Introduction
Type 2 diabetes is one of the most prevalent chronic
diseases worldwide and has serious social and health consequences,
and poses a heavy economic burden. Its clinical characteristics
include insulin resistance, pancreatic β-cell dysfunction and
reduced β-cell numbers (1). In
the pathogenesis of type 2 diabetes, high blood glucose,
inflammatory cytokines, high free fatty acids (FFAs) and amyloid
deposits are the important factors in the progression of diabetes,
all of which lead to β-cell apoptosis (2). The identification of the mechanisms
responsible for β-cell apoptosis are necessary in order to
understand the pathogenesis and to aid in the development of
effective treatments for patients with type 2 diabetes.
Recent studies have demonstrated that advanced
glycation end products (AGEs) may promote β-cell apoptosis in the
pathogenesis of type 2 diabetes (1,3–5).
AGEs are irreversible, complex and ultimately form after a series
of non-enzymatic reactions of proteins, lipids and reducing
glucoses. The production is accelerated when blood glucose is high,
thereby increasing the accumulation of AGEs in the body (6,7).
Previous studies have demonstrated that AGEs are closely related to
diabetic microangiopathy (7–10).
After AGEs bind to the receptor for AGEs (RAGE) on endothelial
cells, they activate the signaling pathways of glycogen synthase
kinase 3β (GSK3β), p38 mitogen-activated protein kinase (p38 MAPK),
extracellular signal-regulated kinase 1 and 2 (ERK1/2), c-Jun
amino-terminal kinase (JNK) and nuclear factor-κB (NF-κB), all of
which lead to endothelial cell dysfunction and diabetic vascular
disease (11–15).
Recent studies have also demonstrated that AGEs play
an important role in β-cell failure. The stimulation of AGEs in
in vitro and in vivo models has been shown to
directly cause the apoptosis of β-cells (3,6,16–18). AGEs stimulate reactive oxygen
species (ROS) generation, and, mediated by their receptor (RAGE),
induce β-cell apoptosis (3,16).
However, these above-mentioned studies have not fully elucidated
the molecular mechanisms of action of AGEs in β-cells. Therefore,
the roles of AGEs in β-cell apoptosis and their mechanisms of
action warrant further investigation.
Tribbles homolog 3 (TRB3) is one of the family
members of tribble homologous proteins. It inhibits mitosis and is
a regulatory factor of the protein kinase B (Akt) pathway (19). Through the inhibition of Akt
activity, TRB3 negatively regulates the insulin-signaling pathway
(20). Our previous studies
demonstrated that TRBs play an important role in β-cell apoptosis.
High blood glucose, high fat and endoplasmic reticulum (ER) stress
upregulate TRB3 expression, which mediates β-cell apoptosis
(21–23). The identification of TRB3
participation in AGE-induced β-cell apoptosis is worthy of
investigation. Studies on cardiomyocytes, epithelial cells and
retinal diabetic nephropathy have shown that the isoform of protein
kinase C (PKC) and PKC β2 (PKCβ2) plays an important role in
AGE-mediated cell damage and kidney damage. By increasing PKCβ2
expression, AGEs enhance PKCβ2 activity, as well as the effects and
displacement of PKCβ2, increasing ROS formation, which ultimately
causes oxidative damage (24–27). Our previous study demonstrated
that TRB3 activated PKCδ and was involved in high-fat-mediated
β-cell apoptosis (22). In this
study, we focused on AGE-mediated β-cell apoptosis. We also
determined whether TRB3 triggered the activation and isoform(s) of
PKC, and whether it mediated the damaging effects of AGEs.
Materials and methods
Cell culture
The rat insulinoma cell line, INS-1 (a gift from Dr
Haiyan Wang, University of Geneva, Geneva, Switzerland), was
maintained in RPMI-1640 containing 10% fetal bovine serum (FBS)
(both from Life Technologies, Waltham, MA, USA), 10 mM HEPES, 2 mM
glutamine and 1 mM sodium pyruvate (all from Sigma-Aldrich, St.
Louis, MO, USA), 50 µM β-mercaptoethanol, 100 U/ml
penicillin and 100 µg/ml streptomycin in an incubator
containing 5% CO2 at 37°C. For the experiments, the
INS-1 cells were cultured with or without 200 µg/ml AGEs
(ab51995; Abcam, Cambridge, MA, USA) for 48 h and collected for
further examination following treatment. INS-1 cells were
pre-incubated with PKC inhibitor, LY-333531 (10 µM) (2362;
Axon Medchem, Groningen, The Netherlands), for 30 min and then
co-treated with AGEs for 48 h.
RNA interference
Lipofectamine 2000 (Invitrogen, Waltham, MA, USA)
was used to transfect TRB3 small interfering RNA (siRNA siTRB3;
purchased from GenePharma Co., Ltd., Shanghai, China) and the
negative control small interference RNA (siNC) and into the INS-1
cells in accordance with the manufacturer's instructions. Target
gene sequences were described in our previous study (23).
Detection of apoptosis
TUNEL staining and flow cytometry were used to
detect the apoptosis of INS-1 cells. A TUNEL kit (Roche
Diagnostics, Indianapolis, IN, USA) was used to detect the
apoptosis of the INS-1 cells seeded in 96-well microplates
following the individual treatments strictly according to the
manufacturer's instructions. Positively stained cells were counted
under a light microscope (DMI6000 B; Leica Microsystems, Wetzlar,
Germany). The apoptotic rate of the INS-1 cells was examined by
flow cytometry using an in situ cell apoptosis detection kit
(BD Biosciences, San Diego, CA, USA), while strictly adhering to
the manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the INS-1 cells after
the corresponding treatments using an RNA extraction kit (Qiagen,
Hilden, Germany). Two micrograms of total RNA were used to
synthesize the cDNA in a reverse transcription reaction (reverse
transcriptase was purchased from Promega, Madison, WI, USA). The
RT-PCR reaction and data were analyzed as previously described
(28). The MyiQ real-time PCR
thermal cycler and SYBR-Green PCR Master Mix kit (both from Bio-Rad
Laboratories, Inc., Hercules, CA, USA) were used for the qPCR
analyses. Target genes were quantified using MyiQ system software.
The specific sequences of the primers used in this study were as
follows: β-actin forward, 5′-GACATCCGTAAAGACCTCTATGCC-3′ and
reverse, 5′-ATAGAGCCACCAATCCACACAGAG-3′; RAGE forward,
5′-GGAAGGACTGAAGCTTGGAAGG-3′ and reverse,
5′-TCCGATAGCTGGAAGGAGGAGT-3′; TRB3 forward,
5′-TGTCTTCAGCAACTGTGAGAGGACGAAG-3′ and reverse,
5′-GTAGGATGGCCGGGAGCTGAGTATC-3′; nicotinamide adenine dinucleotide
phosphate (NADPH) oxidase 4 (NOX4 forward,
5′-TAGCTGCCCACTTGGTGAACG-3′ and reverse,
5′-TGTAACCATGAGGAACAATACCACC-3′.
