Introduction
Hypertrophic cardiomyopathy (HCM: OMIM 192600) is
one of the most common inherited cardiac diseases characterized by
marked thickening of the left ventricle or/and interventricular
septum. HCM may affect groups of all ages and ethnicity, with an
incidence of 1 in 500 individuals in the general population
worldwide (1). In China, at least
one million individuals are expected to be diagnosed with this
disease (2). It has been
considered as the major cause of sudden cardiac death in young
adults and competitive athletes. Most HCM patients experience
obvious clinical symptoms, including shortness of breath,
palpitations, angina and syncope. However, HCM also presents with
high variability in clinical presentation due to the genetic
heterogeneity of the patients. It is predominantly inherited in an
autosomal dominant pattern, but fewer HCM patients present with
X-linked inheritance or Mendelian autosomal recessive disease. The
first mutation highly associated with HCM was discovered in the
β-myosin heavy chain gene (MYH7) in 1990 (3). To date, over 1,400 responsible
mutations have been documented in more than 25 genes (4,5).
Genetic testing can provide more accurate information for clinical
diagnosis, especially for those ambiguous HCM cases, and for
evaluation of the risk of disease occurrence of individuals with
familial HCM (6).
Compared to the traditional genetic testing method,
Sanger sequencing, which is costly and time-consuming, recently
developed next-generation sequencing (NGS) technologies can improve
cost effectiveness. NGS is highly feasible for massive parallel
sequencing and is suitable for inherited disease testing (7–9),
including cardiomyopathies (10–12). Yunnan Province, located in
southwestern China, consists of diverse ethnic groups (13). However, the genetic
characterizations of HCM in this region have been poorly studied
(14,15). In the present study, we used NGS
technology to perform genetic screening of the entire exon sequence
and the flanking regions of 19 most common HCM causative genes in a
Yunnan population. One novel mutation in tafazzin (TAZ,
p.Ile208Vla), a single rare mutation in MYH7 (p.Arg54Gln), and
three double mutations responsible for HCM were respectively
detected in our HCM patients. The prevalence and spectrum of gene
mutations associated with HCM in Yunnan were systematically
described.
Materials and methods
Subjects and clinical evaluation
From September 2013 to December 2015, 18 patients
with HCM, including 8 cases from 6 pedigrees with familial HCM and
10 cases without familial HCM, and 100 healthy controls were
recruited at the Department of Cardiology, The First People's
Hospital of Yunnan Province. Written informed consent was obtained
from all subjects. The demographic data including age, gender, and
history of cardiovascular diseases and other familial diseases were
recorded simultaneously with sample collection. According to the
2011 American College of Cardiology Foundation (ACCF) and the
American Heart Association (AHA) guideline for the diagnosis of HCM
(16), a left ventricular septum
(LVS) or/and interventricular septal thickness (IVST) ≥15 mm in the
absence of any other condition that could explain the hypertrophy
was diagnosed as HCM. This research project was approved by the
Ethics Committee of the First People's Hospital of Yunnan Province,
and performed in compliance with the principles of the Declaration
of Helsinki.
DNA isolation and sequencing
Genomic DNA was isolated from the peripheral whole
blood of 18 HCM patients and 100 healthy controls using a genomic
DNA Miniprep kit (AxyPrep; Axygen, Union City, CA, USA) following
the manufacturer's protocol. OD260/280 of DNA samples
was ~1.8 after purification with AMPure XP reagents (Beckman
Coulter, Fullerton, CA, USA). The DNA concentration was measured
using the Qubit dsDNA HS assay kit (Life Technologies, Carlsbad,
CA, USA). Whole coding exons and in exon-intron boundaries of 19
HCM-related genes were amplified and then sequenced on Ion Torrent
PGM (Life Technologies). These genes were MYH7, myosin
binding protein C (MYBPC3), α-actinin 2 (ACTN2),
desmin (DES), α-galactosidase A (GLA),
lysosome-associated membrane protein 2 (LAMP2), LIM
domain-binding 3 (LDB3), α-myosin heavy chain (MYH6),
regulatory myosine light chain 2 (MYL2), regulatory myosine
light chain 3 (MYL3), TAZ, myopalladin (MYPN),
AMP-activated protein kinase (PRKAG2), sodium channel,
voltage-gated type V α-subunit (SCN5A), Titin-cap
(TCAP), troponin I 3 (TNNI3), troponin T 2
(TNNT2), α-tropomyosin 1 (TPM1), and vinculin
(VCL).
Molecular genetic analysis
Low quality reads of which the read depth was less
than 30× were discarded (17,18), and qualified sequences were mapped
to human reference genome (hg19). The variants of the 19 genes in
each sample were annotated using online software Ion Reporter
(https://ionreporter.lifetechnologies.com/ir/secure/home.html).
The missense variant was considered to be possibly related with HCM
on the basis of the following criteria (19,20): i) the variant has been reported as
an HCM-related mutation according to the documents or Human Gene
Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php), and/or ii) the
predicted amino acid showed a change in a highly conserved
evolution site across many species; iii) the missense variant was
absent in the 100 healthy controls and its minor allele frequency
(MAF) was <5% in the 1000 Genomes Project (http://www.1000genomes.org/), HapMap (http://hapmap.ncbi.nlm.nih.gov/) and/or Exome
Aggregation Consortium (ExAC) databases (http://exac.broadinstitute.org/); iv) it was predicted
as a disease-related mutation by Mutation Taster (21).
All mutations potentially related with HCM were
further confirmed by conventional dideoxy sequencing using the
BigDye Terminator v.3.1 Cycle Sequencing kit (Applied Biosystems,
Foster City, CA, USA), and the obtained sequences were analyzed
using ABI 3130 Genetic Analyzer (Life Technologies). Specific
primers were applied to amplify the fragments of genomic DNA
containing identified candidate variations (Table II).
 | Table IISequences of primers used for
validation of target genes. |
Table II
Sequences of primers used for
validation of target genes.
