Introduction
Osteoarthritis (OA) is a degenerative joint disease
that severly affects the quality of life of patients (1). Traditional treatments only
temporally attenuate the clinical symptoms, but do not effectively
inhibit the pathological progression of OA. It is therefore
important to elucidate the mechanisms responsible for OA and to
identify safe and effective treatments for OA.
Articular cartilage degeneration is the main
pathological change associated with OA (2). A variety of cytokines, growth
factors and enzymes, such as interleukin (IL)-1β (3-5)
and collagenase (6–8) are involved in articular cartilage
degeneration. IL-1β is secreted from synovial cells and
inflammatory cells in OA, and stimulates the production of
proteolytic enzymes, such as stromelysin and collagenase, causing
synovial inflammation and bone resorption (5). Collagenase is upregulated in
OA-affected cartilage and animal models of OA have been
successfully established by an intra-articular injection of
collagenase (6–8). In addition, matrix
metalloproteinases (MMPs) have been shown to play an important role
in the pathogenesis of OA (9),
and the activity of MMPs is regulated by tissue inhibitors of
metalloproteinase (TIMP)-1 in OA (10). In our previous study, we
demonstrated that inflammatory cytokines increase MMP-13 secretion,
which in turn promotes the degradation of type II collagen in
chondrocytes, resulting in the occurrence of OA (11).
The death of chondrocytes is a hallmark of cartilage
degeneration in OA; however, the mechanisms responsible for
chondrocyte death in OA-affected cartilage remain largely unknown.
Autophagy plays a crucial role in maintaining cellular metabolism
and homeostasis (12,13). However, excessive autophagy may
lead to cell death (14,15). Autophagy is regulated by a series
of autophagy-related genes (ATGs) (16), such as Beclin-1 (17) and light chain 3 (LC3) (18). The expression levels of these
genes are commonly used to monitor autophagic activity and flux
(19). LC3 is present in two
forms (LC3-I and LC3-II) and several isoforms (LC3A, LC3B, LC3C).
The conversion of LC3-I to LC3-II is an indicator of autophagosome
formation (20). Accumulating
evidence suggests that the deregulation of autophagy is closely
related to the pathogenesis of OA (21–24). Some researchers have reported that
autophagy is enhanced in OA-affected cartilage (25); however, others have reported that
autophagy is significantly reduced in OA-affected cartilage, and
the age-related loss of autophagy has been linked to cell death in
OA-affected cartilage (26).
In order to elucidate the changes in autophagy
occurring during the progression of OA and the specific role of
autophagy in OA, in this study, we established a cellular model of
OA by treating SW1353 chondrosarcoma cells with IL-1β. In addition,
we created a rabbit model of OA by an intra-articular injection of
collagenase, followed by treatment with the autophagy specific
inhibitor, 3-methyladenine (3-MA). We then observed the
degeneration of cells and cartilage and detected the levels of
autophagy at different time points. The results demonstrated that
autophagy was enhanced during the early stages of experimental OA
and was weakened during the late stages; the inhibition of
autophagy aggravated the degeneration of chondrosarcoma cells and
cartilage. Our results determine the changes of autophagy during
the different stages of OA and the role of impaired autophagy in
the development of OA, suggesting that the regulation of autophagy
may be a potential therapeutic strategy with which to attenuate
OA.
Materials and methods
Cell culture and treatment
SW1353 human chondrosarcoma cells were purchased
from the Institute of Life Science Cell Culture Center (Shanghai,
China) and were cultured in α-minimal essential medium (α-MEM;
HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum
(Gibco, Grand Island, NY, USA), 100 IU/ml penicillin and 100
μg/ml streptomycin (Gibco). The α-MEM contained RNA and DNA,
2 mM L-glutamine and 1 mM sodium pyruvate, but no vitamin C or
glucose. The cells were incubated at 37°C in humidified air
containing 5% CO2. Upon reaching 80% confluence, the
cells were pre-treated with or without the autophagy inhibitor,
3-MA at 5 mmol/l (Sigma-Aldrich, St. Louis, MO, USA). One hour
after pre-treatment, the cells were stimulated with 10 ng/ml IL-1β
(PeproTech, Inc., Rocky Hill, NJ, USA) for 12, 24, 36 and 48 h,
separately. The cells that were not exposed to 3-MA and IL-1β were
used as controls.
Cell viability analysis
The viability of thecells was assessed by MTS assay.
The SW1353 cells were seeded on 96-well plates (5,000 cells/well)
and incubated at 37°C for 24 h, followed by pre-treatment with or
without 3-MA (5 mmol/l) for 1 h, and then stimulation with IL-1β
(10 ng/ml). The wells that were treated without 3-MA and IL-1β were
used as controls. Each sample concentration was replicated 6 times.
Following incubation for 0, 12, 24, 36, and 48 h, 10 μl of
MTS (Sigma-Aldrich) were added to each well and the cells were
incubated at 37°C for a further 2 h. The absorbance was measured
using a multifunctional microplate reader (Molecular Devices,
Sunnyvale, CA, USA) at 490 nm. The relative cell viability (RCV) to
the controls was calculated using the following equation: RCV =
(ODtest/ODcontrol) x100%.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer's instructions. First-strand cDNA was synthesized
using the ReverTra Ace reverse transcriptase cDNA synthesis kit
(Applied Biosystems Inc., Foster City, CA, USA). Quantitative PCR
(qPCR) was performed using a 7500 real-time PCR system with
SYBR-Green (both from Applied Biosystems Inc.). The forward and
reverse primers used were as follows: MMP-13,
5′-CGACTTCTACCCATTTGA-3′ and 5′-TAGCCTTTGGAACTACTTGTC-3′; TIMP-1,
5′-AGATAGCCTGAATCCTGCC-3′ and 5′-CTGGGTGGTAACTCTTTATTTC-3′;
β-actin, 5′-TCGACAACGGCTCCGGCAT-3′ and 5′-AAGGTGTGGTGCCAGATTTTC-3′,
respectively. β-actin was used as an internal control. Each
experiment was repeated 3 times in triplicate. The relative
expression of target genes was calculated using the
2−ΔΔCq method.
Immunofluorescence
The cells were seeded at 1×105/ml on a
glass coverslip into a 6-well plate. Following treatment, the cells
were fixed with 4% cold paraformaldehyde (Solarbio, Beijing, China)
for 30 min and permeabilized with 0.5% T riton X-100
(Sigma-Aldrich) in PBS for 5 min, followed by PBS washing 3 times.
After blocking for 30 min with 2 ml of PBS-0.5% Triton-10% goat
serum (Gibco), the cells were incubated with rabbit anti-LC3B
antibody (1:400; Cat. no. 2775; Cell Signaling Technology, Inc.,
Danvers, MA, USA) at 4°C overnight. Following 3 washes with PBS,
the cells were incubated with Alexa Fluor 555 goat anti-rabbit
secondary antibody (1:1,000 dilution; Cat. no. ZB-2301; Zhongshan
Golden Bridge Biotechnology, Beijing, China) at room temperature
for 2 h followed by 3 separate PBS washes. Subsequently, DAPI
staining (Cat. no. C1006; Beyotime Biotechnology, Shanghai, China)
was used to stain the nuclei, and after rinsing with
ddH2O, the slide was mounted with anti-fading agent and
examined using a confocal microscope (Olympus Corp., Tokyo, Japan)
to quantify the expression of LC3B protein.