Western blot analysis of protein
expression
Following the corresponding treatments of the INS-1
cells, all cellular proteins were lysed in RIPA lysis buffer (Roche
Diagnostics) containing protease inhibitors and the concentration
was measured using a BCA protein assay kit (Beyotime Institute of
Biotechnology, Shanghai, China). Total proteins (20–40
µg)were separated by SDS-polyacrylamide gel electrophoresis
(SDS-PAGE). The separated proteins were then transferred onto a
PVDF membrane followed by blocking the non-specific antigen and
incubating with the corresponding primary antibody overnight. The
primary antibodies used in this study were: a mouse anti-rat
β-actin antibody (A5316; 1:20,000) and a rabbit anti-rat RAGE
antibody (R5278; 1:1,000) (both from Sigma-Aldrich); a rabbit
anti-rat PKCβ2 antibody (07-873-I; 1:1,000) and a mouse anti-rat
TRB3 antibody (ST1032; 1:1,000) (both from Calbiochem, Billerica,
MA, USA), and a rabbit anti-rat NOX4 antibody (ab133303; 1:1,000;
Abcam). The secondary antibodies used in this study were a goat
anti-mouse IgG antibody (A3682) and a goat anti-rabbit IgG antibody
(A0545) (1:20,000; both from Sigma-Aldrich). An analysis of the
protein bands was performed using Quantity One gel analysis
software (Bio-Rad Laboratories, Inc.).
Detection of ROS levels
ROS levels in the INS-1 cells cultured in 96-well
microplates following the corresponding treatments were measured
using a ROS detection assay kit (Shanghai Genmed Gene
Pharmaceutical Technology Co., Ltd., Shanghai, China) with strict
adherence to the manufacturer's instructions. A fluorescence
detection microplate reader was used to measure the fluorescence
intensity of the assay.
Statistical analysis
In this study, data are presented as the means ±
standard error of the mean (means ± SEM). SPSS 16.0 software (SPSS,
Inc., Chicago, IL, USA) was used for statistical analysis. A
comparison between 2 groups was performed using the t-test.
Comparisons among groups were performed using analysis of variance
(ANOVA). A value of P<0.05 was considered to indicate a
statistically significant difference.
Results
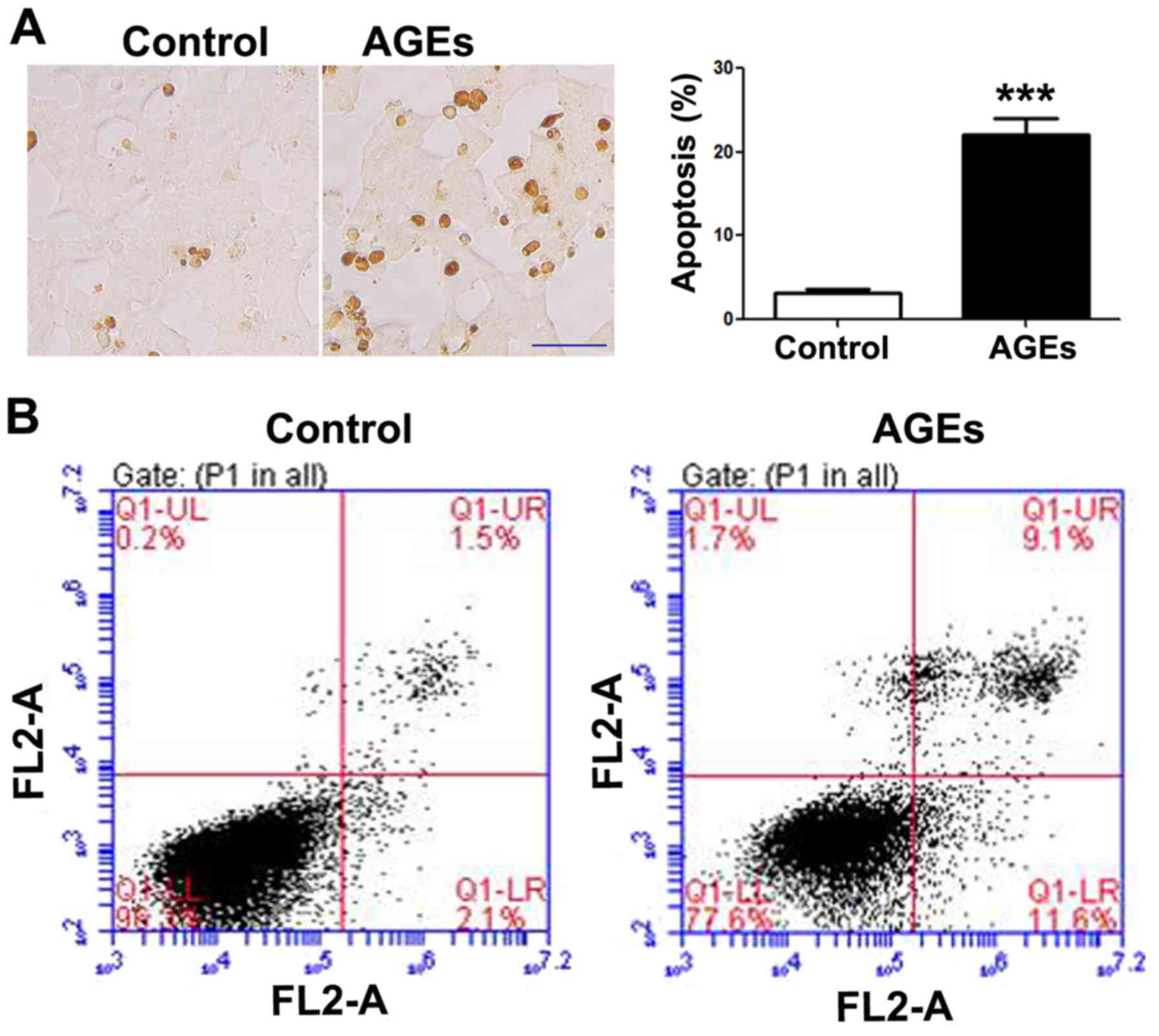
AGEs induce the apoptosis of INS-1
cells
Following exposure to the AGEs (200 µg/ml)
for 48 h, apoptosis was increased in the INS-1 cells as compared to
the control group, as shown by TUNEL staining (Fig. 1A) and flow cytometry (Fig. 1B). A statistically significant
difference in INS-1 cell apoptosis was observed between the
AGE-treated group and the control group (untreated group).
