Introduction
Cdc2, also known as cyclin dependent kinase 1
(CDK1), controls the cell cycle entry from the G2 to the M phase
and promotes the commencement of mitotic phase events (1), the abnormal activation of which
directly causes aberrant cell proliferation, and malignant
transformation and tumorigenesis in prostate cancer cells (2–4).
Cdc2 activation also depends on the phosphorylation of Thr161, and
CDC25-mediated dephosphorylation at Thr14 and Tyr-15, which
exhibits enzymatic activity when only phospho-Thr161 remains
(5).
Purvalanol A is a selective inhibitor of Cdc2, which
strongly inhibits Cdc2 kinase activity at a low concentration of 2
μM (6,7). Investigators have identified that
purvalanol A effectively suppresses Cdc2 activity and the
progression from the G2 phase to mitosis, which leads to the loss
of clonogenicity and cellular apoptosis in both MKN45 and MKN28
X-irradiated gastric cancer cells (8).
Oncoprotein 18 (Op18)/stathmin is a small molecule
weight phosphoprotein which is highly expressed in cancer cells.
Its main functions are to regulate the equilibrium of microtubule
(MT) dynamics and control cell cycle progression, which is closely
associated with the maintenance of tumor malignant phenotypes
(9–12). Op18/stathmin has 4 phosphoserine
sites (p-Ser16, p-Ser25, p-Ser38 and p-Ser63), which integrates and
relays various signals from intra- or extacellular stimuli through
phosphorylated inactivation and dephosphorylated activation
(13–15).
In a previous study, we found that Epstein-Barr
virus-specific protein-latent membrane protein 1 (LMP1) regulates
the Op18/stathmin signaling pathway by mediating Cdc2, which
accelerates cell cycle progression and promotes cell proliferation
(16). In another recent study of
ours, we confirmed that human NCI-H1299 non-small cell lung cancer
cells highly expressing Op18/stathmin were the most highly
resistant to taxol among 5 different cancer cells originating from
epithelia, including CNE1, Hep3B-2, MGC, MCF-7 and NCI-H1299
(17).
In this study, NCI-H1299 cells were employed to
clarify the association between Cdc2 signaling and taxol
resistance, and to elucidate the related molecular mechanisms.
Materials and methods
Cells and cell growth conditions
Both the CNE1 cells and NCI-H1299 cells were a kind
gift from Professor Ya Cao from the Cancer Research Institute of
Central South University and cultured in RPMI-1640 (Gibco-BRL,
Grand Island, NY, USA) with 10% fetal bovine serum (FBS; HyClone,
Logan, UT, USA), 100 IU/ml penicillin and 100 μg/ml
streptomycin at 37°C, 5% CO2.
CNE1 human cancer cells were testified to be the
most sensitive to taxol among 5 various epithelial-deriving tumor
cells in our previous study (17). Thus, they were used in this study
for a comparison to the NCI-H1299 cells.
Antibodies and reagents
The primary antibodies used were anti-stathmin (Cat.
no. 569391; Calbiochem, Billercia, CA, USA), anti-phosphoserine
(Cat. no. 618100; Zymed, San Francisco, CA, USA), anti-Cdc2
(sc-954; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
anti-phospho-Thr161-Cdc2 (Cat. no. 9114; Cell Signaling Technology,
Danvers, MA, USA), anti-cyclin B1 (sc-7393), anti-caspase-3
(sc-7272) (both from Santa Cruz Biotechnology, Inc.),
anti-caspase-8 (Cat. no. 9746), rabbit monoclonal anti-caspase-9
(Cat. no. 9502) (both from Cell Signaling Technology),
anti-phospho-stathmin [phospho-S16 (ab47328), S25 (ab194752), S38
(ab194757), S63 (ab76583); Abcam, Cambridge, MA, USA], anti-Bcl-2
(sc-492; Santa Cruz Biotechnology, Inc.), anti-extracellular
signal-regulated kinase 1 (ERK1; sc-93), anti-phospho-ERK1
(sc-7383) (both from Santa Cruz Biotechnology, Inc.), anti-β-actin
(Cat. no. A2228; Sigma, St. Louis, MO, USA). The secondary
antibodies used where horseradish peroxidase (HRP)-conjugated goat
anti-rabbit IgG (sc-2004) and rabbit anti-mouse IgG (sc-358914)
(both from Santa Cruz Biotechnology, Inc.).
Taxol (Santa Cruz Biotechnology, Inc.) and
purvalanol A (Calbiochem) were both purchased and dissolved in
dimethyl sulfoxide (DMSO) as mother solutions at −20°C for use.
