Introduction
Dysfunction and destruction of the intestinal
epithelial barrier results in an increased mucosal antigen load,
leading to the activation of mucosal immune responses (1,2),
which are associated with the pathogenesis of several intestinal
disorders, including irritable bowel syndrome (IBS) (3–5).
Barrier dysfunction has been documented in many patients with IBS,
and it is especially common in those with post-infectious irritable
bowel syndrome (PI-IBS) (6–8).
Notably, increasing evidence indicates that PI-IBS may result from
a combined process involving barrier dysfunction, low-grade mucosal
inflammation and immune activation (5,9,10).
Intestinal inflammation has attracted increasing
concern in relation to the pathogenesis of PI-IBS. Increased
numbers of immune cells, primarily T cells and mast cells, have
been detected in the colons (11), ilea and duodena (12–14) of subsets of IBS patients.
Additionally, PI-IBS patients have significantly increased
proportions of CD45RO+CD4+ activated/memory T
cells in the lamina propria (15). Interestingly, the number of T
cells that infiltrate into the lamina propria is associated with
the severity of diarrhea (16,17). Some studies have also demonstrated
that the levels of inflammatory cytokines, including IL-1β and
IFN-γ, are increased in the intestines (18,19). Therefore, the proliferation and
activation of T lymphocytes play important roles in the development
and progression of IBS. Intestinal inflammation and immune system
activation, particularly T lymphocyte activation, may contribute to
the sensitization of peripheral or spinal nociceptive pathways and
cause pain and hypersensitivity in PI-IBS patients (20). However, the reason why intestinal
T lymphocytes are activated after gastrointestinal infection
remains unclear. As the primary barrier, the intestine is exposed
to a wide variety of antigens and bacteria, and it plays an
important role in maintaining homeostasis in the body. Abnormal
CD11c+ mononuclear phagocytes, such as dendritic cells
(DCs), macrophages, and monocytes, are involved in the disruption
of immune tolerance in organisms, which can lead to the development
of chronic inflammatory diseases, including Crohn's disease and
others (21,22). Notably, different subtypes of
mononuclear phagocytic cells play different roles.
CD11c+ monocytes/macrophages promote Helicobacter
hepaticus-induced intestinal inflammation through the
production of IL-23 (23). In
addition, in Citrobacter rodentium infection, colonic
CX3CR1+ mononuclear phagocytes have been shown to induce
an innate immune response (24).
Furthermore, phenotypic and functional alterations in lamina
propria dendritic cells (LPDCs) have been suggested to be at least
partly responsible for activation of the effector pathways that
lead to inflammatory bowel disease (25–28). In a previous study, we documented
functional and phenotypic alterations of CD11c+ lamina
propria mononuclear phagocytes (CD11c+ LPMPs) in a
PI-IBS mouse model (29). These
CD11c+ LPMPs were more mature, expressed a greater
number of co-stimulatory molecules, such as CD86 and MHCII,
compared with that in control mice, and they induced T cell
differentiation and cytokine expression. Another study reported
that the number of CD103+ LPDCs was found to be
increased and that IL-4 was secreted to activate mast cells in an
IBS rat model (30). However, the
relationship between changes in CD11c+ LPMPs and
visceral hypersensitivity in the PI-IBS model remains unknown.
Additionally, immune system activation and increased intestinal
permeability often interact and promote each other. Thus, it is
worthwhile to investigate whether CD11c+ LPMPs promote
dysfunction of the mucosal barrier in a PI-IBS model.
Currently, there is little evidence regarding the
role of CD11c+ LPMPs in IBS. The Trichinella
spiralis (T. spiralis) infection mouse model used in our
previous study is an effective model for elucidating the
contribution of these cells to PI-IBS (29). Given the critical functions of
CD11c+ LPMPs in the intestinal immune system and their
putative relationship with mucosal immune activation, we adoptively
transferred these cells from PI-IBS model mice into naïve mice to
test our hypothesis that they contribute to increased intestinal
permeability and visceral hypersensitivity in PI-IBS.
Materials and methods
Mice and Trichinella infection
Male NIH Swiss mice aged 6–8 weeks were obtained
from the Guangdong Medical Laboratory Animal Center (Guangdong,
China). The mice were housed under specific pathogen-free
conditions at the Animal Laboratory Center of Tongji Medical
College (Wuhan, China). They were housed individually at a
temperature of 23–24°C with a light-dark cycle of 12–12 h and were
allowed free access to standard mouse food and water. Each mouse
was gavaged with 350–400 T. spiralis larvae in 0.2 ml
phosphate-buffered saline (PBS). T. spiralis cultures were
acquired from the Department of Parasitology and Microbiology,
Tongji Medical College (Wuhan, China). All experiments were
approved by the Ethics Committee of Tongji Medical College,
Huazhong University of Science and Technology.
Abdominal withdrawal reflex (AWR) in
response to colorectal distension (CRD)
The CRD protocol was performed as previously
described (31). Briefly, a
catheter with a balloon was coated with lubricant and inserted 2 cm
from the anal verge. The behavioral response to CRD was assessed
based on the AWR using a semi-quantitative scoring system (32). AWR scores were recorded following
the application of ascending-limit phasic distension (20, 40, 60
and 80 mmHg) for 20 sec every 4 min. Measurement of the AWR score
was repeated three times at each pressure value. The CRD stimulus
intensity that elicited contraction of the abdominal wall was
recorded as the threshold. Each balloon inflation value was
recorded five times by an observer in a blinded manner to ensure
for accuracy. More than six mice were included in each group.
Pathological characteristics
The mice were sacrificed by cervical dislocation,
and ileal samples were fixed in buffered 10% formalin.
Paraffin-embedded tissues were cut into 5 µm sections and
stained with hematoxylin and eosin (H&E).
Permeability assessment
The ileum was quickly removed from each mouse and
flushed with ice-cold Krebs solution (121 mM NaCl, 25 mM
NaHCO3, 3.8 mM KCl, 1 mM KH2PO4,
1.2 mM CaCl2, 1.2 mM MgSO4 and 11.1 mM
glucose). The external muscle tissue and myenteric plexus were
stripped off of the intestinal specimen. Sections of ileal villus
epithelium were macroscopically identified as previously described
(33,34). Each piece was placed in a Ussing
chamber (Physiology Instruments, Santiago, CA, USA), and both sides
of the chamber were filled with 5 ml Krebs solution, which was
oxygenated, and maintained at 37°C throughout the experiment. The
spontaneous potential difference and short circuit current in the
Ussing chambers were recorded after a 25 min equilibration period.
Transepithelial electrical resistance was calculated with Acquire
and Analyze 2.3 software.
Three milliliters of FITC-dextran (FD4, 1 mg/ml;
Sigma-Aldrich, St. Louis, MO, USA) were added to the mucosal side
of each chamber, and an equivalent volume of Krebs solution was
added to the other side. At 30-min intervals, 100 µl samples
were collected and transferred to 96-well plates in duplicate.
Krebs solution (200 µl) was added to the Ussing chambers
after fluid collection to equalize the volumes. FD4 flux in each
sample was measured at 520 nm with a fluorescence microplate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA). The FD4 concentration
was determined based on standard curves as previously described
(35). The permeability of each
piece of tissue was presented as the calculated FD4 flux over a
60–90 min period. More than six mice were included in each
group.