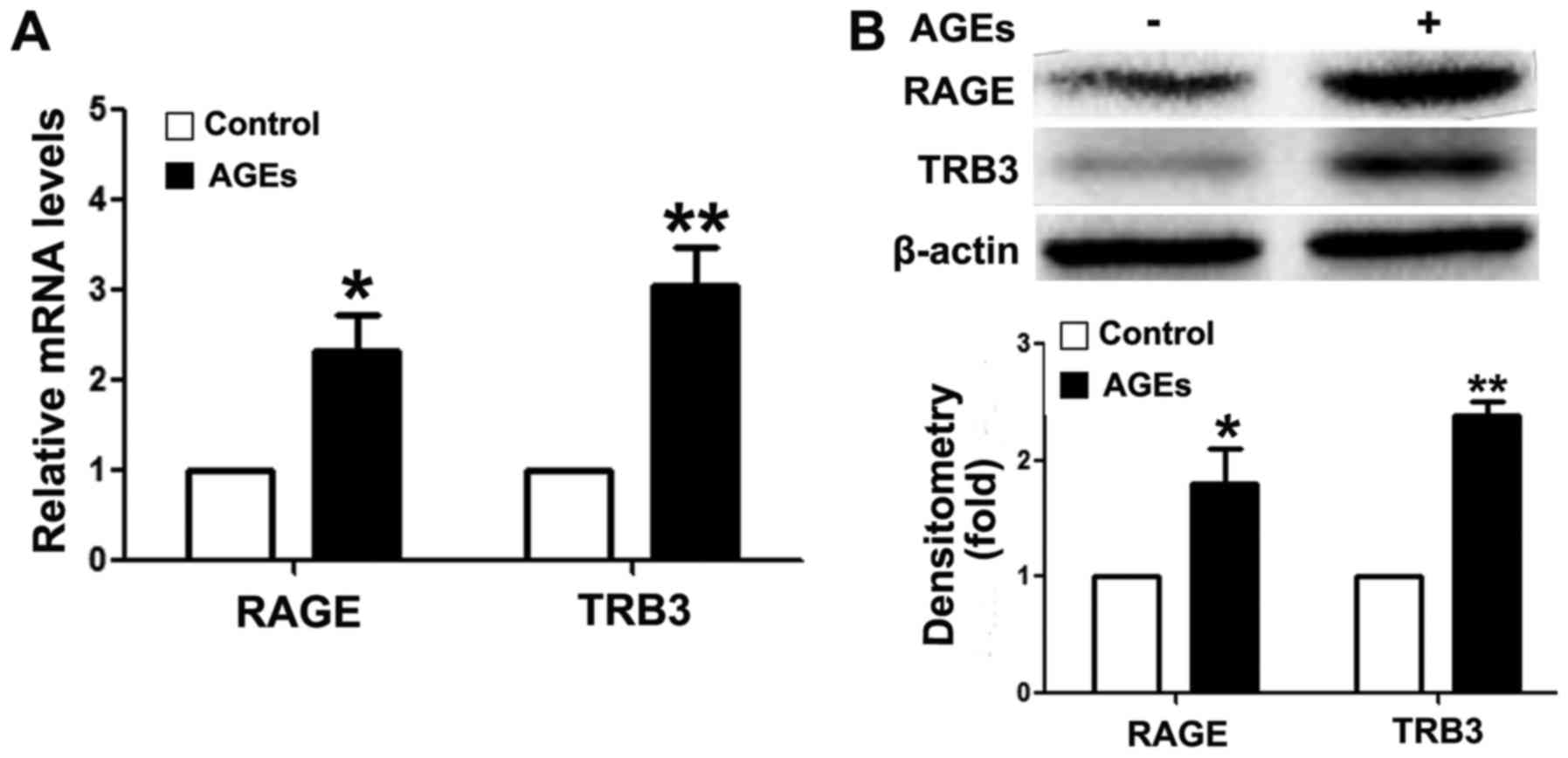
AGEs upregulate intracellular TRB3
expression in INS-1 cells
To analyze the mechanism of action of AGEs, we first
detected RAGE expression in INS-1 cells following exposure to AGEs.
As shown in Fig. 2, the mRNA
(Fig. 2A) and protein expression
(Fig. 2B) levels of RAGE were
upregulated following exposure to AGEs, suggesting that AGEs
mediated the apoptosis of INS-1 cells through RAGE. These findings
further validate the results of previous studies (3,9).
In addition, AGEs upregulated intracellular TRB3 expression levels
at the mRNA and protein level (Fig.
2).
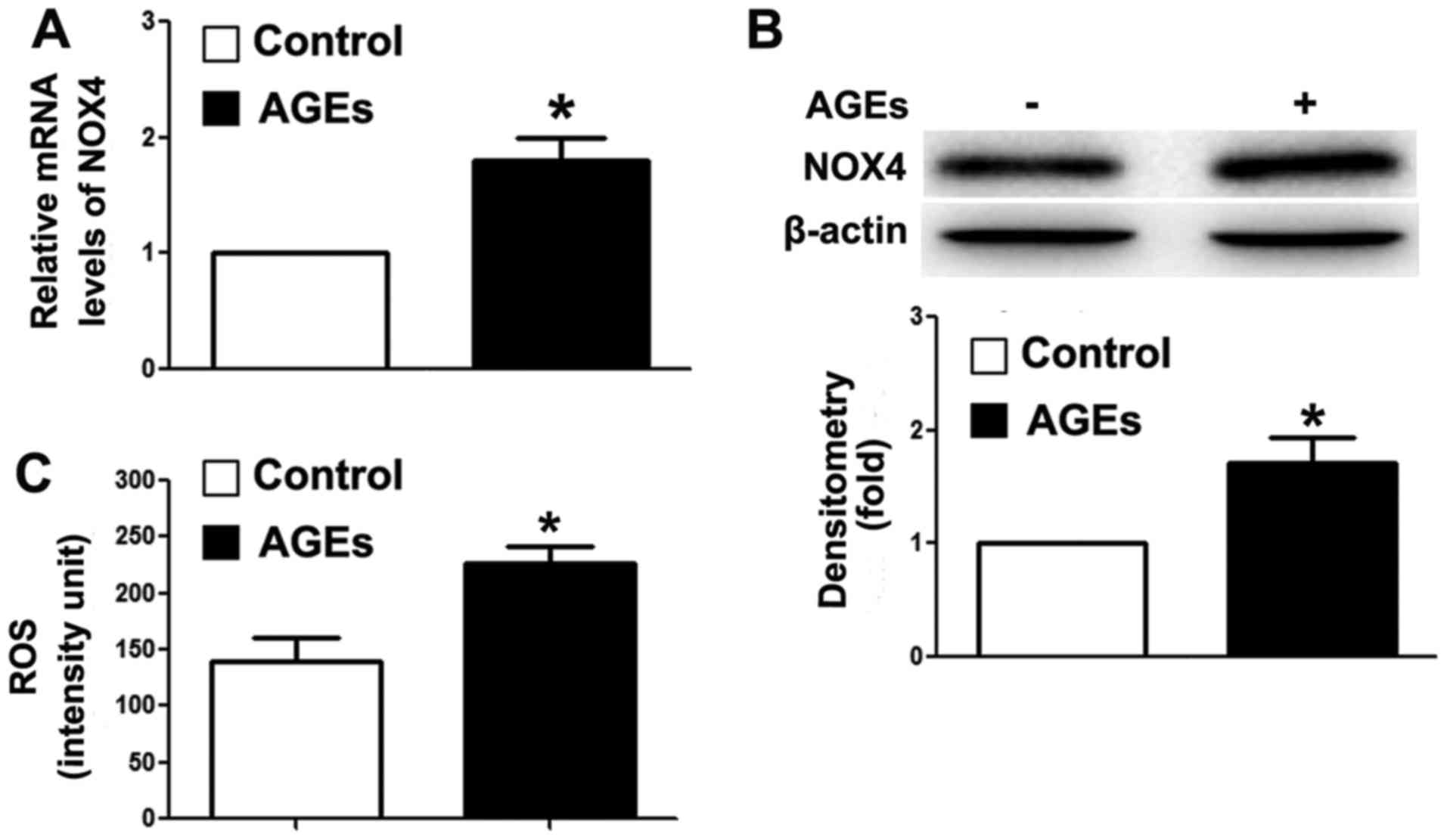
AGEs increase intracellular ROS
levels
Our previous study demonstrated that the
overexpression of TRB3 facilitated high-glucose-induced oxidative
stress (21). Thus, in this
study, we detected intracellular NOX4 expression and ROS levels. As
shown in Fig. 3A and B, AGEs