| Gene symbol | Transcript
name | Exon | Nucleotide
changes | Primer (5′ to
3′) | PCR fragment
(bp) |
|---|
| MYH7 | NM_000257.2 | 3 | c.161G>A | Sense:
CCAAAGCCAGCCTATGGAACTCT | 2,348 |
| Antisense:
GGTCCCCAATGGCTGCAATAAC |
| MYH7 | NM_000257.2 | 8 | c. [730T>C,
732C>T] | Sense:
CTTGCTGGTCTCCAGTAGTATTGT | 536 |
| Antisense:
GGCTGAGCCTAGCAGATTCAT |
| MYH6 | NM_002471 | 12 | c.1087A>T | Sense:
CTGGAGGTGGATGGAGGATGA | 2,155 |
| Antisense:
GGTTGAGGAGTTGGGATTGTGGT |
| SCN5A | SCN5A | 21 | c.3575G>A | Sense:
CATCTCTTCAACCATCCAACCTTCTGC | 275 |
| Antisense:
TCCCTGCCACAACCCTGCATC |
| TNNI3 | NM_000363.4 | 6 | c.370G>C | Sense:
AGGTCTCCCTGTTTTTGGTTCC | 1,076 |
| Antisense:
GGACCTTCATGTACCTCTTTGCTCT |
| GLA | NM_000169 | 1 | c.140G>A | Sense:
CTGGTATGGAAATAGGGCGGGTC | 682 |
| Antisense:
CCTGATTCGGGACAGTTTGCTGG |
| MYBPC3 | NM_000256.3 | 27 | c.2992C>G | Sense:
TATGTGACCAGTGGGCAGTTC | 1,093 |
| Antisense:
GGGTCTTGTGACTGCACAAAG |
| MYH7 | NM_000257.2 | 22 | c.2572C>T | Sense:
GCTAATCAGTGACAAAGCCAGGATC | 1,434 |
| Antisense:
AGGGTGGAAGAGCCAACAGTAGC |
| TNNI3 | NM_000363.4 | 6 | c.433C>G | Sense:
AGGTCTCCCTGTTTTTGGTTCC | 1,076 |
| Antisense:
GGACCTTCATGTACCTCTTTGCTC |
| PRKAG2 | NM_000116.4 | 3 | c.298G>A | Sense:
CAGTCCTGTGTGGTCAGAACTTGG | 907 |
| Antisense:
GGACCAGAAGGATTACGCTTTGAT |
| MYBPC3 | NM_000256.3 | 31 | c.3624delC | Sense:
AGAGGCTCTCGGCATCAGGAAG | 906 |
| Antisense:
ACATAGATGCCCCCGTCAAAGG |
| TAZ | NM_000116.4 | 8 | c.622A>G | Sense:
TCAGGGCCCAGCTTATGCTAAC | 441 |
| Antisense:
TTTAATGTCTCGGTGCCAGGAAG |
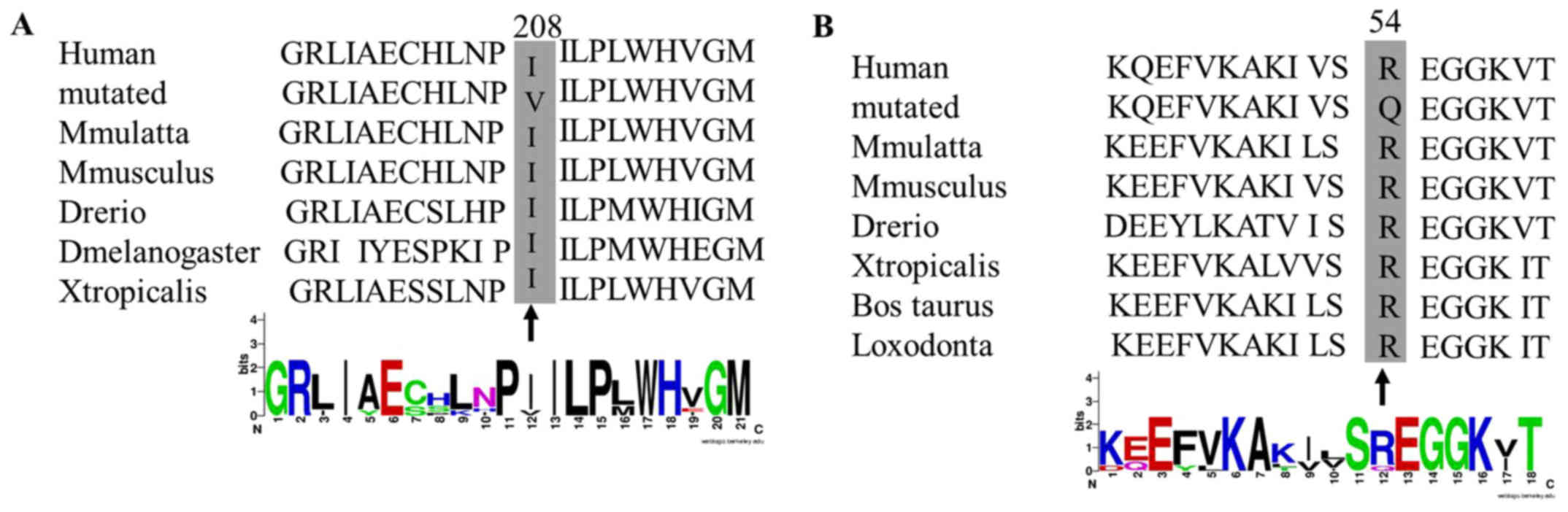
Evolutionary conservation analysis of the rare and
novel mutations, which were performed in many species (including
Macaca mulatta, Mus musculus, Danio rerio,
Drosophila melanogaster, Xenopus tropicalis, Bos
taurus and Loxodonta), was conducted using Clustal W
(http://www.genome.jp/tools/clustalw/)
and the Weblogo (http://weblogo.berkeley.edu/logo.cgi) (15). Based on the lowest energy theory,
the structure of the proteins with a rare mutation found in this
study was predicted by using Robetta (http://robetta.bakerlab.org/) and SWISS-MODEL
(http://www.swissmodel.expasy.org/)
online program, and the result was visualized using the Visual
Molecular Dynamics (VMD) software package (version 1.9.2,
http://www.ks.uiuc.edu/Research/vmd/index.html).
Results
Demographic and clinical
characteristics
From September 2013 to December 2015, a total of 18
HCM patients were recruited, including 11 (61%) males and 7 (39%)
females. Among these, 8 patients were from 6 pedigrees with
familial HCM, and the remaining 10 patients did not have familial
HCM. Their mean age at diagnosis was 45 years (range 23–79) with a
standard deviation (SD) of 16 years. The detailed demographic and
clinical characteristics of the patients are shown in Table I. All the investigated patients
presented typical clinical manifestations of HCM. The mean left
ventricular ejection fraction (LVEF) was increased to 67.1%
(SD=9.9), and the IVST was 18.8±2.7 mm, the left ventricular septum
thickness (LVST) was 26.3±9.7 mm, and the median left ventricular
posterior wall (LVPWT) was 11.7±1.9 mm as measured by doppler
echocardiography.
| Table IClinical characteristics of the HCM
patients as determined by echocardiography. |
Table I
Clinical characteristics of the HCM
patients as determined by echocardiography.
