS-induced
CMVEC dysfunction via downregulation of lncRNA MALAT1 and EZH2
The above results showed that ulinastatin induced
the downregulation of lncRNA MALAT1 in the LPS-induced cells. Thus,
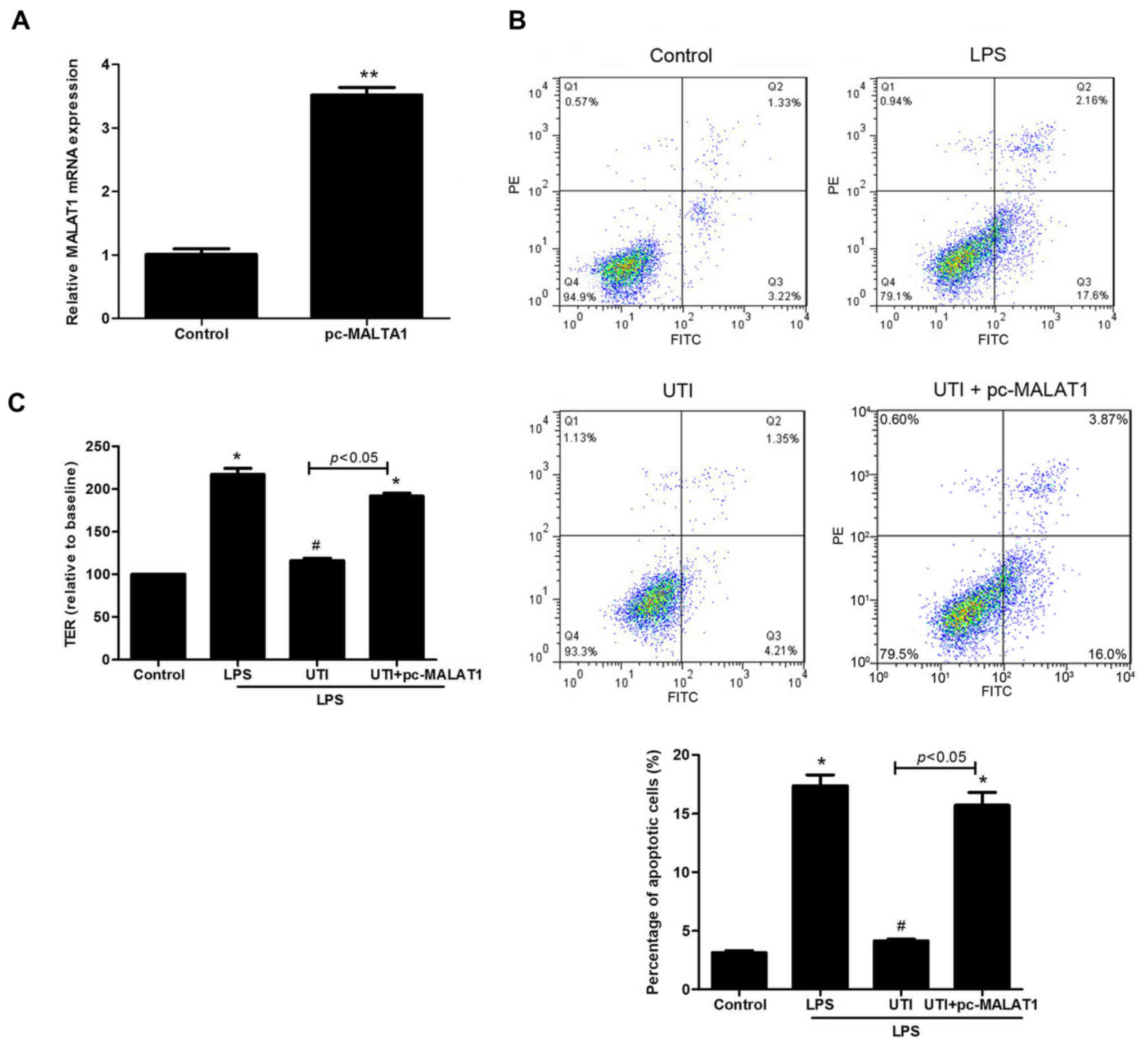
we investigated the effect of overexpression of lncRNA MALAT1 on
CMVEC permeability and apoptosis following treatment with
ulinastatin and LPS. Compared with the untreated cells, the lncRNA
MALAT1 level was significantly increased (p<0.01) (Fig. 5A) in cells transfected with
pc-MALAT1. Then, the results revealed that upon treatment with
ulinastatin, upregulation of lncRNA MALAT1 markedly increased CMEC
permeability (Fig. 5B) and
apoptosis (Fig. 5C) in the
LPS-induced cells (all p<0.05).