upregulated the mRNA and protein expression levels of NOX4. NOX4 is
a major enzyme for the synthesis of intracellular ROS (39). In this study, we detected an
increase in intracellular ROS levels in the cells following
exposure to AGEs (Fig. 3C). Our
findings indicated that AGEs promoted ROS synthesis, and further
induced INS-1 cell damage and apoptosis through TRB3.
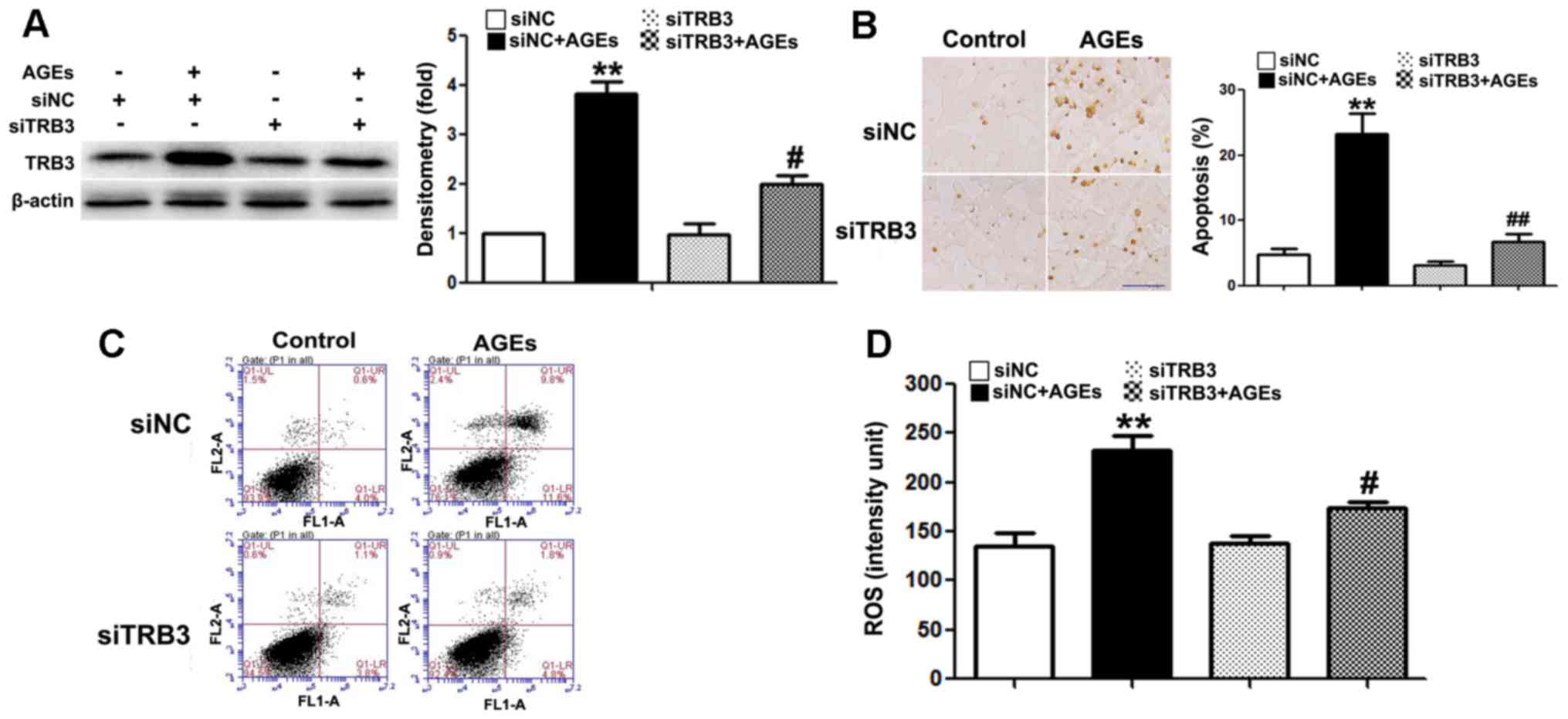
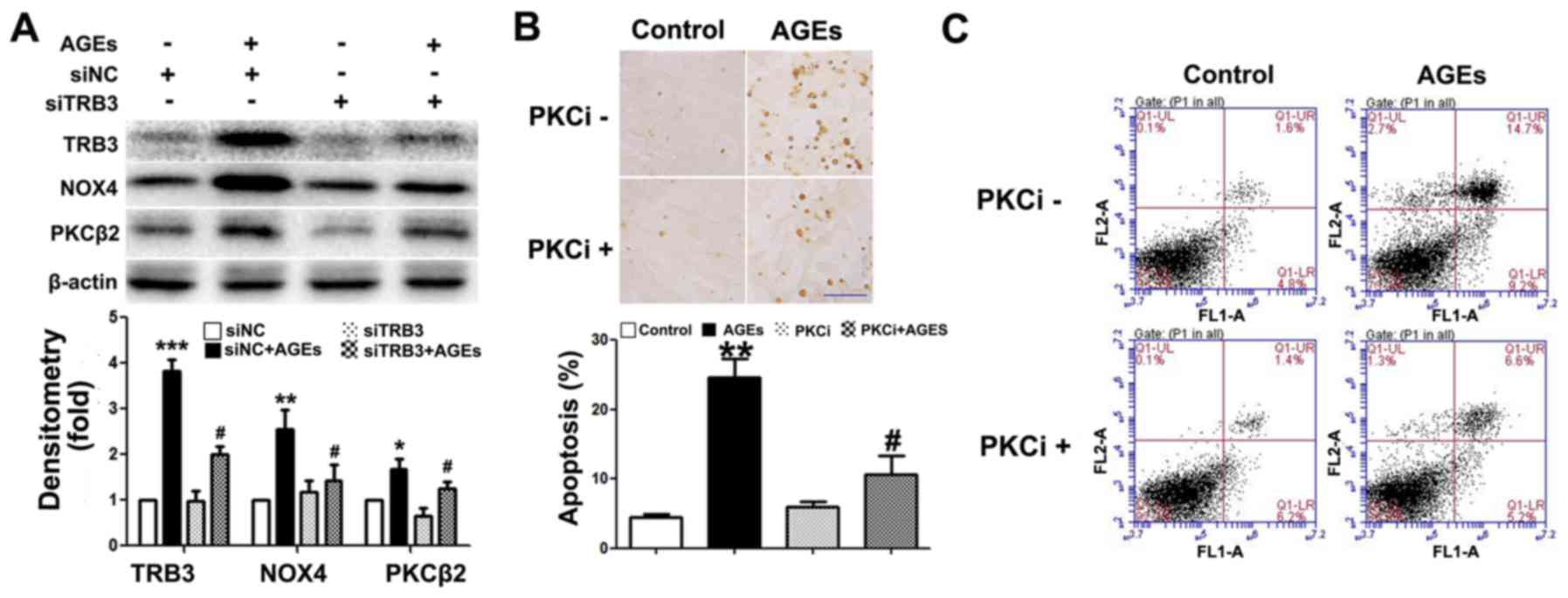
The silencing of TRB3 expression by siRNA
suppresses AGE-induced ROS synthesis and the apoptosis of INS-1
cells
To further determine whether TRB3 participates in
AGE-induced cell damage and apoptosis, we knocked down the
expression of TRB3 in INS-1 cells using siRNA (Fig. 4A). Both AGE-induced cell apoptosis
(Fig. 4B and C) and the
intracellular ROS levels were significantly reduced in the cells in
which TRB3 was knocked down (Fig.
4D). This result suggested that TRB3 is involved in AGE-induced
oxidative damage and the apoptosis of INS-1 cells.
