Introduction
Due to rising prevalence, poor clinical outcome, and
high health care costs, cardiovascular disease (CVD) is becoming a
major public health concern and is the leading cause of death
worldwide. According to the report on Cardiovascular Disease in
China (2014), approximately 290 million individuals in China suffer
from CVD each year, and CVD accounts for two in every five deaths
(1). Clinical and experimental
evidence suggests that the rarefaction of cardiac capillaries,
infiltration of myofibroblasts, and the deposition of collagen and
other extracellular matrix proteins contribute to the progression
of cardiac fibrosis and heart failure, two common clinical outcomes
of CVD (2–4). While exisiting therapies for CVD,
such as pharmacological and invasive therapies, can relieve
symptoms and attenuate disease progression, there is still an
urgent need for novel therapies to effectively treat or even cure
CVD.
Endothelial-mesenchymal transition (EndMT), one
subgroup of cellular phenotypic switching, can induce transcription
factors, which can affect gene expression and promote the loss of
cell-cell adhesion and the change from endothelial morphology and
physiology to a mesenchymal phenotype, a change characterized by
low expression of endothelial markers such as CD31 (Pecam-1) and
vascular endothelial (VE)-cadherin and increased expression of
mesenchymal markers such as α-smooth muscle actin (α-SMA) and
fibroblast-specific protein (FSP)-1. EndMT has been proven to play
a pivotal role in cardiovascular development and disease
progression including cardiac fibrosis (2,5–9).
Recent studies indicate that EndMT can be mediated by transforming
growth factor-β (TGF-β)/bone morphogenetic protein (BMP), Notch and
Wnt-β-catenin and hypoxia signaling pathways (2,10–12). However, the mechanism remains
unclear.
Runt-related transcription factor 3 (RUNX3), a
member of the mammalian runt domain transcription factor family, is
located on human chromosomes 1p36.1. Approximately a decade ago
RUNX3 was claimed to be a tumor suppressor that inhibited the
expression of vascular endothelial growth factor (VEGF) and reduced
the angiogenesis, growth, and metastasis of human gastric cancer
(13). RUNX3 is a direct target
gene of Notch in human endothelial cells and a downstream effector
of the TGF-β signaling pathway, and plays a critical role in
angiogenesis, cell migration and invasion (14,15). Recent studies suggest that RUNX3
knockdown enhances endothelial progenitor cell (EPC) function and
that miR-130a regulates EPC autophagy through RUNX3 (16,17).
This study was designed to investigate the role of
RUNX3 in EndMT as well as its signaling pathways, and to gain
insight into the underlying molecular mechanisms so that novel
therapeutic strategies can be ultimately developed for CVD.
Materials and methods
Cell culture and treatment
Human cardiac microvascular endothelial cells
(HCMECs), purchased from ScienCell Research Laboratories (Carlsbad,
CA, USA), were maintained in endothelial cell medium (ScienCell
Research Laboratories) according to the manufacturer's instructions
and were regularly examined under an inverted microscope (TS100;
Nikon, Tokyo, Japan). After the cells reached 90% confluence, a
subculture of cells was diluted to 40,000 cells/ml, and was seeded
at 10,000 cells/cm2 in a T-25 flask precoated with 5
μg fibronectin (ScienCell Research Laboratories). A healthy
culture displays cobblestone-shaped morphology, and non-granular
cytoplasm while the cell number doubled two to three days in
culture. HCMECs in this study were used between passages 3 and
6.
HCMECs were seeded in a 6-well plate at a density of
10,000 cells/cm2, before they were cultured in normoxic
conditions and were grown to 30–35% confluency. For hypoxia-induced
EndMT, HCMECs were incubated in strictly controlled hypoxic
conditions (1% O2) for 1, 4 and 5 days,
respectively.
HCMECs were cultured under normoxic conditions
before they were divided into 4 groups: i) control group, HCMECs
were cultured under normoxic conditions for 4 days without any
treatment; ii) hypoxia group, HCMECs were cultured under strictly
controlled hypoxic conditions (1% O2) for 4 days; iii)
RUNX3i group, HCMECs were transfected with the lentiviral vector
containing RUNX3 (RUNX3-RNAi lentivirus; Genechem, Shanghai, China)
for 8 h and cultured under strictly controlled hypoxic conditions
(1% O2) for 4 days; iv) green fluoresent protein (GFP)
group, HCMECs were transfected with an empty lentiviral vector
(Genechem) for 8 h and cultured under strictly controlled hypoxic
conditions (1% O2) for 4 days.
Double immunofluorescence staining
HCMECs were grown in 24-well culture plates. After
the cells reached 70–80% confluence, they were washed with cold
phosphate-buffered saline (PBS) 3 times and fixed in 4%
paraformaldehyde (both from Beijing Solarbio Science and
Technology, Co., Ltd., Beijing, China) for 30 min before they were
permeabilized with solution containing 0.1% Trition X-100 (Beijing
Solarbio Science and Technology, Co., Ltd.). After being blocked
with 10% BSA for 1 h at room temperature, HCMECs were further
incubated with two primary antibodies, monoclonal mouse anti-CD31
antibody (Cell Signaling Technology, Inc., Danvers, MA, USA) and
monoclonal rabbit anti-α-SMA antibody (Abcam, Cambridge, UK), at
4°C overnight. Twenty-four hours later, the cells were kept in a
dark room for 1 h at room temperature and mixed with two secondary
antibodies: CY3 red-conjugated goat anti-mouse IgG and FITC
green-conjugated goat anti-rabbit IgG (both from Aspen Bio, Wuhan,
China). After 100 μl DAPI (Aspen Bio) was added into each
well, the mixture was incubated in the dark at room temperature for
another 5 min. In the negative control group, the primary antibody
was replaced with non-immune IgG. The cells were visualized and
mounted using phase contrast fluorescence microscopy (IX51;
Olympus, Tokyo, Japan). All images were measured using Photoshop CS
8.0 software.
The lentivirus (Genechem) in this study expressed
GFP, so HCMECs transduced with lentivirus were incubated with two
secondary antibodies: CY3 red-conjugated goat anti-mouse (Aspen
Bio) and DyLight 405 AffiniPure goat anti-rabbit IgG (H+L)
(Abbkine, Inc., Redlands, CA, USA).
Silencing of RUNX3 with RUNX3-RNAi
lentivirus
The transduction of HCMECs with the lentivirus was
performed according to the manufacturer's instructions with some
modifications. The preliminary study confirmed that the MOI of
HCMECs was 20, and that polybrene (5 μg/ml) could improve
the infection rate. The cells were seeded at 30–40% confluency in
6-well plates with HCMEC culture medium. Twelve hours later, the
cells in each well were incubated with 20 MOI RUNX3-RNAi lentivirus
or control lentivirus (for control group) for 8 h. Lentiviral
infection was confirmed by the visualization of enhanced GFP under
a fluorescence microscope (IX51; Olympus). Then, the culture medium
was replaced with fresh HCMEC supplemented medium and the mixture
was incubated under strictly controlled hypoxic conditions (1%
O2) for an additional 4 days.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from HCMECs using
TRIzol® reagent (Life Technologies, Grand Island, NY,
USA) and the extracted RNA was precipitated with isopropanol
following standard procedures. First-strand complementary DNA
(cDNA) was synthesized with a cDNA synthesis kit (Promega, Beijing,
China) in 20 μl reaction mixture containing 5 μg
total RNA, according to the manufacturer's instructions. The
synthesized cDNA was used for quantitative PCR (qPCR). β-actin was
used as endogenous control and the primer sequences (Generay
Biotech Co, Ltd., Shanghai, China) are listed in Table I. The reaction volume for qPCR was
20 μl, containing 2 μl cDNA. qPCR was performed on a
7500 Real-Time PCR system (Applied Biosystems, Carlsbad, CA, USA)
using the GoScript™ SYBR-Green PCR kit (Promega) according to the
manufacturer's instructions. Relative mRNA expression levels for
the genes of interest were analyzed using the 2−ΔΔCt
method as previously described (18).