| Patient no. | Gender | Age (years) | Family history | IVST (mm) | LVPWT (mm) | LVED (mm) | LVST (mm) | LVEF (%) |
|---|
| A14 | F | 44 | N | 17.6 | 8.8 | 42.7 | 36.6 | 68 |
| 8 | F | 58 | N | 17.3 | 15.9 | 60.7 | 45.3 | 50 |
| 9 | M | 34 | N | NA | NA | NA | NA | NA |
| 15 | M | 50 | N | 15.6 | 12.1 | 44.7 | 27.0 | 70 |
| 16 | M | 57 | N | 16.3 | 13.5 | 49.6 | 26.2 | 78 |
| 24 | M | 44 | N | 15.8 | 9.1 | 41.5 | 30.3 | 53 |
| 25 | F | 58 | N | 19.0 | 14.1 | 43.6 | 22.9 | 79 |
| 26 | F | 79 | N | NA | NA | NA | NA | NA |
| 56 | M | 77 | N | NA | NA | NA | NA | NA |
| 57 | F | 57 | N | 21.0 | 9.0 | 35.0 | NA | 65 |
| Family A,
III:2 | F | 42 | Y (HCM) | 22.6 | 11.2 | 40.9 | 28.3 | NA |
| Family A,
III:4 | F | 36 | Y (HCM) | 22.1 | 12.0 | 42.4 | 28.0 | NA |
| Family A,
III:5 | M | 34 | Y (HCM) | 22.0 | 10.9 | 44.2 | 31.7 | NA |
| Family B, II:1 | M | 23 | N | 15.6 | 12.1 | NA | NA | NA |
| Family C,
III:1 | M | 27 | Y (HCM) | 22.8 | 11.5 | 47.7 | 12.6 | NA |
| Family D,
III:1 | M | 26 | Y (SCD) | 21.1 | 12.0 | 40.3 | 16.0 | 74 |
| Family E, II:1 | M | 47 | Y (HCM) | 16.4 | 11.3 | 53.0 | 13.5 | 73 |
| Family F, II:1 | M | 28 | Y (SCD) | 22.0 | 12.6 | 43.1 | 15.1 | 61 |
| Mean ± SD | NA | 45±16 | NA | 18.8±2.7 | 11.7±1.9 | 44.9±6.2 | 26.3±9.7 | 67.1±9.9 |
Sequence alignment and annotation
analysis
Exons and in exon-intron boundaries of 19
HCM-related genes were sequenced using Ion Torrent PGM (Life
Technologies). There were 383 variants obtained from qualified
reads, with single nucleotide polymorphisms, insertions or/and
deletions. The mean depth of coverage over all missense variants
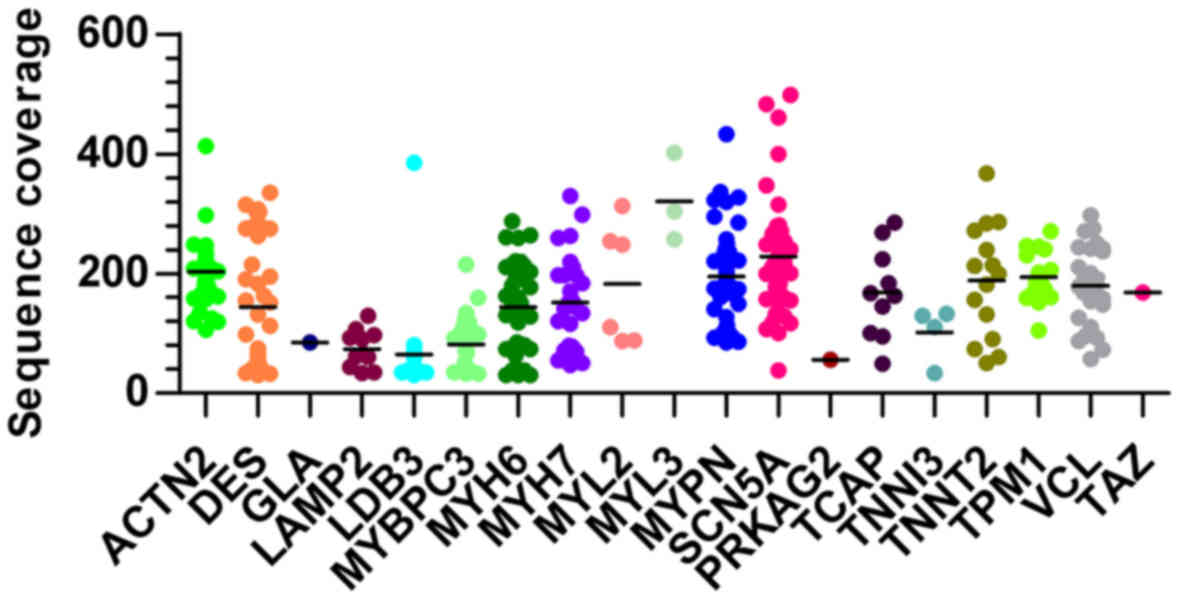
was 165.4-fold (ranged from 30 to 498) as shown in Fig. 1. Those variants were annotated and
the synonymous variants were filtered out using Ion Reporter online
software (https://ionre-porter.lifetechnologies.com/ir/secure/home.html).