Flow cytometric analysis
The cells were plated in 6-well plates, and
pre-treated with 0, 1 and 5 μM purvalanol A for 2 h when
they reached 80% confluency. This was followed by the addition of
100 nM taxol for 12 h. DMSO was used as a solvent control. The
cells were then rinsed with phosphate-buffered saline (PBS) and
digested with 0.25% trypsin.
The cells were cropped by centrifugation and stained
with a mixture of 5 μl propidium iodide (PI) and 10
μl Annexin V-FITC according to the protocol provided with
the Annexin V-FITC Apoptosis Detection kit (Nanjing Biobox Biotech.
Co, Ltd, Nanjing, China).
Cellular apoptosis was assessed by flow cytometry
(FCM) by a specialized agency (the Second Xiangya Hospital
Affiliated Central South University). All experiments were
performed in triplicate.
MTT assay
The cells at the logarithmic phase were seeded at
5,000 cells/well in 96-well plates and pre-treated with various
concentrations of purvalanol A (0, 1 and 5 μM) for 2 h,
followed by the addition of 100 nM taxol. Finally, 10 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl (MTT) were added at the
specified time points of 24, 48 and 72 h. The values of optical
density (OD) at a wavelength 570 nm were then detected using a
microplate reader (BioTek, Winooski, VT, USA). Cell proliferatoin
was calculated using the following formula: relative proliferation
rates (%) = (OD treatment/OD control) ×100%. Six parallel wells
were set in each group.
Colony formation assay
A total of 2,500 cells in a single suspension were
plated per well in a 6-well plate and divided into 6 groups,
including 3 co-treatment groups and 3 gradient groups of purvalanol
A alone. Cell growth was terminated when colonies were observed by
the naked eye after 2 weeks. Colonies containing over 50 cells were
counted using an inverted microscope, and images were acquired
using a DMCI microscope (Leica, Wetzlar, German) at ×200
magnification. All experiments were performed in triplicate.
Assessment of Cdc2 kinase activity
Assays to determine Cdc2 kinase activity were
performed according to the instructions provided with the MESACUP
Cdc2 kinase assay kit (code no. 5235; MBL, Nagoya, Japan). Cells
reaching 80% confluence were treated with various concentrations of
taxol (0, 10 and 100 nM) for 12 h, then lysed in a sample buffer.
The supernatants were then collected for the detection of Cdc2
kinase activity as described in our previous study (16). ELISA was performed according to
the instructions provided with the MESACUP Cdc2 Kinase Assay kit.
Briefly, 100 μl of cell extracts were transferred to a
microwell strip coated with monoclonal antibody 4A4 (4 parallel
wells/sample), incubated at 25°C for 60 min and washed 5 times.
This was followed by the addition of 100 μl peroxidase (POD)
conjugated streptavidin, and incubation at 25°C for 30 min and
washing 5 times. The cells were then treated with 100 μl POD
substrate solution for 5 min, and the reaction was then terminated
with 100 μl Stop Solution (20% phosphoric acid). The OD
value was read at a 490 nm wavelength using a microplate reader
(BioTek). All experiments were carried out in triplicate.
Western blot analysis
Following the removal of the supernatant, the cells
were lysed in cell lysis buffer (50 mM Tris-HCl pH 8.0, 1 mM EDTA,
2% SDS, 5 mM DTT and 10 mM PMSF). Total proteins (50 μg)
were then separated by 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose
membranes and incubated with the specific primary antibodies at 4°C
overnight, followed by the addition of HRP conjugated secondary
antibodies for 2 h at room temperature. An enhanced
chemiluminescence detection kit (Pierce, Rockford, IL, USA) was
applied for immunoblotting.
Immunoprecipitation (IP) - western blot
analysis
In brief, cell extracts were collected in IP lysis
buffer, as described in our previous studies (16,18). Immunoprecipitates of Op18/stathmin
were pulled down by excessive anti-stathmin antibody binding
magnetic immunobeads and separated by 10% SDS-PAGE.
anti-phosphorserine antibody was used to detect the total level of
phosphorylated Op18/stathmin by western blot analysis.
Statistical analysis
Statistical analysis was carried out using SPSS 17.0
statistical software. The specific statistical method applied was
the t-test, and a value of p<0.05 was considered to indicate a
statistically significant difference. All data are presented as the
means ± SD.
Results
Purvalanol A enhances the taxol-induced
apoptosis of NCI-H1299 cells
The cellular apoptotic ratios were 15.13, 19.65 and
31.46% in the NCI-H1299 cells treated with taxol in presence of 0,
1 and 5 μM purvalanol A, respectively. Purvalanol A
augmented taxol-induced cellular apoptosis in a concentration
dependent manner. The images of cellular growth revealed that a
greater number of translucent floating cells appeared in the medium
of the cells treated with purvalanol A (Fig. 1A).