 | Table IRT-PCR primer sequences. |
Table I
RT-PCR primer sequences.
| Genes | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| β-actin |
GCCATGTACGTAGCCATCCA |
GAACCGCTCATTGCCGATAG |
| CD31 |
CCCAGCCCAGGATTTCTTAT |
ACCGCAGGATCATTTGAGTT |
| VE-cadherin |
CCAATCTGACCTGAAAAAGC |
CCACCGTTTTCCGTGTAATA |
| α-SMA |
AAGCACAGAGAGCAAAAGAGGAAT |
ATGTCGTCCCAGTTGGTGAT |
| FSP-1 |
AACTAAAGGAGCTGCTGACCC |
TGTTGCTGTCCAAGTTGCTC |
| Notch1 |
CGCACAAGGTGTCTTCCAG |
AGGATCAGTGGCGTCGTG |
| Hey1 |
GGAGAGGCGCCGCTGTAGTTA |
CAAGGGCGTGCGCGTCAAAGTA |
| Hes1 |
ACAGGGGGTAAAGGCTACTTTG |
CTGCTGCTGCTGCGTTT |
| TGF-β1 |
CACTCCCACTCCCTCTCTC |
GTCCCCTGTGCCTTGATG |
| Smad2 |
GGAGCAGAATACCGAAGGCA |
CTTGAGCAACGCACTGAAGG |
| Smad3 |
ATTCCAGAAACGCCACCTCC |
GCTATTGAACACCAAAATGCAGG |
| Slug | AGATGCATATTCGG
CCCAC |
CCTCATGTTTGTGCAGGAGA |
| Snail |
AATCGGAAGCCTAACTACAGCGAG |
CCTTGGCCTCAGAGAGCTGG |
| RUNX3 |
CATCAAGGTGACCGTGGAC |
GTTCCAGGTCCCCAAAGC |
Total protein extraction and western
blotting
HCMECs were homogenized in RIPA lysis buffer that
contained protease inhibitor PMSF (both from Beijing Solarbio
Science and Technology, Co., Ltd.). After the mixture was
centrifuged at 12,000 × g for 10 min at 4°C, the supernatant was
collected. Total protein quantity was determined using the BCA
protein assay kit (Beyotime Institute of Biotechnology, Haimen,
China) according to the manufacturer's instructions. Protein
samples were mixed with 6X loading buffer (Beijing Solarbio Science
and Technology, Co., Ltd.) and degenerated at 95°C for 10 min.
Forty micrograms of protein from each group was resolved on 4–12%
SDS-PAGE and then transferred onto polyvinylidene fluoride (PVDF)
or nitrocellulose membranes (all from Beijing Solarbio Science and
Technology, Co., Ltd.). The membranes were blocked with 10% non-fat
milk in TBST for 1 h, before they were incubated with the primary
antibodies (Table II) at 4°C
overnight. After being rinsed with TBST 3 times, the membranes were
incubated with the secondary antibodies (Table III) for another hour for 1 h at
room temperature in a shaker and the proteins were visualized using
chemiluminescent kits (Beyotime Institute of Biotechnology).
| Table IIPrimary antibodies. |
Table II
Primary antibodies.
| Primary antibodies
(Cat. no.) | Companies | Dilute
solution | Dilution rate |
|---|
| Rabbit anti-GAPDH
(ab181602) | Abcam | 5% non-fat
milk | 1:10,000 |
| Mouse anti-CD31
(ab24590) | Abcam | 5% non-fat
milk | 1:500 |
| Rabbit
anti-VE-cadherin (#2500) | Cell Signaling
Technology | 5% BSA | 1:500 |
| Rabbit anti-α-SMA
(ab32575) | Abcam | 5% non-fat
milk | 1:5,000 |
| Rabbit anti-FSP-1
(ab41532) | Abcam | 5% BSA | 1:1,000 |
| Rabbit anti-Notch1
(ab52627) | Abcam | 5% non-fat
milk | 1:1,000 |
| Rabbit anti-Slug
(#9585) | Cell Signaling
Technology | 5% BSA | 1:1,000 |
| Rabbit anti-Snail
(#3879) | Cell Signaling
Technology | 5% BSA | 1:1,000 |
| Rabbit anti-Smad2/3
(bs-3484R) | Bioss | 5% non-fat
milk | 1:1,000 |
| Rabbit
anti-p-Smad2/3 (bs-8853R) | Bioss | 5% non-fat
milk | 1:1,000 |
| Rabbit anti-RUNX3
(bs-0378R) | Bioss | 5% non-fat
milk | 1:1,000 |
| Mouse anti-TGF-β2
(ab167655) | Abcam | 5% BSA | 1:1,000 |
| Rabbit anti-VEGFR1
(bs-0170R) | Bioss | 5% non-fat
milk | 1:1,000 |
| Table IIISecondary antibodies. |
Table III
Secondary antibodies.
Secondary
antibodies
(Cat. no.) | Companies |
Goat anti-rabbit
IgG
(ZB-2310) | Zhongshan Golden
Bridge
Biotechnology, Beijing, China |
Goat anti-mouse
IgG
(ZB-2305) | Zhongshan Golden
Bridge
Biotechnology, Beijing, China |
Transwell migration assay
The migration assay was carried out in 24-well
chambers of 6.5-mm diameter with polycarbonate 8-μm pore
membrane filters (Transwell; Corning Life Sciences, Corning, NY,
USA). HCMECs were resuspended in serum-free endothelial cell medium
at 1×105 cells/ml. Two hundred microliters of cells were
placed in the upper Transwell chambers and 500 μl complete
endothelial cell medium was added into each well in the lower
24-well plates. After incubation at 37°C for 24 h, the migrated
cells were stained with crystal violet (Aspen Bio). The images were
obtained using an inverted microscope (IX51; Olympus) and the
migrated cells were counted in 3 random fields (x200) for each
well.
Tube formation assay
The neovascularization assays were performed in
human fibrin matrices. In brief, the Matrigel (BD Biosciences, New
Jersey, NY, USA) was thawed at 4°C overnight before being placed in
a 96-cell plate (50 μl Matrigel/well). The cells were
incubated at 37°C to allow solidification. Then, the cells from
each group were harvested and resuspended (200,000 cells/ml),
before they were seeded in plates (50 μl cells/well) and
incubated at 37°C for 4–6 h. The cell growth was examined under a
phase-contrast microscope (IX51; Olympus). As described in the
previous study (19), tube
formation was the formation of a structure with its length four
times longer than its width. The tube networks were photographed
from 6 randomly chosen fields with a microscope. The results were
analyzed using ImageJ software.
Statistical analysis
All data analysis was performed using GraphPad Prism
7.0 software (GraphPad Software Inc., San Diego, CA, USA).