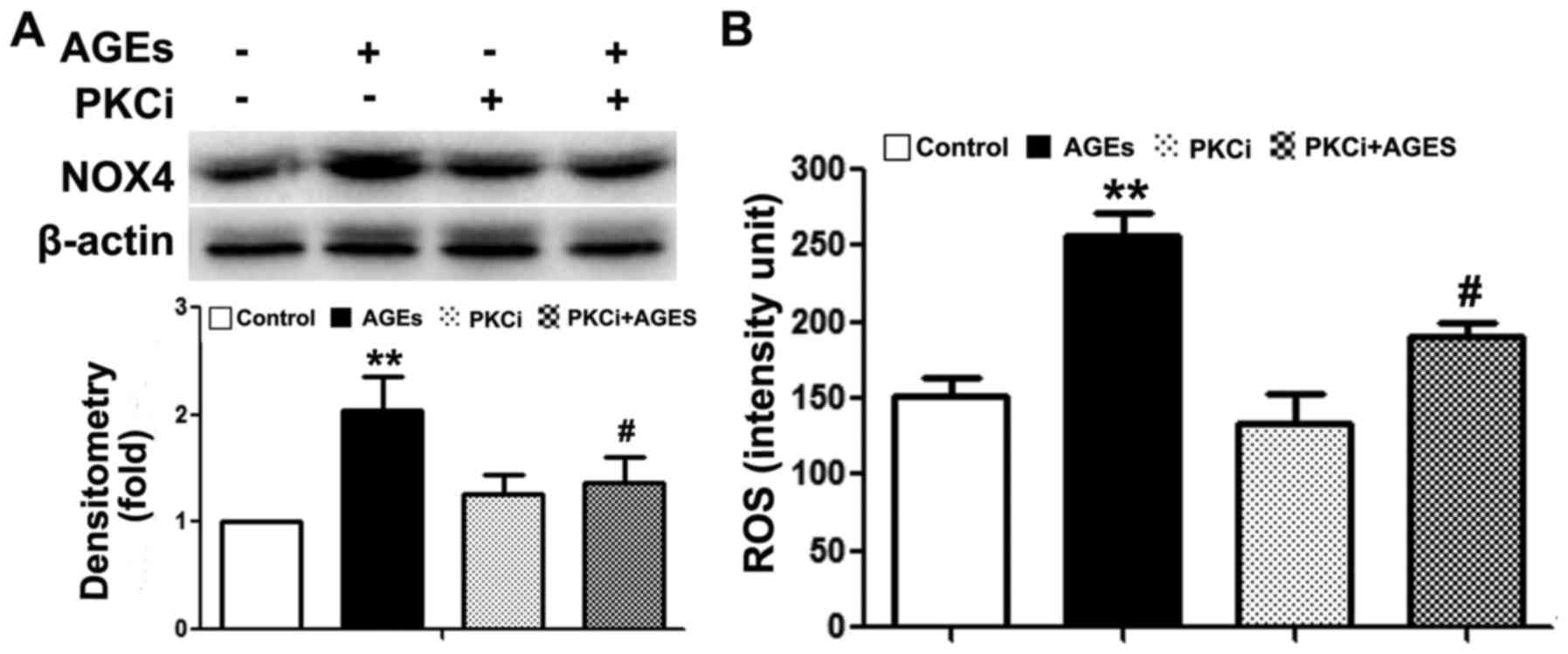
TRB3 regulates AGE-induced ROS synthesis
and the apoptosis of INS-1 cells through the PKCβ2 pathway
Previous studies have demonstrated that the PKCβ2
pathway plays a key role in AGE-induced oxidative damage to
non-islet β-cells (26–29). However, its exact role in β-cells
remains unclear. In this study, we observed an upregulated PKCβ2
expression in INS-1 cells following exposure to AGEs (Fig. 5A). Following the knockdown of TRB3
expression, PKCβ2 and NOX4 expression was downregulated (Fig. 5A). Furthermore, following
pre-treatment with the PKCβ2 specific inhibitor, LY333531,
AGE-induced INS-1 cell apoptosis, the activity of NOX4 and the
intracellular ROS levels were all significantly decreased (Figs. 5B and C, and 6A and B). This result indicated that
TRB3 was involved in AGE-induced oxidative damage and the apoptosis
of INS-1 cells through the upregulation of PKCβ2 activity.
