Introduction
Dihydroartemisinin (DHA), recommended as an
effective antimalarial herbal compound by the World Health
Organization (WHO), is used worldwide to combat the parasite,
Plasmodium falciparum, which causes malaria. Recent studies
have demonstrated the inhibitory effects of DHA on the viability of
various cancer cells (1–4). However, the mechanisms responsible
for the anticancer effects of DHA have not yet been fully
documented. It has been reported that DHA exerts inhibitory effects
on cancer cells through the induction of apoptosis (3,4).
DHA treatment has been shown to result in mitochondrial membrane
depolarization, the release of cytochrome c and caspase
activation (1,5). Bcl-2 family proteins, such as Bax,
Bid and Noxa have also been shown to contribute to DHA-induced
apoptosis (6,7). Moreover, p53 has been reported to
facilitate apoptosis caused by DHA (5,8–10).
These data suggest that the inhibitory effects of DHA on cancer
cells are based on the activation of p53 and the
mitochondrial-related cell apoptosis pathway. Despite these
advances, however, the exact association between upstream signaling
and the downstream activation of the cell death pathway following
treatment with DHA remains unclear.
Caveolin 1 (Cav1) is an important component of
caveolae, and is known to function as a scaffolding protein,
regulating several signaling pathways (11–13). The loss of Cav1 has been
demonstrated to be involved in tumorigenesis in several types of
cancer, and the overexpression of Cav1 has been shown to inhibit
cell and tumor growth (14–18). Thus, Cav1 is regarded as a
potential tumor suppressor. In spite of the fact that a number of
studies have been conducted to investigate the function of Cav1 in
several types of cancer (14–18), studies reporting that Cav1
functions as a tumor suppressor by inhibiting the oxidative stress
response pathway are limited (19). As important mediators of the
apoptotic signaling pathway, reactive oxygen species (ROS) play
important roles in the induction of cancer cell death. DHA has also
reported to induce the generation of ROS as upstream signaling
molecules that initiate the mitochondria-related apoptotic pathway
(20,21). The increased generation of ROS
suggests the inhibition of antioxidant gene expression in response
to oxidative stress; thus, it is possible that proteins which
inhibit the oxidative stress response pathways may function
upstream of the activation of the cell death pathway following
treatment with DHA. Of note, Cav1 has been shown to inhibit
cellular antioxidant capacity through direct interaction with
nuclear factor erythroid 2-related factor 2 (Nrf2) (22,23). Thus, it is reasonable that Cav1
may function upstream of the cell death pathway activated by DHA by
inhibiting the Nrf2-related oxidative stress response pathway.
DHA has also been previously reported to trigger
ROS-mediated Bid activation and mitochondrial translocation
(7,21). Mitochondrial carrier homolog 2
(MTCH2) has been demonstrated to play an important role in
facilitating the mitochondrial recruitment of truncated Bid (t-Bid)
through direct interaction with t-Bid (24,25). In addition to facilitating
apoptosis, the induction of MTCH2 also causes growth and motility
arrest in vitro and the loss of tumorigenicity in
vivo (26). These data
suggest that MTCH2 may be considered as a novel therapeutic
target.
In this study, we evaluated the anticancer effects
of DHA and analyzed the expression of Cav1 and MTCH2 in a cervical
cancer cell line treated with DHA, in an aim to elucidate the
potential mechanisms involved in the anticancer effects of DHA.
Materials and methods
Cell culture
The HeLa cells were obtained from the American Type
Culture Collection (ATCC; Rockville, MD, USA). All cell lines were
grown in Dulbecco's modified Eagle's medium (DMEM) supplemented
with 10% fetal bovine serum (both from HyClone, Logan, UT, USA).
All cell lines were incubated in a humidified atmosphere containing
5% CO2 at 37°C.
Reagents and antibodies
DHA was obtained from Sigma-Aldrich (St. Louis, MO,
USA). Cav1 (polyclonal, rabbit anti-human; cat. no. 16447-1-AP;
dilution 1:1,000), MTCH2 (polyclonal, rabbit anti-human; cat. no.
16888-1-AP; dilution 1:1,000), β-tubulin (monoclonal, mouse
anti-human; cat. no. 66240-1, dilution 1:2,000), β-actin
(polyclonal, rabbit anti-human; cat. no. 23660-1-AP; dilution
1:1,000), GAPDH (polyclonal, rabbit anti-human; cat. no.
10494-1-AP; dilution 1:1,000) and Myc (monoclonal, mouse
anti-human; cat. no. 60003-2-Ig; dilution 1:1,000) antibodies were
obtained from ProteinTech Group, Inc. (Chicago, IL, USA); p53
antibody (monoclonal, mouse anti-human; cat. no. P8999; dilution
1:1,000) was obtained from Sigma-Aldrich; NAD(P)H:quinone
oxidoreductase 1 (NQO1) antibody (monoclonal, mouse anti-human;
cat. no. #3187, dilution 1:1,000) was obtained from Cell Signaling
Technology, Inc. (Beverly, MA, USA) and p84 antibody (monoclonal,
mouse anti-human; cat. no. MA1-23261, dilution 1:1,000) was
obtained from Invitrogen, Waltham, CA, USA. The secondary antibody
(polyclonal, goat anti-rabbit/goat anti-mouse; cat. no.
042-06-15-06/042-06-18-06; dilution 1:10,000) was obtained from
KPL, Inc. (Gaithersburg, MD, USA).
MTT and cell apoptosis assays
The cells were plated into 96-well plates and seeded
at a density of 10,000 cells/well. The cells were treated with DHA
or the vehicle (DMSO) for the indicated periods of time. The final
volume of culture medium in each well was 100 µl. Ten
microliters of MTT solution (concentration, 5 mg/ml) were added to
the 100 µl of medium in each well. The plates were incubated
at 37°C for 4 h, and the supernatant was then removed and 100
µl DMSO were added to each well. The absorbance signals were
measured on a Thermo Scientific Multiskan FC spectrophotometer
(cat. no. 51119000; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at 490 nm. The cell apoptosis-inducing effects of drug
treatment were measured using the CF488A-Annexin V and PI apoptosis
assay kit (Biotium, Inc., Hayward, CA, USA). Samples and assays
were prepared as described in the user manual and then mounted onto
slides. Images were acquired using a Nikon fluorescence microscope
[Nikon Instruments (Shanghai) Co., Ltd., Shanghai, China].
Western blot analysis
The cells were lysed using 1X SDS sample buffer and
were separated by SDS-PAGE. Proteins were transferred onto PVDF
membranes (Millipore Corp., Bedford, MA, USA). The membranes were
first blocked with 5% milk for 1 h, then incubated overnight with
the indicated antibodies. Following 3 washes with TBST (50 mM
Tris-HCl, pH 7.5, 150 mM NaCl and 0.2% Tween-20), the blots were
incubated with secondary antibodies [anti-rabbit secondary antibody
(polyclonal, goat anti-rabbit; cat. no. 042-06-15-06; dilution
1:10,000) and anti-mouse secondary antibody (polyclonal, goat
anti-mouse; cat. no. 042-06-18-06; dilution 1:10,000), KPL, Inc.]
in the dark for 2 h. The blots were then washed again with TBST and
images were acquired using the LI-COR Odyssey infrared imaging
system (LI-COR Biosciences, Lincoln, NE, USA).
Establishment of stable cell lines
For stable Cav1/MTCH2 expression, the HeLa cells
were transduced with Cav1-Myc or MTCH2-Myc lentiviral particles and
stable cell lines were selected with blasticidin (Invitrogen).
Lentiviral particles were our laboratory-made products using
ViraPower Lentiviral Expression system following the manufacturer's
protocol (Invitrogen, Thermo Fisher Scientific, Inc.).