Transmission electron microscopy
Tissues were processed for transmission electron
microscopic observations using modified standard procedures
(36). The tissues were fixed in
2.5% buffered glutaraldehyde for 2 h at 4°C and were subsequently
fixed in 1% osmium tetroxide for 2 h at room temperature. Then,
they were dehydrated in graded alcohol and acetone and embedded in
Epon 812 overnight. Next, ultra-thin 60–80 nm sections were cut
with a diamond knife using a Leica Ultracut UCT (Leica Microsystems
GmbH, Wetzlar, Germany) and stained with uranyl acetate and lead
citrate. The ultrastructures of the tight junctions were observed
using a HITACHI U8010 transmission electron microscope (Hitachi,
Ltd., Tokyo, Japan).
Isolation of CD11c+ LPMPs
At 8 weeks after Trichinella spiralis
infection, CD11c+ LPMPs were isolated from the
intestines of the PI-IBS and normal mice. The entire small
intestine was collected from each mouse, cut open longitudinally,
washed with PBS, and then cut into 0.5-cm pieces. Subsequently, the
tissue pieces were incubated in PBS supplemented with 5% fetal
bovine serum (FBS) (Life Technologies, Gaithersburg, MD, USA), EDTA
(1 mM), DTT (1 mM), and penicillin (100 Units/ml)/streptomycin (100
µg/ml) (1%; Life Technologies) at 37°C for 20 min, followed
by removal of the epithelium. This incubation/epithelial removal
step was repeated twice, and then the tissues were cut into smaller
pieces and subsequently digested with 1 mg/ml collagenase IV (Roche
Diagnostics, Basel, Switzerland) in RPMI-1640 medium (Life
Technologies) at 37°C for 60 min. Next, the cell suspension was
sequentially filtered through 100-µm and 400-µm
filters and washed with RPMI-1640. LPMCs were harvested using a
discontinuous 40%/75% Percoll gradient (GE Healthcare, Chalfont,
UK). After being washed and resuspended in MACS buffer, the LPMCs
were incubated with a microbead-conjugated anti-CD11c antibody
(Miltenyi Biotec GmbH, Bergisch Gladbach, Germany).
CD11c+ LPMPs were selected using MACS columns (Miltenyi
Biotec GmbH). The selected cells were routinely found to contain
~85% CD11c+ cells based on flow cytometry. Cell
viability did not fall below 90% based on trypan blue exclusion
assays.
Adoptive transfer of CD11c+
LPMPs
CD11c+ LPMPs were generated in mice with
or without Trichinella infection and were then adoptively
transferred into NIH Swiss mice. Each mouse was administered
1×106 CD11c+ LPMPs via intravenous injection
into the tail. At 120 h after the transfer of CD11c+
LPMPs, AWR scores were determined. Intestinal tissues were
collected for permeability assays and analysis of intestinal
inflammation. More than six mice were included in each group.
Western blot analysis
Intestinal tissues were homogenized via mechanical
disruption in RIPA buffer with a protease inhibitor cocktail
(EDTA-free; Roche Diagnostics). The samples were then clarified via
centrifugation at 12,000 rpm for 10 min at 4°C. Next, the
supernatants were diluted with loading buffer and heated to 95°C
for 10 min. Subsequently, protein concentrations were determined
using a BCA protein assay kit (Thermo Fisher Scientific, Waltham,
MA, USA). The samples were separated on 10–12% SDS-PAGE gels and
transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). The membranes were then probed overnight at
4°C with antibodies against the TNF-α (goat anti-mouse; AF-410-NA),
IL-4 (rat anti-mouse; MAB404) and IL-10 (rat anti-mouse; MAB417)
cytokines (1 µg/ml, all from R&D Systems, Minneapolis,
MN, USA), occludin-1 (rabbit anti-mouse; 71–1500) and claudin-1
(rabbit anti-mouse; 51–9000) (1.25 µg/ml; both from Life
Technologies) or Gapdh (rabbit anti-mouse; ABS16; 0.5 µg/ml;
Millipore) and were then probed with HRP-conjugated secondary
antibodies [goat anti-rabbit IgG (111-035-003), goat anti-rat IgG
(112-035-003) and rabbit anti-goat IgG (305-035-003); 0.25
µg/ml; Jackson ImmunoResearch Laboratories, Inc., West
Grove, PA, USA] at room temperature for 2 h. The results were
analyzed with Quantity One 4.6.2 software (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). More than six mice were included in each
group.
Data analysis
The AWR scores were compared using the
Kruskal-Wallis one-way analysis of variance (ANOVA) on ranks test,
and significant results (P<0.05) were further analyzed using the
Wilcoxon rank sum test with Bonferroni correction for multiple
comparisons (0.05/3). The other data are expressed as the mean ± SE
and were analyzed via one-way ANOVA, followed by the least
significant difference (LSD) test or Dunnett's T3 multiple range
test. P<0.05 was considered to indicate a statistically
significant difference. Statistical analyses were performed with
SPSS version 19.0 (IBM SPSS, Armonk, NY, USA).
Results
Assessment of intestinal inflammation
after Trichinella infection
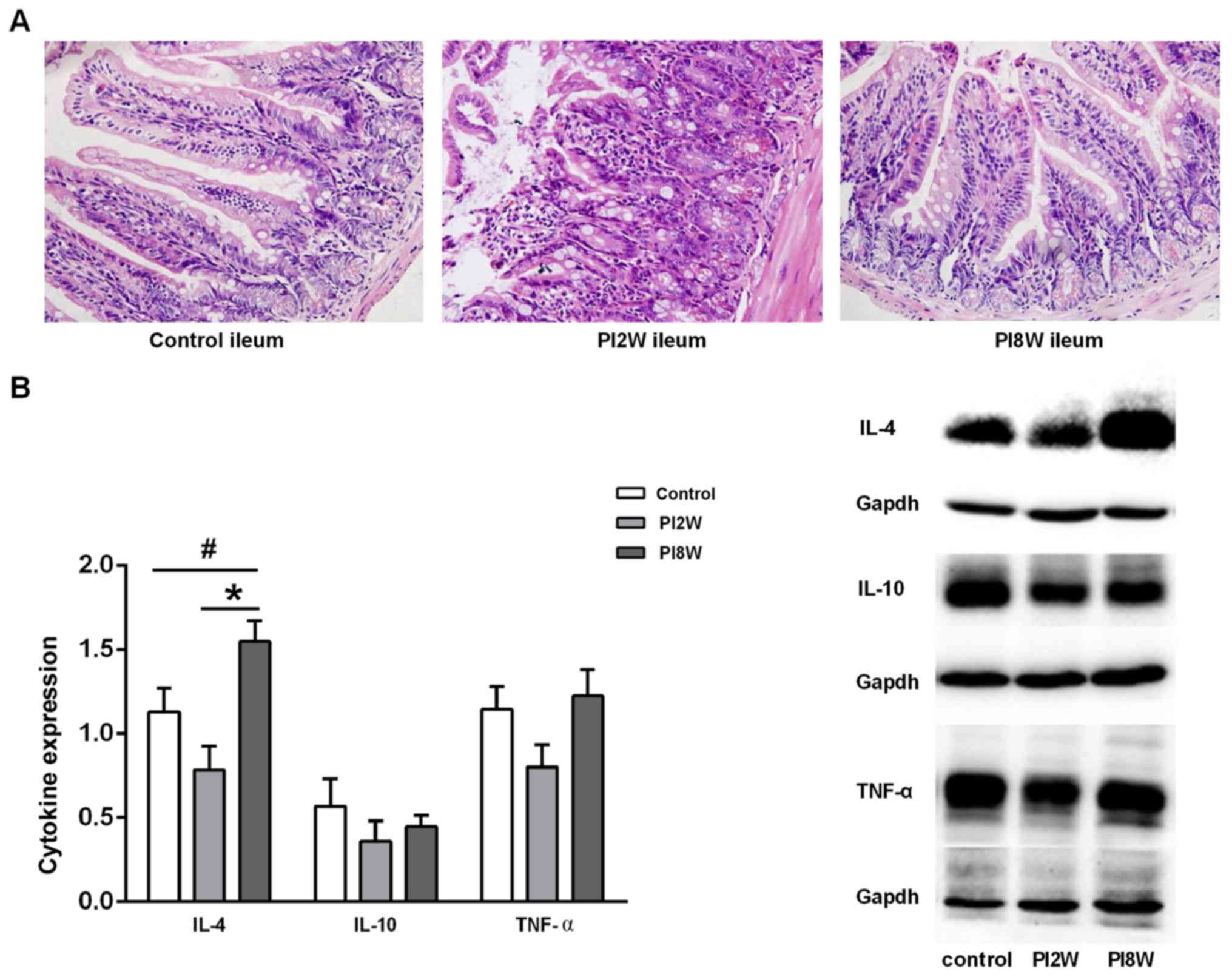
Severe inflammation was observed at 2 weeks
post-infection; however, no discernible inflammation was detected
at 8 weeks post-infection (Fig.
