Introduction
The normal function of the endoplasmic reticulum
(ER) is essential for numerous cellular processes and, ultimately,
cell survival. Conditions that inhibit the protein folding capacity
of the ER lead to the accumulation of misfolded or unfolded
proteins in the ER lumen, generating a potentially toxic state
referred to as ER stress. ER stress is attenuated through the
activation of a complex adaptive cellular response, known as the
unfolded protein response (UPR). Three transmembrane proteins,
inositol-requiring enzyme 1 (IRE1), protein kinase R (PKR)-like ER
kinase (PERK) and activating transcription factor (ATF)6, are
responsible for detecting ER stress and the initiation of the UPR.
Prolonged stress or failure to adapt to ER stress ultimately
culminates in ER stress-induced apoptosis, and ER stress has been
associated with various neurodegenerative, cardiovascular and
orthopedic diseases (1–4). Additionally, ER stress has been
demonstrated to play an important role in cellular differentiation
during developmental processes. Accordingly, characterizing
molecular mediators of the signaling switch between the protective
and apoptotic responses to ER stress, and how these molecules
interact to influence cell differentiation and proliferation, is an
important endeavor which may reveal key aspects of developmental
regulation and cellular pathologies.
Several studies have clearly demonstrated that
physiological stressors influence cell differentiation and survival
during musculoskeletal developmental and reparative processes,
including chondrocyte differentiation, chondrogenesis and
endrochondral ossification (5–7).
Chondrocyte sensitivity to ER stress has been documented and a
clear association between ER stress and several diseases affecting
connective tissue is readily observable through murine genetic
knockout studies and the analysis of diseased human tissues
(8,9); however, the mechanisms through which
ER stress specifically affects differentiation programs in
chondrocytes remain poorly understood.
Of note, bone morphogenetic protein 2 (BMP2), a
pre-eminent cytokine, plays critical roles in embryogenesis, cell
growth, differentiation, bone development and the repair of bone
fractures. It also activates UPR transducers, such as PERK, old
astrocyte specifically-induced substance (OASIS) and ATF6 (10–12). Notably, PERK is a major transducer
of the ER stress response and directly phosphorylates eukaryotic
initiation factor 2α (eIF2α), which specifically promotes the
translation of ATF4. PERK and ATF4 have been shown to play
important roles in osteoblast differentiation and bone formation.
Specifically, Saito et al. as well as others revealed that
ER stress occured during BMP2-induced osteoblast differentiation
and activated the PERK-eIF2α-ATF4 signaling pathway, followed by
the promotion of gene expression essential for osteogenesis
(13–15). In an effort to disentangle the
dual association of PERK/ATF4 signaling with both pro-survival and
pro-apoptotic responses during ER stress, Walter et al, and
others, investigated the association between cell fate and the
temporal activation of PERK/ATF4 in live cells and found that the
shift from cell survival to apoptosis was determined by the timing
of PERK/ATF4 signaling relative to that of IRE1/XBP1, another UPR
signaling pathway (16,17).
However, whether PERK/ATF4 signaling participates in
ER stress-mediated apoptosis during the course of chondrocyte
differentiation, and the potential underlying mechanism(s), remain
unknown. Thus, the current study aimed to better define the
molecular mediators of cell survival during cartilage development
with special regard to molecules associated with chondrocyte
differentiation and ER stress-induced apoptosis. Specifically, the
data presented herein elucidate the involvement of PERK and ATF4 in
cell cycle progression and ER stress-mediated apoptosis during the
course of chondrogenesis. Furthermore, the combined effect(s) of
PERK and ATF4 upon the regulation of the cell cycle and apoptosis
were investigated.
Materials and methods
Ethics statement
All animal experiments were designed in strict
accordance with the recommendations in the Guide for the Care and
Use of Laboratory Animals of the National Science Foundation of
China and conducted with the prior approval of the Chongqing
Medical University Institutional Animal Care and Use Committee
(permit nos. SYXK 2007-0001 and SCXK 2007-0002) and the Committee
on the Ethics of Animal Experiments of Chongqing Medical
University. Mice were housed under controlled temperatures in a 12
h light/dark cycle with easy access to food and water.
Adenoviruses
To generate PERK and ATF4 small interfering RNA
(siRNA; siPERK and siATF4, respectively) expression constructs,
siRNA corresponding to the coding sequence of the PERK and ATF4
genes (siPERK forward, 5′-ACCTCCAAGACCAACCACTTTTTT-3′ and reverse,
5′-AAAGTGGTTGGTCTTGGAGGTTTT-3′; and siATF4 forward,
5′-AGGAGCAAAACAAGACAGCATTTT-3′ and reverse,
5′-ATGCTGTCTTGTTTTGCTCCTTTT-3′) were cloned into the pSES-HUS
vector (an adenoviral shuttle vector for siRNA expression, a gift
from Professor Tangni, Chongqing Medical University) according to
the manufacturer's instructions (18,28). All constructs were verified by
nucleic acid sequencing; subsequent analysis was performed using
BLAST software (National Institutes of Health, available at
http://www.ncbi.nlm.nih.gov/blast/).
Cell culture
To examine the effect of knocking down PERK and ATF4
on chondrogenesis, ATDC5 chondrogenic cells (ATCC®;
PCS-500-051™) and C3H10T1/2 embryonic firbro-blasts (a gift from Dr
Chuanju Liu, New York University School of Medicine, New York, NY,
USA) were infected with adenoviral vector containing siPERK or
siATF4 or siPERK + siATF4 or a control RFP adenovirus before
micromass culture. To examine the effect of the silencing of ATF4
and PERK on chondrogenesis, ATDC5 cells and C3H10T1/2 cells were
infected with siATF4 (MOI=60) or siPERK (MOI=80) adenovirus or
control RFP adenovirus prior to micromass culture. Uninfected cells
were used as the negative controls (NC). Micromass culture was
performed as described previously (19,20). The ATDC5 and C3H10T1/2 cells were
briefly trypsinized and resuspended in Dulbecco's modified Eagle's
medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10%
FBS at a concentration of 106 cells/ml, and 6 drops
(approximately 120 μl) of suspended cells were placed in a
60-mm tissue culture dish (Becton-Dickinson, San Diego, CA, USA).