Establishment of Cav1-knockout cell
line
A Cav1-knockout cell line was generated using the
CRISPER/Cas9 system from Sigma-Aldrich. The HeLa cells were first
transfected with the expression vectors of Cas9 and two gRNAs,
which targeted the AGCCACGGGCCAGCATGTC and TCGCTCAGCTCGTCTGCCA
sequences in exon 1 of Cav1. Twenty-four hours following
transfection, the cells were diluted and seeded in 96-well plates
at 1 cell/well, and monoclonal cell lines without Cav1 expression
were isolated as determined by western blot analysis.
Plasmid construction and cell
transfection
The full-length Cav1 open reading frame (ORF) was
amplified by PCR with a BamHI site-containing 5′ primer:
5′-gatcggatccgccaccatgtctgggggcaaatacgtag-3′, and an XbaI
site-containing 3′ primer: 3′-gtagttgaacgtctttctttatagatctctag-5′,
and cloned into the pcDNA3.1(+)-Myc vector (Invitrogen) to generate
the Cav1-Myc expression construct. Full-length MTCH2 ORF was
amplified by PCR with a HindIII site-containing 5′ primer:
5′-gatcaagctt-gccaccatggcggacgcggccagtcag-3′ and an XbaI
site-containing 3′ primer: 3′-gatctctagaaattaacattttcaggtcac-5′,
and cloned into the pcDNA3.1(+)-Myc vector to generate the
MTCH2-Myc expression construct. For lentiviral particle generation,
the cDNA fragment was transferred into the pLenti6 lentiviral
vector, and the plasmid was then transfected with ViraPower
Lentiviral Packaging mix into 293FT cells (cat. no. R70007;
obtained from Thermo Fisher Scientific, Inc.) using Lipofectamine
2000 (all from Invitrogen). The virus-containing cell culture
medium was harvested 40 h later and used to transduce the HeLa
cells.
Colony formation assay
The HeLa cells were transfected with the Cav1-Myc or
MTCH2 expression vector or empty vector [pcDNA3.1(+)-Myc vector
(Invitrogen)] using Lipofec tamine 2000 (Invitrogen). At 48 h
following transfection, the cells were selected with G418. The
medium with G418 were changed every 3 days. Two weeks later, the
cells were fixed and stained with crystal violet (cat. no.
548-62-9; obtained from Solarbio, Beijing, China).
Crude nuclear fraction isolation
The HeLa cells cultured in a 10-cm plate were
treated with DHA at the indicated concentration for 36 h, and the
cells were then lysed with 0.5% NP-40 lysis buffer (0.5% NP-40, 150
mM NaCl, 10 mM sodium phosphate and pH 7.4) for 20 min on ice, and
cell lysates were collected in an EP tube and followed by
centrifugation at >14,000 × g for 10 min at 4°C. The supernatant
were collected as the cytoplasmic fraction, and the pellet were
re-lysed in 1X SDS sample buffer and collected as the crude nuclear
fraction.
Statistical analysis
Experimental data are expressed as the the means ±
SEM. Data analysis was performed using ImageJ software and Origin
8.0 software. A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
DHA inhibits the viability and induces
the apoptosis of HeLa cells
HeLa cells were used to investigate the anticancer
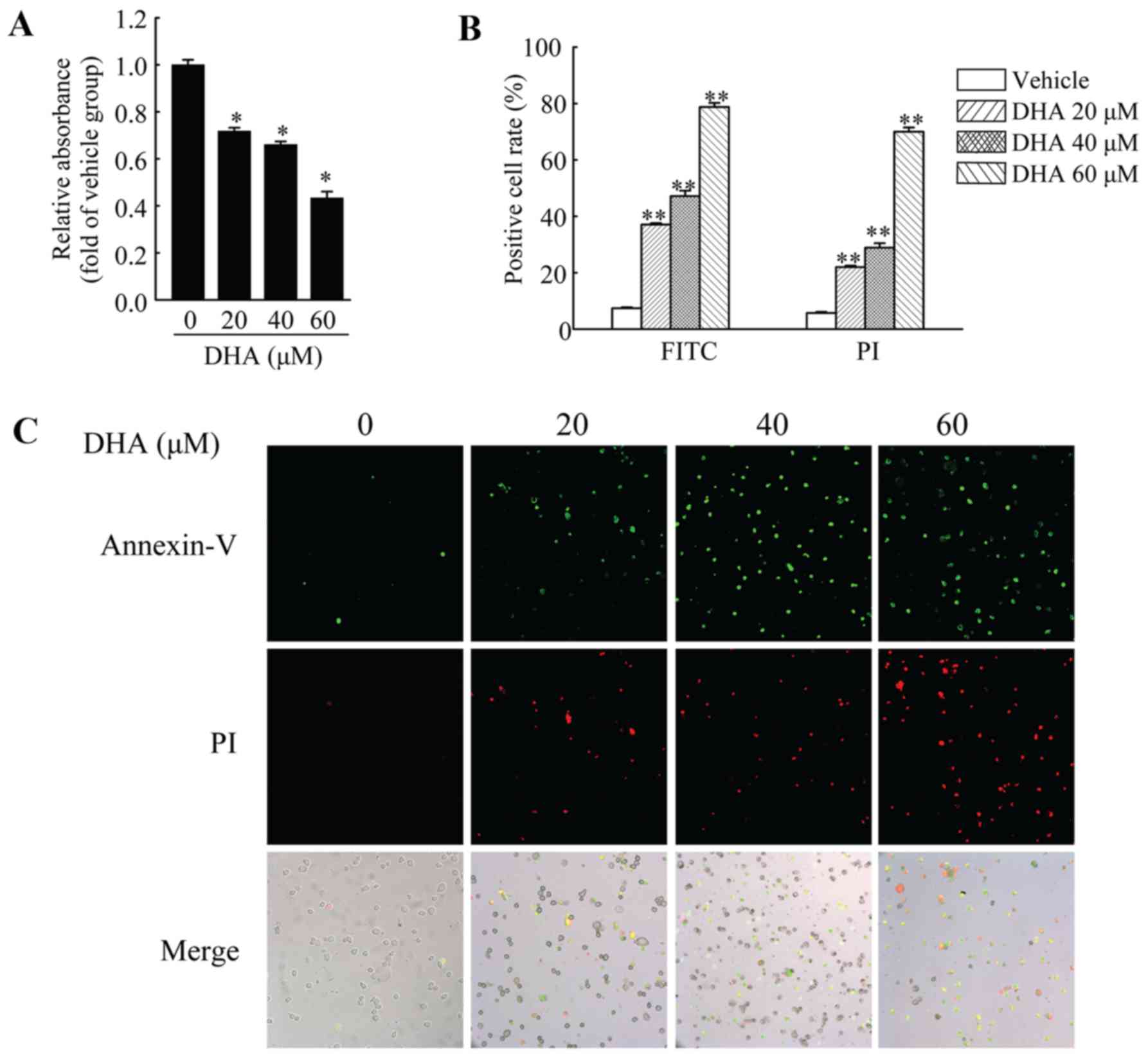
effects of DHA in cervical carcinoma. First, we performed MTT assay
to measure the viability of the HeLa cells. The cells were treated
with DHA at the indicated concentrations for 36 h, and cell
viability was then measured. The results revealed that DHA
significantly inhibited the viability of the HeLa cells, and the
inhibitory effects were dose-dependent (Fig. 1A). In addition, DHA also induced
the apoptosis of the HeLa cells. Annexin V-FITC/PI staining assay
was used to assess the apoptotic HeLa cells following treatment
with DHA at the indicated concentrations. The results revealed that
the number of Annexin V-FITC-positive and PI-positive cells was
significantly increased in a dose-dependent manner following
treatment with DHA (Fig. 1B and
C).