Discussion
Cardiac microvascular dysfunction in sepsis has been
demonstrated (5) and the
protective role of ulinastatin against sepsis has also been
reported (22). Nevertheless, the
effects of ulinastatin on CMVEC dysfunction in sepsis are still
unclear. The present study revealed that compared with LPS-induced
CMVECs, the treatment of ulinastatin significantly reduced CMVEC
permeability and apoptosis. In addition, we found upregulated
lncRNA MALAT1 and EZH2 in the hearts of sepsis rats and LPS-induced
CMVECs, while ulinastatin had an inhibitory effect on the
upregulation of lncRNA MALAT1 and EZH2.
Several studies have confirmed the LPS-induced
sepsis cell or animal model (23,24). CMVEC dysfunction has been reported
to be associated with the occurrence of sepsis (5,25).
This study found that LPS treatment promoted CMVEC dysfunction,
such as hyperpermeability and apoptosis. Consistently, Janicek
et al (26) demonstrated
the hyperpermeability and apoptosis in endothelial cells induced by
LPS. In addition, we found that ulinastatin reduced LPS-induced
CMVEC hyperpermeability and apoptosis. Previous studies have shown
that ulinastatin had inhibitory effects on apoptosis, oxidative
stress and inflammation (9,27).
The protective effect of ulinastatin against vascular endothelial
injury was also reported in patients who underwent heart operation
(28). Moreover, Li et al
(29) reported that ulinastatin
inhibited cell hyperpermeability and apoptosis in oxidant-induced
human umbilical endothelial cells. Lin et al (30) also revealed that ulinastatin
attenuated the hyperpermeability of rat lung microvascular
endothelial cells when treated by shock serum. All these results
collaborate our results. The present study further revealed that
ulinastatin inhibited the levels of ROS, caspase-3 and Bax as well
as increased the level of Bcl-2 in LPS-induced CMVECs. It was well
known that ROS induce mitochondrial oxidative stress and then
promote the release of cytochrome c (31). Oxidative stress plays a vital
regulatory role in mitochondria dependent apoptotic signaling
(32). Mitochondria-mediated
apoptosis could be regulated by anti-apoptotic factor Bcl-2,
pro-apoptotic factor Bax (26),
as well as caspase-3 activation, which increase membrane
permeability (32,33). These results suggest that
ulinastatin inhibits LPS-induced cell hyperpermeability and
apoptosis through a mitochondrial-dependent pathway.
The present study further investigated the role of
lncRNA MALAT1 in CMVEC dysfunction in sepsis. The results revealed
the upregulation of lncRNA MALAT1 and EZH2 in vitro and
in vivo sepsis model, while ulinastatin inhibited the
expression of lncRNA MALAT1 and EZH2. Previous studies have shown
overexpression of MALAT1 in cancers, including non-small cell lung
(34), bladder (35), colorectal (36) and prostate cancer (37). Recently, MALAT1 has been reported
to regulate the production of inflammatory cytokines in cancers