After 2 h of incubation at 37°C, 1 ml of DMEM containing 10% FBS
and BMP2 (300 ng/ml) was added to the culture medium. The medium
was changed every 2–3 days.
RNA extraction and reverse transcription
(RT)-PCR
Total RNA was extracted from the cultured cells
using the RNeasy Mini kit (Qiagen, Hilden, Germany) and reverse
transcribed using the SuperScript pre-amplification system
(Invitrogen, Carlsbad, CA, USA) following the manufacturer's
instructions. The following sequence-specific primers were
synthesized: 5′-AGCACTCAGATGGAGAGAGTCAG-3′ and
5′-GCTATGGGAGTTGTTGGACTGT-3′ for PERK; 5′-TGGCGTCCTCGGCCTTCAC-3′
and 5′-TTCCCTCCCTCCCTTTG ACG-3′ for ATF4. GAPDH was employed as an
internal control using the following of oligonucleotides: 5′-ACC
ACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTG TTGCTGTA-3′. The identity
of each targeted PCR amplification product was confirmed by DNA
sequence analysis of gel-purified bands (Qiagen).
Western blot analysis
Proteins in total cell extract from micromass
cultures of BMP2-treated (300 ng/ml) ATDC5 or C3H10T1/2 cells were
resolved on a 10% SDS-polyacrylamide gel and electroblotted onto
nitrocellulose membranes. After blocking in 10% non-fat dry milk in
Tris-buffered saline Tween-20 [10 mM Tris-HCl (pH 8.0), 150 mM
NaCl, 0.5% Tween-20], the blots were incubated with either mouse
monoclonal anti-PERK antibody (sc-377400; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA) and anti-C/EBP homologous protein (CHOP)
antibody (ab11419; diluted 1:1,000; Abcam, Cambridge, UK) or rabbit
anti-caspase-3 antibody (ab32351; Abcam) and anti-p-c-Jun
N-terminal kinase (JNK) antibody (sc-293138; Santa Cruz
Biotechnology, Inc.) for 1 h. After washing, the respective
secondary antibody [horseradish peroxidase (HRP)-conjugated
anti-mouse immunoglobulin or HRP-conjugated anti-rabbit
immunoglobulin; Sigma. St. Louis, MO, USA; A9044] was added, and
the bound antibody was visualized using an enhanced
chemiluminescence system (Amersham Biosciences, Uppsala,
Sweden).
Culture of fetal mouse bone explants and
immunohistochemistry
Metatarsals were isolated from 8 newborn C57BL/6J
mice and cultured in DMEM (Gibco) containing 1% heat-inactivated
fetal calf serum (Invitrogen) and 100 U/ml penicillin-strep tomycin
in the presence of BMP2 (300 ng/ml) and siPERK, siATF4, or siPERK +
siATF4 for 5 days, followed by histological examination.
Affinity-purified anti-caspase-3 (ab32351; Abcam), caspase-12
(ab62484; Abcam), anti-CHOP (ab11419; Abcam) and anti-p-JNK
(sc-293138; Santa Cruz Biotechnology, Inc.) were diluted at 1:100
and sections were incubated at room temperature for 2 h. For
detection, biotinylated secondary antibody (sc-2364) and
HRP-streptavidin complex (sc-2363) (Santa Cruz Biotechnology, Inc.)
were used. A total of 0.5 mg/ml 3,3′-diami-nobenzidine (DAB) in 50
mM Tris-HCl substrate (Sigma) was used for visualization, and the
sections were counterstained with Mayer's hematoxylin (H9627;
Sigma).
TUNEL assay
Metatarsals from 5 newborn C57BL/6J mice, as well as
micromass cultures of C3H10T1/2 and ATDC5 cells, were cultured for
5 days in the presence of conditioned medium containing BMP2 (300
ng/ml) and siPERK, siATF4, or siATF4 + siPERK followed by the
detection of apoptosis in accordance with the manufacturer's
recommended protocol using the DeadEnd™ Fluorometric TUNEL System
(Promega Corp., Madison, WI, USA). Localized green fluorescence of
apoptotic FITC-labeled TUNEL-positive cells was imaged using a
fluorescence microscope (S/N:2109; Kramer Scientific Corp.,
Amesbury, MA, USA).
Apoptosis analysis by flow cytometry
(FCM)
At 48 h following infection with siPERK, siATF4, or
siPERK + siATF4, ATDC5 and C3H10T1/2 cells in micromass culture
were then cultivated in BMP2 (300 ng/ml) for 1, 3 and 5 days. Each
culture media of BMP2-treated ATDC5 and C3H10T1/2 cells were then
collected for FCM analysis. Briefly, following incubation with
RNase (1 mg/ml; Qiagen) in the dark at 37°C for 1 h, the cells were
stained with propidium iodide (30 mg/ml; Sigma) and analyzed using
a flow cytometer (FACSCalibur; Becton-Dickinson) to determine cell
cycle distribution and detect cellular apoptotic rate. The
experiments were performed in triplicate.
Statistical analysis
Statistical analysis was performed using SPSS 10.0.1
software for Windows. Data are expressed as the means ± SD from at
least 3 independent experiments. The Student's t-test was used to
determine whether two sets of data were significantly different
from each other. Data for multiple variable comparisons were
analyzed by one-way analysis of variance (ANOVA). A value of
P<0.05 was deemed to indicate a statistically significant
difference.
Results
Measuremnt of ATF4 and PERK expression
following transfection with specific siRNA constructs
siPERK and siATF4 adenoviral vectors were
constructed and identified using endonuclease digestion and DNA
sequencing (data not shown). The ATDC5 and C3H10T1/2 cells infected
with siPERK, siATF4, or siPERK + siATF4 were examined by RT-PCR and
western blot analysis. The mRNA level of PERK markedly decreased in
the ATDC5 and C3H10T1/2 cells transfected with siPERK, as compared
with the untransfected controls (Fig.