In the cells treated with purvalanol A alone at
concentrations of 0, 1 and 5 μM, the cellular apoptotic
ratios were 5.44, 6.36 and 7.58%, respectively. These cells almost
reached complete confluence and there were only a few translucent
floating cells (Fig. 1B).
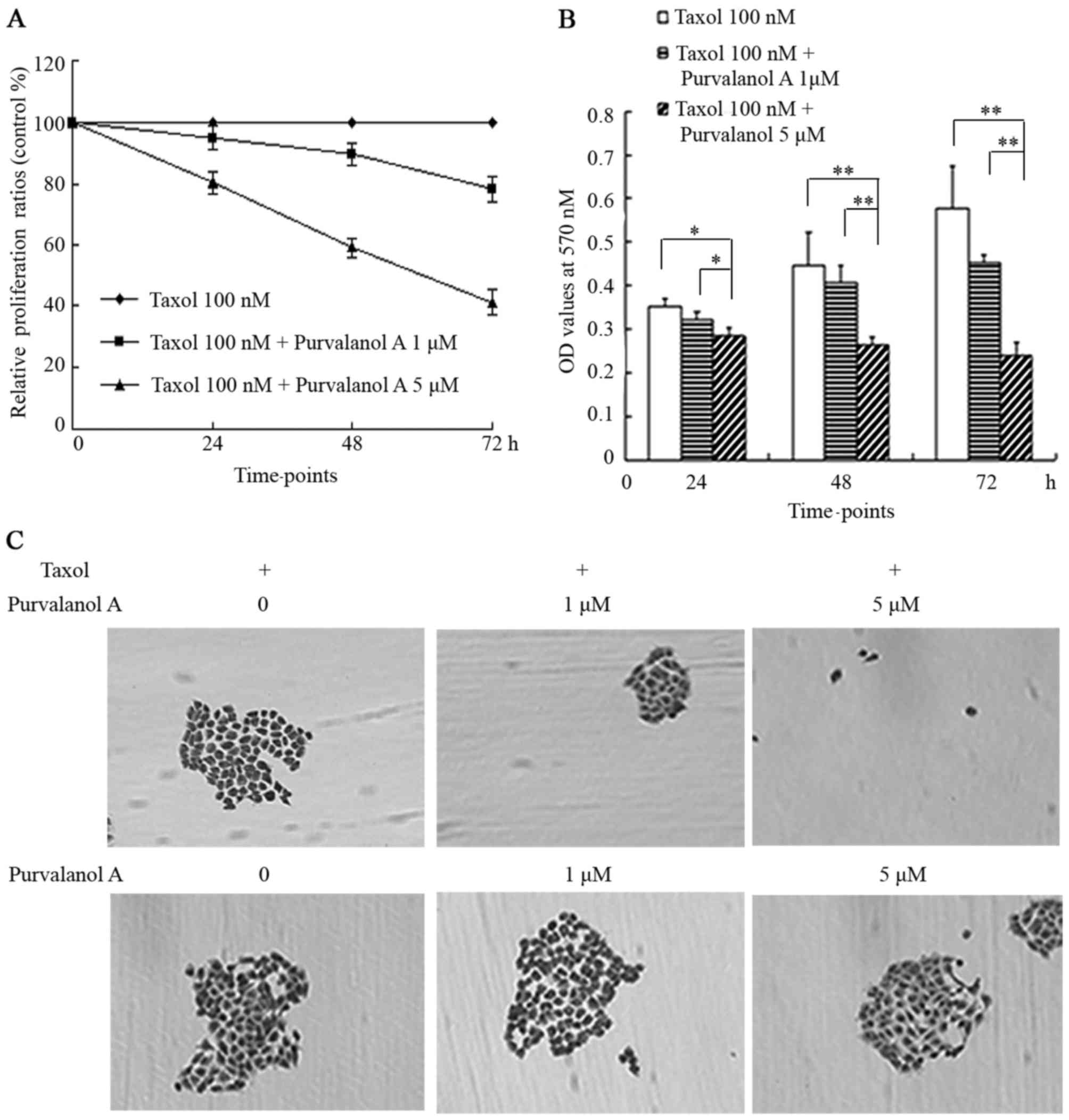
Purvalanol A enhances the inhibitory
effects of taxol on cellular proliferation and colony formation in
NCI-H1299 cells
Purvalanol A enhanced the inhibitory effects of
taxol on cellular proliferation in NCI-H1299 cells. In the cells
co-treated with 5 μM purvalanol A and taxol, the
representative curve steeply descended at the time points of 24, 48
and 72 h; the relative proliferation ratios were 80.53, 59.01 and
41.12%, respectively. In the cells co-treated with 1 μM
purvalanol A and taxol, the representative curve descended slowly,
with proliferation ratios of 94.98, 89.6 and 78.32% at 24, 48 and
72 h, respectively (Fig. 2A).