Experimental data are presented as mean ± SD. Comparisons between
groups were compared using one-way analysis of variance (ANOVA).
P-value <0.05 was considered statistically significant.
Results
Hypoxia induces the transition of HCMECs
to mesenchymal cells
To investigate the potential role of hypoxia in the
induction of EndMT involving HCMECs, we incubated the HCMECs in
strictly controlled hypoxic conditions (1% O2) for 1, 4
and 5 days, respectively. Under hypoxic conditions, the endothelial
cells gradually developed an elongated spindle-shaped structure
(Fig. 1A). The double
immunofluorescence staining results showed that the hypoxia-treated
HCMECs expressed proteins associated with fibroblasts (α-SMA), but
lost proteins associated with endothelial cells (CD31). The changes
became more obviously as cultivation time increased (Fig. 1B).
CD31+/α-SMA+ cells were visualized and
assessed in both the normoxic HCMEC and hypoxic HCMEC groups, and
the results showed that the number of
CD31+/α-SMA+ cells was significantly
increased in the hypoxic HCMEC group at 4 days when compared to the
others (Fig. 1C). These results
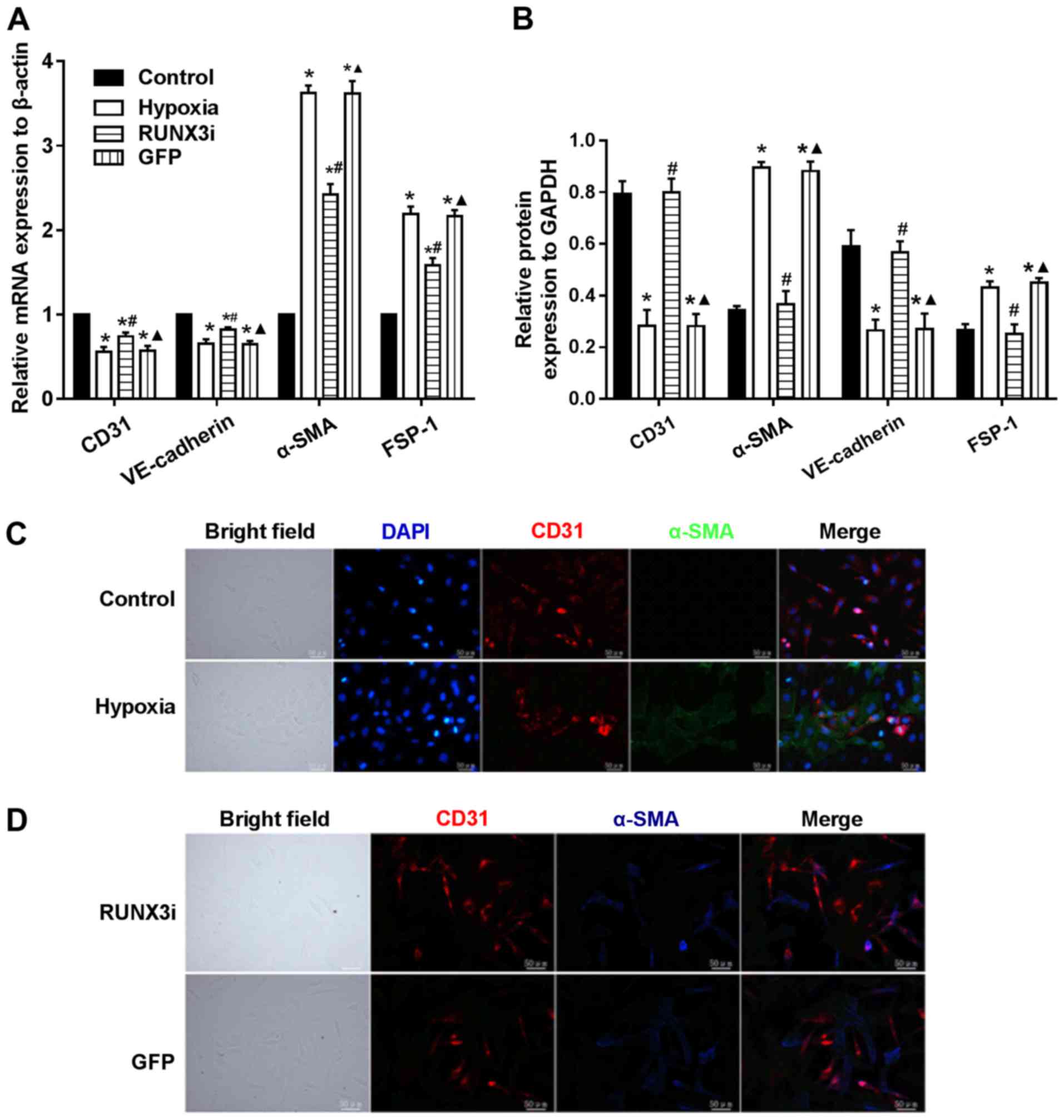
demonstrated that hypoxia induced EndMT. Furthermore, the relative
mRNA expression of endothelial and mesenthymal markers in both
groups were assessed using RT-qPCR, and the results showed that the
endothelial markers, CD31 and VE-cadherin, were downregulated,
while the mesenchymal markers, FSP-1 and α-SMA, were upregulated in
the hypoxic HCMEC group (Fig.
1D).
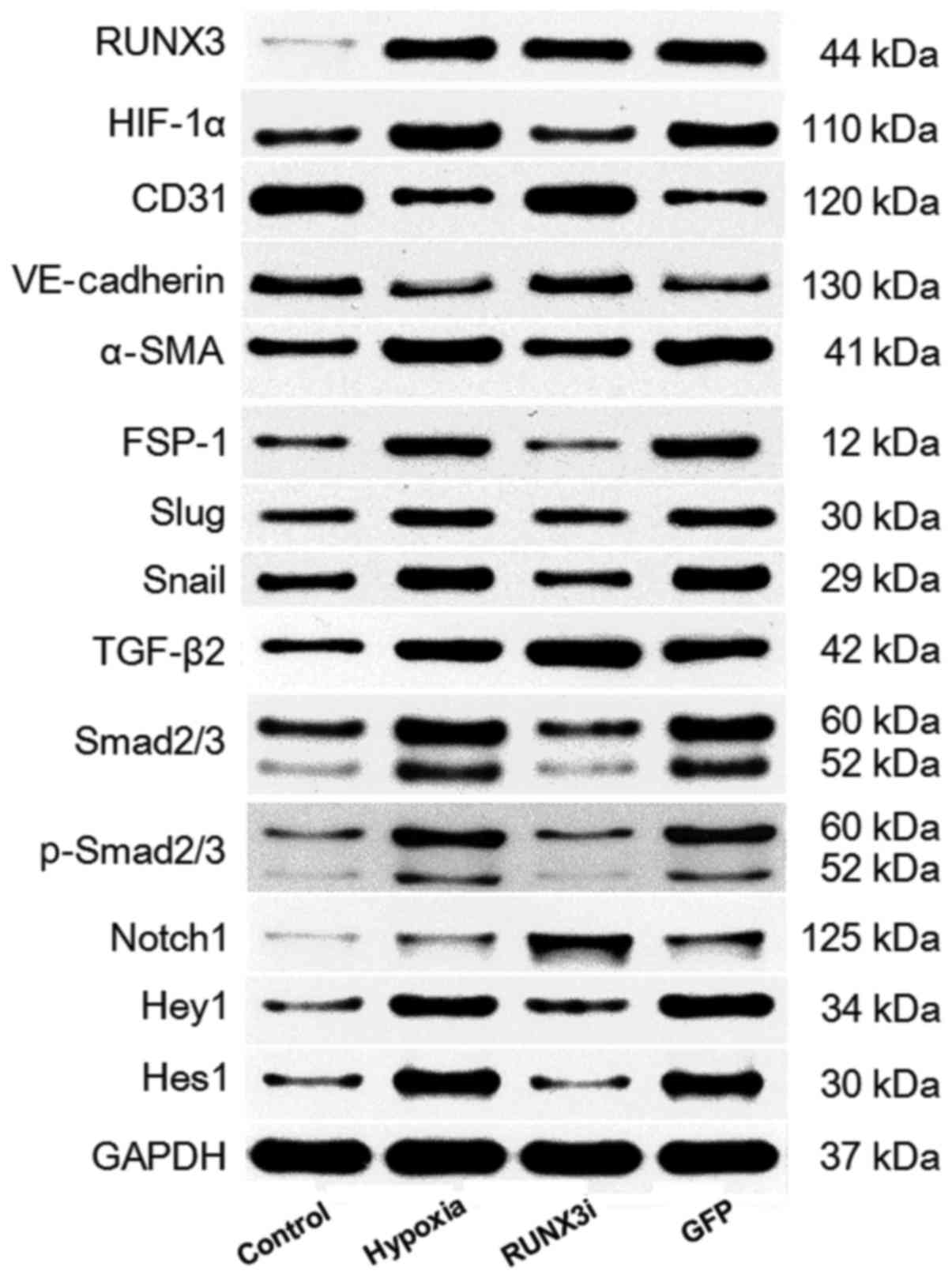
We examined the relative protein expression of RUNX3
and its targeting proteins by western blotting, and the expression
blots are presented in Fig.