DHA induces the increased expression of
endogenous Cav1 and MTCH2
The exact mechanisms involved in the anticancer
effects of DHA are not yet fully understood. Thus, in this study,
we examined the expression of various cancer-related proteins, in
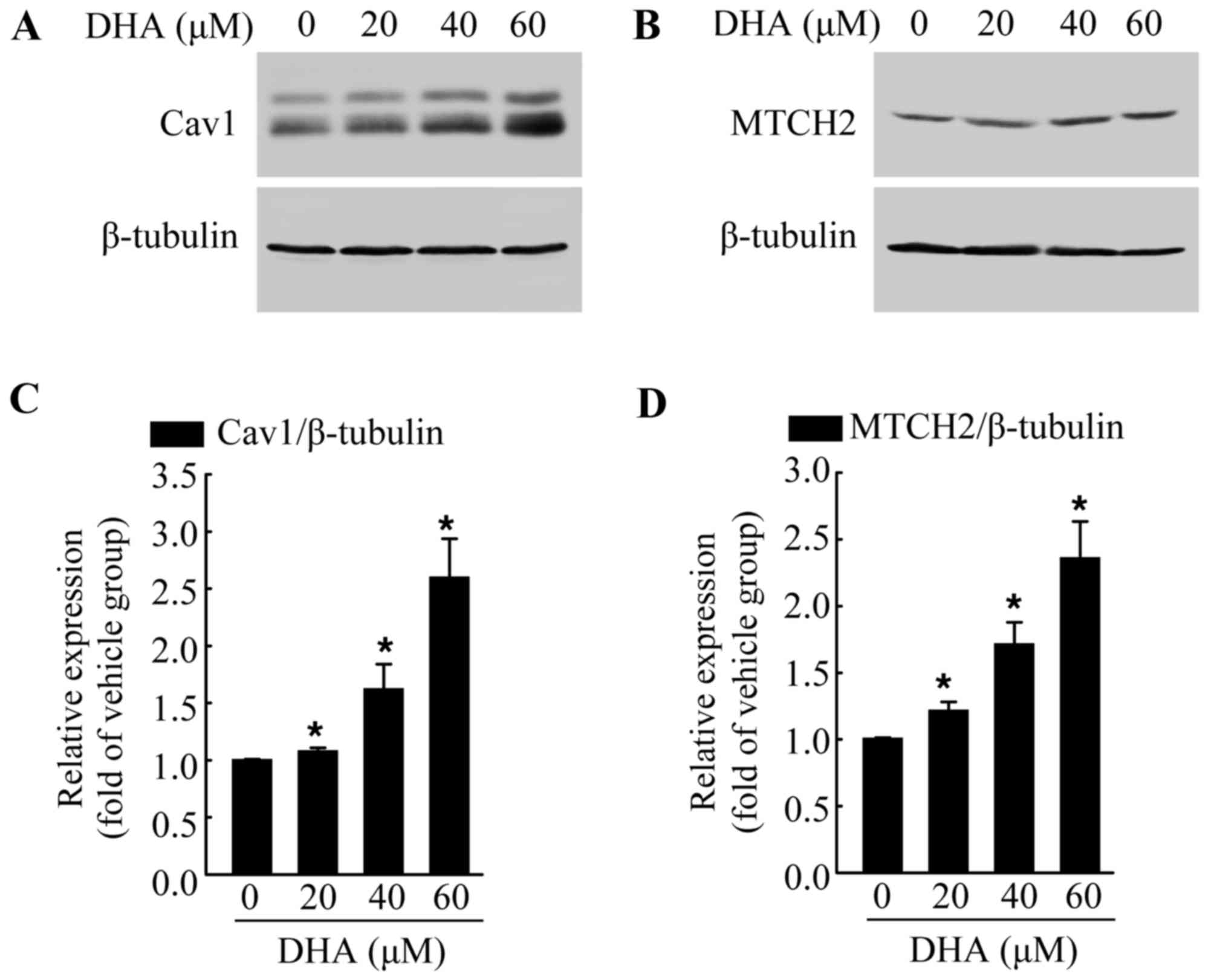
an aim to identify the potential target of DHA. The cells were
exposed to DHA at the indicated concentrations for 36 h, and then
subjected to western blot analysis. We found that DHA significantly
increased the protein levels of Cav1 (Fig. 2A and C) and MTCH2 (Fig. 2B and D) in a dose-dependent manner
when compared with the untreated control group. Cav1 and MTCH2 have
been reported to act as tumor suppressors (27–29). We also detected the increased
expression of Cav1 and MTCH2 following treatment with DHA; thus it
is possible that Cav1 and MTCH2 may mediate the inhibitory effects
of DHA on HeLa cells.
Overexpression of Cav1 and MTCH2 inhibits
colony formation of HeLa cells
To further examine the functional involvement of
Cav1 and MTCH2 in the inhibitory effects of DHA on HeLa cell cell
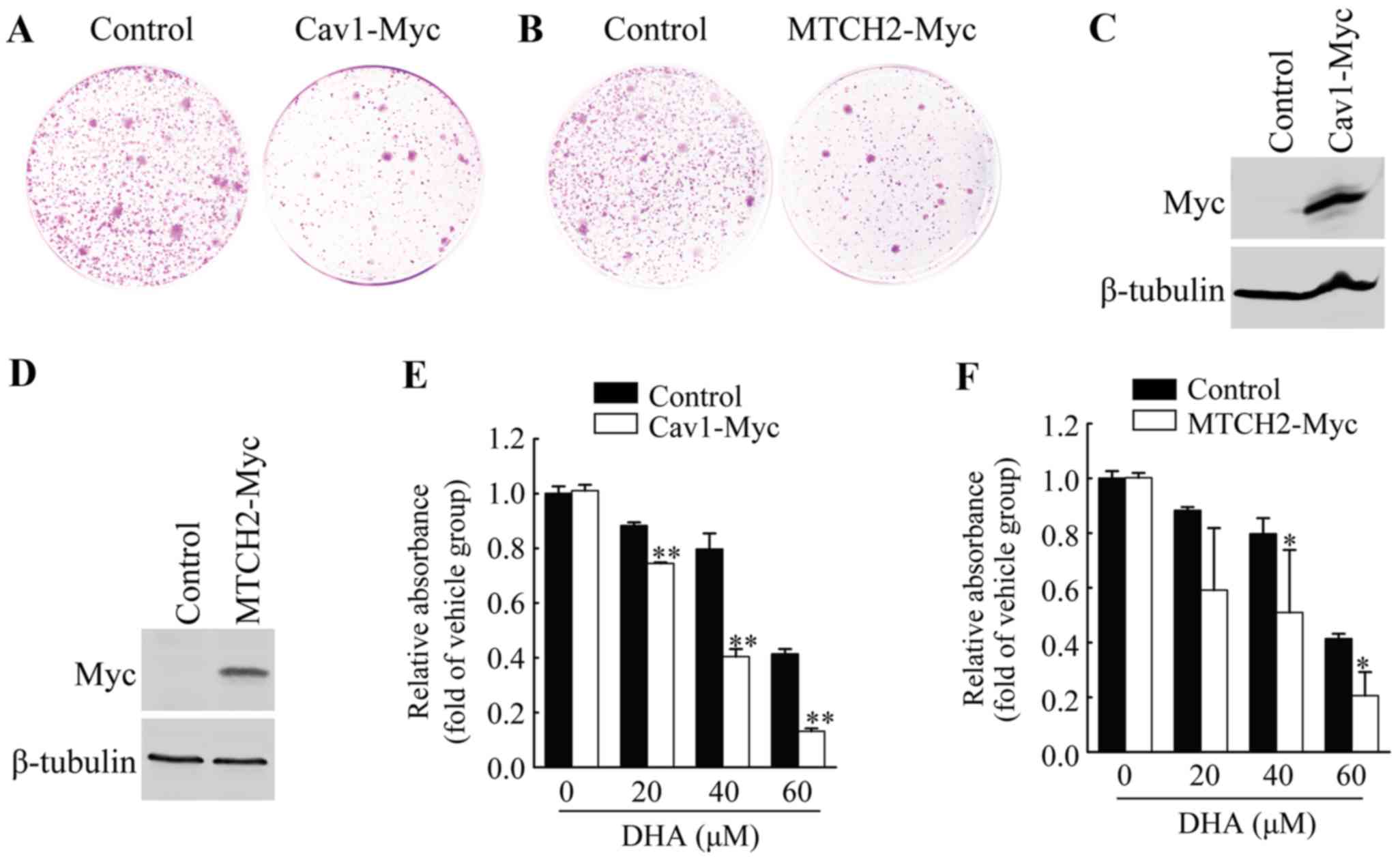
viability, we first performed colony formation assays by
introducing the Cav1-Myc expression vector or MTCH2-Myc expression
vector or control vector into the HeLa cells, and measuring the
growth of G418-resistant colonies. Cav1 overexpression resulted in
the significant inhibition of colony formation (Fig. 3A), and similar results were
observed in the cells overexpressing MTCH2 (Fig. 3B).