1A). The protein level was also significantly decreased in the
siPERK-infected cells, as compared with the control cells (Fig. 1B). Furthermore, as shown in
Fig. 1C and D, the mRNA
expression of ATF4 decreased compared to the controls, in the
siATF4-infected ATDC5 and C3H10T1/2 cells. The ATF4 protein level
was also significantly decreased in the siATF4-infected ATDC5 and
C3H10T1/2 cells compared with the respective control cells. These
results confirm the successful construction of the adenoviral
vectors and the silencing of PERK and ATF4.
 | Figure 1Expression of PERK and ATF4 in ATDC5
and C3H10T1/2 cells after infection with siPERK or siATF4. (A)
Analysis of PERK mRNA level with RT-PCR in (a) ATDC5 and (b)
C3H10T1/2 cells. Lane 1, NC; lane 2, adenovirus control Ad-RFP;
lane 3, siPERK. (B) Determination of PERK protein expression level
by western blot analysis after infection with siPERK in (a) ATDC5
and (b) C3H10T1/2 cells. Lane 1, NC; lane 2, Ad-RFP control group;
lane 3, siPERK group. Proteins were separated by 10% SDS-PAGE and
analyzed with anti-PERK antibody. GAPDH and β-actin were used as
internal controls in RT-PCR and western blotting, respectively. The
levels of PERK mRNA and proteins were markedly increased after
infection with siPERK as compared with control groups. (C) Analysis
of ATF4 mRNA level by RT-PCR in (a) ATDC5 and (b) C3H10T1/2 cells
(b). Lane 1, NC; lane 2, adenovirus control Ad-RFP; lane 3, siATF4.
(D) Determination of ATF4 protein expression level after infection
with siATF4 in (a) ATDC5 and (b) C3H10T1/2 cells. Lane 1, NC; lane
2, adenovirus control group Ad-RFP; lane 3, siATF4 group. Proteins
were separated by 10% SDS-PAGE and analyzed with anti-ATF4
antibody. GAPDH and β-actin were used as internal controls in
RT-PCR and western blot analysis, respectively. The protein and
mRNA levels of ATF4 were markedly reduced after infection with
siATF4 as compared with the control groups. PERK, PKR-like ER
kinase; ATF4, activating transcription factor 4. |
Silencing of ATF4 by transfection with
siATF4 decreases the expression of PERK in chondrogenesis
To further investigate the function of PERK in
chondrogenesis, we examined the expression of PERK in a
BMP2-induced micromass culture of ATDC5 cells by western blot
analysis. As shown in Fig. 2A,
the silencing of PERK by transfection with siPERK markedly
decreased the expression of PERK in the BMP2-stimulated ATDC5 cells
after 3 days of culture. Furthermore, concurrent silencing of the
expression of PERK and ATF4 in the BMP2-stimulated ATDC5 cells
further decreased PERK expression compared to the cells in wich
only PERK was silenced. These results remained consistent after 5
days of the BMP2-induced micromass culture of ATDC5 cells as
assessed by western blot analysis. As shown in Fig. 2B, in the cells treated with BMP2 +
siATF4 + siPERK PERK expression was markedly decreased compared
with the BMP2 + siPERK and BMP2 + siATF4 groups. These data clearly
indicate that the silencing of ATF4 by transfection with siATF4
regulates endogenous PERK protein expression, and further reduces
the expression of PERK inhibited by siPERK. Micromass cultures of
these cells were incubated in the presence of 300 ng/ml BMP2 for
the induction of chondrogenesis.
 | Figure 2siATF4 decreases the expression of
PERK in siPERK-infected ATDC5 cells during chondrogenesis. (A)
Determination of PERK protein expression level after infection with
siPERK, siATF4, and siATF4 + siPERK in BMP2-stimulated cells at 3
days by western blot analysis. Lane 1, NC; lane 2, BMP2 group; lane
3, BMP2 + siPERK group; lane 4, BMP2 + siATF4 group; lane 5, BMP2 +
siPERK + siATF4 group. Proteins were separated by 10% SDS-PAGE and
analyzed with anti-PERK antibody. β-actin was used as internal
control in western blot analysis. The protein levels of PERK were
markedly decreased after infection with BMP2 + siPERK + siATF4
compared with other groups. (B) Determination of PERK protein
expression level after infection with siPERK, siATF4, and siATF4 +
siPERK after 5 days of BMP2-induced culture by western blot
analysis. Lane 1, NC; lane 2, BMP2 group; lane 3, BMP2 + siPERK
group; lane 4, BMP2 + siATF4 group; lane 5, BMP2 + siPERK + siATF4
group. Proteins were separated by 10% SDS-PAGE and analyzed with
anti-PERK antibody. β-actin was used as internal control in western
blot analysis. The protein level of PERK was obviously decreased
after infection with BMP2 + siPERK + siATF4 compared with the other
groups. PERK, PKR-like ER kinase; BMP2, bone morphogenetic protein
2. |
Combined effect of the silencing of ATF4
and PERK on cell growth in chondrocyte differentiation
To examine whether the knockdown of PERK and/or ATF4
can influence the cell cycle profile during chondrogenesis, FCM
analysis was used to determine the cell cycle distributions of
ATDC5 and C3H10T1/2 cells during BMP2-induced chondrogenesis in the
presence of siPERK, siATF4 or siATF4 + siPERK.
At the time points of 1, 3, and 5 days, the
proportion of ATDC5 cells in the S phase following treatment with
BMP2 + siPERK was decreased compared to that of the ATDC5 cells
cultured under the influence of BMP2 alone (Fig. 3A and B). In the ATDC5 cells
treated with BMP2 + siPERK, the cell number in the S phase was
36.03, 34.21 and 28.17% on days 1, 3, and 5, respectively; for the
ATDC5 cells treated with BMP2 + siATF4, 32.17, 30.78, and 29.25% of
the cell population was in the S phase on days 1, 3, and 5,
respectively. The decreased percentage of cells in the S phase was
even more significant in the ATDC5 cells treated with BMP2 + siATF4
+ siPERK, with S phase cells accounting for only 29.78, 26.52 and
17.82% of the cell population at 1, 3, and 5 days,
respectively.
Similarly, in the C3H10T1/2 cells treated with BMP2
+ siPERK, the percentage of cells in the S phase was 42.35, 44.07,
31.21%; and 33.05, 29.01, 27.65% in C3H10T1/2 cells treated by BMP2
+ siATF4 (1, 3, and 5 days, respectively). In addition, 28.91,
27.45 and 18.37% of C3H10T1/2 cells treated with BMP2 + siATF4 +
siPERK were recorded to be in the S phase on 1, 3, and 5 days,
respectively (Fig. 3C and D).