2.
Hypoxia enhances RUNX3 expression in
HCMECs
To investigate the expression of RUNX3 in HCMECs
during hypoxia-induced EndMT, we examined RUNX3 expression by
RT-qPCR and western blotting. Both mRNA and protein levels of RUNX3
were low and increased significantly after the cells were cultured
in hypoxic conditions for 4 days (Figs. 2 and 3A and B). To examine the physical
interaction between the transcriptional factor RUNX3 and EndMT, we
knocked down the expression of RUNX3 in HCMECs using RUNX3-RNAi
lentivirus. As shown in Fig. 3,
the RUNX3-RNAi lentivirus decreased the expression of RUNX3 mRNA
and protein to low levels, while the control lentivirus did not
affect RUNX3 expression. The cultivation of HCMECs in hypoxic
conditions induced intracellular accumulation of hypoxia-inducible
factor-1α (HIF-1α) (Figs. 2 and
3C). Therefore, our results
showed that hypoxia enhanced RUNX3 expression in HCMECs.
Knockdown of RUNX3 attenuates EndMT of
HCMECs
To examine whether RUNX3 plays a pivotal role in
EndMT of HCMECs, we suppressed RUNX3 expression in HCMECs with
RUNX3-RNAi lentivirus and found that RUNX3 knockdown ameliorated
hypoxia-induced EndMT (Fig. 4).
Suppress of RUNX3 downregulated the mRNA expression of α-SMA and
FSP-1 and upregulated the expression of endothelial markers
(Fig. 4A). Hypoxia induced a
notable increase in the expression of α-SMA and FSP-1, while RUNX3
knockdown attenuated α-SMA and FSP-1 protein expression and
increased CD31 and VE-cadherin levels in the HCMECs treated under a
hypoxic condition (Figs. 2 and
4B). Double immunofluorescence
staining assays were carried out to further confirm that knockdown
of RUNX3 attenuated EndMT. CD31 was downregulated while α-SMA was
upregulated in the hypoxia group compared with the control group
(Fig. 4C), while CD31 was
upregulated and α-SMA was downregulated in the RUNX3i group
compared with the GFP group (Fig.
4D). In brief, our data demonstrated that α-SMA expression in
HCMECs under hypoxia was inhibited by RUNX3-RNAi lentivirus, while
the control lentivirus did not affect the protein expression.
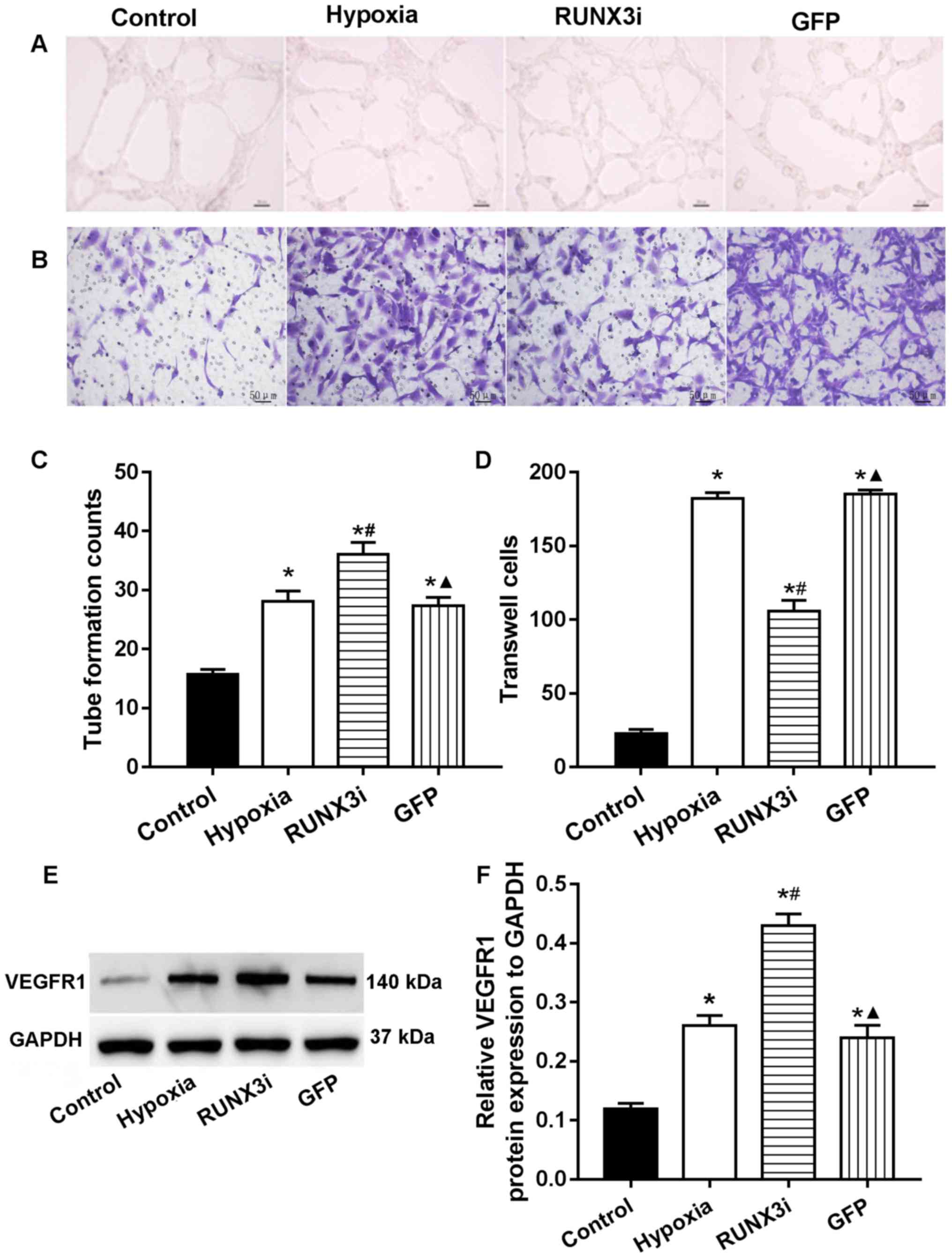
RUNX3 modulates HCMEC function
In human gastric cancer cells, RUNX3 was found to
suppress VEGF expression and reduce angiogenesis. Low RUNX3
expression was associated with increased VEGF expression and
gastric cancer angiogenesis (13). To investigate the role of RUNX3 in
HCMEC function, we examined the angiogenesis and migration ability
via endothelial cell tube formation assay and Transwell migration
assays. Our research showed that knockdown of RUNX3 adversely
affected HCMEC function. Compared with that in the control HCMECs,
hypoxia promoted tube formation and induced cell migration
markedly, while knockdown of RUNX3 attenuated cell migration and
increased angiogenesis in HCMECs treated under a hypoxic condition
(Fig. 5A–D). These results
indicated that the loss of RUNX3 positively affected angiogenic
phenotype and reduced endothelial cell migration. As known, VEGF
receptors in vascular endothelial cells are an important role
during angiogenesis. In this study, we examined the expression of
their proteins, and the results showed that the expression of VEGF
receptor 1 (VEGFR1) in the cells from the hypoxia and RUNX3i groups
were all increased, and the latter was more obvious (Fig. 5E and F).