Rabbit model of OA
A total of 54 healthy New Zealand white male rabbits
(3 months old; weighing 2.30–2.56 kg) were obtained from the Animal
Center of Capital Medical University (Beijing, China). All animal
experiments were performed following the approval of the Ethics
Committee of Capital Medical University. After 4 weeks, the rabbits
were anesthetized with 10% chloral hydrate (2 ml/kg body weight) by
an intraperitoneal injection. Subsequently, the first experimental
group (collagenase-injected group) received an intra-articular
injection in the right knee joint with 0.5 ml saline containing 2.0
mg/ml of collagenase (type II; Sigma-Aldrich), and the second
experimental group (3-MA-treated group) received an intra-articular
injection in the right knee joint with 0.5 ml saline containing 2.0
mg/ml of collagenase combined with 5 mmol/l 3-MA. The injection was
administered on days 1 and 4 (injected twice) as previously
described (6,27). The control group (rabbits injected
twice with 0.5 ml normal saline) contained 6 rabbits, and the
collagenase-injected experimental group and the 3-MA-treated
experimental group each contained 24 rabbits. The rabbits from the
control group were sacrificed at 8 weeks, while the rabbits from
the collagenase-injected experimental group and 3-MA-treated
experimental group were averagely sacrificed at 2, 4, 6 and 8
weeks.
Histological evaluation
Following sacrifice, the lateral femoral condyles of
the rabbits were removed and fixed in 10% neutral buffered formalin
(pH 7.4; VWR International, Radnor, PA, USA) for 48 h and
decalcified with 20% EDTA (Gibco). This was followed by dehydration
through a series of increasing concentrations of ethanol. The
samples were paraffin-embedded, and 5-μm-thick microsections
[created using a microtome (Leica RM2145; Leica, Wetzlar, Germany)]
were stained with Safranin-O (Cat. no. G2540; Solarbio). The
grading of Safranin-O staining was evaluated by blinded individuals
according to the Mankin scoring system (28).
Transmission electron microscopy
The medial femoral condyles were first fixed with
2.5% glutaraldehyde in PBS (pH 7.0) for 48 h. This was followed by
3 PBS washing steps, and decalcification with 20% EDTA. The samples
were further fixed with 1% OsO4 in PBS (pH 7.0) for 2 h
and washed 3 times in PBS, and were then dehydrated with a series
of ethanol concentrations (50, 70, 90, and 100%) for 15 min
intervals. The samples were subsequently incubated in a mixture of
alcohol and isoamyl acetate (v:v=1:1) for 30 min, followed by
incubation with pure isoamyl acetate for 1 h. Finally, the samples
were coated with gold-palladium, cut into ultrathin sections, and
observed using a transmission electron microscope (TEM; Hitachi,
Tokyo, Japan).
Western blot analysis
The cells were washed in ice-cold PBS and lysed in
total protein lysis buffer (Sigma-Aldrich) supplemented with 1
mg/ml protease inhibitor cocktail (Roche, Indianapolis, IN, USA) at
4°C for 30 min. Cartilage from the tibial plateaus was cut into
1-mm-thin slices, and 200 mg of frozen cartilage was pulverized in
liquid nitrogen. The cartilage dry weight was measured, and the
same amount of cartilage was resuspended in total protein lysis
buffer supplemented with 1 mg/ml protease inhibitor cocktail in
order to attain homogenates using the T10 tissue homogenizer (IKA,
Staufen, Germany). The homogenates were incubated at 4°C for 30
min. The samples were then centrifuged at 15,000 rpm for 30 min,
and the supernatants were harvested to measure protein
concentrations using the bicinchoninic acid reagent assay (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Equal amounts of
protein were heated at 99°C for 10 min, and separated using
Tris-glycine gels (Sigma-Aldrich). The proteins were then
transferred onto nitrocellulose membranes (Millipore Corp.,
Billerica, MA, USA). After blocking with 5% dry milk in
Tris-buffered saline Tween-20 (TBST; Merck Millipore) for 2 h, the
membranes were incubated at 4°C (overnight) with rabbit polyclonal
antibody specific for Beclin-1 (Cat. no. 3738) and LC3B (Cat. no.
2775) (Cell Signaling Technology), and mouse monoclonal antibody
specific for β-actin (sc-69879; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). Following three wash steps with TBST, the
membranes were incubated with horseradish peroxidase
(HRP)-conjugated goat anti-rabbit (Cat. no. ZB-2301) or goat
anti-mouse (Cat. no. ZB-5305) IgG (Zhongshan Golden Bridge
Biotechnology Co., Ltd., Beijing, China) for 2 h. The membranes
were washed 3 times with TBST, and subjected to signal development
using enhanced chemiluminescence (ECL) substrate (Thermo Fisher
Scientific, Inc.).
Statistical analysis
The data are expressed as the means ± standard
deviation (SD). Statistical analysis was performed using one-way
ANOVA and the Student's t-test using SPSS statistical software 19.0
(SPSS Inc., Chicago, IL, USA). P-values <0.05 were considered to
indicate statistically significant differences.