1A). H&E staining revealed that hyperemia, swelling and
severe neutrophilic and eosinophilic infiltration were induced in
the whole gut by Trichinella at 2 weeks post-infection.
However, no obvious microscopic changes were observed at 8 weeks
post-infection.
Next, we examined the cytokine expression levels to
determine whether a T helper 1 or T helper 2 immune response was
involved in the intestinal inflammatory response. IL-4 expression
in the ileum was found to be significantly elevated at 8 weeks
post-infection compared with that in the control mice and mice at 2
weeks post-infection (p=0.046; p=0.001); however, no significant
difference in IL-10 or TNF-α expression was observed among these
groups of mice (Fig. 1B). These
results suggested that Trichinella infection in the chronic
stage might be associated with a T helper 2 response.
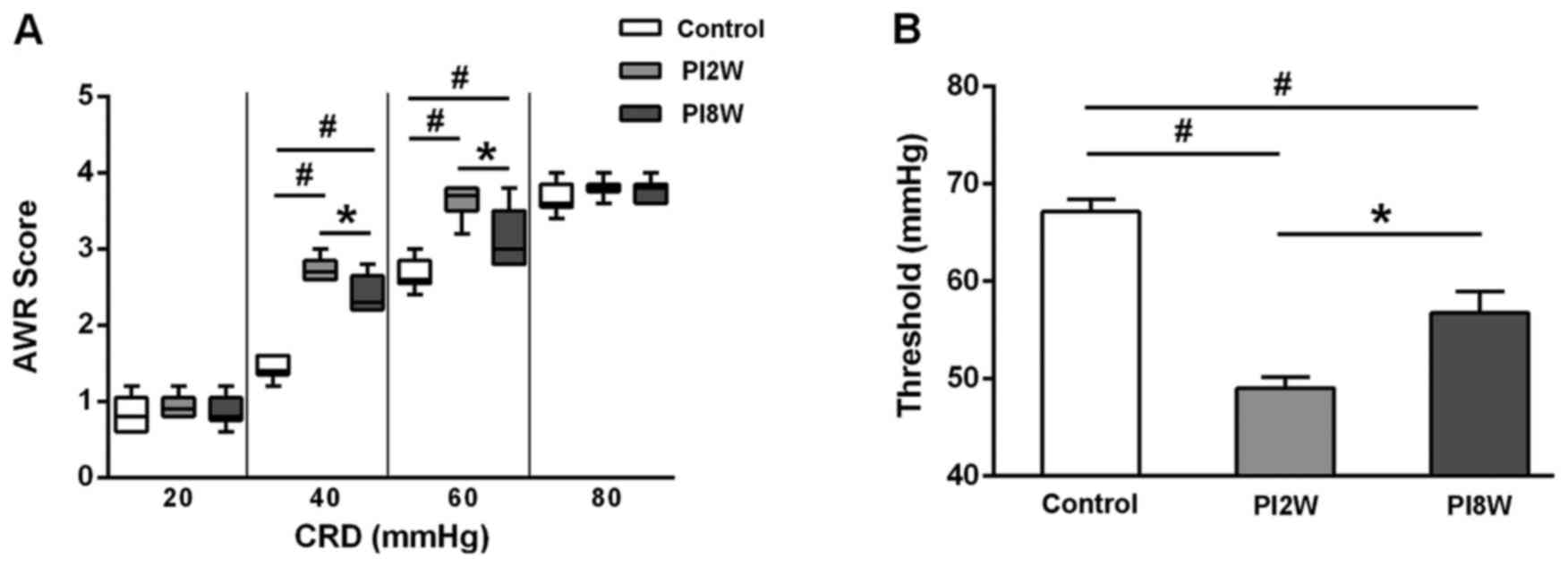
AWR scores after infection
The AWR scores began to increase during the 2nd week
post-infection, and they remained elevated during the 8th week at
CRD intensities of 40 and 60 mmHg (Fig. 2A). Similarly, the thresholds of
the 2- and 8-week post-infection groups were lower than those of
the control group (Fig. 2B).
These results implied that visceral hyperalgesia was sustained even
though intestinal inflammation had subsided by the 8th week after
infection, and they further suggested that the 8-week
post-infection group was a better model for PI-IBS with visceral
hypersensitivity.