As a result, 86 non-synonymous variants were detected in the 18 HCM
patients (data available upon request).
Screening for variants with higher
potential association with HCM
We next focused on the variants which were
potentially related with HCM. Based on the potentially pathogenic
criteria (mentioned in Materials and methods), 12 mutations were
selected from 13 HCM patients and further confirmed by Sanger
sequencing (Table II). There
were 6 single mutations and 3 double mutations which were most
frequently found in MYBPC3 and MYH7 with a prevalence
of 38.5% (5/13) and 23.1% (3/13), respectively. Other variants,
including TNNI3, SCN5A, GLA, TAZ,
PRKAG2 and MYH6, were found in 53.8% (7/13) of the
patients (Figs. 2 and 4). One novel single mutation in TAZ
(p.Ile208Val) was found neither in the 1000 Genomes Project
databases (http://www.1000genomes.org/) nor in the 100 healthy
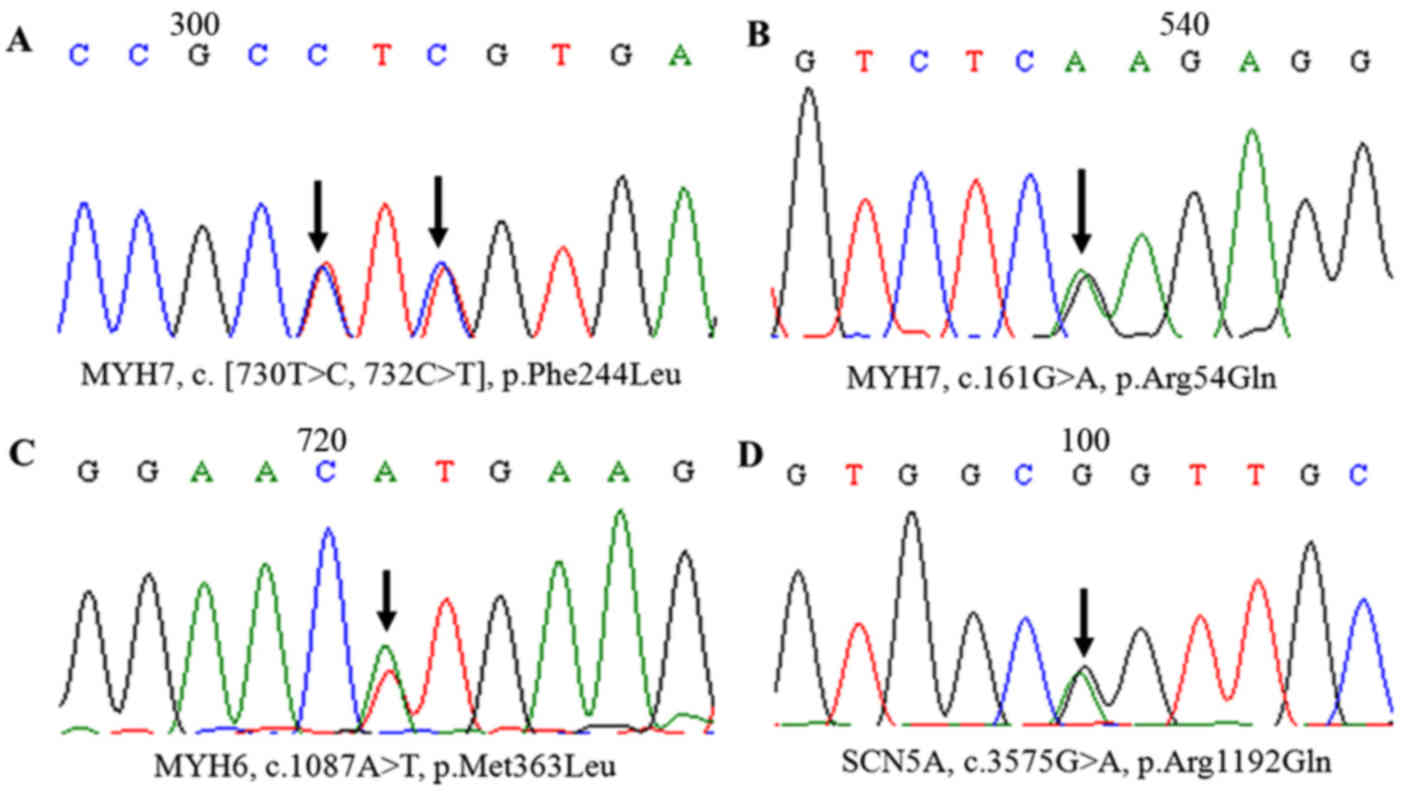
control chromosomes. One reported single mutation in MYH7
(p.Arg54Gln), considered as rare in the general population, was
firstly found in a patient without familial HCM and did not exist
in the databases and the control group mentioned above. The
mutations identified in highly conserved amino acids among many
species may have influenced the structure and function of the
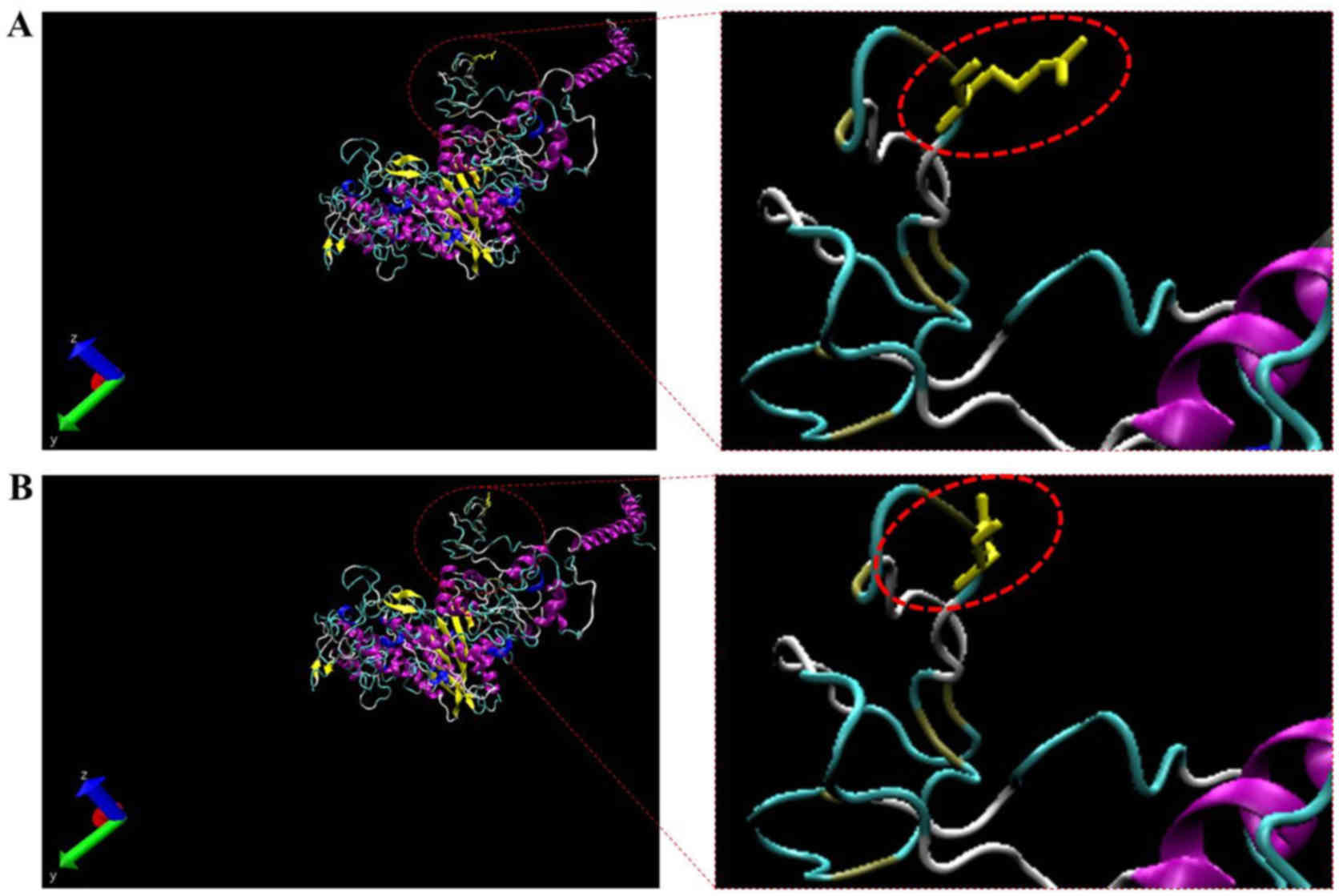
proteins (Fig. 5). Therefore, the
predicted protein structure of a mutant MYH7 (p.Arg54Gln) was
compared to the corresponding wild-type. The homology modeling
analysis showed that the amino acid change resulted in the
structural differences between the wild-type and mutant MYH7
(Figs. 5 and 6). In addition, the 3 double mutations
were found in MYBPC3 (p.Gln998Glu) plus TNNI3 (p.Arg145Gly), PRKAG2
(p.Gly100Ser) plus MYBPC3 (p.Lys1209Serfs*28), and TNNI3
(p.Glu124Gln) plus GLA (p.Trp47*) (Table III and Fig. 2).