Overexpression of Cav1 and MTCH2 enhances
the sensitivity of HeLa cells to DHA
We established HeLa cell lines stably expressing
Cav1 or MTCH2 (Fig. 3C and D).
HeLa cells stably transduced with Cav1-Myc virus (HeLa/Cav1-Myc)
and HeLa cells stably transduced with the empty viral vector
(HeLa/Control) were treated with the vehicle or DHA at the
indicated concentrations for 36 h, and cell viability was then
measured by MTT assay. We found that the stable expression of Cav1
enhanced the inhibitory effects of DHA on cell viability (Fig. 3E). The stable expression of MTCH2
also significantly enhanced the inhibitory effects of DHA on cell
viability (Fig. 3F). These
results suggested that the upregulation of Cav1 and MTCH2 was
responsible for the enhanced inhibitory effects of DHA on cell
viability.
Cav1 regulates p53 activation in HeLa
cells
p53 has been reported to facilitate apoptosis caused
by DHA (5,8–10).
Previous studies have also demonstrated a direct association
between Cav1 and p53 in regulating stress-induced premature
senescence (SIPS) in mouse embryonic fibroblasts (30,31). Thus, in this study, we wished to
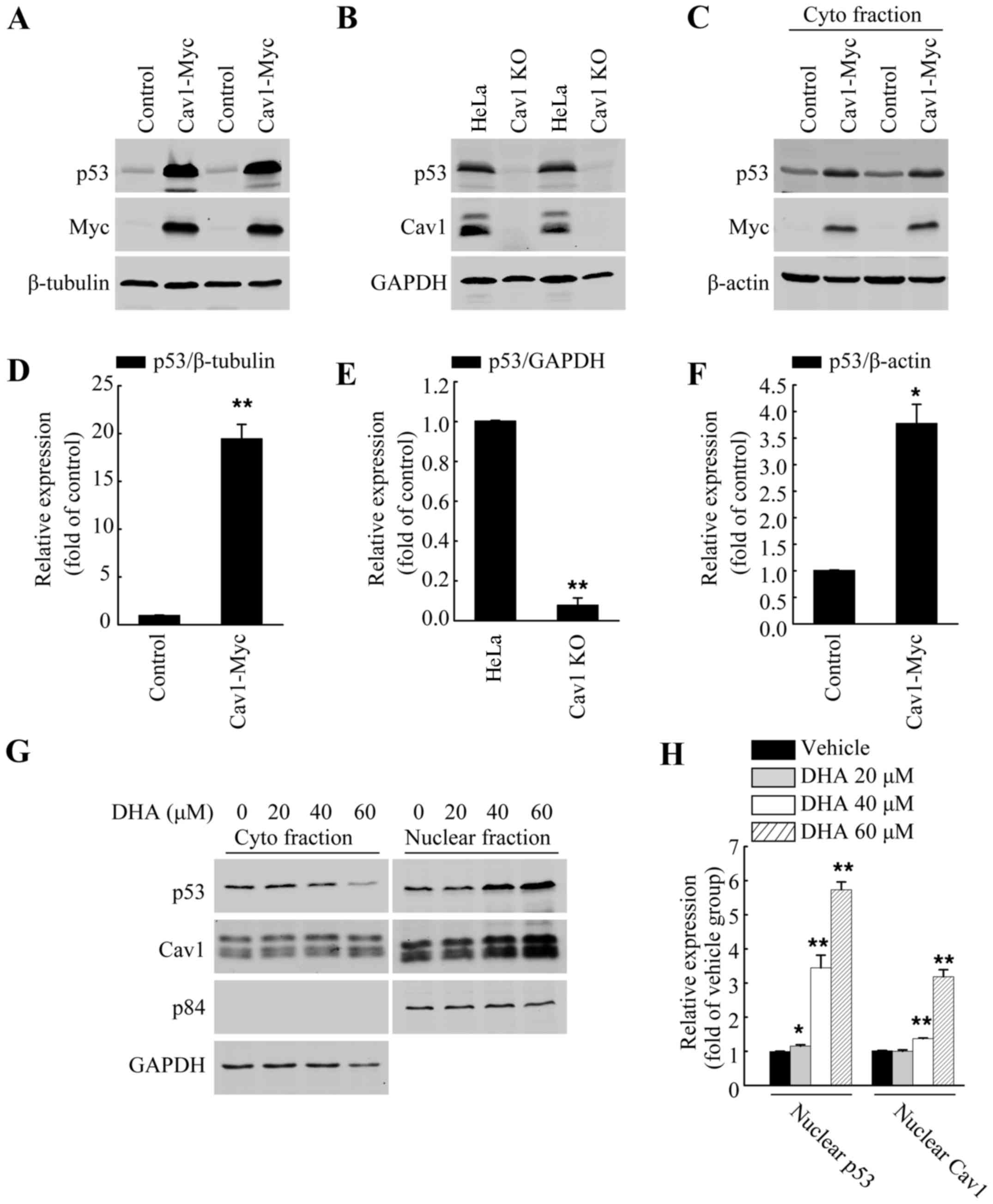
evaluate the role of Cav1 in p53 activation in HeLa cells. Direct
functional analysis in the form of gene knockout and overexpression
analysis were used. After establishing a stable cell line
expressing Cav1 (HeLa/Cav1-Myc) with a stably transduced Cav1-Myc
vector and generated a Cav1-knockout cell line (HeLa/Cav1-KO) using
the CRISPER/cas9 system, we prepared whole cell lysates using SDS
sample buffer, and then performed western blot analysis. Our
results demonstrated that the overexpression of Cav1 in the HeLa
cells was associated with a significant increase (>20-fold) in
the protein level of p53 (Fig. 4A and
D), while the silencing of Cav1 caused a decrease in the
protein level of p53 when compared with the control group (Fig. 4B and E). Of note, when we
harvested the cells with NP40 lysis buffer, we found that the
protein level of p53 in the soluble fraction of HeLa/Cav1-Myc was
significantly increased (Fig. 4C and
F) and the increase in p53 expression was much lower than that
in whole cell lysate fraction. These data suggested that the
majority of the increased p53 protein may exist in the insoluble
nuclear fraction.
Upregulation of Cav1 is associated with
the nuclear localization and stabilization of p53 following
treatment with DHA
Apart from being localized in the plasma membrane,
Cav1 has also been reported to be localized in the nucleus
(23), and the increased nuclear
localization of Cav1 has also observed under
H2O2-induced oxidative stress (32). In this study, to further elucidate
the potential association between Cav1 upregulation and p53
stabilization following treatment with DHA, the HeLa cells were
cultured in a 10-cm plate and treated with DHA at the indicated
concentrations for 36 h, and the crude nuclear fractions and
cytosolic fraction were then collected and subjected to western
blot analysis. We found DHA treatment increased the nuclear
localization of both Cav1 and p53 (Fig. 4G and H). Since we aldready
demonstrated that Cav1 expression was increased following treatment
with DHA (Fig. 2A and C), and
that it directly regulates the stabilization of p53 in HeLa cells
(Fig. 4A–F), these data indicated
that the upregulation and nuclear localization of Cav1 may
contribute to the activation of p53 in HeLa cells treated with
DHA.