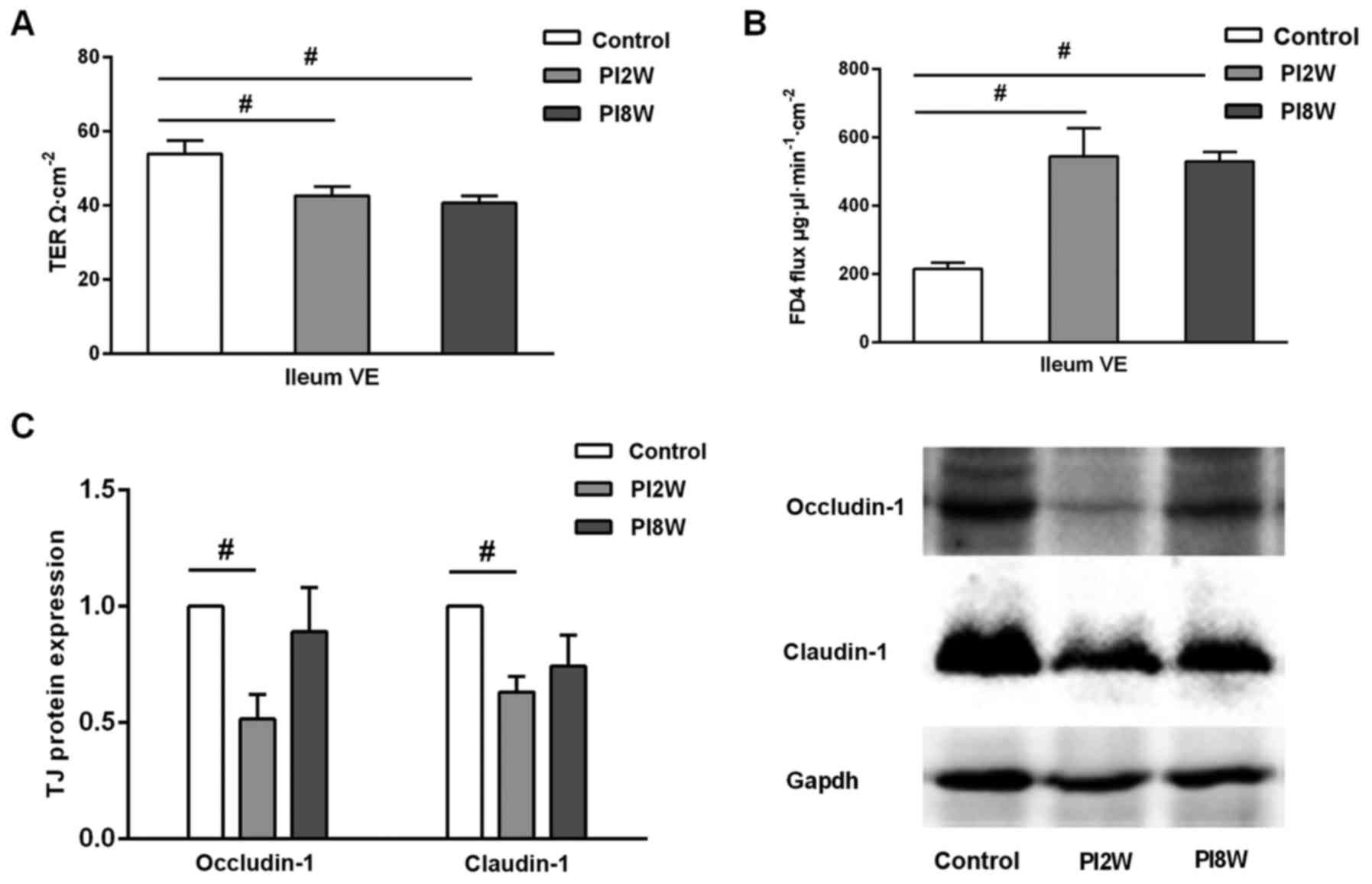
Increased mucosal permeability after
infection
Next, we used a Ussing chamber system to examine
transepithelial electrical resistance and FD4 flux to evaluate
intestinal epithelial barrier function in the ileum after infection
(Fig. 3).
The transepithelial electrical resistance of the
ileal villus epithelium was decreased in both the 2- and 8-week
post-infection groups compared with the control group (42.60±2.51
vs. 53.87±3.55, P=0.008; 40.61±1.95 vs. 3.87±3.55, P=0.002)
(Fig. 3A). In contrast, FD4 flux,
which reflects the barrier function of the paracellular pathways,
exhibited a drastic increase in the ileal villus epithelium
(P=0.001) (Fig. 3B). These
results suggested that intestinal permeability increased
immediately post-infection and that it remained elevated, even at 8
weeks when intestinal inflammation had subsided. To further
identify which paracellular pathway was affected, we focused on the
tight junctions and analyzed the occludin-1 and claudin-1
expression levels. As shown in Fig.
3C, occludin-1 and claudin-1 expression in the ileum were
downregulated in the 2-week post-infection group compared with the
control group (p=0.034; p=0.021); further, their expression was
slightly but non-significantly downregulated in the 8-week group
compared with the control group (Fig.
3C).
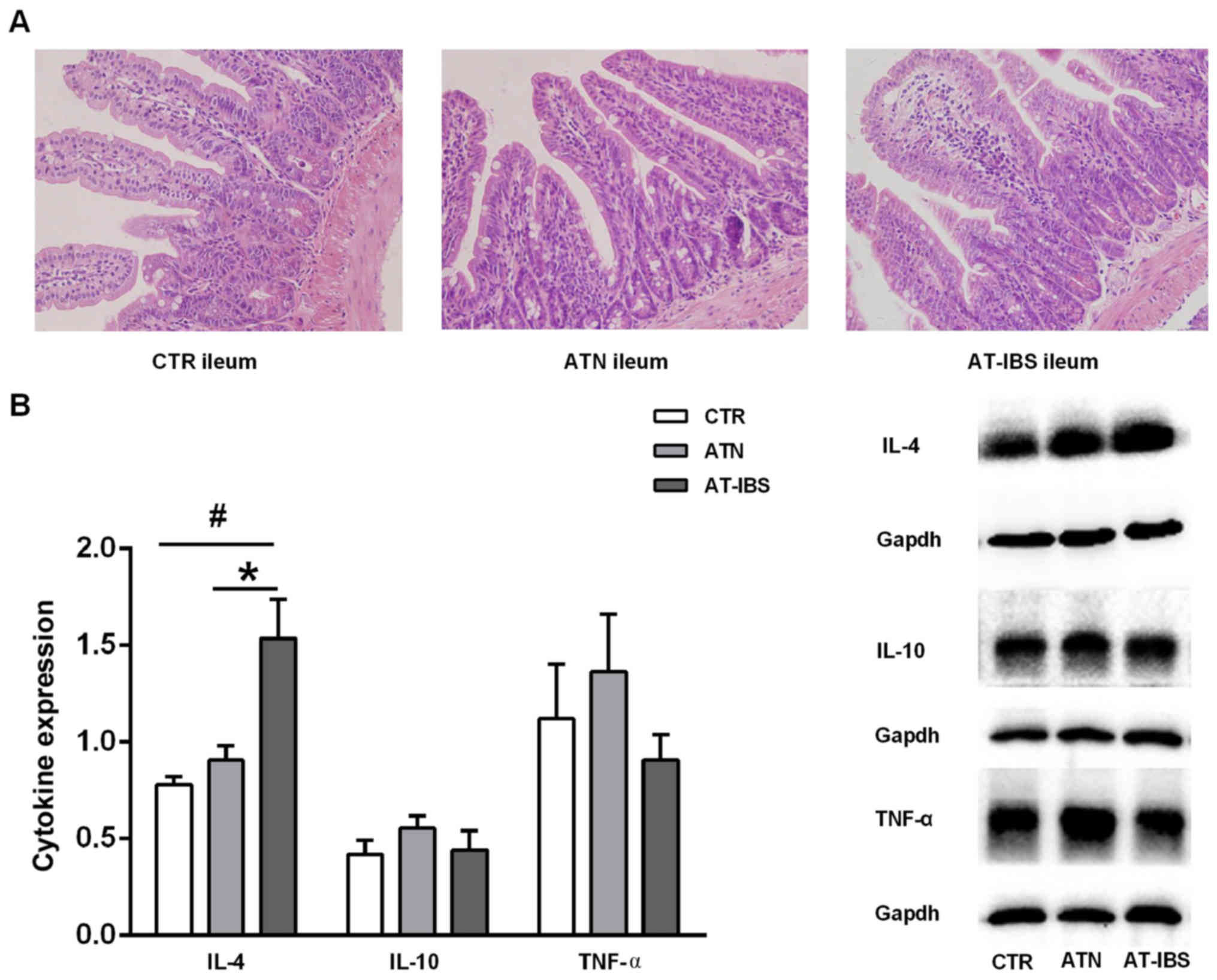
Milder inflammation after adoptive
transfer of CD11c+ LPMPs from PI-IBS mice
CD11c+ LPMPs were isolated from the
intestines of PI-IBS mice and normal mice at 8 weeks after T.