Discussion
Studies using diabetic animal models and clinical
specimens from diabetic patients have demonstrated that, with the
progression of diabetes, the AGE levels in the body gradually
increase (18,26). It has also been demonstrated that
AGEs play an important role in diabetic retinopathy, kidney
diseases, neuropathy and cardiomyopathy (29). Previous studies have shown that
AGEs are the main factors which induce β-cell dysfunction and
apoptosis (3,16,18). Thus, it is important to unravel
the molecular mechanisms of action of AGEs in order to protect
β-cells from injury. In this study, we found that AGEs upregulated
TRB3 expression in INS-1 cells and mediated oxidative damage and
the apoptosis of β-cells through PKCβ2.
AGEs bind with RAGE on cell membranes and trigger
cellular functional response. RAGE is a multi-ligand cell surface
receptor and belongs to the immunoglobulin super-family (30). RAGE can be activated by binding
with different types of ligands, including AGEs, S100 proteins,
HMGB1s and Aβ peptides (31–35). The activation of RAGE is
associated with a number of chronic diseases, including different
types of diabetic complications (e.g., neuropathy and nephropathy),
microvascular disease and chronic inflammation (7). In this study, exposure to AGEs
promoted the apoptosis of INS-1 cells and increased the expression
of their receptor, RAGE; thus, RAGE mediates the damaging effects
of AGEs on β-cells (3,16,18,36).
During the course of diabetes, oxidative stress and
ER stress are the direct factors causing β-cell dysfunction and
apoptosis (37), which results in
insulin resistance in type 2 diabetes and β-cell dysfunction
(38). Factors involved in
oxidative stress include high blood glucose, FFAs and cytokines
(38). In recent studies, AGEs
have been shown to induce β-cell damage through oxidative stress
(3,16,18). In this study, following exposure
to AGEs, the ROS levels in INS-1 cells were significantly elevated.
In addition, NOX expression was downregulated. This result
indicated that AGEs induced oxidative stress in INS-1 cells. NADPH
oxidases are the major sources for intracellular ROS synthesis and
generally have NOX1, NOX2, NOX4 and NOX5 types. A notable feature
of NOX4 is its constitutive activity and preferential generation of
a hydrogen superoxide anion that acts as an oxygen sensor (39). In addition, NOX4 has been
confirmed to play an important role in glucocorticoid-induced INS-1
cell injury (28). Our results
indicated that the AGE-induced oxidative injury to INS-1 cells may
be an important cause of the apoptosis of INS-1 cells.
Many pathways are involved in mediating oxidative
damage in cells. Our previous study showed that TRB3 was associated
with oxidative stress in high-glucose-induced β-cells failure
(21). TRB3 is a homolog of
Drosophila tribbles protein and mammalian protein. TRB3 is
widely expressed in insulin targeted tissues and is closely
associated with insulin resistance and glucose homeostasis
(40). There is recent evidence
to suggest that TRB3 plays an important role in apoptosis. However,
its role remains controversial. Some studies have shown that TRB3
promotes the cytokine-induced apoptosis of pancreatic β-cells, as
well as the ER stress-induced apoptosis of 293 cells, PC-12 cells
(rat neuronal cell line) (41–43). Other studies have shown that TRB3
exerts an anti-apoptotic effect against the nutrient
starvation-induced apoptosis of human prostate carcinoma PC-3
cells, and SaOS2 cells (44,45). These differences may be due to
different cell types and stresses caused by different stimuli.
Relevant studies on β-cell apoptosis have indicated that TRB3 plays
a key role in high blood glucose, high fat, FFA and
cytokine-induced apoptosis in β-cells (21,22,41). In this study, we found that AGEs
stimulated INS-1 apoptosis and increased the expression of TRB3.
The knockdown of TRB3 expression inhibited the apoptosis of INS-1
cells. Moreover, the NOX4 and ROS levels were also decreased,
indicating that TRB3 plays an important role in the AGE-induced
apoptosis of INS-1 cells by affecting ROS levels. The study by
Gorasia et al demonstrated that β-cells were susceptible to
injury caused by oxidative stress and ER stress (46) and an increased effect between
oxidative damage and ER stress (47). TRB3 is an important regulatory
molecule in the ER stress-induced apoptotic pathways (42). In this study, we also demonstrated
that the knockdown of TRB3 expression affected AGE-induced ROS
synthesis and provided evidence of the interaction between
oxidative damage and ER stress in β-cells.
Several studies in the past have indicated that the
PKC path way is associated with oxidative stress induced by ROS
synthesis (24–27,48,49). PKC regulates NADPH oxidase
activity and induces ROS synthesis. In addition, PKC plays an
important role in AGE-induced oxidative damage in cells. Studies
using glomerular microvascular endothelial cells and cardiomyocytes
have demonstrated that AGEs enhanced NADPH oxidase activity through
PKCβ2 and increased ROS synthesis and cell damage (24–27). In this study, INS-1 cells
exhibited an elevated expression of PKCβ2 following exposure to
AGEs. Following the knockdown of TRB3, the expression of PKCβ2 was
decreased and the activity of NADPH oxidase was also decreased. In
addition, the application of specific inhibitors to suppress PKCβ2
activity significantly decreased the intracellular ROS levels and
the apoptosis of INS-1 cells. TRB3 regulated NADPH oxidase and ROS
levels which caused damage to INS-1 cells by affecting the activity
of the PKCβ2 pathway. TRB3, as a related molecule of ER
stress-induced apoptosis, regulates PKC. PKC is the important
regulatory molecule in the pathway of ROS synthesis. Hence, this
study provided a new direction in determining the mechanisms
responsible behind the interaction between oxidative damage and ER
stress.
In conclusion, this study demonstrated that AGE
mediated oxidative stress through TRB3 to damage INS-1 cells and
resulted in the apoptosis of INS-1 cells. TRB3 regulated NADPH
oxidase activity, promoted ROS synthesis and resulted in oxidative
stress in INS-1 cells through the PKCβ2 pathway. Our data provide a
new understanding of the mechanisms responsible for AGE-induced
oxidative injury to β-cells and a new direction for studies aiming
to identify methods with which to protect β-cells from damage.
Acknowledgments
This study was supported by grants from the National
Basic Research Program of China (2012CB966402 to Jinning Lou); and
the National Nature Science Foundation of China (no. 81370873 to
Wenjian Zhang and 81370918 to Xiuli Men).