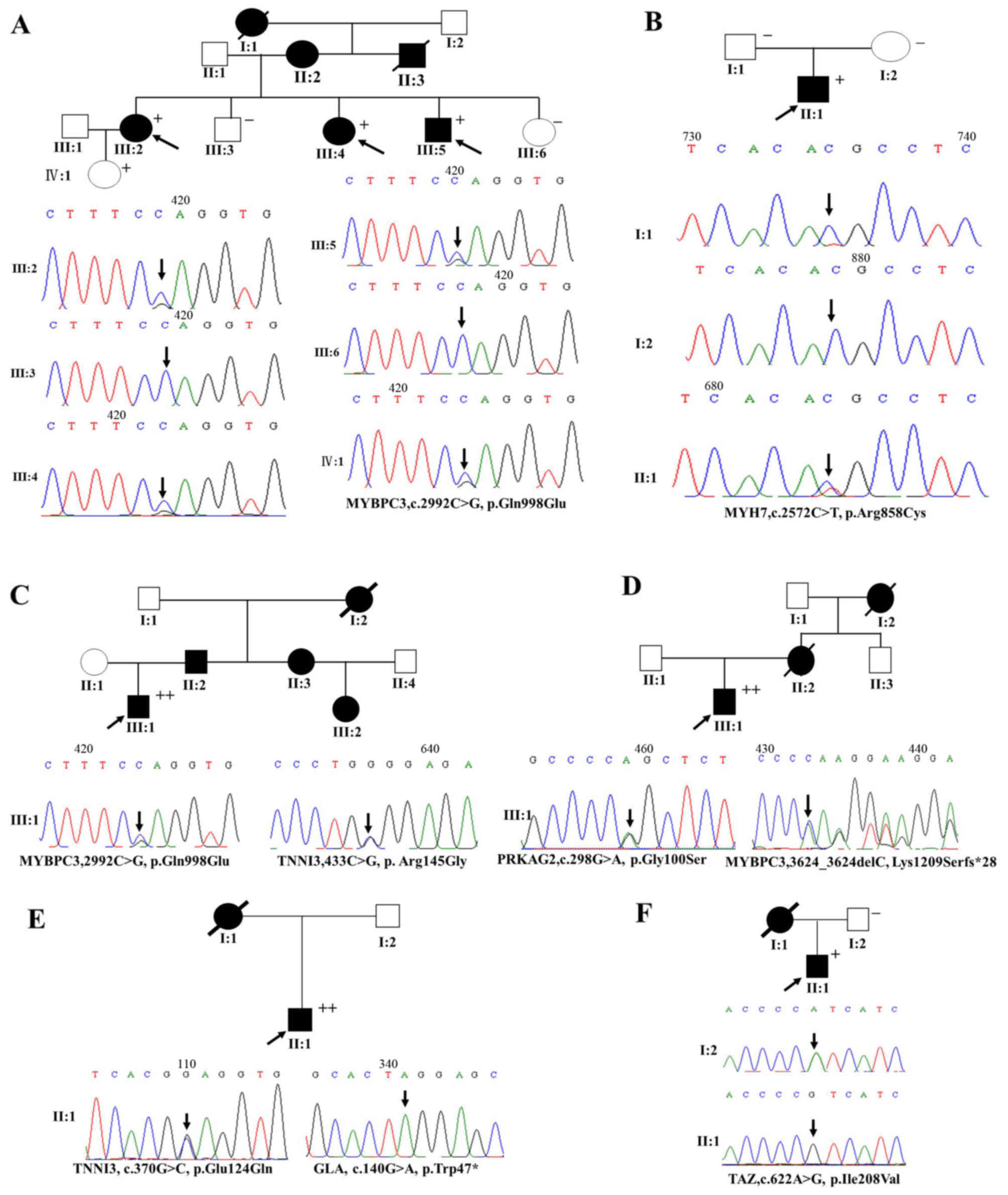 | Figure 2The genetic analyses of target gene
mutations in patients from 6 pedigrees with familial hypertrophic
cardiomyopathy (HCM) (families A, B, C, D, E and F). Squares, male
family members; circles, female family members; open symbols,
normal individuals; solid symbols, affected individuals; black
arrow, proband; plus (+) signs, the presence of disease mutation;
minus (−) signs, the absence of disease mutation. Both (+) and (−)
indicate members for whom the PCR and Sanger sequencing validation
were carried out. |
| Table IIIMutations identified in target genes
of the patients with HCM. |
Table III
Mutations identified in target genes
of the patients with HCM.
| Subjects | Gene name | Amino acid
change | Mutation type | Protein
location | Frequency |
|---|
| A14 | MYH7 | p.Arg54Gln | Missense | Myosin N-terminal
SH3-like domain | 1 |
| 9 | MYH6 | p.Met363Leu | Missense | Class II myosins,
motor domain | 1 |
| 25, 26 | SCN5A | p.Arg1192Gln | Missense | Sodium ion
transport-associated region | 2 |
| 8 | MYH7 | p.Phe244Leu | Missense | Myosin motor
domain | 1 |
| Family A, III:2,
III:4 and III:5; family C, III:1 | MYBPC3 | p.Gln998Glu | Missense | Ig-like C2-type
6 | 4 |
| Family B, II:1 | MYH7 | p.Arg858Cys | Missense | Tropomyosin | 1 |
| Family C,
III:1 | TNNI3 | p.Arg145Gly | Missense | Troponin | 1 |
| Family F, II:1 | TAZ | p.Ile208Val | Missense |
LPLAT_AGPAT-like | 1 |
| Family D,
III:1 | MYBPC3 |
p.Lys1209Serfs*28 | Frame shift | Ig-like C2-type
7 | 1 |
| Family D,
III:1 | PRKAG2 | p.Gly100Ser | Missense | N-terminal
binding | 1 |
| Family E, II:1 | GLA | p.Trp47* | Termination | α-galactohydrolase
activity | 1 |
| Family E, II:1 | TNNI3 | p.Glu124Gln | Missense | Cardiac troponin
C-binding domain | 1 |
Mutations and phenotypes of patients with
familial HCM
Family A, B and F were of the Han ethnic group;
family C, D and E were of the Yi, Naxi and Pumi ethnic groups,
respectively. Our results showed that 3 single mutations and 3
double mutations were found in 6 pedigrees with familial HCM
(Fig. 2). A 28-year-old male HCM
patient, experiencing palpitation and chest tightness, was found to
carry a novel mutation (TAZ, p.Ile208Val). A mutation of
p.Gln998Glu in MYBPC3 was detected in a proband patient (family A,
III:5) who was diagnosed as having HCM at age 34, with intermittent
chest tightness and shortness of breath, and a typical thick IVST
(22 mm). His 2-year-older sisters were respectively diagnosed as
HCM patients at age 36 and 42 years, and his father and grandmother
had undergone sudden death. Another proband (family B, II:1) having
MYH7 p.Arg858Cys was diagnosed as HCM at age 23 years and
experienced chest pain, but his father and mother did not have
disease phenotype. More importantly, the third proband (family C,
III:1) carrying a double mutation (MYBPC3, p.Gln998Glu plus TNNI3,
p.Arg145Gly) had a greater IVST (18.8; >15 mm), and his father,