The histograms indicated that a statistically
significant difference existed between the cells co-treated with 5
μM purvalanol A and taxol, and the other 2 groups (taxol
only, and taxol + 1 μM purvalanol A) at 24 h (p<0.05),
while the differences became more and more significant with the
passing of time (p<0.01) (Fig.
2B).
Only a few and sparse small colonies appeared in the
group treated with 1 μM purvalanol A combined with taxol; no
visibly typical colonies were formed in the group co-treated with 5
μM purvalanol A and taxol, while a large number of colonies
appeared among the group treated with taxol only and in the 3
control groups treated with purvalanol A only at increasing
concentrations, which usually merged into a large colony with
ambiguous borders (Fig. 2C).
Co-treatment with purvalanol A and taxol
inhibits the expression of Op18/stathmin and phosphorylation
The expression of Op18/stathmin was weakened in the
NCI-H1299 cells treated with a combination of purvalanol A and
taxol; the synergistic inhibitory effects were the most evident in
the cells treated with a combination of 5 μM purvalanol A
and taxol in comparison with the other 2 groups (Fig. 3A). Treatment with taxol alone
slightly decreased the expression of Op18/stathmin at the
concentration of 100 nM; however, no notable alternation in the
expression of Op18/stathmin was observed in the cells treated with
purvalanol A at increasing concenrations (Fig. 3B).
IP analysis revealed that the total levels of
phospho-Op18/stathmin were notably decreased in the cells
co-treated with purvalanol A and taxol, compared with the controls
or the cells treated with taxol or purvalanol A alone (Fig. 3C).
Treatment with purvalanol A alone inhibited the
phosphorylation of Op18/stathmin at all 4 serine sites, including
Ser16, Ser25, Ser38 and Ser63 compared with the cells treated with
taxol alone. Treatment with both purvalanol A and taxol mainly
inhibited the phosphorylation of Op18/stathmin at Ser16 and Ser38
compared with the cells treated with purvalanol A alone; however,
the phosphorylation levels of Ser25 and Ser63 sites were similar
between the 2 groups (Fig.
3D).
Treatment with both purvalanol A and
taxol decreases the expression of Bcl-2 and initiates the
activation of caspase-3 and caspase-8
The expression and phosphorylation of ERK was not
altered in the cells treated with both purvalanol A and taxol.
However, combination treatment markedly inhibited the expression of
Bcl-2, with a greater decrease observed with increasing
concentrations of purvalanol A (Fig.
4A). No differences were observed in the expression of these
molecules in the cells treated with purvalanol A alone at
increasing concentrations (Fig.
4B).
Purvalanol A and taxol collectively increased the
expression levels of caspase-3 and caspase-8 with a greater
increase observed with increasing concentrations of purvalanol A.
No obvious changes were observed in the levels of caspase-9
(Fig. 4C). Similarly, treatment
with purvalanol A alone at increasing concentrations did not lead
to any alternations in the levels of caspase-3, caspase-8 and
caspase-9 in the NCI-H1299 cells (Fig. 4D).