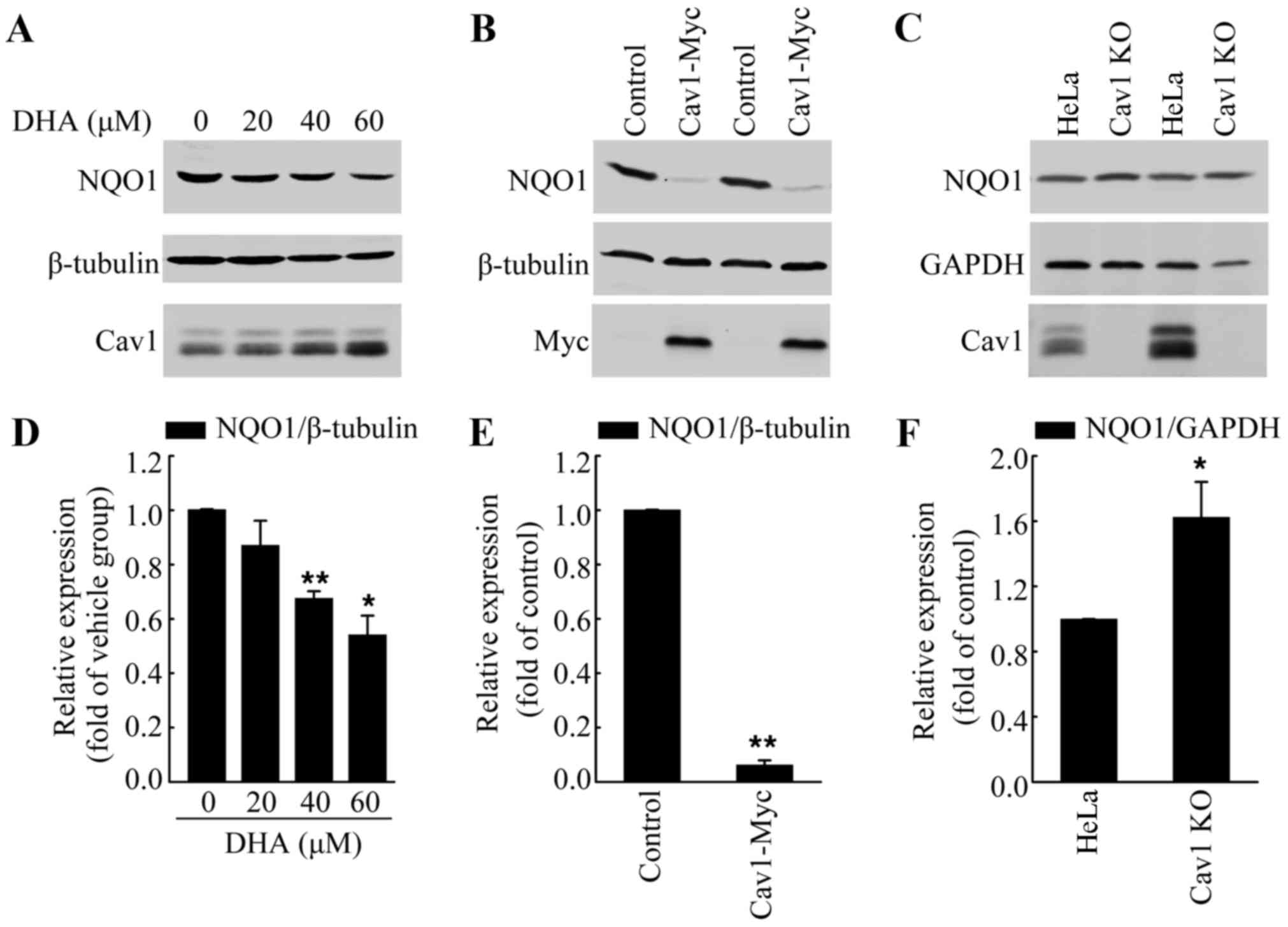
Upregulation of cav1 contributes to the
decreased expression of NQO1 following treatment with DHA
Increased ROS formation has been observed in
response to DHA treatment, which was the main course of DHA-induced
apoptosis (33,34). Notably, Cav1 has been reported to
regulate cellular antioxidant capacity by inhibiting the
transcriptional activity of Nrf2 (22,23). Thus, in this study, we analyzed
the protein level of NQO1, a direct target of Nrf2, in HeLa cells
treated with DHA to determine whether Cav1 is involved in
DHA-induced oxidative stress. We treated the HeLa cells with DHA at
the indicated concentrations for 36 h, and then performed western
blot analysis. Consist with our hypothesis, we found that DHA
treatment led to the downregulation of NQO1, which was associated
with the upregulation of Cav1 (Fig.
5A and D). Moreover, the NQO1 protein level in the
HeLa/Cav1-Myc cells was significantly lower than that in the
HeLa/Control cells (Fig. 5B and
E), while depletion of Cav1 resulted in an increase of NQO1
protein level (Fig. 5C and F).
These results suggested that the increased Cav1 expression was the
possible upstream regulator of NQO1.
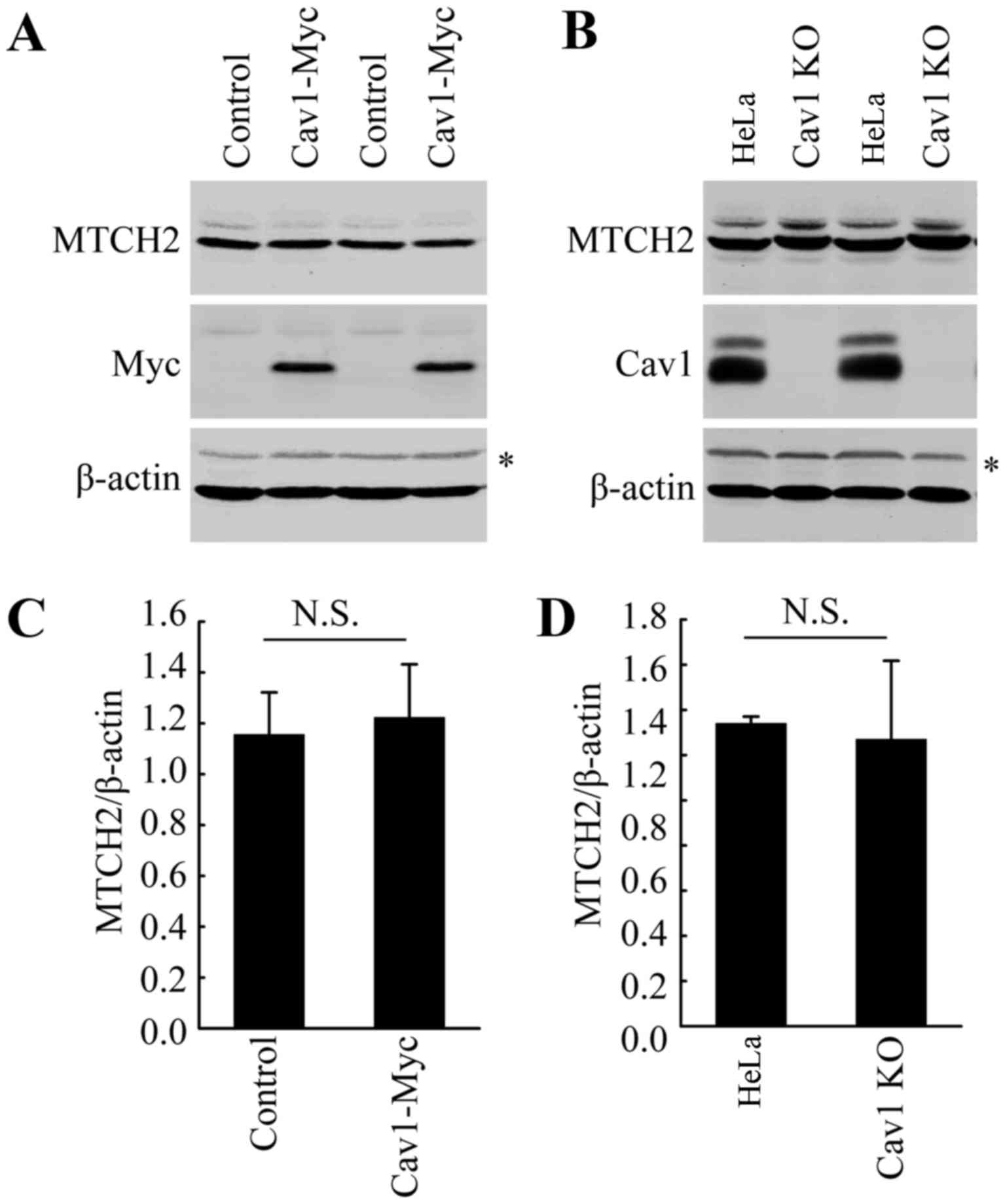
Cav1 does not directly influence the
expression of MTCH2
As indicated above, DHA treatment increased the
expression of both Cav1 and MTCH2 (Fig. 2). Thus, we wished to examine the
potential association between Cav1 and MTCH2. We already
demonstrated the nuclear translocation of Cav1 in the HeLa cells
treated with DHA (Fig. 4). Thus,
we hypothesized that Cav1 may regulate the expression of MTCH2 via
the activation of MTCH2. However, direct functional analysis, such
as gene knockout and overexpression analysis did not support our
hypothesis. Neither Cav1 overexpression nor Cav1 depletion affected
the expression of MTCH2 (Fig.