aunt and female cousin were diagnosed with HCM a fewer years ago. A
proband (family D, III:1) carrying a double mutation (PRKAG2,
p.Gly100Ser plus MYBPC3, p.Lys1209Serfs*28) was diagnosed with HCM
at age 26, and his mother and grandmother had undergone sudden
cardiac death. A proband (family E, II:1) carrying a double
mutation (TNNI3, p.Glu124Gln plus GLA, p.Trp47*) presented with
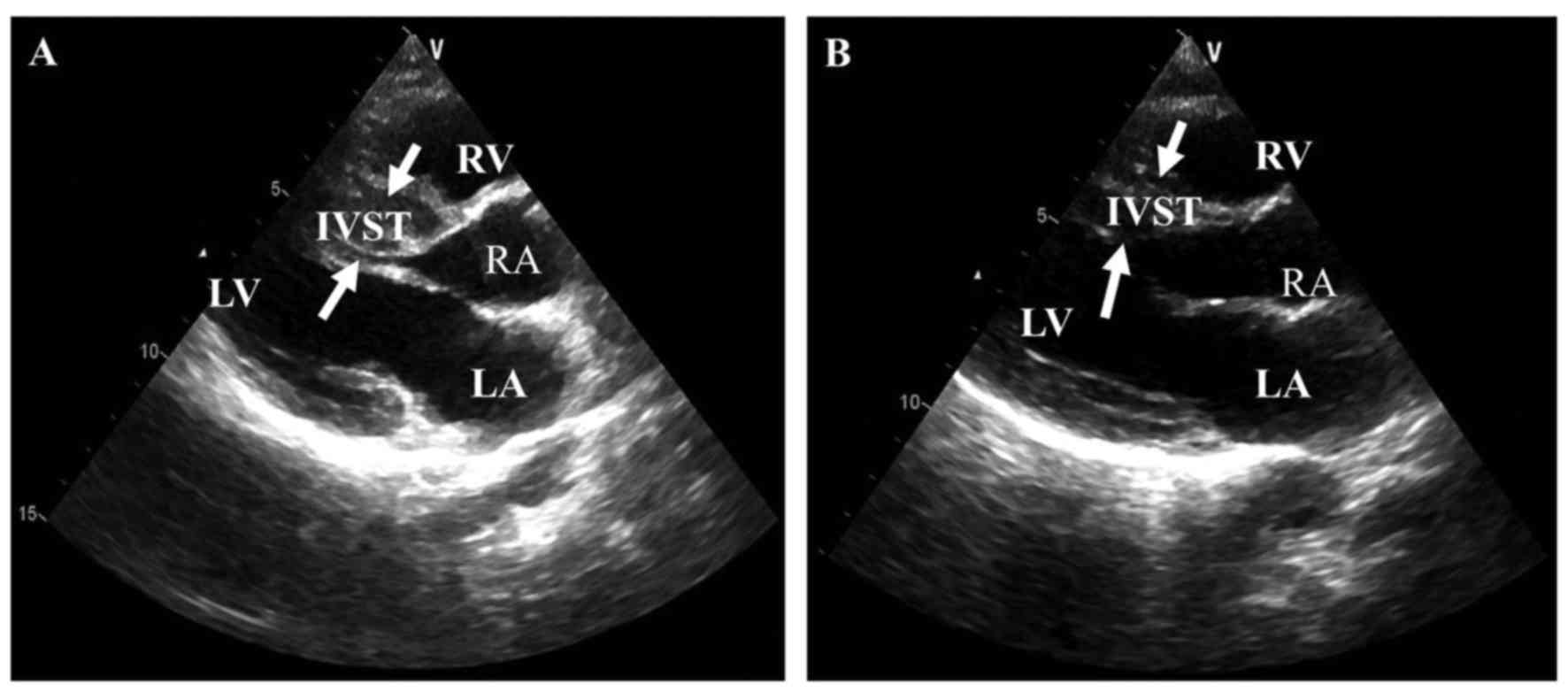
heart failure at age 47 years, and the echocardiography showed that
he had a high IVST (Figs. 2 and
3A).
Discussion
In the present study, we described the molecular
characterization of 18 HCM patients in Yunnan Province via targeted
NGS technology. A total of 383 variants were identified with an
average read depth of 165.4-fold (Fig. 1). Generally, a read of a targeted
nucleotide with read depth ≥30× reads was considered as qualified
(17,18). By filtering out synonymous
variations of the targeted genes, 86 qualified non-synonymous
variations were obtained (data available upon request). This
indicated the feasibility of NGS for genetic testing of 19
HCM-targeted genes.
There were 12 mutations identified in 13 HCM
patients (Table III). The
overall genetic diagnostic rate was 72.2% (13/18) and the frequency
of disease-causing mutations in the HCM cohort were much higher
than previous documentations (22–24), including an American cohort
(54.2%), French cohort (60.6%) and Japanese cohort (67%) (25–27). Mutations in the MYBPC3 gene
were the most prevalent that were detected in 5 of the 13 patients
(38.5%). It has been reported that mutations in the MYBPC3
gene are present in close to 20–30% of HCM cases (4,20,28), and our results were consistent
with these previous studies. The second most prevalent mutations
were found in MYH7 in 3 patients (3/13, 23.1%), which were
followed by mutations in TNNI3 (15.4%). The respective
mutation rate of SCN5A, PRKAG2, MYH6,
TAZ and GLA genes was close to 7.7%.
It is noteworthy that two single mutations were
found for the first time in HCM patients. One in TAZ (p.Ile208Val)
was novel and was found in a familial HCM male patient of 28 years.
The other single mutation was in MYH7 (p.Arg54Gln), which has been
rarely reported in the general population but had not been
previously found in HCM patients. This mutation had a very low
minor allele frequency (1/60,659) in ExAC browser Beta database
(http://exac.broadinstitute.org/). Both
TAZ (p.Ile208Val) and MYH7 (p.Arg54Gln) mutations resulted in
polarity changes in the altered amino acids and these 2 mutations
were not detected in 200 normal chromosomes. It has been speculated
that these mutations may have a significant impact on the structure
and function of the corresponding proteins. Indeed, the homology
modeling analysis showed that, compared to wild-type MYH7, the
structure of mutant MYH7 was altered at the mutated site. More
detailed data are needed to detect whether these mutations
influence the pathogenicity of HCM.