Results
Cell viability
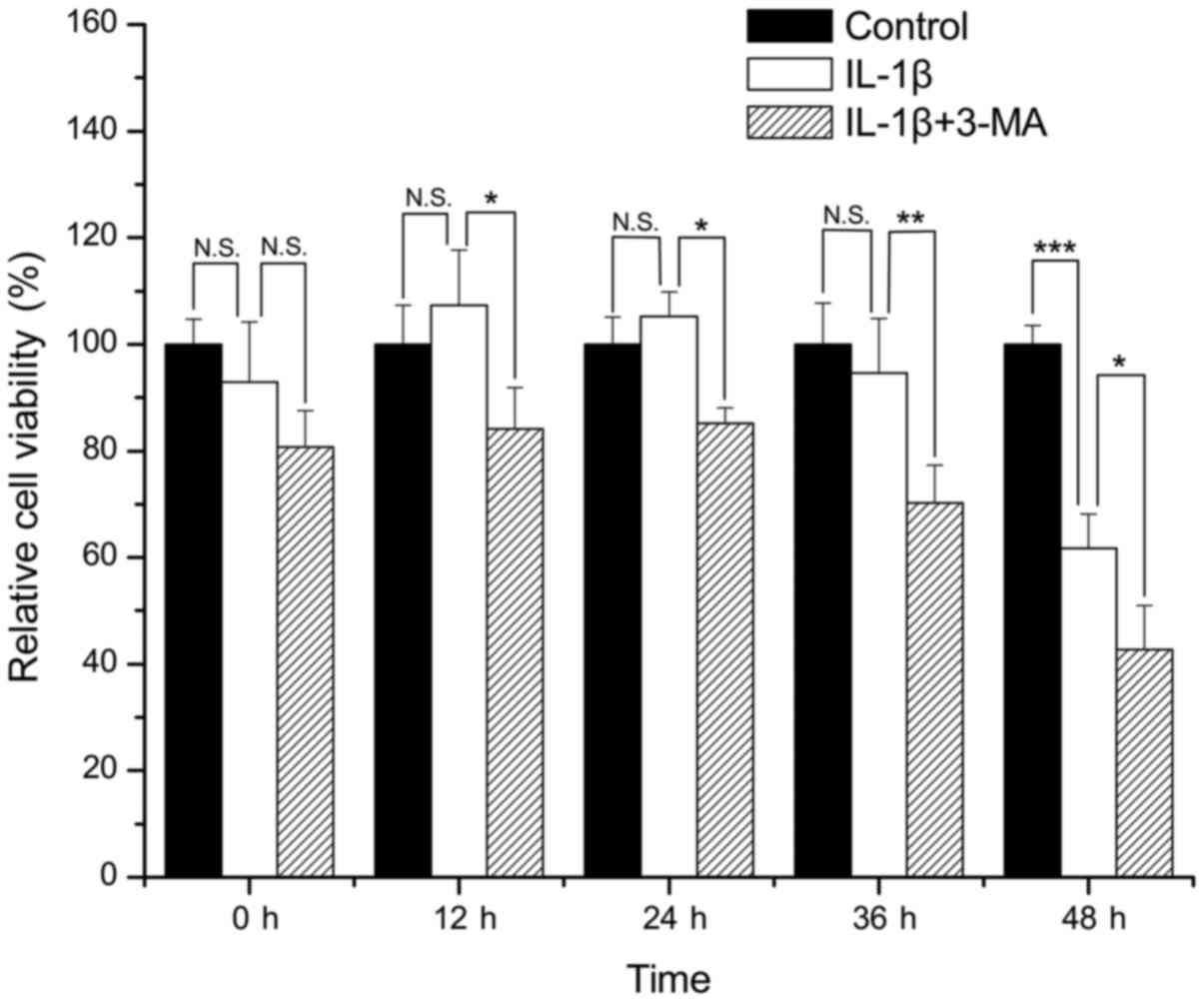
Compared with the control group, there was no
apparent loss of viability in the SW1353 cells treated with IL-1β
for 0, 12, 24 and 36 h (P=0.365, 0.402, 0.600 and 0.582,
respectively). However, treatment of the SW1353 cells with IL-1β
for 48 h resulted in a significant loss of cell viability when
compared with the control group (P<0.001). The viability of the
cells treated with IL-1β in combination with 3-MA was significantly
decreased when compared with that of the cells treated only with
IL-1β at 12, 24, 36 and 48 h (P=0.012, 0.024, 0.007 and 0.043
respectively) (Fig. 1).
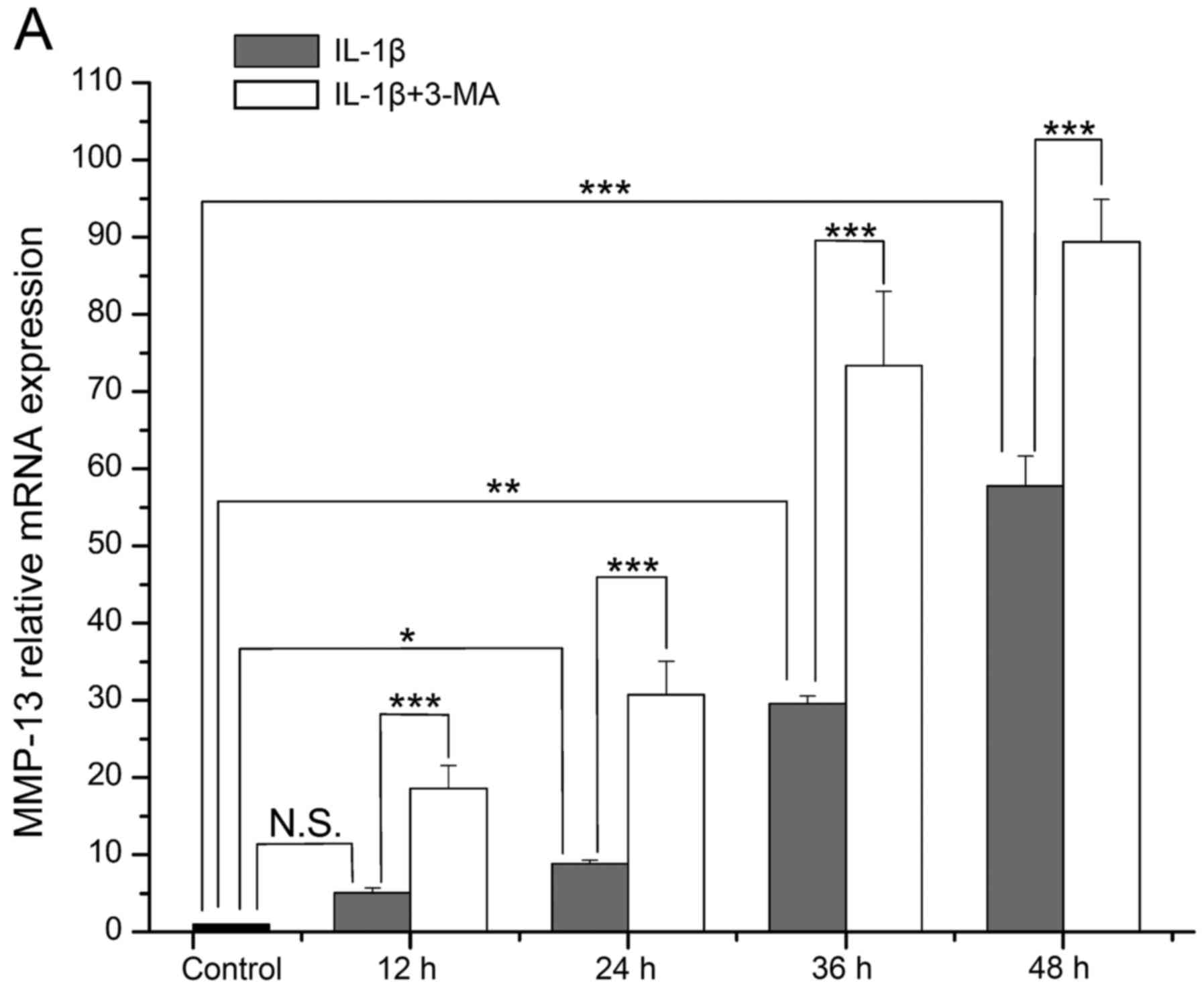
Expression of MMP-13 and TIMP-1
The mRNA level of MMP-13 in the IL-1β-stimulated
SW1353 cells became increasingly higher with time, and it was
increased at each time point when compared with the normal cells
(P=0.078, 0.024, 0.002 and <0.001 respectively). The increase in
MMP-13 expression was significantly enhanced by 3-MA in comparison
with the cells treated with IL-1β only (all P<0.001) (Fig. 2A). The mRNA level of TIMP-1 in the
IL-1β-stimulated SW1353 cells was highest at 12 h, and then it
decreased with time. It was increased at 12 and 24 h (P=0.071 and
0.067), while it was decreased at 36 and 48 h when compared with
the normal cells (P=0.071 and 0.001). In addition, the decrease in
TIMP-1 expression was enhanced by 3-MA in comparison with cells
treated with IL-1β only (P=0.001, <0.001, 0.003 and 0.057
respectively) (Fig. 2B).