6A–D). These data suggested that the induction of Cav1 and
MTCH2 in HeLa cells treated with DHA may not be involved in the
same pathway and further studies are required to elucidate the
mechanisms involved in the upregulation of MTCH2 induced by
DHA.
Discussion
Previous studies have revealed that the
anti-proliferative effects of DHA are based on the
mitochondrial-related activation of cell death pathway induced by
high levels of ROS (33,34), However, the detailed associatoin
between the upstream signaling and downstream activation of cell
death pathway following treatment with DHA remains unclear. Cav1, a
reported cancer suppressor, is an important component of caveolae
and functions as a scaffolding protein in regulating several
signaling pathways (11–13). Recently, Cav1 was also reported to
function as a modulator of oxidative stress by inhibiting the
expression of Nrf2 (22,23). These data suggest a possible role
of Cav1 in mediating elevated ROS generation, which inhibits cell
viability in DHA-treated cells. However, the exact role of Cav1 in
this progression remains unclear. Of note, we found that DHA
treatment increased the expression of Cav1 in HeLa cells and the
overexpression of Cav1 enhanced the sensitivity to DHA. These data
suggested a potential link between the activation of the cell death
pathway triggered by the upregulation of Cav1 and the inhibition of
the Nrf2-mediated signaling pathway following treatment with
DHA.
The Nrf2-mediated signaling pathway plays a critical
role in protecting cells from oxidative stress induced cell death
by transcriptionally activating antioxidant genes, including NQO1.
NQO1 expression is increased in response to oxidative stress and an
elevated NQO1 expression is essential for reducing ROS levels and
maintaining the function of the mitochondria under oxidative stress
(35). However, to the best of
our knowledge, the involvement of NQO1 in the generation of high
levels of ROS induced by DHA treatment has not been reported to
date. Notably, the current study indicated that a potential
association may exist between NQO1 and the DHA-induced inhibition
of cell viability. DHA has been reported to induce high levels of
ROS (33,34), which indicates the inhibition of
the ability to remove free radicals following treatment with DHA.
In other words, the elevated generation of ROS may indicate
decreased antioxidant gene expression. In support of this
assumption, we found that DHA treatment inhibited the expression of
NQO1 which was associated with decreased cell viability. These data
suggested that the downregulation of NQO1 following treatment with
DHA may contribute to the elevated ROS generation and to the
eventual inhibition of cell viability. Moreover, we also
demonstrated that Cav1 may be responsible for the decreased
expression of NQO1 following treatment wih DHA. The decreased
expression of NQO1 was associated with the elevated expression of
Cav1 following treatment with DHA. Functional analysis, namely Cav1
overexpression and Cav1 depletion also indicated that Cav1
regulated the protein level of NQO1 in HeLa cells. Since previous
researchers have demonstrated the direct association between Cav1
upregulation and the inhibition of Nrf2 activation (22,23), our results suggested that Cav1 may
play an important role in triggering the activation of the cell
death pathway by inhibiting the Nrf2-mediated signaling pathway
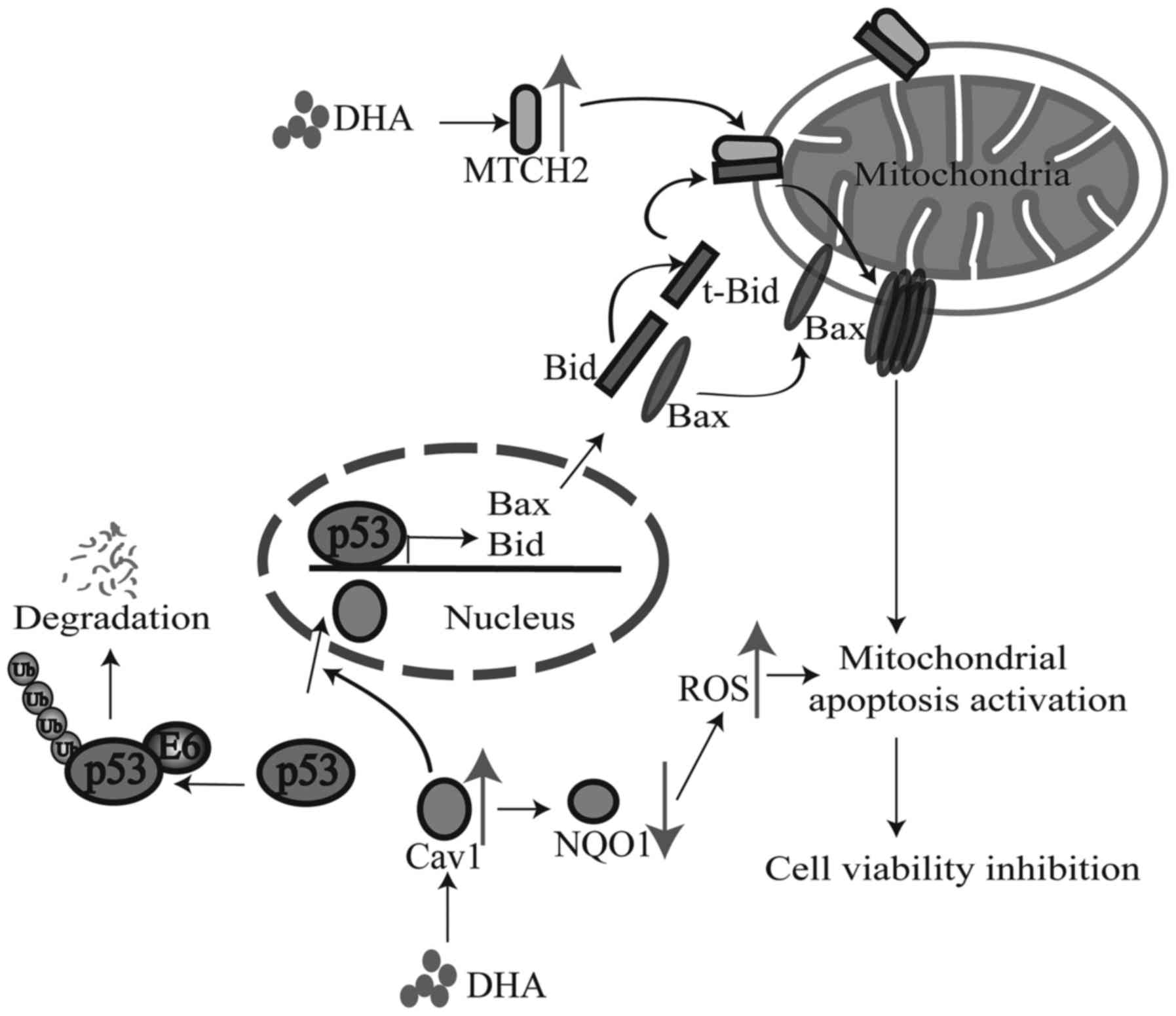
following treatment with DHA (Fig.