In the 6 pedigrees with familial HCM, 3 of them
(50%) were found to carry double mutations, which were firstly
discovered in this study. These double mutations were in MYBPC3
(p.Gln998Glu) plus TNNI3 (p.Arg145Gly), PRKAG2 (p.Gly100Ser) plus
MYBPC3 (p.Lys1209Serfs*28), and TNNI3 (p.Glu124Gln) plus GLA
(p.Trp47*), respectively. According to previous literature, ~15% of
familial HCM patients carry a double heterozygous mutation
(29), which was far less than
our results. This implies that special molecular genetic mechanisms
exist in Yunnan patients with familial HCM. There are 26 ethnic
groups in Yunnan Province and consanguineous marriages are widely
acceptable. The higher consanguineous marriage percentage than that
of the same groups in developed regions of China (30) has led to the accumulation of
defective gene mutations making the offspring at higher disease
risks.
Of these 18 patients, the relationships between the
phenotype and the genetics of HCM in familial HCM patients carrying
double mutations were further analyzed. The 27-year-old patient
(family C, III:1) showed a severe phenotype (IVST=22.8 mm), and had
double mutations in MYBPC3 (p.Gln998Glu) plus TNNI3 (p.Arg145Gly).
A 'double dose effect' of gene mutation (15) was speculated to lead to a more
malignant clinical phenotype and an early onset of HCM. The patient
(family D, III:1) with the PRKAG2 (p.Gly100Ser) plus MYBPC3
(p.Lys1209Serfs*28) mutations was characterized as having
relatively severe hypertrophy and an early onset of HCM, the same
feature of the patient from family E (TNNI3, p.Glu124Gln plus GLA,
p.Trp47*). The TNNI3 (p.Glu124Gln) mutation was originally reported
in Taiwanese patients with familial HCM (29). The Taiwanese are the descendants
of early settlers from the southeast coast of China during the last
few centuries (31). The present
study showed its second appearance in Chinese HCM patient.
Moreover, the children of HCM probands (family C, III:1; family D,
III:1; and family E, II:1) would have a higher risk rate of this
disease and similar mutations. According to our results, it is
recommended that these probands should implement prenatal screening
for the mutations of MYBPC3, p.Gln998Glu plus TNNI3, p.Arg145Gly;
PRKAG2, p.Gly100Ser plus MYBPC3, p.Lys1209Serfs*28; and TNNI3,
p.Glu124Gln plus GLA, p.Trp47*, respectively. For family B, the
proband (II:1, 23 years of age) with the MYH7, p.Arg858Cys mutation
did not show a severe phenotype, but his parents had no disease
phenotype and the genetic testing was negative. This suggests that
MYH7 (p.Arg858Cys) is a de novo mutation.
This study described the mutational spectrum of
patients with HCM in Yunnan, China. However, the number of HCM
patients was limited and the obtained clinical data were not
consistent for all subjects. A large-scale study of HCM patients in
Yunnan is needed to further confirm our results.
In conclusion, in the present study, 2 single and 3
double mutations were firstly found in HCM patients. The 3 double
mutations were detected in different ethnic groups and a novel
single mutation was found in TAZ (p.Ile208Val). The mutation in
MYH7 (p.Arg54Gln), previously reported as being rare in the general
population and having a very low minor allele frequency, was found
in a female patient without familial HCM. Our results add new data
to the mutational spectrum of Yunnan HCM patients. It provides
useful information for the presymptomatic intervention of HCM and
the management of patients with familial HCM.
Acknowledgments
The authors would like to thank the staff of the
First People's Hospital of Yunnan Province for providing their
support. The present study was supported by the New Products
Project of Yunnan Province, China (grant no. 2016BC003) and Major
Program of Applied Basic Research of Yunnan Province, China (grant
no. 2013FC007).
References
|
1
|
Ackerman MJ, Priori SG, Willems S, Berul
C, Brugada R, Calkins H, Camm AJ, Ellinor PT, Gollob M, Hamilton R,
et al Heart Rhythm Society (HRS); European Heart Rhythm Association
(EHRA): HRS/EHRA expert consensus statement on the state of genetic
testing for the channelopathies and cardiomyopathies: This document
was developed as a partnership between the Heart Rhythm Society
(HRS) and the European Heart Rhythm Association (EHRA). Europace.
13:1077–1109. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H,
Lu S, Wu P, Zhang Y, Shen L, et al: Prevalence of idiopathic
hypertrophic cardiomyopathy in China: A population-based
echocardiographic analysis of 8080 adults. Am J Med. 116:14–18.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Geisterfer-Lowrance AA, Kass S, Tanigawa
G, Vosberg HP, McKenna W, Seidman CE and Seidman JG: A molecular
basis for familial hypertrophic cardiomyopathy: A beta cardiac
myosin heavy chain gene missense mutation. Cell. 62:999–1006. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pinto YM, Wilde AA, van Rijsingen IA,
Christiaans I, Deprez RH and Elliott PM: Clinical utility gene card
for: Hypertrophic cardiomyopathy (type 1–14). Eur J Hum Genet.
19:192011. View Article : Google Scholar
|
|
5
|
Seidman CE and Seidman JG: Identifying
sarcomere gene mutations in hypertrophic cardiomyopathy: A personal
history. Circ Res. 108:743–750. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brauch KM, Karst ML, Herron KJ, de Andrade
M, Pellikka PA, Rodeheffer RJ, Michels VV and Olson TM: Mutations
in ribonucleic acid binding protein gene cause familial dilated
cardiomyopathy. J Am Coll Cardiol. 54:930–941. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao Y, Feng Y, Zhang YM, Ding XX, Song
YZ, Zhang AM, Liu L, Zhang H, Ding JH and Xia XS: Targeted
next-generation sequencing of candidate genes reveals novel
mutations in patients with dilated cardiomyopathy. Int J Mol Med.
36:1479–1486. 2015.PubMed/NCBI
|
|
8
|
Gómez J, Reguero JR, Morís C, Martín M,
Alvarez V, Alonso B, Iglesias S and Coto E: Mutation analysis of
the main hyper-trophic cardiomyopathy genes using multiplex
amplification and semiconductor next-generation sequencing. Circ J.
78:2963–2971. 2014. View Article : Google Scholar
|
|
9
|
Wu W, Lu CX, Wang YN, Liu F, Chen W, Liu
YT, Han YC, Cao J, Zhang SY and Zhang X: Novel phenotype-genotype
correlations of restrictive cardiomyopathy with myosin-binding
protein C (MYBPC3) gene mutations tested by next-generation
sequencing. J Am Heart Assoc. 4:42015.
|
|
10
|
Mango R, Luchetti A, Sangiuolo R,
Ferradini V, Briglia N, Giardina E, Ferrè F, Helmer Citterich M,
Romeo F, Novelli G, et al: Next heneration sequencing and linkage
analysis for the molecular diagnosis of a novel overlapping
syndrome characterized by hypertrophic cardiomyopathy and typical
electrical instability of Brugada syndrome. Circ J. 80:938–949.