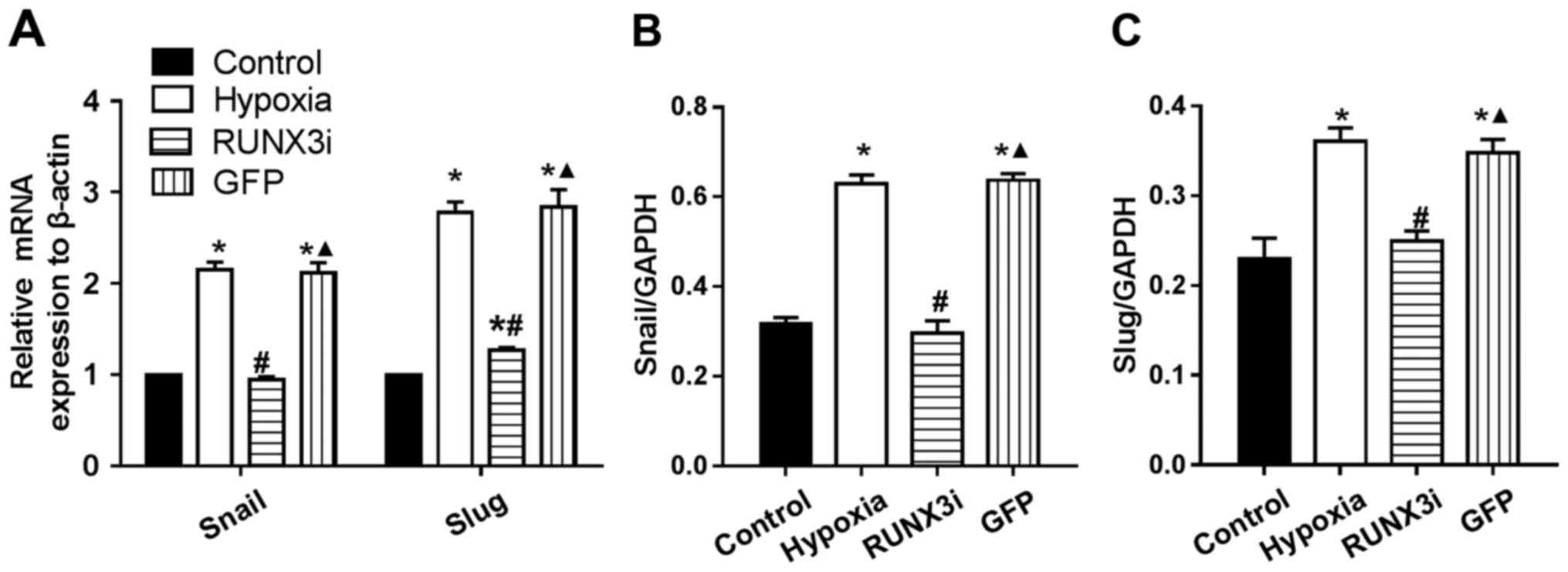
RUNX3 regulates the expression of
principal EndMT transcriptional factors
Several transcription factors induce EndMT. The
principal EndMT transcriptional factors include zinc-finger binding
transcription factors Snail and Slug (12,20–22). As shown in Fig. 6, principal EndMT transcriptional
factors Snail and Slug were upregulated under hypoxia, suggesting
that hypoxia triggered the phenotypic changes of EndMT in HCMECs.
Fu et al demonstrated that overexpression of RUNX3 induced
EndMT in endothelial cells and upregulated the expression of Slug
and Snail (14). To further
investigate the interactions between these two positive regulators
of EndMT and RUNX3, we found that the knockdown of RUNX3 in HCMECs
upregulated the mRNA expression of endothelial markers and
downregulated the expression of mesenchymal markers (Fig. 4A) such as Slug and Snail (Fig. 6A). The downregulation of Slug and
Snail was further confirmed by western blotting (Figs. 2 and 6B and C). Thus, our data demonstrated
that low expression of RUNX3 alleviated EndMT in HCMECs.