7).
DHA has been reported to induce the activation of
the cell death pathway, including Bid activation and mitochondria
translocation (7,21). The tumor suppressor, p53, plays an
important role in mediating the cell death pathway. It has also
been reported that p53 regulates the expression of Bid (36) and facilitates the apoptosis caused
by DHA (5,8–10).
However, the exact role of p53 in the DHA-mediated inhibition of
cell viability remains unclear. Previous studies have shown a
direct association between Cav1 and p53 in regulating SIPS in mouse
embryonic fibroblasts (30,31). Thus, it is possible that Cav1 may
also regulate p53 activation following treatment with DHA. In line
with our hypothesis, we found that although the basal level of p53
in HeLa cells was low, the overexpression of Cav1 significantly
increased the protein level of p53, whereas the depletion of Cav1
induced the downregulation of p53 in HeLa cells. Of note, previous
studies have indicated that Cav1 regulates the stabilization of p53
by inhibiting the interaction of p53 and its ubiquitin ligase,
mouse double minute 2 homolog (Mdm2); however, in HeLa cells, the
proteasome-dependent degradation of p53 is mainly promoted by HPV
E6 protein and inhibiting the interaction of p53 and Mdm2 cannot
lead to the stabilization of p53 (37,38). These data suggested that Cav1 may
regulate the stabilization of p53 in a different manner, rather
than disassociating p53 from Mdm2 in HeLa cells. The mechanisms
through which Cav1 induces the stabilization of p53 in HeLa cells
are not yet clear. In the current study, we found that DHA
treatment induced the accumulated nuclear localization of both Cav1
and p53. It has been reported that inhibiting the nuclear export of
p53 with small molecule nuclear export inhibitors, such as
leptomycin B and actinomycin D leads to the activation of p53 in
HeLa cells (37,38). Combined with our findings, these
data suggest that Cav1 may facilitate the nuclear translocation or
inhibit the nuclear export of p53 to regulate the stability of p53
(Fig. 7); further studies are
required to elucidate the complete mechanisms involved.
Recent studies have indicated MTCH2 as a potential
cancer suppressor (26) and
apoptosis facilitator via its interaction with t-Bid (24,25). We also found that the protein
level of MTCH2 was increased in HeLa cells treated with DHA,
indicating that MTCH2 may also contribute to the DHA-induced
inhibition of cell viability in cancer cells. MTCH2 facilitates the
mitochondrial translocation of t-Bid (24,25). We also demonstrated the nuclear
translocation of p53 following treatment with DHA; thus, we assumed
that DHA may induce the nuclear translocation and activation of p53
by upregulating Cav1. The activation of p53 then leads to
upregulation of Bid and MTCH2 facilitates the translocation of
cleaved Bid to the mitochondria, and eventually leads to the
activation of the mitochondrial cell death pathway (Fig. 7). Despite the positive correlation
between Cav1 and MTCH2 expression following treatment with DHA, it
is worth noting that Cav1 cannot directly regulate the expression
of MTCH2, as demonstrated by the functional overexpression of Cav1
and the depletion of Cav1. Further studies are required in order to
elucidate the detailed mechanisms involved in the upregulation of
MTCH2 in cells treated with DHA.
In conclusion, according to our findings, we made a
hypothesis that Cav1 and MTCH2 may function coordinately as
upstream signaling sensors and the upregulation of Cav1 and MTCH2
enhances the inhibitory effects of DHA on cell viability by
inducing the nuclear activation of p53, the down-regulation of NQO1
and the mitochondrial translocation of tBid [although we did not
examine the exact role of Bid in this study, we hypothesize that it
may play a role according to previous studies (24,25) and ours], which eventually leads to
the activation of the downstream cell death pathway (Fig. 7). Our study not only elucidated
the potential mechanisms responsible for the anticancer effects of
DHA, but also provide promising targets for cancer therapy.
Acknowledgments
The present study was supported by research grants
from the Natural Science Foundation of Chengdu University (no.
2011XJZ14) and the Natural Science Foundation of China (no.
51402027).
References
|
1
|
Cao L, Duanmu W, Yin Y, Zhou Z, Ge H, Chen
T, Tan L, Yu A, Hu R, Fei L, et al: Dihydroartemisinin exhibits
anti-glioma stem cell activity through inhibiting p-AKT and
activating caspase-3. Pharmazie. 69:752–758. 2014.
|
|
2
|
Lucibello M, Adanti S, Antelmi E, Dezi D,
Ciafrè S, Carcangiu ML, Zonfrillo M, Nicotera G, Sica L, De Braud
F, et al: Phospho-TCTP as a therapeutic target of
Dihydroartemisinin for aggressive breast cancer cells. Oncotarget.
6:5275–5291. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu M, Sun L, Zhou J, Zhao Y and Deng X:
Dihydroartemisinin-induced apoptosis is associated with inhibition
of sarco/endoplasmic reticulum calcium atpase activity in
colorectal cancer. Cell Biochem Biophys. 73:137–145. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liao K, Li J and Wang Z:
Dihydroartemisinin inhibits cell proliferation via
AKT/GSK3β/cyclinD1 pathway and induces apoptosis in A549 lung
cancer cells. Int J Clin Exp Pathol. 7:8684–8691. 2014.
|
|
5
|
Zhang CZ, Zhang H, Yun J, Chen GG and Lai
PBS: Dihydroartemisinin exhibits antitumor activity toward
hepatocellular carcinoma in vitro and in vivo. Biochem Pharmacol.
83:1278–1289. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhao X, Zhong H, Wang R, Liu D, Waxman S,
Zhao L and Jing Y: Dihydroartemisinin and its derivative induce
apoptosis in acute myeloid leukemia through Noxa-mediated pathway
requiring iron and endoperoxide moiety. Oncotarget. 6:5582–5596.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu YY, Chen TS, Wang XP and Li L:
Single-cell analysis of dihydroartemisinin-induced apoptosis
through reactive oxygen species-mediated caspase-8 activation and
mitochondrial pathway in ASTC-a-1 cells using fluorescence imaging
techniques. J Biomed Opt. 15:0460282010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cabello CM, Lamore SD, Bair WB III, Qiao
S, Azimian S, Lesson JL and Wondrak GT: The redox antimalarial
dihydroartemisinin targets human metastatic melanoma cells but not
primary melanocytes with induction of NOXA-dependent apoptosis.
Invest New Drugs. 30:1289–1301. 2012. View Article : Google Scholar
|
|
9
|
Ji Y, Zhang YC, Pei LB, Shi LL, Yan JL and
Ma XH: Anti-tumor effects of dihydroartemisinin on human
osteosarcoma. Mol Cell Biochem. 351:99–108. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang CZ, Pan Y, Cao Y, Lai PB, Liu L,
Chen GG and Yun J: Histone deacetylase inhibitors facilitate
dihydroartemisinin-induced apoptosis in liver cancer in vitro and
in vivo. PLoS One. 7:e398702012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Simmons GE Jr, Taylor HE and Hildreth JE:
Caveolin-1 suppresses human immunodeficiency virus-1 replication by
inhibiting acetylation of NF-κB. Virology. 432:110–119. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garrean S, Gao XP, Brovkovych V, Shimizu
J, Zhao YY, Vogel SM and Malik AB: Caveolin-1 regulates NF-kappaB
activation and lung inflammatory response to sepsis induced by
lipopolysaccharide. J Immunol. 177:4853–4860. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang XM, Kim HP, Song R and Choi AM:
Caveolin-1 confers anti-inflammatory effects in murine macrophages
via the MKK3/p38 MAPK pathway. Am J Respir Cell Mol Biol.