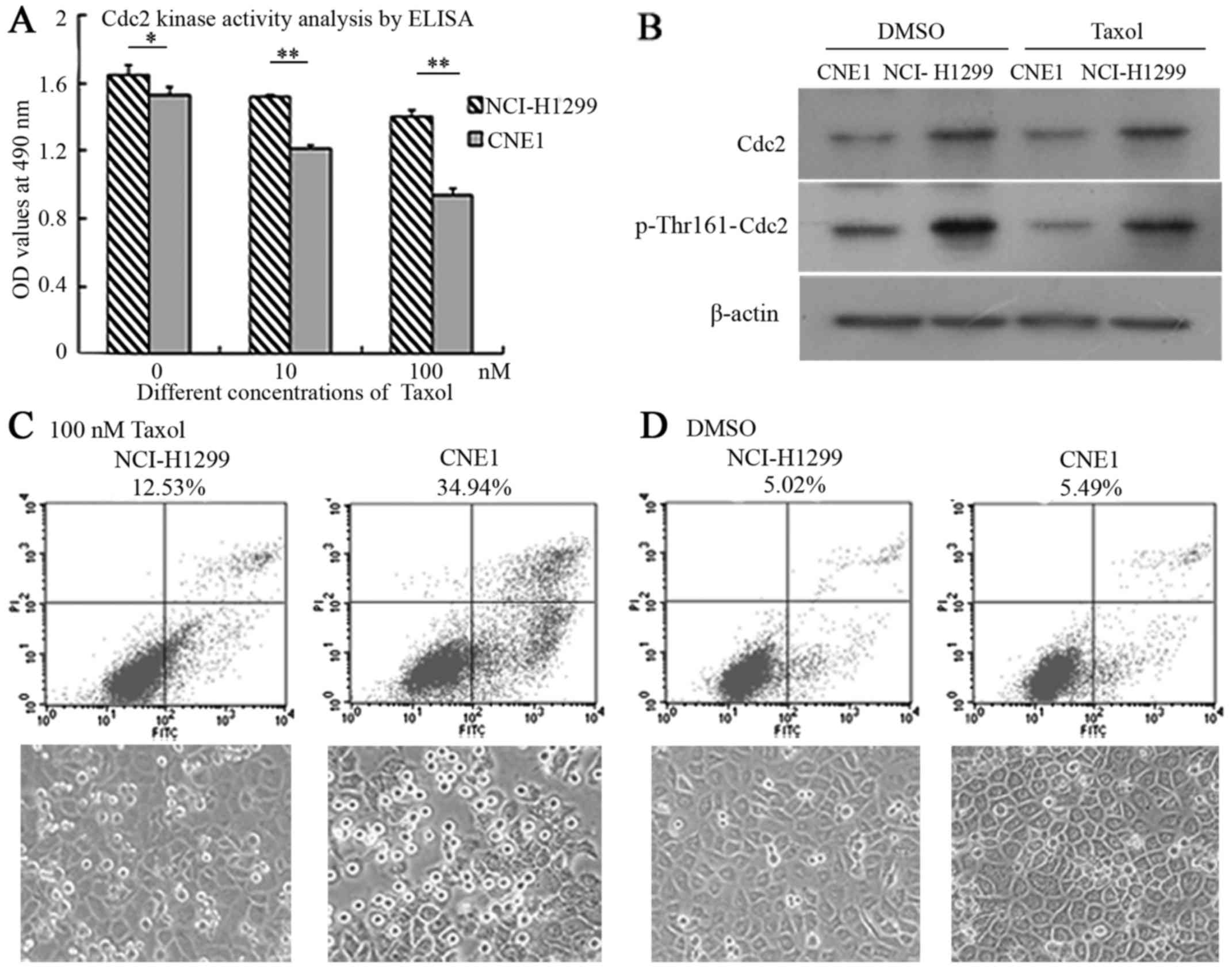
Cdc2 is positively associated with the
development of taxol resistance in different epithelia-derived
tumor cells
The histograms indicated that Cdc2 kinase activity
was markedly higher in the NCI-H1299 cells compared with the CNE1
cells before and after treatment with taxol. Taxol also
downregulated the activity of Cdc2 to a certain extent in a
concentration-dependant manner in both cell lines (NCI-H1299 and
CNE1), although this decrease in the levels Cdc2 was more evident
in the CNE1 cells in comparison to the NCI-H1299 cells; the
differences became more significant with the addition of Taxol
(p<0.01) (Fig. 5A).
The results of western blot analysis revealed that
the expression levels of Cdc2 and phosporylation at the Thr161 site
were markedly higher in the NCI-H1299 cells than in the CNE1 cells
before and after treatment with taxol. Treatment with taxol alone
did not affect the expression of Cdc2, but slightly decreased the
phosphorylation of Cdc2-Thr161 in both cell lines, which was
coincident with the analysis of Cdc2 kinase activity by
enzyme-linked immunosorbent assay (ELISA) (Fig. 5B).
FCM analysis revealed that the cellular apoptotic
ratios were 12.53 and 34.94% in the NCI-H1299 cells and CNE1 cells
treated with taxol, while the cellular apoptotic ratios were 5.02
and 5.49% in the controls treated with solvent DMSO; the status of
cell growth shown in the images correlated with the findings of
apoptotic analysis (Fig. 5C and
D).