(38). Polycomb protein EZH2 was
also reported to be overexpressed in cancers (39). EZH2 was considered as an oncogenic
gene that interacts with EED and SUZ12 by forming the polycomb
repressive complex-2 (PRC2) (40). H3K27me3 is a key functional
component of EZH2 (40). This
study revealed that lncRNA MALAT1 could bind to EZH2, and MALAT1
silencing inhibited the expression of EZH2 target genes
DAB2IP and Brachyury in cells precipitated with EZH2
or H3K27me3. These results indicated the interaction of MALAT1 and
EZH2 in CMVECs, which was consistent with the study of Wang et
al (41). Increasing evidence
has shown that both MALAT1 and EZH2 are associated with cell
proliferation, apoptosis and metastasis in cancer cells (42,43). All these results indicate that
ulinastatin may inhibit LPS-induced CMVEC dysfunction by inhibiting
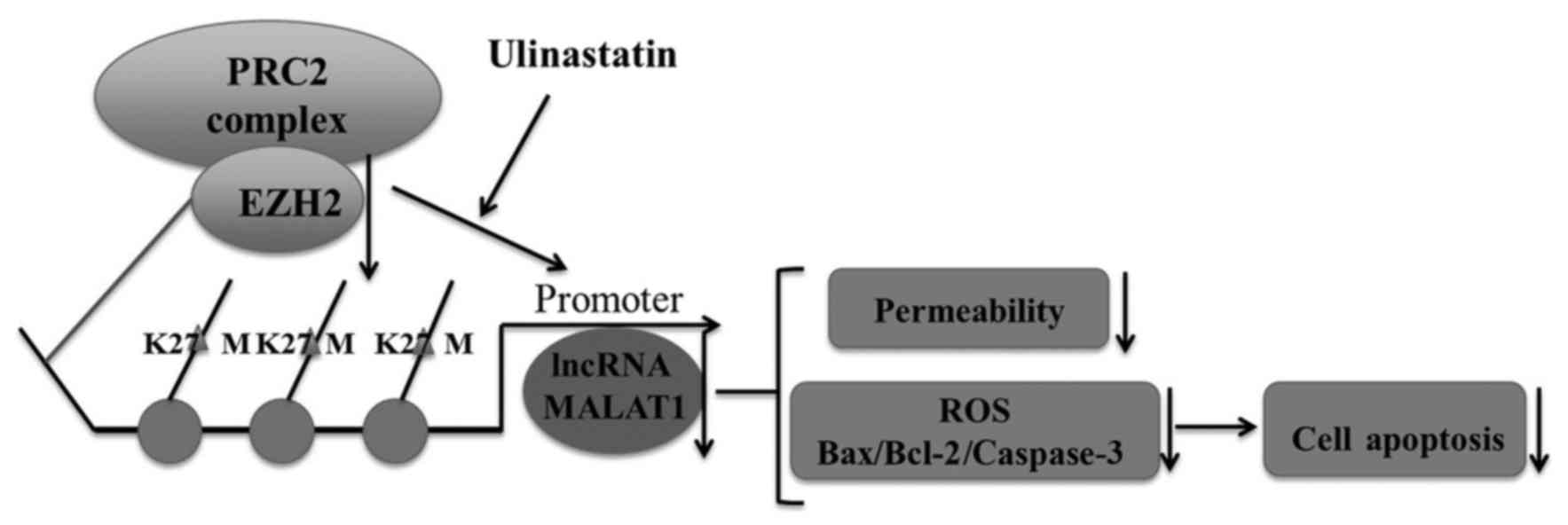
lncRNA MALAT1 and EZH2. The diagram illustrating the mechanism of
ulinastatin in LPS-induced CMVECs is shown in Fig. 6.
Ulinastatin protects against LPS-induced CMVEC
hyperpermeability and apoptosis via downregulation of lncRNA MALAT1
and EZH2 in sepsis.
Acknowledgments
This study was supported by the Graduate Innovation
Fund of Xinjiang Medical University (grant no. CXCY002).
References
|
1
|
Sprung CL and Reinhart K: Definitions for
sepsis and septic shock. JAMA. 316:456–457. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brun-Buisson C, Meshaka P, Pinton P and
Vallet B; EPISEPSIS Study Group: EPISEPSIS: A reappraisal of the
epidemiology and outcome of severe sepsis in French intensive care
units. Intensive Care Med. 30:580–588. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gaieski DF, Edwards JM, Kallan MJ and Carr
BG: Benchmarking the incidence and mortality of severe sepsis in
the United States. Crit Care Med. 41:1167–1174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zaky A, Deem S, Bendjelid K and Treggiari
MM: Characterization of cardiac dysfunction in sepsis: An ongoing
challenge. Shock. 41:12–24. 2014. View Article : Google Scholar
|
|
5
|
Lush CW and Kvietys PR: Microvascular
dysfunction in sepsis. Microcirculation. 7:83–101. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Scarabelli T, Stephanou A, Rayment N,
Pasini E, Comini L, Curello S, Ferrari R, Knight R and Latchman D:
Apoptosis of endothelial cells precedes myocyte cell apoptosis in
ischemia/reperfusion injury. Circulation. 104:253–256. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salmon AH and Satchell SC: Endothelial
glycocalyx dysfunction in disease: Albuminuria and increased
microvascular permeability. J Pathol. 226:562–574. 2012. View Article : Google Scholar
|
|
8
|
Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao
C, Xin C, Zhu D, Li Y, Yan W, et al: TXNIP mediates NLRP3
inflammasome activation in cardiac microvascular endothelial cells
as a novel mechanism in myocardial ischemia/reperfusion injury.