The difference between the S phase cell distribution
data for each treatment group and that of the corresponding control
groups reached statistical significance (P<0.05). These data
indicate that the knockdown of PERK inhibits cell cycle
distribution during chondrogenesis, and that the silencing of ATF4
enhances the inhibitory effect of the silencing of PERK on cell
growth during chondrocyte differentiation.
The FCM data also revealed that in the ATDC5 cells
treated with BMP2 + siATF4 + siPERK, the percentage of cells in the
G2 phase was 18.82±0.91, 9.31±1.02, 6.07±0.85% on culture days 1,
3, and 5 days, respectively. In the C3H10T1/2 cells treated with
BMP2 + siATF4 + siPERK, 17.75±0.92, 8.51±0.92, and 5.87±0.94% of
cells were in the G2 phase at 1, 3, and 5 days of culture,
respectively (Fig. 3, P<0.05).
Collectively, these results indicate that the silencing of PERK
affects cell cycle distribution by reducing the number of cells in
the S phase cells during chondrogenesis. Specifically,the knockdown
of PERK inhibited cell proliferation during chondrocyte development
with arrest in the G1 phase, a decrease in the number of cells in
the S phase and the delay of the progression to the G2-M phase.
Furthermore, the silencing of ATF4 enhanced the inhibitory effect
of the silencing of PERK on cell cycle progression during
chondrocyte differentiation.
Combined effect of the silencing of ATF4
and PERK on ER stress-mediated apoptosis
We then sought to determine whether the silencing of
PERK (using siPERK), ATF4 (using siATF4) or both (using siATF4 +
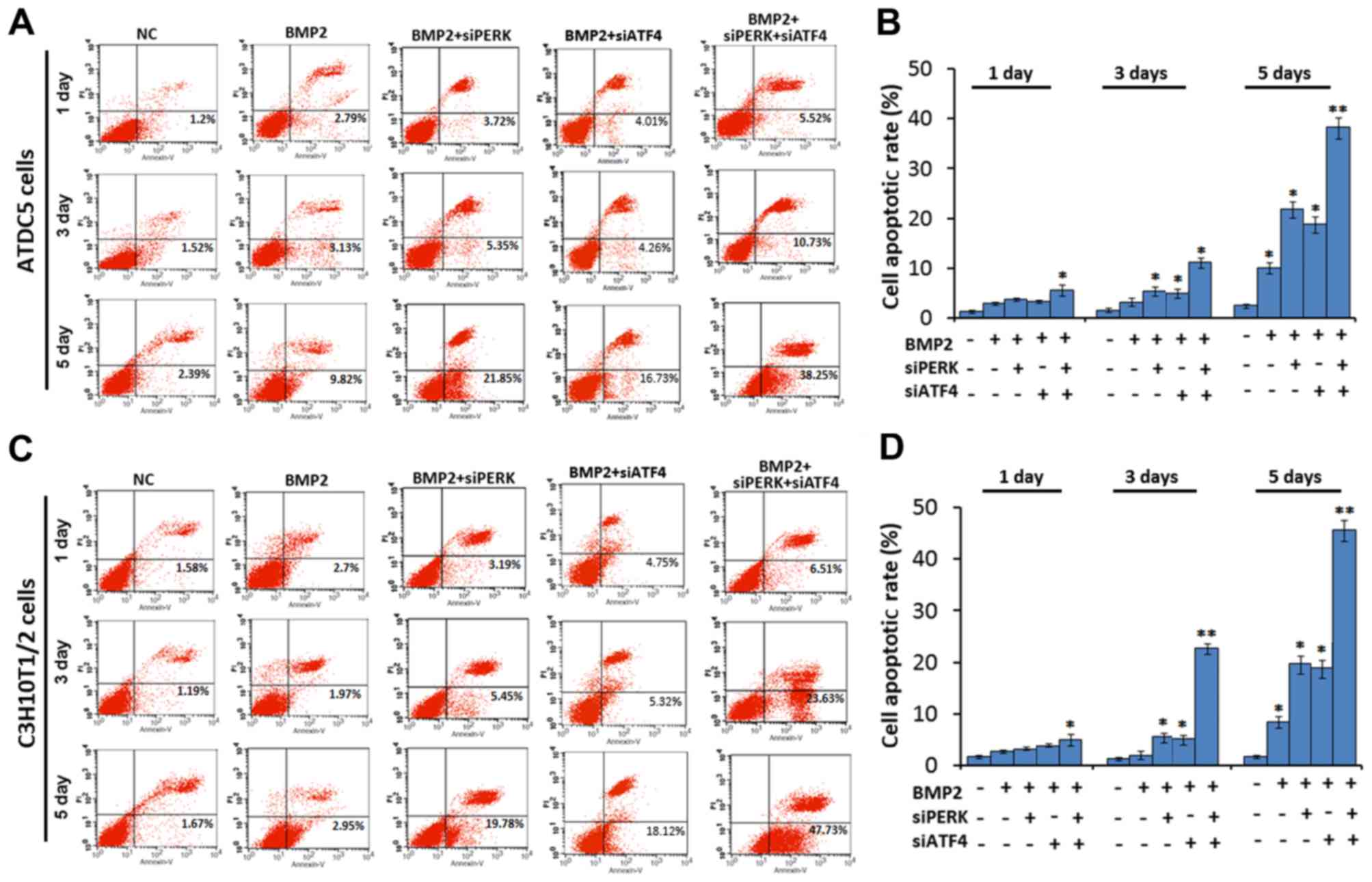
siPERK) affects cell apoptosis. As shown in Fig. 4, the cell apoptotic rate was
markedly increased after micromass culture of the ATDC5 cells for
1, 3 and 5 days following treatment with BMP2 + siATF4 + siPERK, as
compared with the BMP2 + siPERK, BMP2 + siATF4 and BMP2 treatment
groups. The cell apoptotic rate was 3.72, 5.35 and 21.85% in the
BMP2 + siPERK-treated ATDC5 cells, and 4.01, 4.26 and 16.73% in the
BMP2 + siATF4-treated ATDC5 cells on 1, 3 and 5 days, which clearly
reflects a larger population of apoptotic cells as compared with
the cells treated with BMP2 alone. In the BMP2 + siATF4 +
siPERK-treated ATDC5 cells, the cell apoptotic rate was markedly
increased to 5.52, 10.73 and 38.25% on days 1, 3 and 5,
respectively. The differences between each treatment group and the
BMP2 control group reached statistical significance (P<0.05 and
P<0.01, Fig. 4A and B).