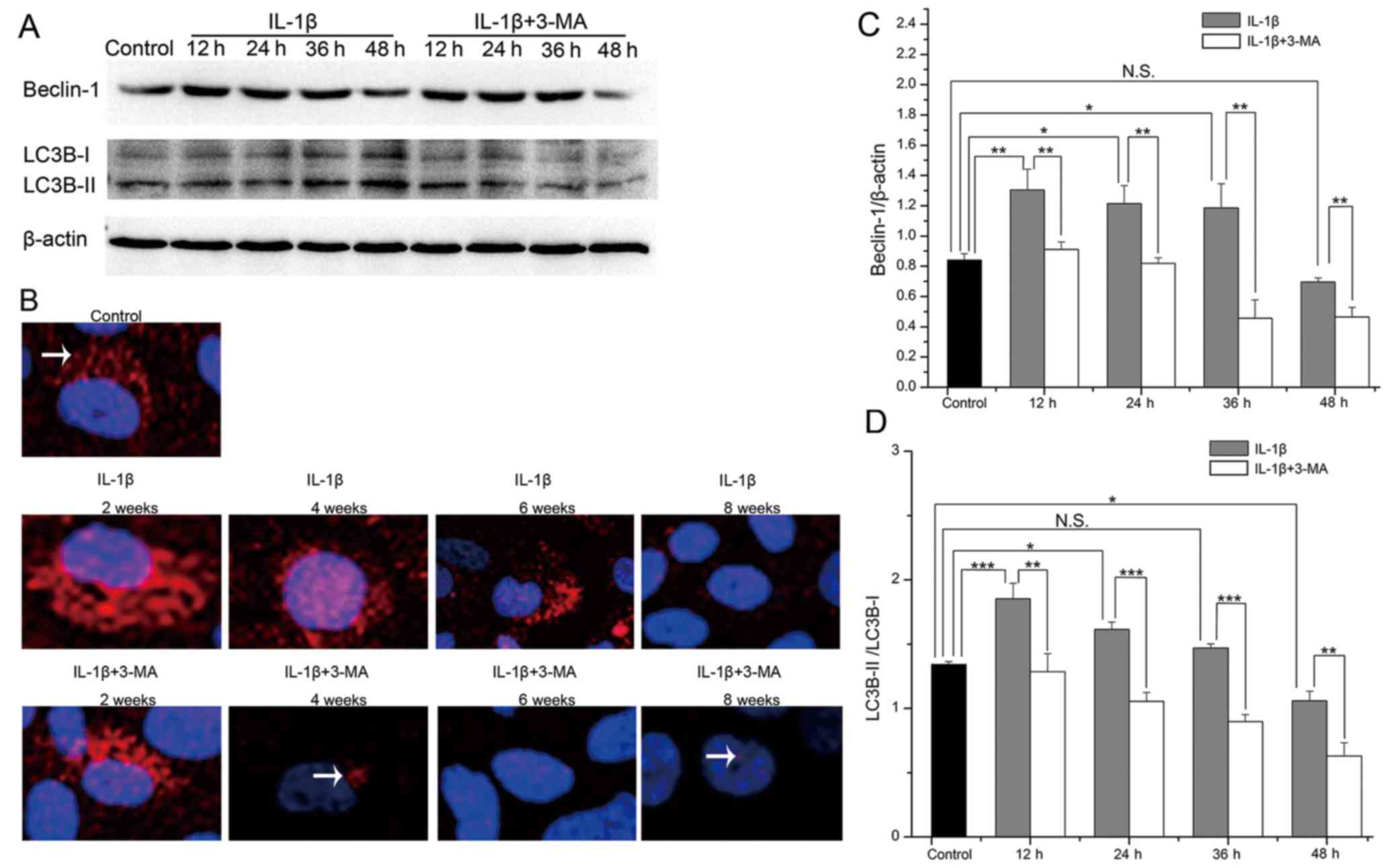
Expression of Beclin-1 and LC3B in
IL-1β-stimulated SW1353 cells
To detect the levels of autophagy in the
IL-1β-stimulated SW1353 cells, we evaluated the protein expression
levels of Beclin-1 and LC3B by western blot analysis (Fig. 3A). In the IL-1β-stimulated SW1353
cells, the expression levels of Beclin-1 and the LC3B-II/LC3B-I
ratio were both highest at 12 h, and they then decreased with time.
Both of these were increased at 12 (P=0.007 and <0.001), 24
(P=0.029 and 0.013) and 36 h (P=0.045 and 0.364), while they were
decreased at 48 h (P=0.631 and 0.010) in the IL-1β-stimulated
SW1353 cells when compared with the normal cells. At each time
point, the expression levels of Beclin-1 (P=0.009, 0.005, 0.003 and
0.005 respectively) and the LC3B-II/LC3B-I ratio (P=0.006,
<0.001, <0.001 and 0.004 respectively) were both decreased in
the cells treated with 3-MA when compared with the cells treated
only with IL-1β (Fig. 3C and D).
We further evaluated the protein expression of LC3B by
immunofluorescence and found that the results observed were the
same as those of western blot analysis (Fig. 3B). These results demonstrated that
autophagy was first enhanced and then weakened in the
IL-1β-stimulated SW1353 cells, and the expression levels of
Beclin-1 and the LC3B-II/LC3B-I ratio were inhibited by 3-MA
treatment.