spiralis infection. The mice that received CD11c+
LPMPs from the PI-IBS or normal mice displayed inconspicuous
microscopic inflammation, as visualized by H&E staining
(Fig. 4A). No significant
differences in the histological characteristics were observed
between the mice that received CD11c+ LPMPs from the
PI-IBS mice (AT-IBS group), those that received CD11c+
LPMPs from the normal mice (ATN group), and the untreated mice (CTR
group). The ATN group was included in this study to ensure that
CD11c+ LPMPs were the only variable in the experiment
and to eliminate effects of the adoptive transfer on the
experimental results. Although no inflammation or damage was
observed in the microscopic examinations, increased IL-4 expression
was detected in the ilea of the AT-IBS mice compared with that in
the ilea of the CTR and ATN mice (P=0.019; P=0.048). There was no
difference in IL-10 or TNF-α expression in the ileum between the
CTR, ATN and AT-IBS mice (Fig.
4B).
Increased intestinal permeability after
adoptive transfer of CD11c+ LPMPs from PI-IBS mice
The transfer of CD11c+ LPMPs from PI-IBS
mice resulted in decreased transepithelial electrical resistance in
the ileal villus epithelium compared with that in the CTR and ATN
mice (31.68±5.41 vs. 55.23±3.81, P=0.001; 31.68±5.41 vs.
49.23±4.54, P=0.008) (Fig. 5A).
In contrast, the FD4 flux in the ileal villus epithelium was
increased following CD11c+ LPMP transfer (P=0.043)
(Fig. 5B). These results
suggested that intestinal permeability was increased in the ileal
villus epithelium. Furthermore, tight junction protein expression
was slightly but non-significantly downregulated in the AT-IBS mice
compared with that in the CTR and ATN mice (Fig. 5C).
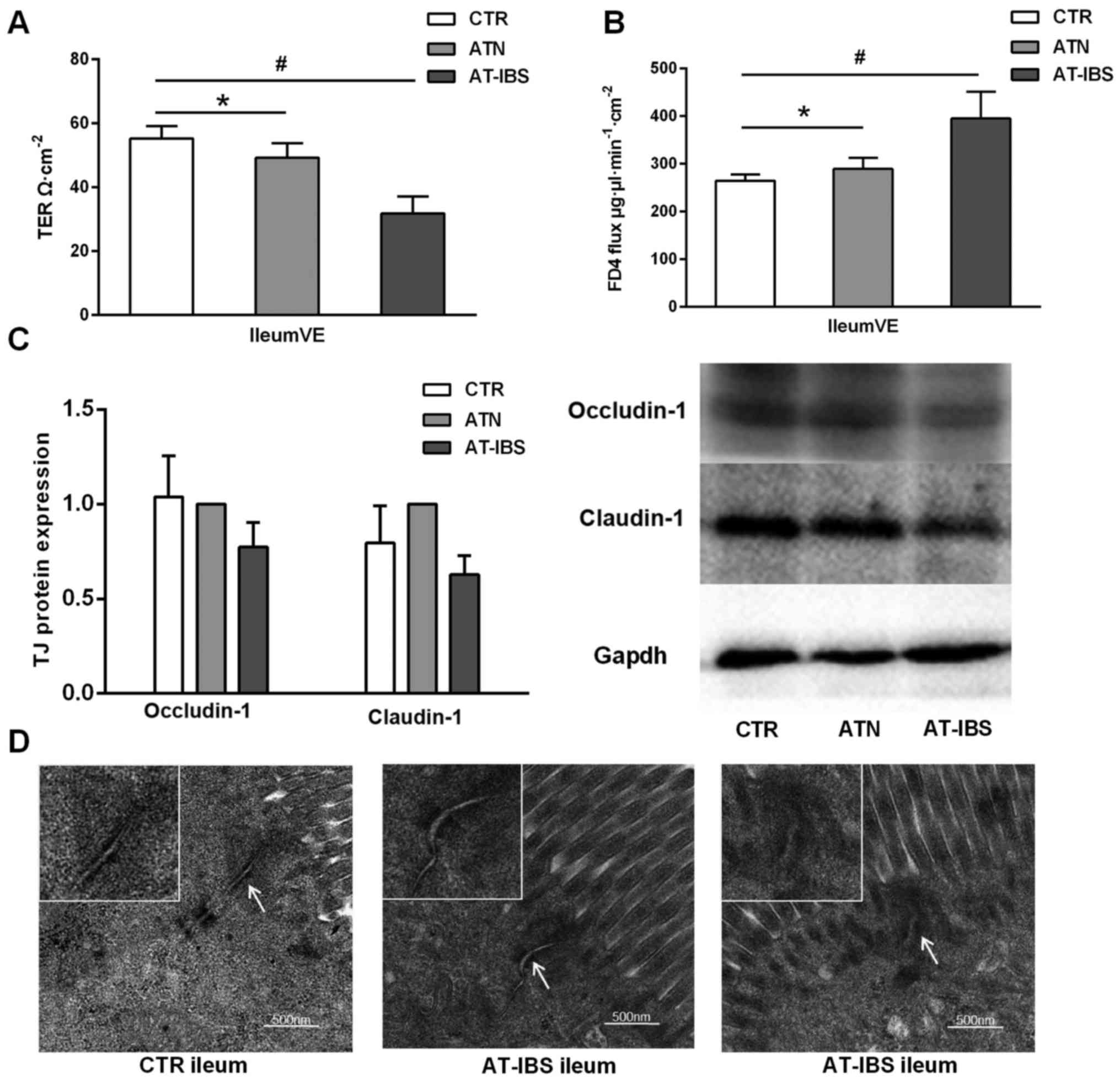 | Figure 5Increased intestinal permeability
after adoptive transfer of CD11c+ LPMPs from PI-IBS
mice. (A) TER of the ileal VE in the CTR, ATN and AT-IBS groups.
(B) FD4 fluxes in the three groups. (C) Expression of the tight
junction (TJ) proteins occludin-1 and claudin-1 in the ileum. All
data are presented as the mean ± SE; n≥6 mice per group. (D) Tight
junction ultrastructure, as determined by transmission electron
microscopy. The arrows indicate tight junctions. The mice in the
AT-IBS group exhibited increases in the apical intercellular
distance and proportion of dilated junctions, as well as
perijunctional cytoskeletal condensation. ATN, adoptive transfer of
CD11c+ LPMPs from normal mice; AT-IBS, adoptive transfer
of CD11c+ LPMPs from PI-IBS mice; CTR, control mice
injected with the same volume of saline. #P<0.05 for
the ATN and AT-IBS groups vs. the CTR group; *P<0.05
for the ATN group vs. the AT-IBS group. The ATN group was included
in this study to ensure that CD11c+ LPMPs were the only
variables in the experiment and to eliminate effects of the
adoptive transfer on the experimental results. LPMPs, lamina
propria mononuclear phagocytes; PI-IBS, post-infectious irritable
bowel syndrome; TER, transepithelial electrical resistance; VE,
villus epithelium; SE, standard error. |
However, ultrastructural alterations of tight
junction proteins were detected in the mice that received
CD11c+ LPMPs from the PI-IBS mice. As previously
described, the routine H&E staining performed in the present
study revealed no differences in epithelial architecture between
the control and adoptive transfer groups. Transmission electron
microscopy imaging showed that the functional alterations described
above were associated with ultrastructural changes in the
inter-cellular junctions, including disassembly (Fig. 5D). The mice that received
CD11c+ LPMPs from the PI-IBS mice exhibited increases in
the apical intercellular distance and proportion of dilated
junctions compared with the controls. These mice also displayed a
greater number of junctions with perijunctional cytoskeletal
condensation (Fig. 5D).