References
|
1
|
Nowotny K, Jung T, Höhn A, Weber D and
Grune T: Advanced glycation end products and oxidative stress in
type 2 diabetes mellitus. Biomolecules. 5:194–222. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Volchuk A and Ron D: The endoplasmic
reticulum stress response in the pancreatic β-cell. Diabetes Obes
Metab. 12(Suppl 2): 48–57. 2010. View Article : Google Scholar
|
|
3
|
Lim M, Park L, Shin G, Hong H, Kang I and
Park Y: Induction of apoptosis of β cells of the pancreas by
advanced glycation end-products, important mediators of chronic
complications of diabetes mellitus. Ann N Y Acad Sci. 1150:311–315.
2008. View Article : Google Scholar
|
|
4
|
Lee BW, Chae HY, Kwon SJ, Park SY, Ihm J
and Ihm SH: RAGE ligands induce apoptotic cell death of pancreatic
β-cells via oxidative stress. Int J Mol Med. 26:813–818.
2010.PubMed/NCBI
|
|
5
|
Jung H, Joo J, Jeon Y, Lee J, In J, Kim D,
Kang E, Kim Y, Lim Y, Kang J, et al: Advanced glycation end
products downregulate glucokinase in mice. Diabetes Metab Res Rev.
27:557–563. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Puddu A, Storace D, Durante A, Odetti P
and Viviani GL: Glucagon-like peptide-1 counteracts the detrimental
effects of advanced glycation end-products in the pancreatic beta
cell line HIT-T 15. Biochem Biophys Res Commun. 398:462–466. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jakus V and Rietbrock N: Advanced
glycation end-products and the progress of diabetic vascular
complications. Physiol Res. 53:131–142. 2004.PubMed/NCBI
|
|
8
|
Baumann M: Role of advanced glycation end
products in hypertension and cardiovascular risk: human studies. J
Am Soc Hypertens. 6:427–435. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chilelli NC, Burlina S and Lapolla A:
AGEs, rather than hyperglycemia, are responsible for microvascular
complications in diabetes: a 'glycoxidation-centric' point of view.
Nutr Metab Cardiovasc Dis. 23:913–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yamagishi S, Nakamura N, Suematsu M,
Kaseda K and Matsui T: Advanced glycation end products: a molecular
target for vascular complications in diabetes. Mol Med. 21(Suppl
1): S32–S40. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li BY, Li XL, Gao HQ, Zhang JH, Cai Q,
Cheng M and Lu M: Grape seed procyanidin B2 inhibits advanced
glycation end product-induced endothelial cell apoptosis through
regulating GSK3β phosphorylation. Cell Biol Int. 35:663–669. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun C, Liang C, Ren Y, Zhen Y, He Z, Wang
H, Tan H, Pan X and Wu Z: Advanced glycation end products depress
function of endothelial progenitor cells via p38 and ERK 1/2
mitogen-activated protein kinase pathways. Basic Res Cardiol.
104:42–49. 2009. View Article : Google Scholar
|
|
13
|
Adamopoulos C, Piperi C, Gargalionis AN,
Dalagiorgou G, Spilioti E, Korkolopoulou P, Diamanti-Kandarakis E
and Papavassiliou AG: Advanced glycation end products upregulate
lysyl oxidase and endothelin-1 in human aortic endothelial cells
via parallel activation of ERK1/2-NF-κB and JNK-AP-1 signaling
pathways. Cell Mol Life Sci. 73:1685–1698. 2016. View Article : Google Scholar
|
|
14
|
Morita M, Yano S, Yamaguchi T and Sugimoto
T: Advanced glycation end products-induced reactive oxygen species
generation is partly through NF-kappa B activation in human aortic
endothelial cells. J Diabetes Complications. 27:11–15. 2013.
View Article : Google Scholar
|
|
15
|
Sang HQ, Gu JF, Yuan JR, Zhang MH, Jia XB
and Feng L: The protective effect of Smilax glabra extract on
advanced glycation end products-induced endothelial dysfunction in
HUVECs via RAGE-ERK1/2-NF-κB pathway. J Ethnopharmacol.
155:785–795. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lin N, Zhang H and Su Q: Advanced
glycation end-products induce injury to pancreatic beta cells
through oxidative stress. Diabetes Metab. 38:250–257. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu Y, Shu T, Lin Y, Wang H, Yang J, Shi Y
and Han X: Inhibition of the receptor for advanced glycation
endproducts (RAGE) protects pancreatic β-cells. Biochem Biophys Res
Commun. 404:159–165. 2011. View Article : Google Scholar
|
|
18
|
Coughlan MT, Yap FY, Tong DC,
Andrikopoulos S, Gasser A, Thallas-Bonke V, Webster DE, Miyazaki J,
Kay TW, Slattery RM, et al: Advanced glycation end products are
direct modulators of β-cell function. Diabetes. 60:2523–2532. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng WP, Wang BW, Lo HM and Shyu KG:
Mechanical stretch induces apoptosis regulator TRB3 in cultured
cardiomyocytes and volume-overloaded heart. PLoS One.
10:e01232352015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Du K, Herzig S, Kulkarni RN and Montminy
M: TRB3: a tribbles homolog that inhibits Akt/PKB activation by
insulin in liver. Science. 300:1574–1577. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Qian B, Wang H, Men X, Zhang W, Cai H, Xu
S, Xu Y, Ye L, Wollheim CB and Lou J: TRIB3 [corrected] is
implicated in glucotoxicity- and endoplasmic
reticulum-stress-induced [corrected] beta-cell apoptosis. J
Endocrinol. 199:407–416. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qin J, Fang N, Lou J, Zhang W, Xu S, Liu
H, Fang Q, Wang Z, Liu J, Men X, et al: TRB3 is involved in free
fatty acid-induced INS-1-derived cell apoptosis via the protein
kinase C δ pathway. PLoS One. 9:e960892014. View Article : Google Scholar
|
|
23
|
Fang N, Zhang W, Xu S, Lin H, Wang Z, Liu
H, Fang Q, Li C, Peng L and Lou J: TRIB3 alters endoplasmic
reticulum stress-induced β-cell apoptosis via the NF-κB pathway.
Metabolism. 63:822–830. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scivittaro V, Ganz MB and Weiss MF: AGEs
induce oxidative stress and activate protein kinase C-β(II) in
neonatal mesangial cells. Am J Physiol Renal Physiol.
278:F676–F683. 2000.PubMed/NCBI
|
|
25
|
Li L and Renier G: Activation of
nicotinamide adenine dinucleotide phosphate (reduced form) oxidase
by advanced glycation end products links oxidative stress to
altered retinal vascular endothelial growth factor expression.