2016. View Article : Google Scholar
|
|
11
|
Glotov AS, Kazakov SV, Zhukova EA,
Alexandrov AV, Glotov OS, Pakin VS, Danilova MM, Poliakova IV,
Niyazova SS, Chakova NN, et al: Targeted next-generation sequencing
(NGS) of nine candidate genes with custom AmpliSeq in patients and
a cardiomyopathy risk group. Clin Chim Acta. 446:132–140. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Millat G, Chanavat V and Rousson R:
Evaluation of a new NGS method based on a custom AmpliSeq library
and Ion Torrent PGM sequencing for the fast detection of genetic
variations in cardiomyopathies. Clin Chim Acta. 433:266–271. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang J, He J, Zeng XH, Ge SJ, Huang Y, Su
J, Ding XM, Yang JQ, Cao YJ, Chen H, et al: Genetic heterogeneity
of the β-globin gene in various geographic populations of Yunnan in
southwestern China. PLoS One. 10:e01229562015. View Article : Google Scholar
|
|
14
|
Zhao Y, Feng Y, Zhang YM, Ding XX, Song
YZ, Zhang AM, Liu L, Zhang H, Ding JH and Xia XS: Targeted
next-generation sequencing reveals hot spots and doubly
heterozygous mutations in chinese patients with familial
Cardiomyopathy. BioMed Res Int. 2015:5618192015.PubMed/NCBI
|
|
15
|
Zhao Y, Cao H, Song Y, Feng Y, Ding X,
Pang M, Zhang Y, Zhang H, Ding J and Xia X: Identification of novel
mutations including a double mutation in patients with inherited
cardiomyopathy by a targeted sequencing approach using the Ion
Torrent PGM system. Int J Mol Med. 37:1511–1520. 2016.PubMed/NCBI
|
|
16
|
Gersh BJ, Maron BJ, Bonow RO, Dearani JA,
Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, et
al American College of Cardiology Foundation/American Heart
Association Task Force on Practice Guidelines: 2011 ACCF/AHA
Guideline for the Diagnosis and Treatment of Hypertrophic
Cardiomyopathy: a report of the American College of Cardiology
Foundation/American Heart Association Task Force on Practice
Guidelines. Developed in collaboration with the American
Association for Thoracic Surgery, American Society of
Echocardiography, American Society of Nuclear Cardiology, Heart
Failure Society of America, Heart Rhythm Society, Society for
Cardiovascular Angiography and Interventions, and Society of
Thoracic Surgeons. J Am Coll Cardiol. 58:e212–e260. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sikkema-Raddatz B, Johansson LF, de Boer
EN, Almomani R, Boven LG, van den Berg MP, van Spaendonck-Zwarts
KY, van Tintelen JP, Sijmons RH, Jongbloed JD, et al: Targeted
next-generation sequencing can replace Sanger sequencing in
clinical diagnostics. Hum Mutat. 34:1035–1042. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tarabeux J, Zeitouni B, Moncoutier V,
Tenreiro H, Abidallah K, Lair S, Legoix-Né P, Leroy Q, Rouleau E,
Golmard L, et al: Streamlined ion torrent PGM-based diagnostics:
BRCA1 and BRCA2 genes as a model. Eur J Hum Genet. 22:535–541.
2014. View Article : Google Scholar :
|
|
19
|
Maron BJ, Maron MS and Semsarian C:
Genetics of hypertrophic cardiomyopathy after 20 years: Clinical
perspectives. J Am Coll Cardiol. 60:705–715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Jiang T, Piao C, Li X, Guo J, Zheng
S, Zhang X, Cai T and Du J: Screening Mutations of MYBPC3 in 114
Unrelated Patients with Hypertrophic Cardiomyopathy by Targeted
Capture and Next-generation Sequencing. Sci Rep. 5:114112015.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zou Y, Wang J, Liu X, Wang Y, Chen Y, Sun
K, Gao S, Zhang C, Wang Z, Zhang Y, et al: Multiple gene mutations,
not the type of mutation, are the modifier of left ventricle
hypertrophy in patients with hypertrophic cardiomyopathy. Mol Biol
Rep. 40:3969–3976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Millat G, Bouvagnet P, Chevalier P,
Dauphin C, Jouk PS, Da Costa A, Prieur F, Bresson JL, Faivre L,
Eicher JC, et al: Prevalence and spectrum of mutations in a cohort
of 192 unrelated patients with hypertrophic cardiomyopathy. Eur J
Med Genet. 53:261–267. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arai S, Matsuoka R, Hirayama K, Sakurai H,
Tamura M, Ozawa T, Kimura M, Imamura S, Furutani Y, Joho K, et al:
Missense mutation of the beta-cardiac myosin heavy-chain gene in
hypertrophic cardiomyopathy. Am J Med Genet. 58:267–276. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Van Driest SL, Vasile VC, Ommen SR, Will
ML, Tajik AJ, Gersh BJ and Ackerman MJ: Myosin binding protein C
mutations and compound heterozygosity in hypertrophic
cardiomyopathy. J Am Coll Cardiol. 44:1903–1910. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Richard P, Charron P, Carrier L, Ledeuil
C, Cheav T, Pichereau C, Benaiche A, Isnard R, Dubourg O, Burban M,
et al: EUROGENE Heart Failure Project: Hypertrophic cardiomyopathy:
Distribution of disease genes, spectrum of mutations, and
implications for a molecular diagnosis strategy. Circulation.
107:2227–2232. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kubo T, Kitaoka H, Okawa M, Baba Y, Hirota
T, Hayato K, Yamasaki N, Matsumura Y, Otsuka H, Arimura T, et al:
Genetic screening and double mutation in Japanese patients with
hypertrophic cardiomyopathy. Circ J. 75:2654–2659. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Emrahi L, Tabrizi MT, Gharehsouran J,
Ardebili SM and Estiar MA: Spectrum of MYBPC3 gene mutations in
patients with hypertrophic cardiomyopathy, reporting two novel
mutations from north-west of Iran. Clin Lab. 62:757–764. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chiou KR, Chu CT and Charng MJ: Detection
of mutations in symptomatic patients with hypertrophic
cardiomyopathy in Taiwan. J Cardiol. 65:250–256. 2015. View Article : Google Scholar
|
|
30
|
Honglin W: An investigation on
consanguineous marriage in nine ethnic groups of Yunnan province.
Acta Anthropologica Sinica. 4:0121998.
|
|
31
|
Lin M, Chu CC, Chang SL, Lee HL, Loo JH,
Akaza T, Juji T, Ohashi J and Tokunaga K: The origin of Minnan and
Hakka, the so-called 'Taiwanese', inferred by HLA study. Tissue
Antigens. 57:192–199. 2001. View Article : Google Scholar : PubMed/NCBI
|