Additionally, in the micromass culture of C3H10T1/2
cells, the cell apoptotic rate was also increased in the BMP2 +
siPERK group (3.19, 5.45 and 19.78%, days 1, 3 and 5) and BMP2 +
siATF4 group (4.75, 5.32 and 18.12%, days 1, 3 and 5) compared to
the BMP2 group (2.7, 1.97 and 2.95%, days 1, 3 and 5). In the
C3H10T1/2 cells transfected with both siPERK and siATF4, the
apoptotic rate was 6.51, 23.63 and 47.73% on 1, 3 and 5 days,
respectively. The differences between each treatment group and the
BMP2 control group reached statistical significance (P<0.05 and
P<0.01, Fig. 4C and D).
Taken together, these data demonstrate that the
knockdown of PERK using siPERK or ATF4 using siAT4 enhances ER
stress-mediated apoptosis in BMP2-induced chondrocyte
differentiation. Further, the combined application of siPERK and
siATF4 further promoted ER stress-mediated apoptosis in chondrocyte
differentiation induced by BMP2, generating an additive 'push'
toward apoptosis.
To confirm the influence of ER stress-mediated
apoptosis by transfection with siATF4 and siPERK in BMP2-stimulated
ATDC5 cells, the expression of ER stress-mediated apoptotic
molecules, including CHOP, caspase-3, caspase-12 and p-JNK, was
detected by western blot analysis in the ATDC5 cells stimulated
with BMP2 for 3 and 5 days. The expression of CHOP, caspase-3,
caspase-12 and p-JNK was markedly increased in the ATDC5 cells
following infection with siPERK, siATF4 and siATF4 + siPERK, and
BMP2 treatment over 3 and 5 days. As shown in Fig. 5, both siPERK and siATF4 induced a
marked increase in the expression levels of p-JNK, active (cleaved)
caspase-3, caspase-12 and CHOP in the ATDC5 cells transfected with
siPERK. Furthermore, the expression levels of p-JNK, cleaved
caspase-3, caspase-12 and CHOP were markedly increased following 3
and 5 days of BMP2 induction in the siATF4 + siPERK-infected ATDC5
cells. These results indicated that transfection with siPERK,
siATF4 and siPERK + siATF4 increased the expression of ER
stress-mediated apoptosis signaling pathway molecules during
chondrocyte differentiation. The individual effects of the
silencing of PERK and ATF4 exerted a more robust, additive combined
effect as observed with the implementation of the siPERK + siATF4
treatment condition.
The silencing of both ATF4 and PERK
(siATF4 + siPERK) increases ER stress-mediated apoptosis in
vitro
The high-density culture system was then incubated
in the absence (CTR) or presence of 300 ng/ml BMP2, BMP2 + siPERK,
BMP2 + siATF4 and BMP2 + siATF4 + siPERK for 3 days, at which
point, a TUNEL assay was performed to examine the effects of siPERK
and siATF4 + siPERK on apoptosis during chondrogenesis. As shown in
Fig. 6, during BMP2-induced
chondrocyte differentiation, the number of TUNEL-positive cells
increased significantly in the ATDC5 cells transfected with BMP2 +
siPERK (34.36%), BMP2 + siATF4 (42.85%) compared with the ATDC5
cells treated with BMP2 only (15.53%), and the number of
TUNEL-positive cells was further enhanced in the ATDC5 cells
treated with BMP2 + siA TF4 + siPERK (69.71%).
In addition, in the C3H10T1/2 cells treated with
BMP2 + siPERK, and those treated with BMP2 + siATF4, the number of
TUNEL-positive cells increased (32.83 and 39.36%) compared with
that of the C3H10T1/2 BMP2-treated cells (17.68%). The number of
TUNEL-positive C3H10T1/2 cells treated with BMP2 + siATF4 + siPERK
(66.52%) was also significantly greater than that recorded from
either the C3H10T1/2 BMP2 + siPERK-treated cells or BMP2 +
siATF4-treated cells. TUNEL assay was repeated in triplicate. The
differences between the BMP2 + siATF4 + siPERK, BMP2 + siPERK, BMP2
+ siATF4 and BMP2 groups reached statistical significance
(P<0.05, Fig. 6). It should be
noted that the silencing of PERK and ATF4 activated ER
stress-mediated apoptosis during chondrocyte differentiation
induced by BMP2; the silencing of both ATF4 and PERK (siATF4 +
siPERK) enhanceed ER stress-mediated apoptosis to a level exceeding
that induced by the silencing of ATF4 or PERK alone.
In order to verify whether the silecing of PERK
(using siPERK), ATF4 (using siATF4) or both (siATF4 + siPERK)
affects growth plate chondrocytes in developing tissue, TUNEL assay
was undertaken to determine the effect of siPERK, siATF4 and siATF4
+ siPERK on apoptosis in cartilage tissue (Fig. 7). The result show that the
TUNEL-positive cells in the BMP2 + siATF4 + siPERK group were
significantly increased compared with the BMP2 + siPERK, BMP2 +
siATF4 and the BMP2 group. These results further demonstrate that a
cumulative effect of siATF4 and siPERK in pushing chondrocytes
toward an apoptotic cell fate.
Silencing of ATF4 and PERK induces ER
stress-mediated caspase activation in chondrocyte tissue
To further understand the molecular events of ER
stress-mediated apoptosis induced by the silencing of ATF4 and PERK
(siATF4 + siPERK) in chondrogenesis, the effect of transfection
with siATF4 + siPERK on endochondral bone formation was examined by
implementing cultures of metatarsals isolated from newborn mice as
an ex vivo model of bone formation. Firstly, the metatarsals
were cultured for 5 days in the presence of conditioned medium
containing 300 ng/ml BMP2 (control), BMP2 + siPERK, BMP2 + siATF4
or BMP2 + siATF4 + siPERK adenovirus. Western blot analysis was
then used to examine the expression of ATF4 and PERK in the
metatarsal culture extracts. The protein level of ATF4 was markedly
decreased in the siATF4 and siPERK + siATF4-infected culture
extracts, as compared with the protein level of ATF4 in the BMP2
and BMP2 + siPERK-treated culture extracts. Likewise, the protein
level of PERK was markedly decreased in the siPERK and siPERK +
siATF4-infected culture extracts, as compared to the BMP2 and BMP2
+ siATF4 treatment group (Fig.