Discussion
Taxol is an effective antitumor chemotherapeutic
agent derived from plants, which induces cell cycle arrest and
cellular apoptosis by promoting MT polymerization (19–21). With the wide application of taxol
in clinical practice, a large number of resistant tumors have
emerged; therefore, high doses of taxol are increasingly
prescribed, which results in severe side-effects. To date, no
antagonizing agents have been identified (22). To minimize the dosage of taxol,
the combined use of taxol with other traditional drugs has been
used in an attempt to improve taxol sensitivity (23,24). These strategies have achieved some
success, but also lead to new risks of acquiring multidrug
resistance.
In this study, we demonstrated that the Cdc2/CDK1
inhibitor, purvalanol A, effectively enhanced the sensitivity of
NCI-H1299 cells to taxol and inhibited cellular proliferation and
colony formation. Other studies have also confirmed that CDK1
inhibition significantly enhances drug-induced colony formation and
apoptosis in breast cancer cells and colon carcinoma cells and
glioma cells (25–27).
Op18/stathmin is a downstream molecular target of
Cdc2, and tumor cells which highly express Op18/Stathmin are
resistant to taxol (17,28). This study certified that the
combination of purvalanol A and taxol mainly inhibited the
expression of Op18/stathmin and phosphorylation at the Ser16 and
Ser38 sites, while purvalanol A alone uniformly inhibited the
phosphorylation of Op18/stathmin at all 4 serine sites. Other
studies have shown that Cdc2 predominantly phosphorylates
Op18/stathmin at Ser25 and Ser38 sites, which is partly different
from our results, which may implicate other inhibitory activities
of purvalanol A (6,29).
Bcl-2 is an anti-apoptotic factor involved in the
resistance of conventional drugs. The overexpression of Bcl-2
potentially induces the development of taxol resistance in lung
cancer cells (30). ERK is also
an upstream kinase of Op18/stathmin, and ERK-mediated Op18/stathmin
signaling also complicates to cell cycle progression and taxol
sensitivity (18,31). Cellular apoptosis is closely
associated with the activation of caspase-3, caspase-8 and
caspase-9. As previously demonstrated, the blocking of ERK and
taxol jointly induced exogenous cellular apoptosis via the
upregulation of the expression of caspase-3 and caspase-9 in
NCI-H1299 cells; the activation of caspase-3 and caspase-8 promoted
cellular apoptosis and autophagy in A549 human lung cancer cells
exposed to Paris polyphylla steroidal saponins (PPSS);
paclitaxel and Op18 silencing collectively induced cell death
through initiating caspase-3 and caspase-9 activations in
nasopharyngeal carcinoma CNE1 cells (18,32,33). This study indicated that
co-treatment with purvalanol A and taxol decreased the expression
of Bcl-2 and initiated the activation of the extrinsic cell death
pathway through the activation of caspase-3 and caspase-8, but did
not markedly affect the expression of ERK and phosphorylation.
We further found that both the expression of Cdc2
and the level of p-Thr161-Cdc2 was markedly higher in the NCI-H1299
cells in contrast with the CNE1 cells, which was consistent with
the detection of Cdc2 kinase activity, while the NCI-H1299 cells
with a high Cdc2 kinase activity exhibited obvious resistance to
taxol in comparison with the relatively sensitive CNE1 cells, which
implied that Cdc2 kinase activity is positively associated with the
development of taxol resistance in different epithelia-derived
cancer cells. Another study also demonstrated that a high
expression of Cdc2 was negatively associated with the curative
effects of chemotherapeutics and was a poor prognostic factor in
epithelial-derived ovarian cancer and laryngeal squamous cell
carcinoma (34,35).
In conclusion, the findings of our study suggest
that purvalanol A enhances the cytotoxic effects of taxol through
Op18/stathmin in non-small cell lung cancer. Our findings may prove
to be helpful in reducing the doses of taxol applied clinically and
to alleviate the side-effects.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (no. 81272274) and the
Key Project of Hunan Province Scientific Research of Colleges and
Universities (no. 12A018). The authors would like to thank Dr Liu
Sufang from the Second Xiangya Hospital Affiliated Central South
University for cellular apoptosis detection and Dr Tao Yongguang
from the Cancer Research Institute of Central South University for
providing valuable suggestions for the manuscript.
References
|
1
|
Zhao XF and Gartenhaus RB: Phospho-p70S6K
and cdc2/cdk1 as therapeutic targets for diffuse large B-cell
lymphoma. Expert Opin Ther Targets. 13:1085–1093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu P, Kao TP and Huang H: CDK1 promotes
cell proliferation and survival via phosphorylation and inhibition
of FOXO1 transcription factor. Oncogene. 27:4733–4744. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pérez de Castro I, de Cárcer G and
Malumbres M: A census of mitotic cancer genes: New insights into
tumor cell biology and cancer therapy. Carcinogenesis. 28:899–912.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tripathi A and Chaube SK: Reduction of
phosphorylated Thr-161 Cdk1 level participates in
roscovitine-induced Fas ligand-mediated apoptosis in rat eggs
cultured in vitro. In Vitro Cell Dev Biol Anim. 51:174–182. 2015.