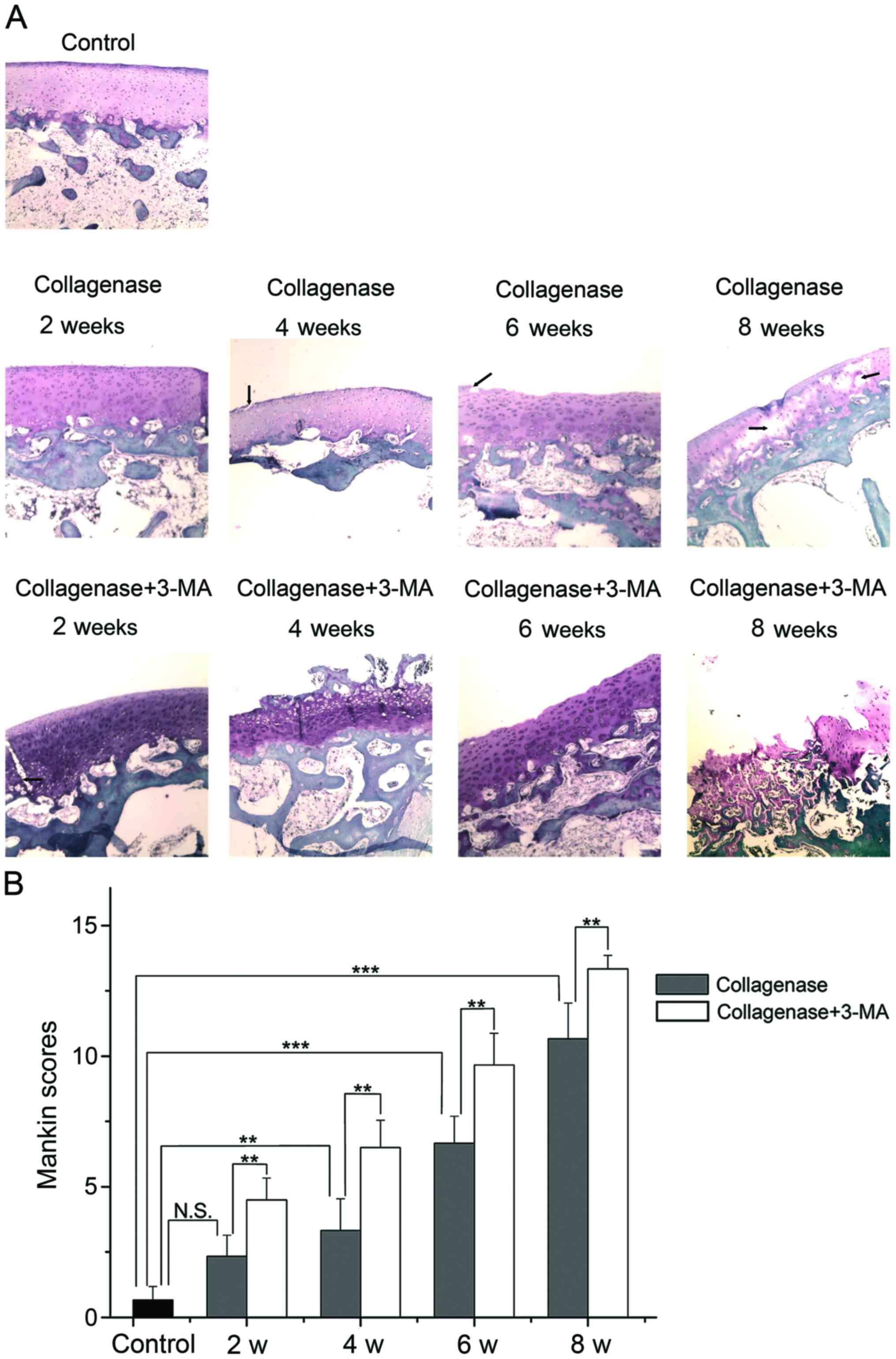
Histological evaluation
The cartilage of the lateral femoral condyles was
stained with Safranin-O and examined under a light microscope
(Fig. 4A), and the average scores
of Safranin-O staining were evaluated by blinded observers
(Fig. 4B). Photomicrographs of
the cartilage revealed that the cartilage of the control group was
not degenerated. In the collagenase-injected group, the cartilage
surface was slightly irregular and the slight loss of Safranin-O
staining was observed at 2 weeks after the injection. At 4 weeks,
clefts were observed in the transitional zone, and cell
proliferation and relatively reduced Safranin-O staining ability
were observed in the transitional and radial zone. At 6 weeks,
clefts and the disappearance of cells in the transitional zone, and
reduced Safranin-O staining were evident. At 8 weeks, the loss of
cartilage extended to the radial zone, and clefts and reduced
staining of Safranin-O were apparent. The extent of OA was
presented as Mankin scores (higher score, greater degeneration of
cartilage). According to the summed score, the collagenase-injected
group developed OA in a time-dependent manner and had a
significantly higher score than the control group at 4, 6 and 8
weeks (P=0.004, <0.001 and <0.001 respectively). The score in
the 3-MA-treated group was significantly higher than the
collagenase-injected group at each time point (all P=0.001). These
results indicated that the inhibition of autophagy by 3-MA
significantly enhanced the collagenase-induced cartilage
degeneration of OA.
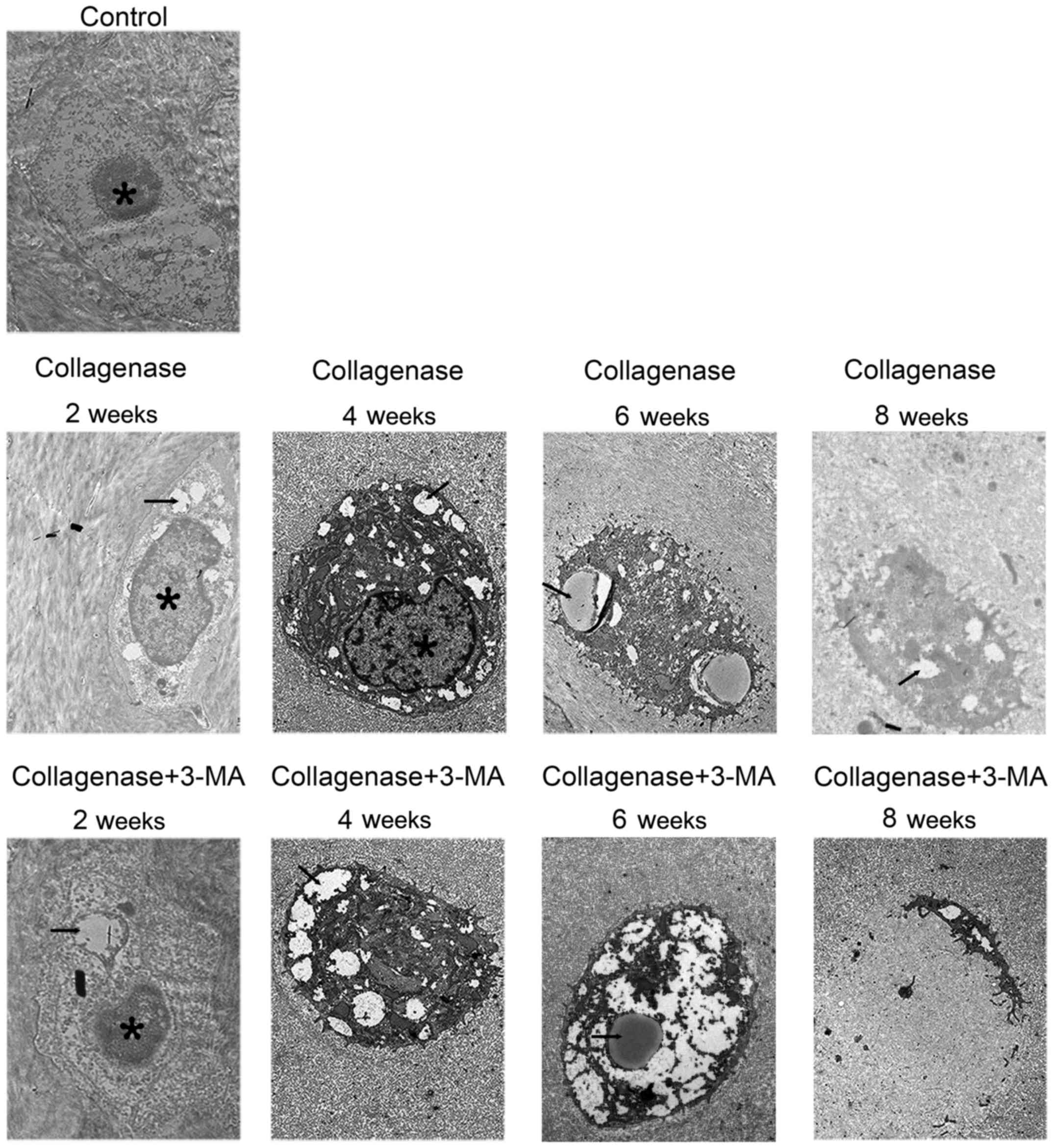
Transmission electron microscopy
The cartilage of the medial femoral condyles was
examined by a TEM to observe autophagosomes and chondrocyte
degeneration (Fig. 5). In the
control group, chondrocytes containing round nuclei were observed
in the lacunae. In the collagenase-injected group, much more
autophagosomes in chondrocytes were observed at 2 and 4 weeks, and
the autophagosomes became less at 6 and 8 weeks. Furthermore,
chondrocyte degeneration increased with time. In addition, abundant
rough endoplasmic reticulum and other organelles were observed in
the cytoplasm. In the cartilage from the collagenase-injected group
at 8 weeks, condensed chondrocytes with several autophagosomes were
observed. In the 3-MA-treated group at all 4 time points, autophagy
in chondrocytes was less and chondrocyte degeneration was greater
than the collagenase-injected group. In the cartilage from the
3-MA-treated group at 8 weeks, apparent chondrocyte lysis and cell
debris were observed in the lacunae of cellular materials.