8A).
 | Figure 8Expression of cleaved caspase-3,
CHOP, p-JNK and caspase-12 in the growth plate chondrocytes in
vivo. Metatarsals were explanted from newborn mouse embryos and
cultured in the presence of conditioned medium of BMP2 (300 ng/ml),
BMP2 + siATF4 + siPERK for 5 days. (A) Western blot analysis was
used to detect the expression of ATF4 and PERK in metatarsals
cultured in the presence of conditioned medium of BMP2 (300 ng/ml),
BMP2 + siPERK, BMP2 + siATF4 or BMP2 + siPERK + siATF4 for 5 days.
(B, a and e) Immunohistochemistry staining was observed in
low-power microphotograph of a section stained with anti-active
caspase-3 monoclonal antibody (brown) and counterstained with
Mayer's hematoxylin (blue). (b and f) Immunohistochemistry staining
was observed in low-power microphotograph of a section stained with
anti-CHOP monoclonal antibody (brown) and counterstained with
Mayer's hematoxylin (blue). (c and g) Immunohistochemistry staining
was observed in low-power microphotograph of a section stained with
anti-p-JNK monoclonal antibody (brown) and counterstained with
Mayer's hematoxylin (blue). (d and h) Immunohistochemistry staining
was observed in low-power microphotograph of a section stained with
anti-caspase-12 monoclonal antibody (brown) and counterstained with
Mayer's hematoxylin (blue) and the scale bars represent 100
μm. CHOP, C/EBP homologous protein; BMP2, bone morphogenetic
protein 2; ATF4, activating transcription factor 4; PERK, PKR-like
ER kinase. |
We then detected the expression of ER
stress-specific caspases. At the time of explantation, these
explants consisted of undifferentiated cartilage. Over a 5-day
culture period, these explants underwent all sequential stages of
endochondral bone formation. As shown in Fig. 8B, treatment with siATF4 + siPERK
increased the expression of apoptosis-related proteins, such as
cleaved caspase-3, CHOP, p-JNK and caspase-12.
These results demonstrated the activation of
caspase-3, p-JNK, CHOP and caspase-12 by ER stress during
chondro-genesis and that the silecing of ATF4 and PERK increased
the expression of ER stress-mediated apoptosis signaling pathway
molecules. Taken together, these data demonstrated that the
combined silencing of ATF4 and PERK enhanced ER stress-mediated
apoptosis in BMP2-induced chondrogenesis.
Discussion
In eukaryotic cells, signaling pathways relay
information between the ER, cytosol and nuclei to restrict the
accumulation of unfolded proteins in the ER. A number of studies
have shown that factors influencing cell fate and/or
differentiation are activated during ER stress. In mammalian cells,
the UPR plays a fundamental role in maintaining cellular
homeostasis and is therefore at the center of many normal
physiological responses and pathologies (21–24).
Cells respond to ER stress via ER stress sensors,
leading to the UPR. PERK is a major transducer of the ER stress
response and directly phosphorylates eIF2α, resulting in
translational attenuation (16,25,26). Whether and how PERK/ATF4
participates in ER stress-mediated apoptosis in the process of
chondrocyte differentiation, and the mechanisms of how ER
stress-mediated apoptosis is regulated in chondrogenesis remain
unknown.
Our current study aimed to address the combined
effect of the silencing of PERK and ATF4 on ER stress-mediated
apoptosis during the process of chondrogenesis, as well as to
elucidate the molecular mechanisms involved. To define the
influence of these molecules, we first adenoviral vectors carrying
siPERK and siATF4, and infected the ATDC5 and C3H10T1/2 cells.
Protein analysis of whole cell extracts validated our approach, as
the expression of PERK and ATF4 was markedly decreased in each of
the cells expressing the relevant adenoviral vectors (Fig. 1). Furthermore, we demonstrated
that the silencing of ATF4 was able to regulate endogenous PERK
gene expression, evidenced by the further reduction in PERK
expression in the cells co-transfected with siATF4 and siPERK, as
compared to the cells transfected with siPERK alone (Fig. 2).
We previously reported that BMP2 mediates mild ER
stress during chondrogenesis and activates the IRE1α-XBP1 pathway;
X-box binding protein 1 spliced (XBP1s) in turn enhances
chondrocyte hypertrophy by functioning as a co-factor of RUNX2. We
also previously found that BMP2 activates UPR-signaling molecules
in chondrogenesis, such as XBP1s, BiP and IRE1α (27,28). Herein, we expanded upon our
previous findings by defining the role of PERK/ATF4 in ER
stress-mediated apoptosis during chondrocyte differentiation. Our
current data indicate that PERK/ATF4 influences cell cycle
distribution in chondrogenesis. Firstly, the application of siPERK
and siATF4 inhibited cell proliferation in chondrocyte development
with G1 phase arrest, a reduction in the number of cells in the S
phase and the delay of G2-M phase progression. The joint
application of siATF4 and siPERK resulted in the enhanced
disruption of cell cycle distribution (Fig. 3). FCM analysis illustrated that
siPERK and siATF4 enhanced ER stress-mediated apoptosis in
chondrogenesis induced by BMP2, and siATF4 also enhanced the
apoptotic effect of siPERK (Fig.
4).
ER stress-induced cell death is a new, exciting
apoptotic pathway, the full impact of which, particularly in
development and the pathology of disease, remains undetermined
(29,30). It is known that caspases, a family
of cysteine proteases including caspase-3, -9, and -12, act as a
common death effect or molecules in various forms of apoptosis.
Caspase-12 is an ER-associated proximal effector in the caspase
activation cascade, and cells lacking this enzyme are partially
resistant to inducers of ER stress (31–33). Three pathways have been identified
as being involved in ER stress-mediated apoptosis: the
caspase-12/caspase-4 pathway, the CHOP pathway and the IRE1-JNK
pathway. Caspase-12 and -4 have been proposed as caspases that
initiate ER stress-induced cell death with caspase-12 reported to
directly cleave pro-caspase-9 and induce apoptosis (34–37). CHOP induces ER stress-induced cell
death, at least in part, by suppressing the expression of Bcl-2 and
inducing Bim expression. It has been reported that IRE1a also
participates in ER stress-induced cell death by activating JNK.