Changes in the TGF-β signaling pathway in
hypoxia-induced EndMT
Since hypoxia induces EndMT, we speculated that
hypoxia may induce EndMT by activating TGF-β signaling. We examined
the expression of receptors and target genes involved in the
signaling pathway, and found that hypoxia upregulated the mRNA
expression of TGF-β1, Smad2 and Smad3, while the knockdown of RUNX3
increased TGF-β1 mRNA expression and reduced the mRNA expression of
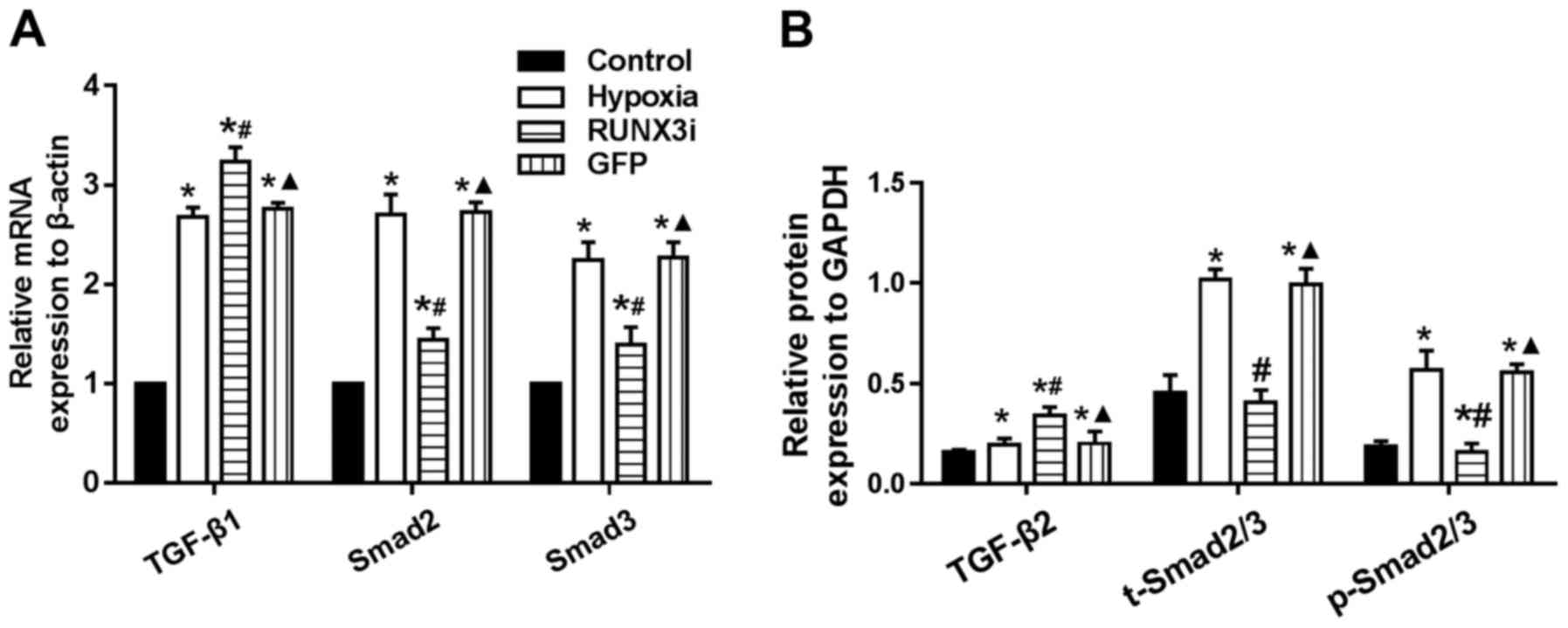
Smad2 and Smad3 (Fig. 7A).
Upregulation of TGF-β2, Smad2/3 and phosphorylation of Smad2/3
(p-Smad2/3) in HCMECs in the hypoxia group was further confirmed by
western blotting and the levels of Smad2/3 and p-Smad2/3 in the
HCMECs in the RUNX3i group were lower than those in the hypoxia
group, while the level of TGF-β2 was increased (Figs. 2 and 7B). Therefore, our data demonstrated
that hypoxia-induced EndMT of HCMECs activated TGF-β signaling and
RUNX3 is a downstream target of TGF-β signaling.
Changes in the Notch signaling pathway in
hypoxia-induced EndMT
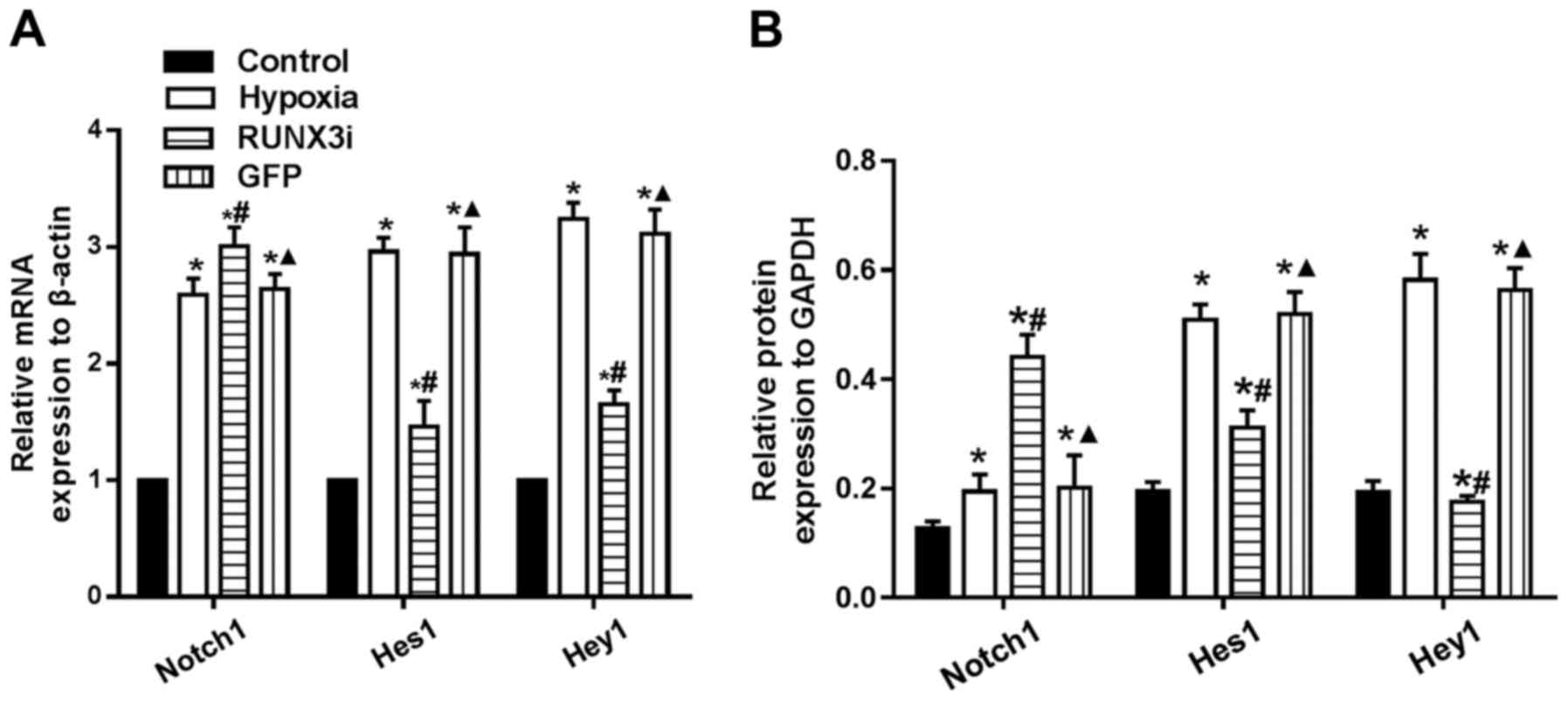
To examine whether hypoxia induces EndMT by
activating Notch signaling through a positive feedback mechanism,
we assessed the expression of Notch1, Hes1 and Hey1, and found that
hypoxia enhanced Notch1, Hes1 and Hey1 mRNA expression. Knockdown
of RUNX3 markedly upregulated Notch-1 mRNA expression, while mRNA
expression of Hes1 and Hey1 was significantly downregulated
(Fig. 8A). Similarly, changes in
the protein levels in HCMECs in each group were further confirmed
by western blotting (Figs. 2 and
8B). Our research demonstrated
that hypoxia-induced EndMT of HCMECs activated Notch signaling and
RUNX3 is likely to be the downstream target of Notch signaling
pathway.
Discussion
Endothelial cells play an important role in cardiac
functions. Previous research has shown that disorders in
endothelial function are associated with adverse cardiac remodeling
(12,21). HCMECs can modulate vascular tone
by releasing several endothelium-derived contracting and relaxing
factors, by regulating and degradating vasoactive peptides, and
through enzymes located on the endothelial surface. Therefore, they
exert physiological and pathophysiological effects on the function
of cardiac myocytes. In addition, HCMECs can grow rapidly in
vitro with the cell number doubling in two to three days in
culture, making the in vitro experiment feasible.