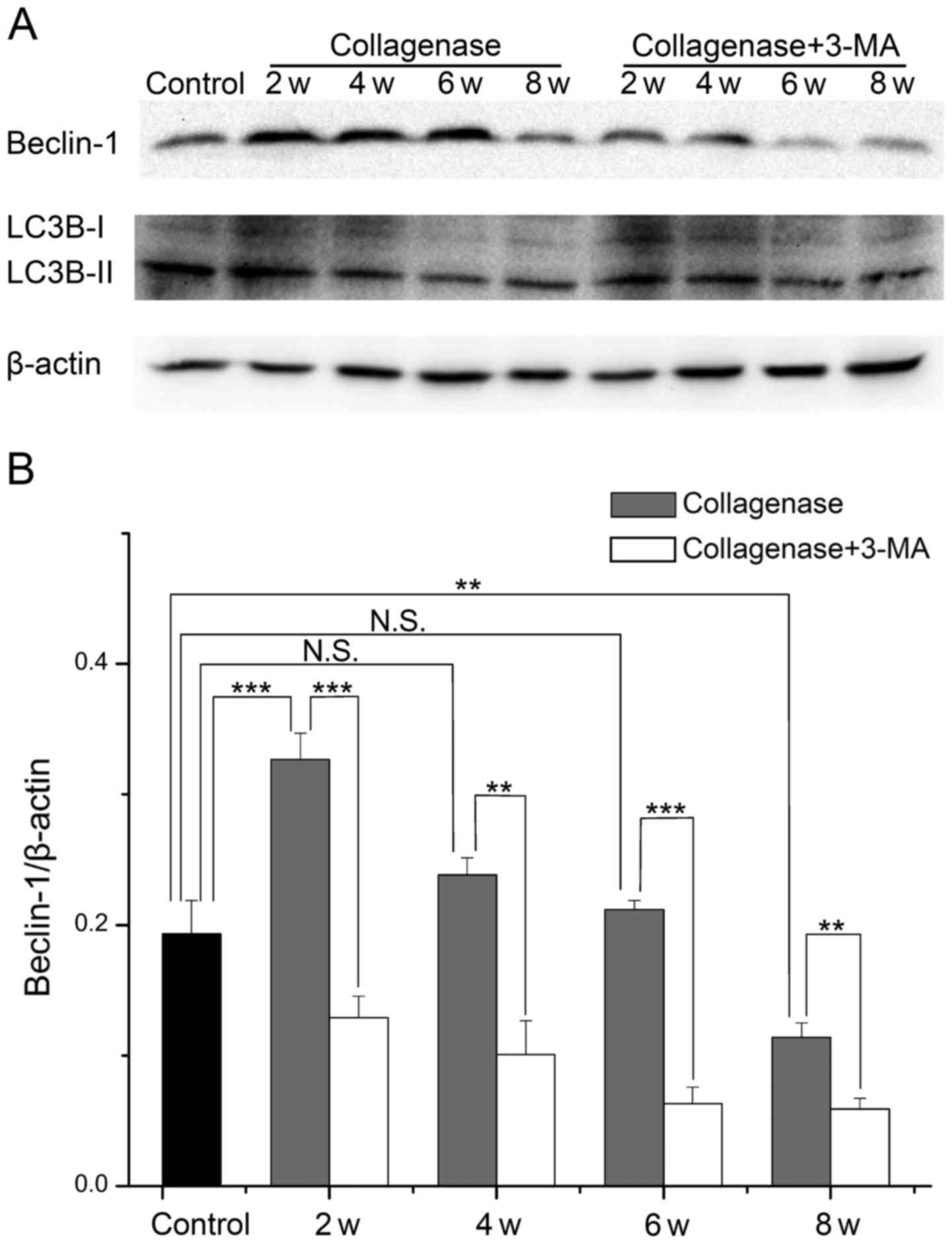
Expression of Beclin-1 and LC3B in
cartilage from rabbit with OA
To further investigate the changes in autophagy and
the role of impaired autophagy in the pathological process of OA,
we evaluated the protein expression levels of Beclin-1 and LC3B in
cartilage from rabbits with OA by western blot analysis (Fig. 6A). In cartilage from the
collagenase-injected group, the expression levels of Beclin-1 and
the LC3B-II/LC3B-I ratio were both highest at 2 weeks, and they
then decreased with time. The expression level of Beclin-1 was
increased at 2, 4 and 6 weeks (P<0.001, 0.094 and 0.768,
respectively), and it was decreased at 8 weeks (P=0.003) compared
with the cartilage from the control group. The LC3B-II/LC3B-I ratio
was only increased at 2 weeks (P=0.007), and decreased at 4, 6 and
8 weeks (P=0.437, 0.002 and <0.001, respectively). At each time
point, the expression levels of Beclin-1 and the LC3B-II/LC3B-I
ratio were decreased in cartilage from the 3-MA-treated group
compared to cartilage from the collagenase-injected group. These
results demonstrated that autophagy was first enhanced and then
weakened in cartilage from rabbits with collagenase-induced OA, and
treatment with 3-MA enhanced the decrease in Beclin-1 expression
and the LC3B-II/LC3B-I ratio.
Discussion
OA is a complex joint disorder with multiple
etiologies, including aging, obesity, mechanical and biochemical
factors (29,30). The degeneration of cartilage is
the main pathology of OA and is associated with many inflammatory
cytokines, such as IL-1β (31,32) and collagenase (33,34). IL-1β stimulates the expression of
collagenase in chondrocytes and is often applied to produce
cellular OA models for in vitro studies (35), and a number of of studies have
shown that SW1353 cells can take the place of human chondrocytes
for research (36–38). In this study, IL-1β was applied to
SW1353 cells to establish a cellular model of OA. We found that the
expression of MMP-13 was significantly promoted, while the
expression of TIMP-1 and cell viability were significantly
suppressed at 48 h, further indicating that IL-1β induced the
degeneration of chondrocytes.