View Article : Google Scholar
|
|
6
|
Gray NS, Wodicka L, Thunnissen AM, Norman
TC, Kwon S, Espinoza FH, Morgan DO, Barnes G, LeClerc S, Meijer L,
et al: Exploiting chemical libraries, structure, and genomics in
the search for kinase inhibitors. Science. 281:533–538. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mori Y, Inoue Y, Taniyama Y, Tanaka S and
Terada Y: Phosphorylation of the centrosomal protein, Cep169, by
Cdk1 promotes its dissociation from centrosomes in mitosis. Biochem
Biophys Res Commun. 468:642–646. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Iizuka D, Inanami O, Kashiwakura I and
Kuwabara M: Purvalanol A enhances cell killing by inhibiting
up-regulation of CDC2 kinase activity in tumor cells irradiated
with high doses of X rays. Radiat Res. 167:563–571. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu HP, Li CF, Lee SW, Wu WR, Chen TJ,
Chang KY, Liang SS, Tsai CJ and Shiue YL: Overexpression of
stathmin 1 confers an independent prognostic indicator in
nasopharyngeal carcinoma. Tumour Biol. 35:2619–2629. 2014.
View Article : Google Scholar
|
|
10
|
Baquero MT, Hanna JA, Neumeister V, Cheng
H, Molinaro AM, Harris LN and Rimm DL: Stathmin expression and its
relationship to microtubule-associated protein tau and outcome in
breast cancer. Cancer. 118:4660–4669. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Akhtar J, Wang Z, Jiang WP, Bi MM and
Zhang ZP: Stathmin overexpression identifies high risk for
lymphatic metastatic recurrence in pN0 esophageal squamous cell
carcinoma patients. J Gastroenterol Hepatol. 29:944–950. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wegiel B, Wang Y, Li M, Jernigan F and Sun
L: Novel indolyl-chalcones target stathmin to induce cancer cell
death. Cell Cycle. 15:1288–1294. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yip YY, Yeap YY, Bogoyevitch MA and Ng DC:
Differences in c-Jun N-terminal kinase recognition and
phosphorylation of closely related stathmin-family members. Biochem
Biophys Res Commun. 446:248–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu Y, Liu C, Xu YF, Cheng H, Shi S, Wu CT
and Yu XJ: Stathmin destabilizing microtubule dynamics promotes
malignant potential in cancer cells by epithelial-mesenchymal
transition. Hepatobiliary Pancreat Dis Int. 13:386–394. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akhtar J, Wang Z, Yu C, Li CS, Shi YL and
Liu HJ: STMN-1 is a potential marker of lymph node metastasis in
distal esophageal adenocarcinomas and silencing its expression can
reverse malignant phenotype of tumor cells. BMC Cancer. 14:282014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin X, Liu S, Luo X, Ma X, Guo L, Li L, Li
Z, Tao Y and Cao Y: EBV-encoded LMP1 regulates Op18/stathmin
signaling pathway by cdc2 mediation in nasopharyngeal carcinoma
cells. Int J Cancer. 124:1020–1027. 2009. View Article : Google Scholar
|
|
17
|
Lin X, Liao Y, Xie J, Liu S, Su L and Zou
H: Op18/stathmin is involved in the resistance of Taxol among
different epithelial carcinoma cell lines. Cancer Biother
Radiopharm. 29:376–386. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin X, Liao Y, Chen X, Long D, Yu T and
Shen F: Regulation of oncoprotein 18/stathmin signaling by ERK
concerns the resistance to Taxol in nonsmall cell lung cancer
cells. Cancer Biother Radiopharm. 31:37–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Laurie SA, Solomon BJ, Seymour L, Ellis
PM, Goss GD, Shepherd FA, Boyer MJ, Arnold AM, Clingan P, Laberge
F, et al: Randomised, double-blind trial of carboplatin and
paclitaxel with daily oral cediranib or placebo in patients with
advanced non-small cell lung cancer: NCIC Clinical Trials Group
study BR29. Eur J Cancer. 50:706–712. 2014. View Article : Google Scholar
|
|
20
|
Wang T, Lv JH, Zhang XF, Li CJ, Han X and
Sun YJ: Tissue inhibitor of metalloproteinase-1 protects MCF-7
breast cancer cells from paclitaxel-induced apoptosis by decreasing
the stability of cyclin B1. Int J Cancer. 126:362–370. 2010.