Basic Res Cardiol. 109:4152014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu CE, Zhang MY, Zou CW and Guo L:
Evaluation of the pharmacological function of ulinastatin in
experimental animals. Molecules. 17:9070–9080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang N, Liu X, Zheng X, Cao H, Wei G, Zhu
Y, Fan S, Zhou H and Zheng J: Ulinastatin is a novel candidate drug
for sepsis and secondary acute lung injury, evidence from an
optimized CLP rat model. Int Immunopharmacol. 17:799–807. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cao YZ, Tu YY, Chen X, Wang BL, Zhong YX
and Liu MH: Protective effect of Ulinastatin against murine models
of sepsis: Inhibition of TNF-α and IL-6 and augmentation of IL-10
and IL-13. Exp Toxicol Pathol. 64:543–547. 2012. View Article : Google Scholar
|
|
12
|
Huang N, Wang F, Wang Y, Hou J, Li J and
Deng X: Ulinastatin improves survival of septic mice by suppressing
inflammatory response and lymphocyte apoptosis. J Surg Res.
182:296–302. 2013. View Article : Google Scholar
|
|
13
|
Karnad DR, Bhadade R, Verma PK, Moulick
ND, Daga MK, Chafekar ND and Iyer S: Intravenous administration of
ulinastatin (human urinary trypsin inhibitor) in severe sepsis: A
multicenter randomized controlled study. Intensive Care Med.
40:830–838. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bashir F, Rather MA, Saleem B and Hamid A:
a prospective, randomized study using ulinastatin for the treatment
of patients with severe sepsis. JEMDS. 3:12241–12246. 2014.
View Article : Google Scholar
|
|
15
|
Shi X, Sun M, Liu H, Yao Y and Song Y:
Long non-coding RNAs: A new frontier in the study of human
diseases. Cancer Lett. 339:159–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Johnson R: Long non-coding RNAs in
Huntington's disease neurodegeneration. Neurobiol Dis. 46:245–254.
2012. View Article : Google Scholar
|
|
17
|
Congrains A, Kamide K, Oguro R, Yasuda O,
Miyata K, Yamamoto E, Kawai T, Kusunoki H, Yamamoto H, Takeya Y, et
al: Genetic variants at the 9p21 locus contribute to
atherosclerosis through modulation of ANRIL and CDKN2A/B.
Atherosclerosis. 220:449–455. 2012. View Article : Google Scholar
|
|
18
|
Spizzo R, Almeida MI, Colombatti A, Calin
GA and Martin L: Long non-coding RNAs and cancer: A new frontier of
translational research? Oncogene. 31:4577–4587. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guo F, Li Y, Liu Y, Wang J, Li Y and Li G:
Inhibition of metastasis-associated lung adenocarcinoma transcript
1 in CaSki human cervical cancer cells suppresses cell
proliferation and invasion. Acta Biochim Biophys Sin (Shanghai).
42:224–229. 2010. View Article : Google Scholar
|
|
20
|
Brooks HF, Moss RF, Davies NA, Jalan R and
Davies DC: Caecal ligation and puncture induced sepsis in the rat
results in increased brain water content and perimicrovessel
oedema. Metab Brain Dis. 29:837–843. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia JB, Liu GH, Chen ZY, Mao CZ, Zhou DC,
Wu HY, Park KS, Zhao H, Kim SK, Cai DQ, et al: Hypoxia/ischemia
promotes CXCL10 expression in cardiac microvascular endothelial
cells by NFκB activation. Cytokine. 81:63–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang FY, Fang B, Qiang XH, Yu TO, Zhong
JR, Cao J and Zhou LX: The efficacy and immunomodulatory effects of
ulinastatin and thymosin α1 for sepsis: A systematic review and
meta-analysis. Biomed Res Int. 2016:95084932016.
|
|
23
|
Tsoyi K, Lee TY, Lee YS, Kim HJ, Seo HG,
Lee JH and Chang KC: Heme-oxygenase-1 induction and carbon
monoxide-releasing molecule inhibit lipopolysaccharide
(LPS)-induced high-mobility group box 1 release in vitro and
improve survival of mice in LPS-and cecal ligation and
puncture-induced sepsis model in vivo. Mol Pharmacol. 76:173–182.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Djoumerska-Alexieva I, Pashova S, Vassilev
T and Pashov A: The protective effect of modified intravenous
immunoglobulin in LPS sepsis model is associated with an increased
IRA B cells response. Autoimmun Rev. 12:653–656. 2013. View Article : Google Scholar
|
|
25
|
Lee WL and Slutsky AS: Sepsis and
endothelial permeability. N Engl J Med. 363:689–691. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Janicek A, Tharakan B, Sawant DA, Hunter
FA and Childs EW: Lipopolysaccharide-induced endothelial cell
hyperpermeability: Role of mitochondrial apoptotic signaling
pathway. J Surg Res. 172:3302012. View Article : Google Scholar
|
|
27
|
Hu CL, Xia JM, Cai J, Li X, Liao XX, Li H,
Zhan H, Dai G and Jing XL: Ulinastatin attenuates oxidation,
inflammation and neural apoptosis in the cerebral cortex of adult
rats with ventricular fibrillation after cardiopulmonary
resuscitation. Clinics (Sao Paulo). 68:1231–1238. 2013. View Article : Google Scholar
|
|
28
|
Miura M, Sugiura T, Aimi Y, Yasuda K, Ito
S, Baba E and Katsuya H: Effects of ulinastatin on PMNL and
vascular endothelial injury in patients undergoing open heart
surgery with CPB. Masui. 47:29–35. 1998.In Japanese. PubMed/NCBI
|
|
29
|
Li G, Li T, Li Y, Cai S, Zhang Z, Zeng Z,
Wang X, Gao Y, Li Y and Chen Z: Ulinastatin inhibits
oxidant-induced endothelial hyperpermeability and apoptotic
signaling. Int J Clin Exp Pathol. 7:7342–7350. 2014.