34:434–442. 2006. View Article : Google Scholar
|
|
14
|
Huertas-Martínez J, Rello-Varona S,
Herrero-Martín D, Barrau I, García-Monclús S, Sáinz-Jaspeado M,
Lagares-Tena L, Núñez-Álvarez Y, Mateo-Lozano S, Mora J, et al:
Caveolin-1 is down-regulated in alveolar rhabdomyosarcomas and
negatively regulates tumor growth. Oncotarget. 5:9744–9755. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bender FC, Reymond MA, Bron C and Quest
AF: Caveolin-1 levels are down-regulated in human colon tumors, and
ectopic expression of caveolin-1 in colon carcinoma cell lines
reduces cell tumorigenicity. Cancer Res. 60:5870–5878.
2000.PubMed/NCBI
|
|
16
|
Bélanger MM, Roussel E and Couet J:
Caveolin-1 is down-regulated in human lung carcinoma and acts as a
candidate tumor suppressor gene. Chest. 125(Suppl): 106S2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang X, Pan L, Pu H, Wang Y, Zhang X, Li
C and Yang Z: Loss of caveolin-1 promotes endothelial-mesenchymal
transition during sepsis: a membrane proteomic study. Int J Mol
Med. 32:585–592. 2013.PubMed/NCBI
|
|
18
|
Wang R, He W, Li Z, Chang W, Xin Y and
Huang T: Caveolin-1 functions as a key regulator of
17β-estradiol-mediated autophagy and apoptosis in BT474 breast
cancer cells. Int J Mol Med. 34:822–827. 2014.PubMed/NCBI
|
|
19
|
Trimmer C, Sotgia F, Whitaker-Menezes D,
Balliet RM, Eaton G, Martinez-Outschoorn UE, Pavlides S, Howell A,
Iozzo RV, Pestell RG, et al: Caveolin-1 and mitochondrial SOD2
(MnSOD) function as tumor suppressors in the stromal
microenvironment: a new genetically tractable model for human
cancer associated fibroblasts. Cancer Biol Ther. 11:383–394. 2011.
View Article : Google Scholar :
|
|
20
|
Benhar M, Engelberg D and Levitzki A: ROS,
stress-activated kinases and stress signaling in cancer. EMBO Rep.
3:420–425. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mao H, Gu H, Qu X, Sun J, Song B, Gao W,
Liu J and Shao Q: Involvement of the mitochondrial pathway and
Bim/Bcl-2 balance in dihydroartemisinin-induced apoptosis in human
breast cancer in vitro. Int J Mol Med. 31:213–218. 2013.
|
|
22
|
Volonte D, Liu Z, Musille PM, Stoppani E,
Wakabayashi N, Di YP, Lisanti MP, Kensler TW and Galbiati F:
Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by
caveolin-1 promotes stress-induced premature senescence. Mol Biol
Cell. 24:1852–1862. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li W, Liu H, Zhou JS, Cao JF, Zhou XB,
Choi AM, Chen ZH and Shen HH: Caveolin-1 inhibits expression of
antioxidant enzymes through direct interaction with nuclear
erythroid 2 p45-related factor-2 (Nrf2). J Biol Chem.
287:20922–20930. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Katz C, Zaltsman-Amir Y, Mostizky Y,
Kollet N, Gross A and Friedler A: Molecular basis of the
interaction between proapoptotic truncated BID (tBID) protein and
mitochondrial carrier homologue 2 (MTCH2) protein: key players in
mitochondrial death pathway. J Biol Chem. 287:15016–15023. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zaltsman Y, Shachnai L, Yivgi-Ohana N,
Schwarz M, Maryanovich M, Houtkooper RH, Vaz FM, De Leonardis F,
Fiermonte G, Palmieri F, et al: MTCH2/MIMP is a major facilitator
of tBID recruitment to mitochondria. Nat Cell Biol. 12:553–562.
2010. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Leibowitz-Amit R, Tsarfaty G, Abargil Y,
Yerushalmi GM, Horev J and Tsarfaty I: Mimp, a mitochondrial
carrier homologue, inhibits Met-HGF/SF-induced scattering and
tumorigenicity by altering Met-HGF/SF signaling pathways. Cancer
Res. 66:8687–8697. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yu K, Ganesan K, Tan LK, Laban M, Wu J,
Zhao XD, Li H, Leung CH, Zhu Y, Wei CL, et al: A precisely
regulated gene expression cassette potently modulates metastasis
and survival in multiple solid cancers. PLoS Genet. 4:e10001292008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arigoni M, Barutello G, Riccardo F, Ercole
E, Cantarella D, Orso F, Conti L, Lanzardo S, Taverna D, Merighi I,
et al: miR-135b coordinates progression of ErbB2-driven mammary
carcinomas through suppression of MID1 and MTCH2. Am J Pathol.
182:2058–2070. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han F, Gu D, Chen Q and Zhu H: Caveolin-1
acts as a tumor suppressor by down-regulating epidermal growth
factor receptor-mitogen-activated protein kinase signaling pathway
in pancreatic carcinoma cell lines. Pancreas. 38:766–774. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bartholomew JN, Volonte D and Galbiati F:
Caveolin-1 regulates the antagonistic pleiotropic properties of
cellular senescence through a novel Mdm2/p53-mediated pathway.
Cancer Res. 69:2878–2886. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Volonte D, Zou H, Bartholomew JN, Liu Z,
Morel PA and Galbiati F: Oxidative stress-induced inhibition of
Sirt1 by caveolin-1 promotes p53-dependent premature senescence and
stimulates the secretion of interleukin 6 (IL-6). J Biol Chem.
290:4202–4214. 2015. View Article : Google Scholar :
|
|
32
|
Chrétien A, Piront N, Delaive E, Demazy C,
Ninane N and Toussaint O: Increased abundance of cytoplasmic and
nuclear caveolin 1 in human diploid fibroblasts in H(2)O(2)-induced
premature senescence and interplay with p38alpha(MAPK). FEBS Lett.
582:1685–1692. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hosoya K, Murahari S, Laio A, London CA,
Couto CG and Kisseberth WC: Biological activity of
dihydroartemisinin in canine osteosarcoma cell lines. Am J Vet Res.
69:519–526. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Z, Hu W, Zhang JL, Wu XH and Zhou HJ:
Dihydroartemisinin induces autophagy and inhibits the growth of
iron-loaded human myeloid leukemia K562 cells via ROS toxicity.
FEBS Open Bio. 2:103–112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kim J, Kim SK, Kim HK, Mattson MP and Hyun
DH: Mitochondrial function in human neuroblastoma cells is
up-regulated and protected by NQO1, a plasma membrane redox enzyme.
PLoS One. 8:e690302013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sax JK, Fei P, Murphy ME, Bernhard E,
Korsmeyer SJ and El-Deiry WS: BID regulation by p53 contributes to
chemosensitivity. Nat Cell Biol. 4:842–849. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Koivusalo R, Mialon A, Pitkänen H,
Westermarck J and Hietanen S: Activation of p53 in cervical cancer
cells by human papillomavirus E6 RNA interference is transient, but
can be sustained by inhibiting endogenous nuclear export-dependent
p53 antagonists. Cancer Res. 66:11817–11824. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hietanen S, Lain S, Krausz E, Blattner C
and Lane DP: Activation of p53 in cervical carcinoma cells by small
molecules. Proc Natl Acad Sci USA. 97:8501–8506. 2000. View Article : Google Scholar : PubMed/NCBI
|