Various diseases, such as fibrosis and cancer,
develop EndMT under hypoxia as a result of ischemic conditions. One
previous study showed that the lack of oxygen inhibited prolyl
hydroxylases, which are important in degrading HIF-1α under
normoxic conditions (23).
Previous studies have also shown that HIF-1α could induce the
expression of EndMT-associated transcriptional factors, such as
Snail, Slug and TGF-β (20,24). In the present study, we
demonstrated that hypoxia induced the transition of HCMECs to
mesenchymal cells. However, the underlying molecular mechanism
remains unknown.
In our study, we found that RUNX3 mRNA expression
was low in HCMECs but increased when stimulated by hypoxia, which
modulates intracellular signaling pathways and plays an important
role in vascular endothelial cells. Several studies have
demonstrated that RUNX3 is a downstream effector of TGF-β and Notch
signaling pathways, and plays critical roles in regulating cell
functions, such as angiogenesis, cell migration and apoptosis
(14,15,17,25). Our results showed that TGF-β and
Notch signaling were activated during the hypoxia-induced EndMT of
HCMECs. Knockdown of RUNX3 by RUNX3-RNAi lentivirus suppressed the
mRNA and protein expression of mesenchymal markers and induced
endothelial markers, suggesting that RUNX3 contributed to EndMT.
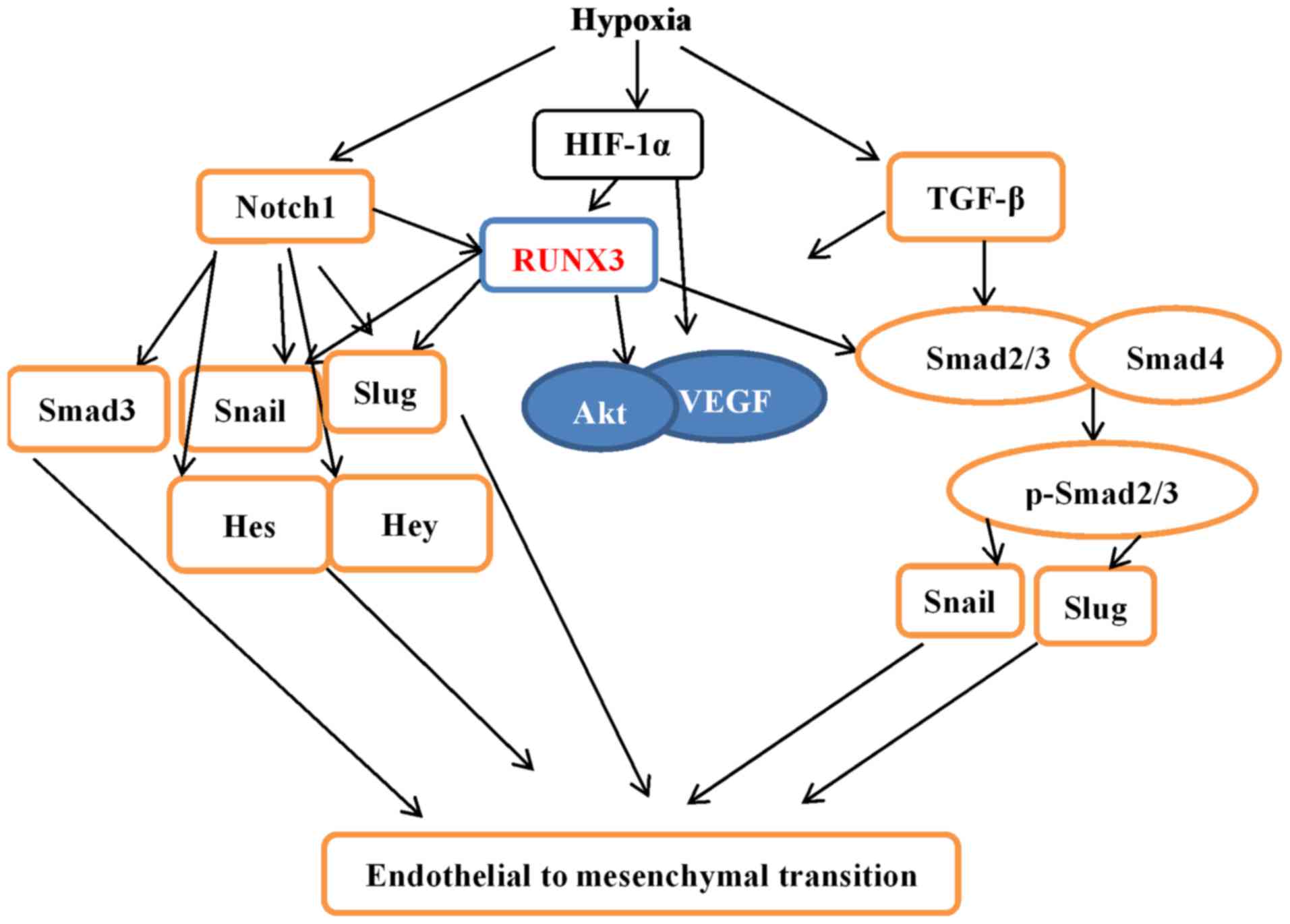
Our research not only demonstrated that hypoxia induced EndMT in
HCMECs but also provides a novel molecular mechanism (Fig. 9).
Several studies have demonstrated that cell
migration ability is enhanced and angiogenesis is suppressed during
EndMT (2,17,26,27). However, our research showed that
hypoxia markedly promoted tube formation and induced cell migration
and that hypoxia stabilized HIF-1α and induced the expression of
VEGF. HIF-1α also increases VEGF expression (28). Therefore, a possible explanation
would be that these factors enhanced angiogenesis and cell
migration, while EndMT partially offset the effects.
Recent research shows that hypoxia and the
TGF-β/Smad signaling pathways synergistically induce EndMT
(20). TGF-β and Notch signaling
pathways were found to be the most common signaling mechanisms
promoting EndMT participation in cardiovascular development and
disease progression (2,5,10,29,30). EndMT is a key point in
cardiovascular development and disease. Our present study confirmed
that the knockdown of RUNX3 attenuated EndMT of HCMECs, positively
impacted the angiogenic phenotype, and reduced endothelial cell
migration. The expression levels of TGF-β1 and Notch1 were
upregulated, while downstream genes were downregulated, suggesting
that RUNX3 is a common downstream target of TGF-β and Notch
signaling pathways. Since our study was carried out in
vitro, and the in vivo environment is more complex,
further studies are warranted to gain a better understanding.
In conclusion, the present study demonstrated that
RUNX3 plays an important role in endothelial cells. Our findings
lay the foundation for further in vitro experiments. In
addition, our study provided a novel molecular mechanism for CVD
and further underscored the importance of RUNX3 during EndMT.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (81041097 and 81460046) and the Natural
Science Foundation of Jiangxi Province (20142BAB205040). We thank
Dr Wan Zhang and Dr Junyi Zeng for expert technical assistance.
References
|
1
|
Chen W, Gao R, Liu L, Zhu M, Wang W, Wang
Y, Wu Z, Li H, Zheng Z, Jiang L and Hu S: Outline of the report on
cardiovascular disease in China 2014. Chin Circ J. 30:617–622.