Changes in visceral sensation after
adoptive transfer of CD11c+ LPMPs
Finally, we examined visceral sensation after
CD11c+ LPMP transfer. As illustrated in Fig. 6, the mice that received
CD11c+ LPMPs from PI-IBS mice exhibited greater visceral
sensitivity. Significant increases in the AWR scores were observed
at pressures of 40 and 60 mmHg compared with those of the control
mice (Fig. 6A). Similarly, the
thresholds were lower following the adoptive transfer of
CD11c+ LPMPs from the PI-IBS mice (Fig. 6B); consequently, the mice that
received these cells exhibited visceral hypersensitivity.
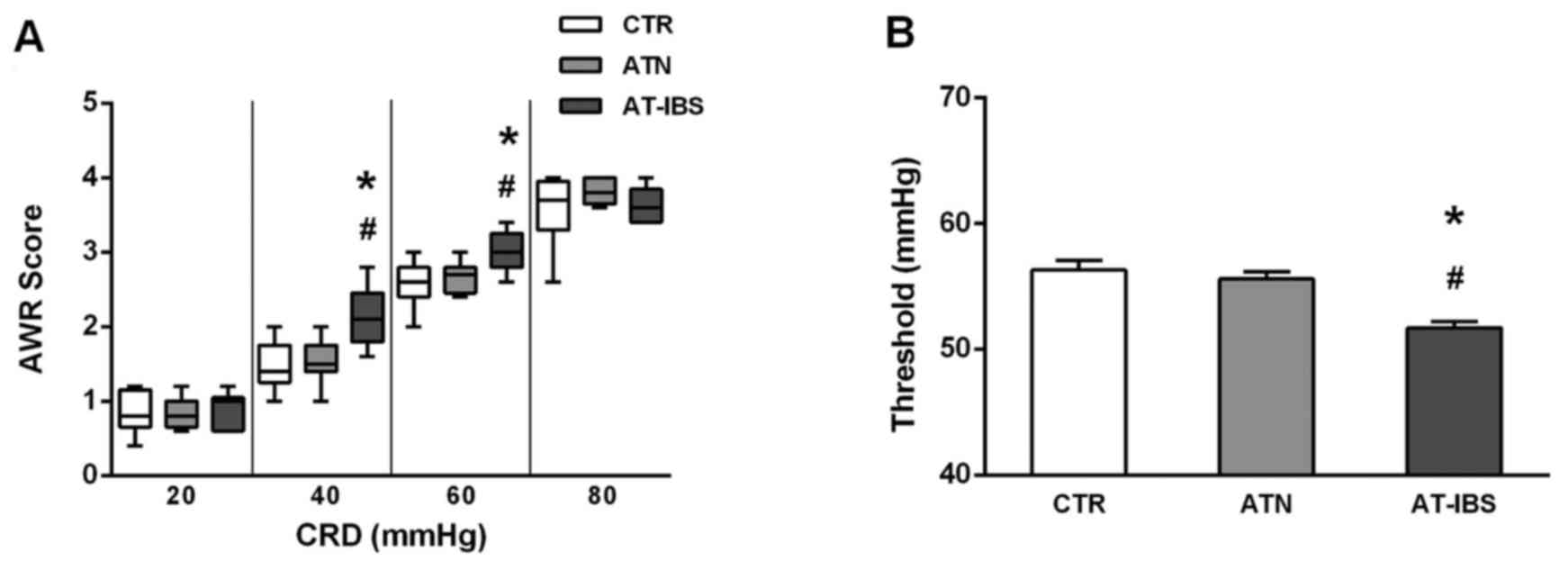 | Figure 6Changes in visceral sensation after
adoptive transfer of CD11c+ LPMPs. (A) Box plot of the
AWR scores obtained at 20, 40, 60 and 80 mmHg. The lines in the
boxes represent the medians, and the lines at the ends of the boxes
represent the 25th and 75th percentiles. The error bars denote the
5th and 95th percentiles; n≥6 mice per group. (B) Thresholds at
various CRD intensities. The means ± SEs are plotted; n≥6 mice per
group. ATN, adoptive transfer of CD11c+ LPMPs from
normal mice; AT-IBS, adoptive transfer of CD11c+ LPMPs
from PI-IBS mice; CTR, control mice injected with the same volume
of saline. #P<0.05 for the ATN and AT-IBS groups vs.
the CTR group; *P<0.05 for the ATN group vs. the
AT-IBS group. The ATN group was included in this study to ensure
that CD11c+ LPMPs were the only variable in the
experiment and to eliminate effects of the adoptive transfer on the
experimental results. LPMPs, lamina propria mononuclear phagocytes;
AWR, abdominal withdrawal reflex; CRD, colorectal distension; SE,
standard error; PI-IBS, post-infectious irritable bowel
syndrome;. |
Discussion
In the present study, we demonstrated that increased
intestinal permeability, visceral hypersensitivity and intestinal
inflammation were present during both the acute and chronic stages
of Trichinella infection. Even after expulsion of the
parasite, these changes persisted in the PI-IBS stage. The transfer
of CD11c+ LPMPs from PI-IBS mice into naïve mice not
only resulted in the transfer of enteric inflammation but also
caused abnormal intestinal function, characterized by epithelial
barrier disruption and visceral hyperalgesia.
To better understand the role of CD11c+
LPMPs in sustained inflammation associated with PI-IBS, we employed
a direct method of adoptive transfer. As previously described,
CD11c+ LPMPs in PI-IBS mice secrete several cytokines to
induce T-cell differentiation into the Th1, Th2 and Th17 subtypes
(29). Therefore, to identify the
immune responses induced by CD11c+ LPMPs, these cells
were adoptively transferred from PI-IBS mice into naïve mice.
Although microscopic examinations did not reveal any obvious
inflammation following adoptive transfer, mild inflammation was
noted based on the cytokine profiles in the small intestine. An
increase in IL-4 expression, implicating the T helper 2 response,
was detected in the ilea of the mice that received
CD11c+ LPMPs from the PI-IBS mice compared with that in
the ilea of the controls. Previous studies have shown that certain
subtypes of mononuclear phagocytes can be transferred to
inflammatory bowel disease model mice to promote or relieve colitis
(37–39); however, the adoptive transfer of
CD11c+ LPMPs from PI-IBS mice has not been previously
reported. Thus, our study is the first to report that the
relatively mild inflammation in PI-IBS mice can be transferred to
naïve mice via CD11c+ LPMPs.