These findings support the notion that ER stress leads to several
redundant pathways for caspase activation (38–40).
In order to gauge the activation of
ER-stress-mediated apoptotic pathways, we detected the expression
of phosphorylated JNK, cleaved caspase-3, CHOP and caspase-12
following treatment with BMP2/BMP2 + siPERK/BMP2 + siATF4/BMP2 +
siPERK + siATF4. The expression levels of phosphorylated JNK,
cleaved caspase-3, CHOP and caspase-12 were increased in the cells
treated with BMP2 + siPERK + siATF4 as compared with those treated
with BMP2 + siPERK or BMP2 + siATF4 or BMP2 alone (Fig. 5). Additionally, the expression of
ER stress-mediated apoptosis signaling pathway-associated molecules
was also increased in the BMP2 + siPERK group and BMP2 + siATF4
group as compared to the BMP2 treatment control group. Accordingly,
we demonstrated that transfection with siPERK and siATF4 increased
the expression of ER stress-mediated apoptotic signaling pathway
molecules during chondrogenesis and that co-transfection with
siATF4 enhanced the upregulated expression of apoptotic molecules
induced upon treatment with siPERK.
Furthermore, the results of TUNEL assay and
immunohistochemistry revealed that the BMP2 + siATF4 + siPERK group
featured many more apoptotic cells as compared with the BMP2 +
siPERK group, BMP2 + siATF4 group and the BMP2 group, demonstrating
that the silencing of PERK and ATF4 increased the expression of ER
stress-mediated apoptosis signaling pathway molecules during
chondrogenesis (Figs. 6Figure 7–8).
In a word, our data indicate that the silencing of
PERK and ATF4 enhance ER stress-mediated apoptosis during
chondrogenesis and that the joint silencing of ATF4 and PERK leads
to a more profound promotion of apoptotic signaling that is
observed following the silencing of either PERK or ATF4 alone.
In conclusion, this study provides novel insight
into the role of PERK and ATF4 in regulating ER stress-mediated
apoptosis during chondrocyte differentiation. Our study supports
the notion that the knockdown of PERK, a key regulator of the
mammalian UPR, decreases the growth of chondrocytes and induces ER
stress-mediated apoptosis during chondrogenesis. The knockdown of
ATF4 using siATF4 similarly resulted in the decreased growth of
chondrocytes and greater ER stress-mediated apoptosis during
chondrogenesis. The simultaneous knockdown of PERK and ATF4
augmented ER stress-mediated apoptosis, surpassing the levels
observed under the knockdown conditions of ATF4 or PERK alone,
during chondrocyte differentiation. Continued research in this
field is necessary to clarify the complexities of this cell-death
pathway. New insight into the mechanistic basis of stress responses
will open new perspectives for the development of molecular
target-based treatment approaches, and may thus have great
potential for use in the treatment of cartilage disorders and
arthritic conditions.
Abbreviations:
|
BMP2
|
bone morphogenetic protein 2
|
|
ATF6
|
activating transcription factor 6
|
|
ERS
|
ER stress
|
|
UPR
|
unfolded protein response
|
|
PERK
|
protein kinase R-like endoplasmic
reticulum kinase
|
|
IRE1α
|
inositol-re quiring enzyme 1α
|
|
XBP1s
|
X-box binding protein 1 spliced
|
Acknowledgments
The authors would like to thank Aubryanna
Hettinghouse (Department of Orthopaedic Surgery and Cell Biology,
New York University School of Medicine) for critically reading the
manuscript. This study was supported by the National Natural
Science Foundation of China (81371928 and 81171697); New Century
Excellent Talent Support Project of Education Ministry of China
(NCET-12-1090).
References
|
1
|
Pluquet O, Pourtier A and Abbadie C: The
unfolded protein response and cellular senescence. A review in the
theme: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. Am J Physiol Cell Physiol.
308:C415–C425. 2015. View Article : Google Scholar
|
|
2
|
Grootjans J, Kaser A, Kaufman RJ and
Blumberg RS: The unfolded protein response in immunity and
inflammation. Nat Rev Immunol. 16:469–484. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Karali E, Bellou S, Stellas D, Klinakis A,
Murphy C and Fotsis T: VEGF signals through ATF6 and PERK to
promote endothelial cell survival and angiogenesis in the absence
of ER stress. Mol Cell. 54:559–572. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Murakami T, Saito A, Hino S, Kondo S,
Kanemoto S, Chihara K, Sekiya H, Tsumagari K, Ochiai K, Yoshinaga
K, et al: Signalling mediated by the endoplasmic reticulum stress
transducer OASIS is involved in bone formation. Nat Cell Biol.
11:1205–1211. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Horiuchi K, Tohmonda T and Morioka H: The
unfolded protein response in skeletal development and homeostasis.
Cell Mol Life Sci. 73:2851–2869. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zuscik MJ, Hilton MJ, Zhang X, Chen D and
O'Keefe RJ: Regulation of chondrogenesis and chondrocyte
differentiation by stress. J Clin Invest. 118:429–438. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Saito A and Imaizumi K: Endoplasmic
reticulum stress response in osteogenesis. Clin Calcium.
23:1569–1575. 2013.In Japanese. PubMed/NCBI
|
|
8
|
Saito A, Hino S, Murakami T, Kanemoto S,
Kondo S, Saitoh M, Nishimura R, Yoneda T, Furuichi T, Ikegawa S, et
al: Regulation of endoplasmic reticulum stress response by a
BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat
Cell Biol. 11:1197–1204. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rajpar MH, McDermott B, Kung L, Eardley R,
Knowles L, Heeran M, Thornton DJ, Wilson R, Bateman JF, Poulsom R,
et al: Targeted induction of endoplasmic reticulum stress induces
cartilage pathology. PLoS Genet. 5:e10006912009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rosen V: BMP2 signaling in bone
development and repair. Cytokine Growth Factor Rev. 20:475–480.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Canalis E, Economides AN and Gazzerro E:
Bone morpho-genetic proteins, their antagonists, and the skeleton.