View Article : Google Scholar
|
|
21
|
Feng W, Xiaoyan X, Xuan Y, Xiangke L,
Zichang Y, Ran Z, Liuxing W and Qingxia F: Silencing
stathmin-modulating efficiency of chemotherapy for esophageal
squamous cell cancer with paclitaxel. Cancer Gene Ther. 22:115–121.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yared JA and Tkaczuk KH: Update on taxane
development: New analogs and new formulations. Drug Des Devel Ther.
6:371–384. 2012.PubMed/NCBI
|
|
23
|
Chen LK, Liang Y, Yang QY, Xu F, Zhou NN,
Xu GC, Liu GZ and Wei WD: Triplet platinum-based combination
sequential chemotherapy improves survival outcome and quality of
life of advanced non-small cell lung cancer patients. Asian Pac J
Cancer Prev. 13:1863–1867. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee HH, Ye S, Li XJ, Lee KB, Park MH and
Kim SM: Combination treatment with paclitaxel and doxorubicin
inhibits growth of human esophageal squamous cancer cells by
inactivation of Akt. Oncol Rep. 31:183–188. 2014.
|
|
25
|
Kang J, Sergio CM, Sutherland RL and
Musgrove EA: Targeting cyclin-dependent kinase 1 (CDK1) but not
CDK4/6 or CDK2 is selectively lethal to MYC-dependent human breast
cancer cells. BMC Cancer. 14:322014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hayashi T, Adachi K, Ohba S and Hirose Y:
The Cdk inhibitor flavopiridol enhances temozolomide-induced
cytotoxicity in human glioma cells. J Neurooncol. 115:169–178.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Meng F, Bhupathi D, Sun JD, Liu Q,
Ahluwalia D, Wang Y, Matteucci MD and Hart CP: Enhancement of
hypoxia-activated prodrug TH-302 anti-tumor activity by Chk1
inhibition. BMC Cancer. 15:4222015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Balasubramani M, Nakao C, Uechi GT,
Cardamone J, Kamath K, Leslie KL, Balachandran R, Wilson L, Day BW
and Jordan MA: Characterization and detection of cellular and
proteomic alterations in stable stathmin-overexpressing,
Taxol-resistant BT549 breast cancer cells using offgel IEF/PAGE
difference gel electrophoresis. Mutat Res. 722:154–164. 2011.
View Article : Google Scholar :
|
|
29
|
Chen PW, Lin SJ, Tsai SC, Lin JH, Chen MR,
Wang JT, Lee CP and Tsai CH: Regulation of microtubule dynamics
through phosphorylation on stathmin by Epstein-Barr virus kinase
BGLF4. J Biol Chem. 285:10053–10063. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Han ZX, Wang HM, Jiang G, Du XP, Gao XY
and Pei DS: Overcoming paclitaxel resistance in lung cancer cells
via dual inhibition of stathmin and Bcl-2. Cancer Biother
Radiopharm. 28:398–405. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin X, Tang M, Tao Y, Li L, Liu S, Guo L,
Li Z, Ma X, Xu J and Cao Y: Epstein-Barr virus-encoded LMP1
triggers regulation of the ERK-mediated Op18/stathmin signaling
pathway in association with cell cycle. Cancer Sci. 103:993–999.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He H, Sun YP, Zheng L and Yue ZG:
Steroidal saponins from Paris polyphylla induce apoptotic cell
death and autophagy in A549 human lung cancer cells. Asian Pac J
Cancer Prev. 16:1169–1173. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu Y, Tang M, Wu Y, Weng X, Yang L, Xu W,
Yi W, Gao J, Bode AM, Dong Z, et al: A combination of paclitaxel
and siRNA-mediated silencing of Stathmin inhibits growth and
promotes apoptosis of nasopharyngeal carcinoma cells. Cell Oncol
(Dordr). 37:53–67. 2014. View Article : Google Scholar
|
|
34
|
Xi Q, Huang M, Wang Y, Zhong J, Liu R, Xu
G, Jiang L, Wang J, Fang Z and Yang S: The expression of CDK1 is
associated with proliferation and can be a prognostic factor in
epithelial ovarian cancer. Tumour Biol. 36:4939–4948. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang JQ, Liu HX, Liang Z, Sun YM and Wu M:
Over-expression of p53, p21 and Cdc2 in histologically negative
surgical margins is correlated with local recurrence of laryngeal
squamous cell carcinoma. Int J Clin Exp Pathol. 7:4295–4302.
2014.PubMed/NCBI
|