More importantly, the increased permeability and
inflammation observed in the PI-IBS mouse model could also be
transferred by CD11c+ LPMPs. The mice that received
CD11c+ LPMPs from PI-IBS mice exhibited decreased
transepithelial electrical resistance and increased FD4
fluorescence intensity in the ileal villus epithelium. Although no
significant changes in the expression of tight junction proteins
were observed, ultrastructural alterations of tight junctions were
detected in the ileum by transmission electron microscopy. Previous
studies have shown that a number of cytokines, including IL-4,
TNF-α and IL-6, cause changes in tight junction permeability
(35,40–44). Therefore, the low-grade
inflammation marked by increased IL-4 expression observed in our
study may have contributed to the barrier dysfunction in the PI-IBS
mice.
All of these findings indicate that mild
inflammation caused by the adoptive transfer of CD11c+
LPMPs results in increased epithelial permeability. The decreased
transepithelial electrical resistance and increased FD4
fluorescence intensity in the ileal villus epithelium observed in
the PI-IBS and AT-IBS mice are suggestive of increased permeability
of the intestinal epithelial cell barrier that is dependent on the
villus epithelial pathway.
In addition to barrier dysfunction, the visceral
hypersensitivity of the PI-IBS mice was also found to be
transferred by CD11c+ LPMPs. Significant increases in
the AWR scores were observed at intensities of 40 and 60 mmHg, and
the thresholds decreased following the adoptive transfer of
CD11c+ LPMPs from the PI-IBS mice. The changes in
visceral sensation observed in the present study may have resulted
from a combined process of barrier dysfunction and mild mucosal
inflammation induced by CD11c+ LPMP transfer. Our
results showed that the increases in visceral sensitivity,
intestinal permeability and IL-4 expression, as well as the
histological characteristics, were consistent in both the PI-IBS
and AT-IBS animal model groups. Therefore, our findings strongly
imply that CD11c+ LPMPs play an important role in the
development of PI-IBS.
In the present study, in addition to obvious
visceral hypersensitivity and increased mucosal permeability,
severe inflammation was detected in the intestinal mucosa during
the acute infection stage. After expulsion of the parasites,
persistent low-grade inflammation was present, as indicated by the
increased cytokine levels. Furthermore, visceral hyperalgesia and
barrier dysfunction were sustained. The major characteristics of
IBS include visceral hypersensitivity, altered secretion,
intestinal sensory nerve abnormalities, barrier dysfunction and
alterations in intestinal immune function. Consequently, mouse
models of PI-IBS based on T. spiralis infection are widely
used to investigate both the immunological and functional changes
associated with gut inflammation, as described in previous studies
(45,46).
In conclusion, we demonstrated that increased
mucosal permeability and visceral hypersensitivity were maintained,
even under conditions of mild inflammation, in the studied PI-IBS
mouse model. CD11c+ LPMPs from these mice were able to
transfer not only enteric inflammation but also abnormal intestinal
function, characterized by epithelial barrier disruption and
visceral hyperalgesia, to normal mice. These findings may
contribute to the current understanding of the role of mild
inflammation in the pathophysiology of PI-IBS.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81500415 and
81330014). The website is http://www.nsfc.gov.cn/.
References
|
1
|
Shen L and Turner JR: Role of epithelial
cells in initiation and propagation of intestinal inflammation.
Eliminating the static: Tight junction dynamics exposed. Am J
Physiol Gastrointest Liver Physiol. 290:G577–G582. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Turner JR: Intestinal mucosal barrier
function in health and disease. Nat Rev Immunol. 9:799–809. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martínez C, Lobo B, Pigrau M, Ramos L,
González-Castro AM, Alonso C, Guilarte M, Guilá M, de Torres I,
Azpiroz F, et al: Diarrhoea-predominant irritable bowel syndrome:
An organic disorder with structural abnormalities in the jejunal
epithelial barrier. Gut. 62:1160–1168. 2013. View Article : Google Scholar
|
|
4
|
Keszthelyi D, Troost FJ, Jonkers DM, van
Eijk HM, Lindsey PJ, Dekker J, Buurman WA and Masclee AA:
Serotonergic reinforcement of intestinal barrier function is
impaired in irritable bowel syndrome. Aliment Pharmacol Ther.
40:392–402. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cheng P, Yao J, Wang C, Zhang L and Kong
W: Molecular and cellular mechanisms of tight junction dysfunction
in the irritable bowel syndrome. Mol Med Rep. 12:3257–3264.
2015.PubMed/NCBI
|
|
6
|
Barbara G: Mucosal barrier defects in
irritable bowel syndrome. Who left the door open? Am J
Gastroenterol. 101:1295–1298. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Camilleri M and Gorman H: Intestinal
permeability and irri table bowel syndrome. Neurogastroenterol
Motil. 19:545–552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dunlop SP, Hebden J, Campbell E, Naesdal
J, Olbe L, Perkins AC and Spiller RC: Abnormal intestinal
permeability in subgroups of diarrhea-predominant irritable bowel
syndromes. Am J Gastroenterol. 101:1288–1294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piche T, Barbara G, Aubert P, Bruley des
Varannes S, Dainese R, Nano JL, Cremon C, Stanghellini V, De
Giorgio R, Galmiche JP, et al: Impaired intestinal barrier
integrity in the colon of patients with irritable bowel syndrome:
Involvement of soluble mediators. Gut. 58:196–201. 2009. View Article : Google Scholar
|
|
10
|
Thabane M and Marshall JK: Post-infectious
irritable bowel syndrome. World J Gastroenterol. 15:3591–3596.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Spiller RC, Jenkins D, Thornley JP, Hebden
JM, Wright T, Skinner M and Neal KR: Increased rectal mucosal
enteroendocrine cells, T lymphocytes, and increased gut
permeability following acute Campylobacter enteritis and in
post-dysenteric irritable bowel syndrome. Gut. 47:804–811. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guilarte M, Santos J, de Torres I, Alonso
C, Vicario M, Ramos L, Martínez C, Casellas F, Saperas E and
Malagelada JR: Diarrhoea-predominant IBS patients show mast cell
activation and hyperplasia in the jejunum. Gut. 56:203–209. 2007.
View Article : Google Scholar
|
|
13
|
Walker MM, Talley NJ, Prabhakar M,
Pennaneac'h CJ, Aro P, Ronkainen J, Storskrubb T, Harmsen WS,
Zinsmeister AR and Agreus L: Duodenal mastocytosis, eosinophilia
and intraepithelial lymphocytosis as possible disease markers in
the irritable bowel syndrome and functional dyspepsia. Aliment
Pharmacol Ther. 29:765–773. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ohman L and Simrén M: Pathogenesis of IBS:
Role of inflammation, immunity and neuroimmune interactions. Nat
Rev Gastroenterol Hepatol. 7:163–173. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sundin J, Rangel I, Kumawat AK,
Hultgren-Hörnquist E and Brummer RJ: Aberrant mucosal lymphocyte
number and subsets in the colon of post-infectious irritable bowel
syndrome patients. Scand J Gastroenterol. 49:1068–1075. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bhuiyan MR, Majumder TK, Raihan AA, Roy
PK, Farha N and Kamal M: Histopathological alterations in
post-infectious irritable bowel syndrome in Bangladeshi population.