Endocr Rev. 24:218–235. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jang WG, Kim EJ, Kim DK, Ryoo HM, Lee KB,
Kim SH, Choi HS and Koh JT: BMP2 protein regulates osteocalcin
expression via Runx2-mediated Atf6 gene transcription. J Biol Chem.
287:905–915. 2012. View Article : Google Scholar :
|
|
13
|
Saito A, Ochiai K, Kondo S, Tsumagari K,
Murakami T, Cavener DR and Imaizumi K: Endoplasmic reticulum stress
response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved
in osteoblast differentiation induced by BMP2. J Biol Chem.
286:4809–4818. 2011. View Article : Google Scholar
|
|
14
|
Liu CY, Schröder M and Kaufman RJ:
Ligand-independent dimerization activates the stress response
kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J
Biol Chem. 275:24881–24885. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsai SF, Tao M, Ho LI, Chiou TW, Lin SZ,
Su HL and Harn HJ: Isochaihulactone-induced DDIT3 causes ER
stress-PERK independent apoptosis in glioblastoma multiforme cells.
Oncotarget. 2016.
|
|
16
|
Walter F, Schmid J, Düssmann H, Concannon
CG and Prehn JH: Imaging of single cell responses to ER stress
indicates that the relative dynamics of IRE1/XBP1 and PERK/ATF4
signalling rather than a switch between signalling branches
determine cell survival. Cell Death Differ. 22:1502–1516. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo FJ, Liu Y, Zhou J, Luo S, Zhao W, Li X
and Liu C: XBP1S protects cells from ER stress-induced apoptosis
through Erk1/2 signaling pathway involving CHOP. Histochem Cell
Biol. 138:447–460. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Miyake S, Makimura M, Kanegae Y, Harada S,
Sato Y, Takamori K, Tokuda C and Saito I: Efficient generation of
recombinant adenoviruses using adenovirus DNA-terminal protein
complex and a cosmid bearing the full-length virus genome. Proc
Natl Acad Sci USA. 93:1320–1324. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu CJ, Prazak L, Fajardo M, Yu S, Tyagi N
and Di Cesare PE: Leukemia/lymphoma-related factor a POZ
domain-containing transcriptional repressor, interacts with histone
deacetylase-1 and inhibits cartilage oligomeric matrix protein gene
expression and chondrogenesis. J Biol Chem. 279:47081–47091. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Y, Kong L, Carlson CS and Liu CJ:
Cbfa1-dependent expression of an interferon-inducible p204 protein
is required for chondrocyte differentiation. Cell Death Differ.
15:1760–1771. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fulda S, Gorman AM, Hori O and Samali A:
Cellular stress responses: Cell survival and cell death. Int J Cell
Biol. 2010:2140742010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo F, Lin EA, Liu P, Lin J and Liu C:
XBP1U inhibits the XBP1S-mediated upregulation of the iNOS gene
expression in mammalian ER stress response. Cell Signal.
22:1818–1828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Harding HP, Calfon M, Urano F, Novoa I and
Ron D: Transcriptional and translational control in the Mammalian
unfolded protein response. Annu Rev Cell Dev Biol. 18:575–599.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ron D: Translational control in the
endoplasmic reticulum stress response. J Clin Invest.
110:1383–1388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lumley EC, Osborn AR, Scott JE, Scholl AG,
Mercado V, McMahan YT, Coffman ZG and Brewster JL: Moderate
endoplasmic reticulum stress activates a PERK and p38-dependent
apoptosis. Cell Stress Chaperones. Oct 20–2016.Epub ahead of print.
PubMed/NCBI
|
|
26
|
Shah A and Kumar A:
Methamphetamine-mediated endoplasmic reticulum (ER) stress induces
type-1 programmed cell death in astrocytes via ATF6, IRE1α and PERK
pathways. Oncotarget. Jun 14–2016.Epub ahead of print.
|
|
27
|
Guo FJ, Xiong Z, Han X, Liu C, Liu Y,
Jiang R and Zhang P: XBP1S, a BMP2-inducible transcription factor,
accelerates endochondral bone growth by activating GEP growth
factor. J Cell Mol Med. 18:1157–1171. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han X, Zhou J, Zhang P, Song F, Jiang R,
Li M, Xia F and Guo FJ: IRE1α dissociates with BiP and inhibits ER
stress-mediated apoptosis in cartilage development. Cell Signal.
25:2136–2146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Han D, Lerner AG, Vande Walle L, Upton JP,
Xu W, Hagen A, Backes BJ, Oakes SA and Papa FR: IRE1alpha kinase
activation modes control alternate endoribonuclease outputs to
determine divergent cell fates. Cell. 138:562–575. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pincus D, Chevalier MW, Aragón T, van
Anken E, Vidal SE, El-Samad H and Walter P: BiP binding to the
ER-stress sensor Ire1 tunes the homeostatic behavior of the
unfolded protein response. PLoS Biol. 8:e10004152010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Degterev A, Boyce M and Yuan J: A decade
of caspases. Oncogene. 22:8543–8567. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Samali A, Zhivotovsky B, Jones D, Nagata S
and Orrenius S: Apoptosis: Cell death defined by caspase
activation. Cell Death Differ. 6:495–496. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hitomi J, Katayama T, Eguchi Y, Kudo T,
Taniguchi M, Koyama Y, Manabe T, Yamagishi S, Bando Y, Imaizumi K,
et al: Involvement of caspase-4 in endoplasmic reticulum
stress-induced apoptosis and Abeta-induced cell death. J Cell Biol.
165:347–356. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakagawa T and Yuan J: Cross-talk between
two cysteine protease families. Activation of caspase-12 by calpain
in apoptosis. J Cell Biol. 150:887–894. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by down-regulating Bcl2 and perturbing the
cellular redox state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakanishi K, Sudo T and Morishima N:
Endoplasmic reticulum stress signaling transmitted by ATF6 mediates
apoptosis during muscle development. J Cell Biol. 169:555–560.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hassler J, Cao SS and Kaufman RJ: IRE1, a
double-edged sword in pre-miRNA slicing and cell death. Dev Cell.
23:921–923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View Article : Google Scholar : PubMed/NCBI
|