|
|
30
|
Lin B, Liu Y, Li T, Zeng K, Cai S, Zeng Z,
Lin C, Chen Z and Gao Y: Ulinastatin mediates protection against
vascular hyperpermeability following hemorrhagic shock. Int J Clin
Exp Pathol. 8:7685–7693. 2015.PubMed/NCBI
|
|
31
|
Atlante A, Calissano P, Bobba A, Azzariti
A, Marra E and Passarella S: Cytochrome c is released from
mitochondria in a reactive oxygen species (ROS)-dependent fashion
and can operate as a ROS scavenger and as a respiratory substrate
in cerebellar neurons undergoing excitotoxic death. J Biol Chem.
275:37159–37166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Childs EW, Tharakan B, Hunter FA, Tinsley
JH and Cao X: Apoptotic signaling induces hyperpermeability
following hemorrhagic shock. Am J Physiol Heart Circ Physiol.
292:H3179–H3189. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ji P, Diederichs S, Wang W, Böing S,
Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et
al: MALAT-1, a novel noncoding RNA, and thymosin beta4 predict
metastasis and survival in early-stage non-small cell lung cancer.
Oncogene. 22:8031–8041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ying L, Chen Q, Wang Y, Zhou Z, Huang Y
and Qiu F: Upregulated MALAT-1 contributes to bladder cancer cell
migration by inducing epithelial-to-mesenchymal transition. Mol
Biosyst. 8:2289–2294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu C, Yang M, Tian J, Wang X and Li Z:
MALAT-1: a long non-coding RNA and its important 3′ end functional
motif in colorectal cancer metastasis. Int J Oncol. 39:169–175.
2011.PubMed/NCBI
|
|
37
|
Ren S, Liu Y, Xu W, Sun Y, Lu J, Wang F,
Wei M, Shen J, Hou J, Gao X, et al: Long noncoding RNA MALAT-1 is a
new potential therapeutic target for castration resistant prostate
cancer. J Urol. 190:2278–2287. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gutschner T, Hämmerle M and Diederichs S:
MALAT1 - a paradigm for long noncoding RNA function in cancer. J
Mol Med (Berl). 91:791–801. 2013. View Article : Google Scholar
|
|
39
|
Bachmann IM, Halvorsen OJ, Collett K,
Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP and
Akslen LA: EZH2 expression is associated with high proliferation
rate and aggressive tumor subgroups in cutaneous melanoma and
cancers of the endometrium, prostate, and breast. J Clin Oncol.
24:268–273. 2006. View Article : Google Scholar
|
|
40
|
Cao Q, Yu J, Dhanasekaran SM, Kim JH, Mani
RS, Tomlins SA, Mehra R, Laxman B, Cao X, Yu J, et al: Repression
of E-cadherin by the polycomb group protein EZH2 in cancer.
Oncogene. 27:7274–7284. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang D, Ding L, Wang L, Zhao Y, Sun Z,
Karnes RJ, Zhang J and Huang H: LncRNA MALAT1 enhances oncogenic
activities of EZH2 in castration-resistant prostate cancer.
Oncotarget. 6:41045–41055. 2015.PubMed/NCBI
|
|
42
|
Xie L, Zhang Z, Tan Z, He R, Zeng X, Xie
Y, Li S, Tang G, Tang H and He X: MicroRNA-124 inhibits
proliferation and induces apoptosis by directly repressing EZH2 in
gastric cancer. Mol Cell Biochem. 392:153–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiao F, Hu H, Yuan C and Wang L, Jiang W,
Jin Z, Guo Z and Wang L: Elevated expression level of long
noncoding RNA MALAT-1 facilitates cell growth, migration and
invasion in pancreatic cancer. Oncol Rep. 32:2485–2492.
2014.PubMed/NCBI
|