Previous studies have successfully established
models of OA using rabbits following the intra-articular injection
of collagenase (39,40). Moreover, it has been reported that
the injection of 1.0 mg of collagenase is sufficient to induce
OA-like changes for 6 weeks (6).
Accordingly, 1.0 mg of collagenase was injected into the right
knees of rabbits in this study, and the results observed by light
microscopy and TEM were roughly consistent with those of previous
studies, and further demonstrated that collagenase may be involved
in cartilage degeneration in OA. However, the time needed for
OA-like changes to occur was not 6 weeks but 8 weeks in this study,
which may be due to various reasons, such as individual differences
between animals. Other methods such as the one described by Hulth
et al (41) involve the
opening of the joint, exposing the articular surface, resulting in
swelling, edema and congestion, which may affect the experimental
results. The intra-articular injection of drugs does not involve
the exposure of the articular surface, and this thus avoids
side-effects such as swelling, edema and congestion caused by
surgery affecting the experimental results. Thus, this method is
suitable for the investigation of the effects of drugs on OA.
Recent studies have demonstrated that autophagy is
involved in certain bone and cartilage diseases, such as cervical
disc degeneration (42),
cartilage degeneration of the temporomandibular joint (23), degradation of Meckel's cartilage
(43) and OA (44). However, the results regarding
changes in autophagy and the specific role of autophagy in the
progression of OA are sometimes contradictory (25,26). In this study, we found that the
expression of Beclin-1 and LC3B was first increased and then
decreased both in the IL-1β-stimulated SW1353 cells and in
cartilage from rabbits with collagenase-induced OA. TEM observation
revealed that much more autophagosomes in chondrocytes were
observed at the early time points and less were observed at later
time points in cartilage from rabbits with collagenase-induced OA.
These results suggest that autophagy is enhanced during the early
stages and is weakened during the late stages of experimental OA,
which determines the changes in autophagy in different stages of OA
and provides the possible cause of the contradictory reports.
During the early period, there was no apparent loss
of cell viability in the IL-1β-stimulated SW1353 cells; the mRNA
level of MMP-13 was increased at 12 h and that of TIMP-1 was
increased at 12 and 24 h without statistical significance.
Moreover, the OA score of cartilage from the collagenase-injected
group at 2 weeks showed no significance compared to the control
group. These results may be related to the enhancement of autophagy
during the early period of experimental OA.
To further investigate the role of the loss of
autophagy in the pathogenesis of OA, a chemical inhibitor of the
autophagic process, 3-MA, was applied to treat IL-1β-stimulated
SW1353 cells and was directly injected into the knee joints of
rabbits (45). We found that 3-MA
treatment significantly inhibited the expression of Beclin-1 and
LC3B in cells and caitilage, and the autophagosomes in chondrocytes
were reduced. In addition, the loss of cell viability, the
upregulation of MMP-13 and the downregulation of TIMP-1 induced by
IL-1β were significantly enhanced in the SW1353 cells treated with
3-MA. Importantly, the degeneration of cartilage and chondrocytes
in rabbit joints with collagenase-induced OA was aggravated by
treatment with 3-MA. These results demonstrated that 3-MA
aggravated the severity of experimental OA by inhibiting
autophagy.
In conclusion, our results demonstrated that
autophagy was first enhanced and then weakened in IL-1β-stimulated
SW1353 cells and in rabbits with collagenase-induced OA
degenerative cartilage. 3-MA aggravated the severity of
experimental OA via the inhibition of autophagy, suggesting that
the regulation of autophagy may be a potential therapeutic strategy
for the treatment of OA.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 31171672). We are thankful to the
Liver Research Center of Beijing Friendship Hospital of Capital
Medical University for providing technical assistance.
References
|
1
|
Buckland J: Osteoarthritis: targeting
cartilage erosion in OA. Nat Rev Rheumatol. 6:642010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lahm A, Kasch R, Mrosek E, Spank H,
Erggelet C, Esser J and Merk H: Semiquantitative analysis of ECM
molecules in the different cartilage layers in early and advanced
osteoarthritis of the knee joint. Histol Histopathol. 27:609–615.
2012.PubMed/NCBI
|
|
3
|
Wang JH, Shih KS, Wu YW, Wang AW and Yang
CR: Histone deacetylase inhibitors increase microRNA-146a
expression and enhance negative regulation of interleukin-1β
signaling in osteoarthritis fibroblast-like synoviocytes.
Osteoarthritis Cartilage. 21:1987–1996. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hui W, Young DA, Rowan AD, Xu X, Cawston
TE and Proctor CJ: Oxidative changes and signalling pathways are
pivotal in initiating age-related changes in articular cartilage.
Ann Rheum Dis. 75:449–458. 2016. View Article : Google Scholar :
|
|
5
|
Kim HJ, So HS, Lee JH, Park C, Lee JB,
Youn MJ, Kim SJ, Yang SH, Lee KM, Kwon KB, et al: Role of
proinflammatory cytokines in cisplatin-induced vestibular hair cell
damage. Head Neck. 30:1445–1456. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kikuchi T, Sakuta T and Yamaguchi T:
Intra-articular injection of collagenase induces experimental
osteoarthritis in mature rabbits. Osteoarthritis Cartilage.
6:177–186. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Adães S, Ferreira-Gomes J, Mendonça M,
Almeida L, Castro-Lopes JM and Neto FL: Injury of primary afferent
neurons may contribute to osteoarthritis induced pain: an
experimental study using the collagenase model in rats.
Osteoarthritis Cartilage. 23:914–924. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Adães S, Mendonça M, Santos TN,
Castro-Lopes JM, Ferreira-Gomes J and Neto FL: Intra-articular
injection of collagenase in the knee of rats as an alternative
model to study nociception associated with osteoarthritis.
Arthritis Res Ther. 16:R102014. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klatt AR, Klinger G, Paul-Klausch B, Kühn
G, Renno JH, Wagener R, Paulsson M, Schmidt J, Malchau G and
Wielckens K: Matrilin-3 activates the expression of
osteoarthritis-associated genes in primary human chondrocytes. FEBS
Lett. 583:3611–3617. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Naito K, Takahashi M, Kushida K, Suzuki M,
Ohishi T, Miura M, Inoue T and Nagano A: Measurement of matrix
metalloproteinases (MMPs) and tissue inhibitor of
metalloproteinases-1 (TIMP-1) in patients with knee osteoarthritis:
comparison with generalized osteoarthritis. Rheumatology (Oxford).
38:510–515. 1999. View Article : Google Scholar
|
|
11
|
Yang L, Guo A and Gu JC: c-Jun N-terminal
kinase and nuclear factor κB mediate nitric oxide-induced
expression of matrix metalloproteinase-13. Int Orthop.