Mymensingh Med J. 19:275–281. 2010.PubMed/NCBI
|
|
17
|
Kirsch R and Riddell RH: Histopathological
alterations in irritable bowel syndrome. Mod Pathol. 19:1638–1645.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gwee KA, Collins SM, Read NW, Rajnakova A,
Deng Y, Graham JC, McKendrick MW and Moochhala SM: Increased rectal
mucosal expression of interleukin 1beta in recently acquired
post-infectious irritable bowel syndrome. Gut. 52:523–526. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen J, Zhang Y and Deng Z: Imbalanced
shift of cytokine expression between T helper 1 and T helper 2
(Th1/Th2) in intestinal mucosa of patients with post-infectious
irritable bowel syndrome. BMC Gastroenterol. 12:912012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Feng B, La JH, Schwartz ES and Gebhart GF:
Irritable bowel syndrome: Methods, mechanisms, and pathophysiology.
Neural and neuro-immune mechanisms of visceral hypersensitivity in
irritable bowel syndrome. Am J Physiol Gastrointest Liver Physiol.
302:G1085–G1098. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Scott CL, Henri S and Guilliams M:
Mononuclear phagocytes of the intestine, the skin, and the lung.
Immunol Rev. 262:9–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang H, Liu JQ, Yu Y, Mo LH, Ge RT, Zhang
HP, Liu ZG, Zheng PY and Yang PC: Regulation of TWIK-related
potassium channel-1 (Trek1) restitutes intestinal epithelial
barrier function. Cell Mol Immunol. 13:110–118. 2016. View Article : Google Scholar :
|
|
23
|
Arnold IC, Mathisen S, Schulthess J, Danne
C, Hegazy AN and Powrie F: CD11c(+) monocyte/macrophages promote
chronic Helicobacter hepaticus-induced intestinal inflammation
through the production of IL-23. Mucosal Immunol. 9:352–363. 2016.
View Article : Google Scholar
|
|
24
|
Aychek T, Mildner A, Yona S, Kim KW, Lampl
N, Reich-Zeliger S, Boon L, Yogev N, Waisman A, Cua DJ, et al:
IL-23-mediated mononuclear phagocyte crosstalk protects mice from
Citrobacter rodentium-induced colon immunopathology. Nat Commun.
6:65252015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hart AL, Al-Hassi HO, Rigby RJ, Bell SJ,
Emmanuel AV, Knight SC, Kamm MA and Stagg AJ: Characteristics of
intestinal dendritic cells in inflammatory bowel diseases.
Gastroenterology. 129:50–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silva MA, López CB, Riverin F, Oligny L,
Menezes J and Seidman EG: Characterization and distribution of
colonic dendritic cells in Crohn's disease. Inflamm Bowel Dis.
10:504–512. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Krajina T, Leithäuser F, Möller P,
Trobonjaca Z and Reimann J: Colonic lamina propria dendritic cells
in mice with CD4+ T cell-induced colitis. Eur J Immunol.
33:1073–1083. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Karlis J, Penttila I, Tran TB, Jones B,
Nobbs S, Zola H and Flesch IE: Characterization of colonic and
mesenteric lymph node dendritic cell subpopulations in a murine
adoptive transfer model of inflammatory bowel disease. Inflamm
Bowel Dis. 10:834–847. 2004. View Article : Google Scholar
|
|
29
|
Long Y, Wang W, Wang H, Hao L, Qian W and
Hou X: Characteristics of intestinal lamina propria dendritic cells
in a mouse model of postinfectious irritable bowel syndrome. J
Gastroenterol Hepatol. 27:935–944. 2012. View Article : Google Scholar
|
|
30
|
Li M, Zhang L, Lu B, Chen Z, Chu L, Meng L
and Fan Y: Role of dendritic cell-mediated abnormal immune response
in visceral hypersensitivity. Int J Clin Exp Med. 8:13243–13250.
2015.PubMed/NCBI
|
|
31
|
Jones RC III, Otsuka E, Wagstrom E, Jensen
CS, Price MP and Gebhart GF: Short-term sensitization of colon
mechanoreceptors is associated with long-term hypersensitivity to
colon distention in the mouse. Gastroenterology. 133:184–194. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Al-Chaer ED, Kawasaki M and Pasricha PJ: A
new model of chronic visceral hypersensitivity in adult rats
induced by colon irritation during postnatal development.
Gastroenterology. 119:1276–1285. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Velin AK, Ericson AC, Braaf Y, Wallon C
and Söderholm JD: Increased antigen and bacterial uptake in
follicle associated epithelium induced by chronic psychological
stress in rats. Gut. 53:494–500. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Keita AV, Söderholm JD and Ericson AC:
Stress-induced barrier disruption of rat follicle-associated
epithelium involves corticotropin-releasing hormone, acetylcholine,
substance P, and mast cells. Neurogastroenterol Motil. 22:770–778.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Overman EL, Rivier JE and Moeser AJ: CRF
induces intestinal epithelial barrier injury via the release of
mast cell proteases and TNF-α. PLoS One. 7:e399352012. View Article : Google Scholar
|
|
36
|
Pozzo Miller LD and Landis DM: Cytoplasmic
structure in organotypic cultures of rat hippocampus prepared by
rapid freezing and freeze-substitution fixation. Synapse.
13:195–205. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Siddiqui KR, Laffont S and Powrie F:
E-cadherin marks a subset of inflammatory dendritic cells that
promote T cell-mediated colitis. Immunity. 32:557–567. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McDole JR, Wheeler LW, McDonald KG, Wang
B, Konjufca V, Knoop KA, Newberry RD and Miller MJ: Goblet cells
deliver luminal antigen to CD103+ dendritic cells in the
small intestine. Nature. 483:345–349. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cascio JA, Haymaker CL, Divekar RD,
Zaghouani S, Khairallah MT, Wan X, Rowland LM, Dhakal M, Chen W and
Zaghouani H: Antigen-specific effector CD4 T lymphocytes school
lamina propria dendritic cells to transfer innate tolerance. J
Immunol. 190:6004–6014. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Poritz LS, Garver KI, Tilberg AF and
Koltun WA: Tumor necrosis factor alpha disrupts tight junction
assembly. J Surg Res. 116:14–18. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McDermott JR, Bartram RE, Knight PA,
Miller HR, Garrod DR and Grencis RK: Mast cells disrupt epithelial
barrier function during enteric nematode infection. Proc Natl Acad
Sci USA. 100:7761–7766. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Edelblum KL and Turner JR: The tight
junction in inflammatory disease: Communication breakdown. Curr
Opin Pharmacol. 9:715–720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Al-Sadi R, Ye D, Boivin M, Guo S, Hashimi
M, Ereifej L and Ma TY: Interleukin-6 modulation of intestinal
epithelial tight junction permeability is mediated by JNK pathway
activation of claudin-2 gene. PLoS One. 9:e853452014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Al-Sadi R, Boivin M and Ma T: Mechanism of
cytokine modulation of epithelial tight junction barrier. Front
Biosci (Landmark Ed). 14:2765–2778. 2009. View Article : Google Scholar
|
|
45
|
Bercík P, Wang L, Verdú EF, Mao YK,
Blennerhassett P, Khan WI, Kean I, Tougas G and Collins SM:
Visceral hyperalgesia and intestinal dysmotility in a mouse model
of postinfective gut dysfunction. Gastroenterology. 127:179–187.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fu Y, Wang W, Tong J, Pan Q, Long Y, Qian
W and Hou X: Th17 cells influence intestinal muscle contraction
during Trichinella spiralis infection. J Huazhong Univ Sci
Technolog Med Sci. 29:481–485. 2009. View Article : Google Scholar : PubMed/NCBI
|