Metabolism. 55:1516–1523. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Jiang YW, Zhang WJ, Xu SQ, Liu HL,
Yang WY and Lou JN: Differential activations of PKC/PKA related to
microvasculopathy in diabetic GK rats. Am J Physiol Endocrinol
Metab. 302:E173–E182. 2012. View Article : Google Scholar
|
|
27
|
Zhang L, Huang D, Shen D, Zhang C, Ma Y,
Babcock SA, Chen B and Ren J: Inhibition of protein kinase C βII
isoform ameliorates methylglyoxal advanced glycation
endproduct-induced cardiomyocyte contractile dysfunction. Life Sci.
94:83–91. 2014. View Article : Google Scholar
|
|
28
|
Guo B, Zhang W, Xu S, Lou J, Wang S and
Men X: GSK-3β mediates dexamethasone-induced pancreatic β cell
apoptosis. Life Sci. 144:1–7. 2016. View Article : Google Scholar
|
|
29
|
Singh VP, Bali A, Singh N and Jaggi AS:
Advanced glycation end products and diabetic complications. Korean
J Physiol Pharmacol. 18:1–14. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bierhaus A, Humpert PM, Morcos M, Wendt T,
Chavakis T, Arnold B, Stern DM and Nawroth PP: Understanding RAGE,
the receptor for advanced glycation end products. J Mol Med (Berl).
83:876–886. 2005. View Article : Google Scholar
|
|
31
|
Neeper M, Schmidt AM, Brett J, Yan SD,
Wang F, Pan YC, Elliston K, Stern D and Shaw A: Cloning and
expression of a cell surface receptor for advanced glycosylation
end products of proteins. J Biol Chem. 267:14998–15004.
1992.PubMed/NCBI
|
|
32
|
Hofmann MA, Drury S, Fu C, Qu W, Taguchi
A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, et al: RAGE
mediates a novel proinflammatory axis: a central cell surface
receptor for S100/calgranulin polypeptides. Cell. 97:889–901. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hori O, Brett J, Slattery T, Cao R, Zhang
J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al: The
receptor for advanced glycation end products (RAGE) is a cellular
binding site for amphoterin. Mediation of neurite outgrowth and
co-expression of rage and amphoterin in the developing nervous
system. J Biol Chem. 270:25752–25761. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deane R, Du Yan S, Submamaryan RK, LaRue
B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, et al:
RAGE mediates amyloid-β peptide transport across the blood-brain
barrier and accumulation in brain. Nat Med. 9:907–913. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mackic JB, Stins M, McComb JG, Calero M,
Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, et al:
Human blood-brain barrier receptors for Alzheimer's amyloid-beta
1–;40. Asymmetrical binding, endocytosis, and transcytosis at the
apical side of brain microvascular endothelial cell monolayer. J
Clin Invest. 102:734–743. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lo MC, Chen MH, Lee WS, Lu CI, Chang CR,
Kao SH and Lee HM: Nε-(carboxymethyl) lysine-induced mitochondrial
fission and mitophagy cause decreased insulin secretion from
β-cells. Am J Physiol Endocrinol Metab. 309:E829–E839.
2015.PubMed/NCBI
|
|
37
|
Hasnain SZ, Prins JB and McGuckin MA:
Oxidative and endoplasmic reticulum stress in β-cell dysfunction in
diabetes. J Mol Endocrinol. 56:R33–R54. 2016. View Article : Google Scholar
|
|
38
|
Evans JL, Goldfine ID, Maddux BA and
Grodsky GM: Are oxidative stress-activated signaling pathways
mediators of insulin resistance and β-cell dysfunction? Diabetes.
52:1–8. 2003. View Article : Google Scholar
|
|
39
|
Nisimoto Y, Diebold BA, Cosentino-Gomes D
and Lambeth JD: Nox4: a hydrogen peroxide-generating oxygen sensor.
Biochemistry. 53:5111–5120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Prudente S, Sesti G, Pandolfi A, Andreozzi
F, Consoli A and Trischitta V: The mammalian tribbles homolog
TRIB3, glucose homeostasis, and cardiovascular diseases. Endocr
Rev. 33:526–546. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Humphrey RK, Newcomb CJ, Yu SM, Hao E, Yu
D, Krajewski S, Du K and Jhala US: Mixed lineage kinase-3
stabilizes and functionally cooperates with TRIBBLES-3 to
compromise mitochondrial integrity in cytokine-induced death of
pancreatic beta cells. J Biol Chem. 285:22426–22436. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ohoka N, Yoshii S, Hattori T, Onozaki K
and Hayashi H: TRB3, a novel ER stress-inducible gene, is induced
via ATF4-CHOP pathway and is involved in cell death. EMBO J.
24:1243–1255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zou CG, Cao XZ, Zhao YS, Gao SY, Li SD,
Liu XY, Zhang Y and Zhang KQ: The molecular mechanism of
endoplasmic reticulum stress-induced apoptosis in PC-12 neuronal
cells: the protective effect of insulin-like growth factor I.
Endocrinology. 150:277–285. 2009. View Article : Google Scholar
|
|
44
|
Schwarzer R, Dames S, Tondera D, Klippel A
and Kaufmann J: TRB3 is a PI 3-kinase dependent indicator for
nutrient starvation. Cell Signal. 18:899–909. 2006. View Article : Google Scholar
|
|
45
|
Ord D, Meerits K and Ord T: TRB3 protects
cells against the growth inhibitory and cytotoxic effect of ATF4.
Exp Cell Res. 313:3556–3567. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gorasia DG, Dudek L, Veith PD, Shankar R,
Safavi-Hemami H, Williamson NA, Reynolds EC, Hubbard MJ and Purcell
AW: Pancreatic beta cells are highly susceptible to oxidative and
ER stresses during the development of diabetes. J Proteome Res.
14:688–699. 2015. View Article : Google Scholar
|
|
47
|
Zhang K: Integration of ER stress,
oxidative stress and the inflammatory response in health and
disease. Int J Clin Exp Med. 3:33–40. 2010.PubMed/NCBI
|
|
48
|
Pérez LM, Milkiewicz P, Ahmed-Choudhury J,
Elias E, Ochoa JE, Sánchez Pozzi EJ, Coleman R and Roma MG:
Oxidative stress induces actin-cytoskeletal and tight-junctional
alterations in hepatocytes by a Ca2+-dependent,
PKC-mediated mechanism: protective effect of PKA. Free Radic Biol
Med. 40:2005–2017. 2006. View Article : Google Scholar
|
|
49
|
Pérez LM, Milkiewicz P, Elias E, Coleman
R, Sánchez Pozzi EJ and Roma MG: Oxidative stress induces
internalization of the bile salt export pump, Bsep, and bile salt
secretory failure in isolated rat hepatocyte couplets: a role for
protein kinase C and prevention by protein kinase A. Toxicol Sci.
91:150–158. 2006. View Article : Google Scholar : PubMed/NCBI
|