35:1261–1266. 2011. View Article : Google Scholar
|
|
12
|
Ryter SW, Cloonan SM and Choi AM:
Autophagy: a critical regulator of cellular metabolism and
homeostasis. Mol Cells. 36:7–16. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murrow L and Debnath J: Autophagy as a
stress-response and quality-control mechanism: implications for
cell injury and human disease. Annu Rev Pathol. 8:105–137. 2013.
View Article : Google Scholar
|
|
14
|
Wang SY, Yu QJ, Zhang RD and Liu B: Core
signaling pathways of survival/death in autophagy-related cancer
networks. Int J Biochem Cell Biol. 43:1263–1266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang J, Wang W, Zhang H, Hu Y, Wang M and
Yin Z: The dual role of autophagy in chondrocyte responses in the
pathogenesis of articular cartilage degeneration in osteoarthritis.
Int J Mol Med. 32:1311–1318. 2013.PubMed/NCBI
|
|
16
|
Klionsky DJ, Cregg JM, Dunn WA Jr, Emr SD,
Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M,
et al: A unified nomenclature for yeast autophagy-related genes.
Dev Cell. 5:539–545. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Caramés B, Olmer M, Kiosses WB and Lotz
MK: The relationship of autophagy defects to cartilage damage
during joint aging in a mouse model. Arthritis Rheumatol.
67:1568–1576. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barranco C: Osteoarthritis: activate
autophagy to prevent cartilage degeneration? Nat Rev Rheumatol.
11:1272015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang M, Zhang J, Lu L, Qiu ZY, Zhang X,
Yu SB, Wu YP and Wang MQ: Enhancement of chondrocyte autophagy is
an early response in the degenerative cartilage of the
temporomandibular joint to biomechanical dental stimulation.
Apoptosis. 18:423–434. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Lang W, Ye H, Yu F, Li H, Chen J,
Cai L, Chen W, Lin R, Huang Y, et al: Tougu Xiaotong capsule
inhibits the tidemark replication and cartilage degradation of
papain-induced osteoarthritis by the regulation of chondrocyte
autophagy. Int J Mol Med. 31:1349–1356. 2013.PubMed/NCBI
|
|
25
|
Almonte-Becerril M, Navarro-Garcia F,
Gonzalez-Robles A, Vega-Lopez MA, Lavalle C and Kouri JB: Cell
death of chondrocytes is a combination between apoptosis and
autophagy during the pathogenesis of Osteoarthritis within an
experimental model. Apoptosis. 15:631–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Benito MJ, Veale DJ, FitzGerald O, van den
Berg WB and Bresnihan B: Synovial tissue inflammation in early and
late osteoarthritis. Ann Rheum Dis. 64:1263–1267. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mankin HJ, Johnson ME and Lippiello L:
Biochemical and metabolic abnormalities in articular cartilage from
osteoarthritic human hips. III. Distribution and metabolism of
amino sugar-containing macromolecules. J Bone Joint Surg Am.
63:131–139. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
van der Kraan PM: Understanding
developmental mechanisms in the context of osteoarthritis. Curr
Rheumatol Rep. 15:3332013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Goldring MB and Goldring SR:
Osteoarthritis. J Cell Physiol. 213:626–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu X, Kondragunta V, Kornman KS, Wang HY,
Duff GW, Renner JB and Jordan JM: IL-1 receptor antagonist gene as
a predictive biomarker of progression of knee osteoarthritis in a
population cohort. Osteoarthritis Cartilage. 21:930–938. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Santangelo KS, Nuovo GJ and Bertone AL: In
vivo reduction or blockade of interleukin-1β in primary
osteoarthritis influences expression of mediators implicated in
pathogenesis. Osteoarthritis Cartilage. 20:1610–1618. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bucknor MD, Nardo L, Joseph GB, Alizai H,
Srikhum W, Nevitt MC, Lynch JA, McCulloch CE and Link TM:
Association of cartilage degeneration with four year weight gain -
3T MRI data from the Osteoarthritis Initiative. Osteoarthritis
Cartilage. 23:525–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Botter SM, van Osch GJ, Waarsing JH, van
der Linden JC, Verhaar JA, Pols HA, van Leeuwen JP and Weinans H:
Cartilage damage pattern in relation to subchondral plate thickness
in a collagenase-induced model of osteoarthritis. Osteoarthritis
Cartilage. 16:506–514. 2008. View Article : Google Scholar
|
|
35
|
Roman-Blas JA, Contreras-Blasco MA, Largo
R, Alvarez-Soria MA, Castañeda S and Herrero-Beaumont G:
Differential effects of the antioxidant n-acetylcysteine on the
production of catabolic mediators in IL-1beta-stimulated human
osteoarthritic synoviocytes and chondrocytes. Eur J Pharmacol.
623:125–131. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schaefer JF, Millham ML, de Crombrugghe B
and Buckbinder L: FGF signaling antagonizes cytokine-mediated
repression of Sox9 in SW1353 chondrosarcoma cells. Osteoarthritis
Cartilage. 11:233–241. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu YC, Jayakumar T, Duann YF, Chou YC,
Hsieh CY, Yu SY, Sheu JR and Hsiao G: Chondroprotective role of
sesamol by inhibiting MMPs expression via retaining NF-κB signaling
in activated SW1353 cells. J Agric Food Chem. 59:4969–4978. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tetsunaga T, Nishida K, Furumatsu T,
Naruse K, Hirohata S, Yoshida A, Saito T and Ozaki T: Regulation of
mechanical stress-induced MMP-13 and ADAMTS-5 expression by RUNX-2
transcriptional factor in SW1353 chondrocyte-like cells.
Osteoarthritis Cartilage. 19:222–232. 2011. View Article : Google Scholar
|
|
39
|
Kwon DR, Park GY and Lee SU: The effects
of intra-articular platelet-rich plasma injection according to the
severity of collagenase-induced knee osteoarthritis in a rabbit
model. Ann Rehabil Med. 36:458–465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim SB, Kwon DR, Kwak H, Shin YB, Han HJ,
Lee JH and Choi SH: Additive effects of intra-articular injection
of growth hormone and hyaluronic acid in rabbit model of
collagenase-induced osteoarthritis. J Korean Med Sci. 25:776–780.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hulth A, Lindberg L and Telhag H:
Experimental osteoarthritis in rabbits. Preliminary report. Acta
Orthop Scand. 41:522–530. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu H, Xiong S, Wang H, Zhang M and Yu Y:
The evidence and the possible significance of autophagy in
degeneration model of human cervical end-plate cartilage. Exp Ther
Med. 7:537–542. 2014.PubMed/NCBI
|
|
43
|
Yang RT, Zhang C, Liu Y, Zhou HH and Li
ZB: Autophagy prior to chondrocyte cell death during the
degeneration of Meckel's cartilage. Anat Rec (Hoboken).
295:734–741. 2012. View
Article : Google Scholar
|
|
44
|
Zhang Y, Vasheghani F, Li YH, Blati M,
Simeone K, Fahmi H, Lussier B, Roughley P, Lagares D, Pelletier JP,
et al: Cartilage-specific deletion of mTOR upregulates autophagy
and protects mice from osteoarthritis. Ann Rheum Dis. 74:1432–1440.
2015. View Article : Google Scholar
|
|
45
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|