2015.In Chinese.
|
|
2
|
Zeisberg EM, Tarnavski O, Zeisberg M,
Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT,
Roberts AB, et al: Endothelial-to-mesenchymal transition
contributes to cardiac fibrosis. Nat Med. 13:952–961. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeisberg EM and Kalluri R: Origins of
cardiac fibroblasts. Circ Res. 107:1304–1312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oka T, Akazawa H, Naito AT and Komuro I:
Angiogenesis and cardiac hypertrophy: maintenance of cardiac
function and causative roles in heart failure. Circ Res.
114:565–571. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kovacic JC, Mercader N, Torres M, Boehm M
and Fuster V: Epithelial-to-mesenchymal and
endothelial-to-mesenchymal transition: from cardiovascular
development to disease. Circulation. 125:1795–1808. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ranchoux B, Antigny F, Rucker-Martin C,
Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F,
Planté S, et al: Endothelial-to-mesenchymal transition in pulmonary
hypertension. Circulation. 131:1006–1018. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Richards J, El-Hamamsy I, Chen S, Sarang
Z, Sarathchandra P, Yacoub MH, Chester AH and Butcher JT:
Side-specific endothelial-dependent regulation of aortic valve
calcification: interplay of hemodynamics and nitric oxide
signaling. Am J Pathol. 182:1922–1931. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yao Y, Jumabay M, Ly A, Radparvar M,
Cubberly MR and Boström KI: A role for the endothelium in vascular
calcification. Circ Res. 113:495–504. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hashimoto N, Phan SH, Imaizumi K, Matsuo
M, Nakashima H, Kawabe T, Shimokata K and Hasegawa Y:
Endothelial-mesenchymal transition in bleomycin-induced pulmonary
fibrosis. Am J Respir Cell Mol Biol. 43:161–172. 2010. View Article : Google Scholar
|
|
10
|
Liu J, Dong F, Jeong J, Masuda T and Lobe
CG: Constitutively active Notch1 signaling promotes endothelial
mesenchymal transition in a conditional transgenic mouse model. Int
J Mol Med. 34:669–676. 2014.PubMed/NCBI
|
|
11
|
Aisagbonhi O, Rai M, Ryzhov S, Atria N,
Feoktistov I and Hatzopoulos AK: Experimental myocardial infarction
triggers canonical Wnt signaling and endothelial-to-mesenchymal
transition. Dis Model Mech. 4:469–483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu X, Tan X, Tampe B, Sanchez E, Zeisberg
M and Zeisberg EM: Snail is a direct target of hypoxia-inducible
factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal
transition of human coronary endothelial cells. J Biol Chem.
290:16653–16664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peng Z, Wei D, Wang L, Tang H, Zhang J, Le
X, Jia Z, Li Q and Xie K: RUNX3 inhibits the expression of vascular
endothelial growth factor and reduces the angiogenesis, growth, and
metastasis of human gastric cancer. Clin Cancer Res. 12:6386–6394.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fu Y, Chang AC, Fournier M, Chang L,
Niessen K and Karsan A: RUNX3 maintains the mesenchymal phenotype
after termination of the Notch signal. J Biol Chem.
286:11803–11813. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen F, Liu X, Bai J, Pei D and Zheng J:
The emerging role of RUNX3 in cancer metastasis (Review). Oncol
Rep. 35:1227–1236. 2016.
|
|
16
|
Xu Q, Meng S, Liu B, Li MQ, Li Y, Fang L
and Li YG: Micro-RNA-130a regulates autophagy of endothelial
progenitor cells through Runx3. Clin Exp Pharmacol Physiol.
41:351–357. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng S, Cao J, Zhang X, Fan Y, Fang L,
Wang C, Lv Z, Fu D and Li Y: Downregulation of microRNA-130a
contributes to endothelial progenitor cell dysfunction in diabetic
patients via its target Runx3. PLoS One. 8:e686112013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang J, Li B, Zheng Z, Kang T, Zeng M,
Liu Y and Xia B: Protective effects of Notch1 signaling activation
against high glucose-induced myocardial cell injury: analysis of
its mechanisms of action. Int J Mol Med. 36:897–903.
2015.PubMed/NCBI
|
|
19
|
Ma FX, Zhou B, Chen Z, Ren Q, Lu SH,
Sawamura T and Han ZC: Oxidized low density lipoprotein impairs
endothelial progenitor cells by regulation of endothelial nitric
oxide synthase. J Lipid Res. 47:1227–1237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu X, Tan X, Hulshoff MS, Wilhelmi T,
Zeisberg M and Zeisberg EM: Hypoxia-induced endothelial-mesenchymal
transition is associated with RASAL1 promoter hypermethylation in
human coronary endothelial cells. FEBS Lett. 590:1222–1233. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SW, Won JY, Kim WJ, Lee J, Kim KH,
Youn SW, Kim JY, Lee EJ, Kim YJ, Kim KW, et al: Snail as a
potential target molecule in cardiac fibrosis: paracrine action of
endothelial cells on fibroblasts through snail and CTGF axis. Mol
Ther. 21:1767–1777. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Frías A, Lambies G, Viñas-Castells R,
Martínez-Guillamon C, Dave N, García de Herreros A and Díaz VM: A
switch in Akt isoforms is required for Notch-induced Snail1
expression and protection from cell death. Mol Cell Biol.
36:923–940. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Watson CJ, Collier P, Tea I, Neary R,
Watson JA, Robinson C, Phelan D, Ledwidge MT, McDonald KM, McCann
A, et al: Hypoxia-induced epigenetic modifications are associated
with cardiac tissue fibrosis and the development of a
myofibroblast-like phenotype. Hum Mol Genet. 23:2176–2188. 2014.
View Article : Google Scholar
|
|
25
|
Zheng Z, Zhu L, Zhang X, Li L, Moon S, Roh
MR and Jin Z: RUNX3 expression is associated with sensitivity to
pheophorbide a-based photodynamic therapy in keloids. Lasers Med
Sci. 30:67–75. 2015. View Article : Google Scholar
|
|
26
|
Tang RN, Lv LL, Zhang JD, Dai HY, Li Q,
Zheng M, Ni J, Ma KL and Liu BC: Effects of angiotensin II receptor
blocker on myocardial endothelial-to-mesenchymal transition in
diabetic rats. Int J Cardiol. 162:92–99. 2013. View Article : Google Scholar
|
|
27
|
Zhou X, Chen X, Cai JJ, Chen LZ, Gong YS,
Wang LX, Gao Z, Zhang HQ, Huang WJ and Zhou H: Relaxin inhibits
cardiac fibrosis and endothelial-mesenchymal transition via the
Notch pathway. Drug Des Devel Ther. 9:4599–4611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vasconcelos RC, Costa AL, Freitas RA,
Bezerra BA, Santos BR, Pinto LP and Gurgel BC: Immunoexpression of
HIF-1α and VEGF in periodontal disease and healthy gingival
tissues. Braz Dent J. 27:117–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Garside VC, Chang AC, Karsan A and
Hoodless PA: Co-ordinating Notch, BMP, and TGF-β signaling during
heart valve development. Cell Mol Life Sci. 70:2899–2917. 2013.
View Article : Google Scholar
|
|
30
|
Niessen K and Karsan A: Notch signaling in
cardiac development. Circ Res. 102:1169–1181. 2008. View Article : Google Scholar : PubMed/NCBI
|