Introduction
Viral myocarditis (VMC) is defined as inflammation
of the heart muscles. It is a common illness that can lead to
severe consequences even death in infants and young adults
(1,2). In a recent multicenter analysis of
624 patients with histologically confirmed myocarditis, the
presence of various viral genomes was confirmed in 239 biopsy
samples (38%) (3). Among the
identified viral genomes, adenovirus, enterovirus and
cytomegalovirus were the most common groups. In particular,
coxsackievirus B is consistently among the most common.
Coxsackieviruses are members of the Picornaviridae family,
enterovirus genus. Coxsackievirus B, particularly coxsackievirus B3
(CVB3), has been recognized as the most common cause of VMC
associated with heart failure in infants, children and young adults
(4). CVB3 infects cells via the
coxsackievirus and adenovirus receptor (CAR). The entering of CVB3
induces a direct cytopathic effect (CPE) and even cell death in
host cells. It also provokes an immune response. Both of these
effects play an important role in the pathogenesis of VMC (1,5).
The immune response has a double effect on host cells, and the
balance between these positive and negative effects may ultimately
determine the course of disease after CVB3 infection (1,6).
To date, several pathogenic mechanisms causing tissue injury and
fibrosis have been unraveled in CVB3 infection, including direct
CVB3-induced damage to the heart tissue, host-cell inflammatory
responses to the viral infection or a mixture of these two, which
may synergistically promote cardio-toxicity. CVB3-induced acute
myocarditis is most likely the consequence of direct virus-induced
myocyte damage followed by the host inflammatory response that is
associated with persistent CVB3 infection (7). The possible pathogenic mechanisms of
CVB3 include activation of caspase-3, phosphorylation and
activation of extracellular signal-regulated kinase (ERK)1/2,
activation of protein kinase B/Akt (PKB/Akt) and severe endoplasmic
reticulum (ER) stress (8–11).
Autophagy is a significant cellular catabolic
process in which long-lived proteins and damaged organelles are
degraded in lysosomes. Autophagy is a tightly regulated process and
defects in autophagy are associated with many human diseases,
including cancer, myopathy and neurodegeneration (12,13). Autophagy has also been implicated
in the clearance of pathogens and antigen presentation (14–16). In recent years, mounting evidence
indicates that autophagy is involved in the process of CVB3-related
VMC (17,18). CVB3 induces the formation of
double-membrane vesicles (autophagosomes) and the conversion of
non-modified microtubule-associated protein light chain 3 (LC3)-I
to lipidated LC3-II elevated in vitro (19–21). In a model of 3-week-old BALB/c
mice with CVB3 infection, electron microscopic observation showed
that autophagosome-like vesicles were induced in the cardiac
myocytes of mice at 3, 5, and 7 days after CVB3 infection. LC3-I
and LC3-II were also significantly increased in the myocardium and
the cardiac myocytes extracted from the ventricles of mice infected
with CVB3. Moreover, viral protein synthesis was significantly
decreased in primary cardiac myocytes following treatment with
3-methyladenine, an inhibitor of autophagy (20). CVB3 may exploit the autophagic
response to promote viral replication (22) via inhibiting the fusion of
autophagosomes with lysosomes to provide sufficient membrane
structures for viral RNA replication (23,24).
Mammalian target of rapamycin (mTOR) is described as
a key homeostatic regulator of cell growth, proliferation, and
survival as well as metabolism by upregulating protein, lipid
synthesis and inhibiting excessive autophagy. mTOR is closely
related with autophagy induced by viral infection. Sindbis virus
and avian influenza viruses can induce autophagy by suppressing
mTOR signaling (25,26). Vesicular stomatitis virus induces
autophagy by regulating the phosphatidylinositol 3-kinase
(PI3K)/Akt signaling pathway (27), and hepatitis C virus induces
autophagy by inactivating the Akt/TSC/mTOR pathway based on ER
stress (28). Avibirnavirus VP2
can induce autophagy through Akt/mTOR pathway inactivation mediated
by the HSP90AA1-VP2 complex (29). The function of mTOR is primarily
mediated by mTOR complex 1 (mTORC1) and mTORC2. The initiation of
autophagy requires the Unc-51-like kinase (ULK) complex and is
ultimately regulated by mTORC1. Under fed conditions, the ULK
complex is bound to mTORC1, whereas nutrient depletion results in
dissociation of the ULK complex from mTORC1. In the unbound state,
the ULK complex induces autophagy (30). Recent studies have also
established mTORC2 as an independent positive regulator of
autophagy (31,32).
Although the number of autophagosomes increases
after CVB3 infection, the exact mechanism is unclear. Earlier
studies suggested that CVB3 may directly or indirectly induce
autophagy via the AMPK/MEK/ERK and Ras/Raf/MEK/ERK signaling
pathways in host cells (20,33). In mammalian cells, the
PI3K/Akt/mTOR signaling pathway is the primary pathway that
regulates autophagy when cells are exposed to certain conditions,
such as starvation, oxidative stress, infection and tumor
suppression (34). Our previous
study demonstrated that the PI3K/Akt/mTOR signaling pathway is
associated with apoptosis in CVB3-infected cells (8,35,36). Whether the PI3K/Akt/mTOR signaling
pathway is involved in the process of autophagy in CVB3-infected
cells is unknown. The present study seeks to examine whether the
PI3K/Akt/mTOR signaling pathway participates in the autophagic
process after CVB3 infection in HeLa cells.
Materials and methods
Antibodies and chemical reagents
Polyclonal antibody against LC3-I/II (Sigma-Aldrich,
St. Louis, MO, USA), antibody against β-actin and SQSTM1 rabbit
polyclonal antibody/p62 (both from ProteinTech Group, Inc.,
Chicago, IL, USA) were used at a dilution of 1:1,000. Monoclonal
anti-enterovirus antibody (Dako, Carpinteria, CA, USA) was used at
a dilution of 1:100. Goat anti-mouse IgG horseradish
peroxidase-conjugated secondary antibody and goat anti-rabbit IgG
horseradish peroxidase-conjugated secondary antibody (both from
CWBIO, Beijing, China) were used at a dilution of 1:2,000.
Rapamycin, ZSTK474, MK2206, and chloroquine phosphate were
purchased from Selleck Chemicals (Houston, TX, USA).
Cell culture and virus propagation
HeLa cells were obtained from the Institute of
Oncology, Central South University, China. The stably transfected
cell lines containing the plasmid pcDNA3.1-myc-HisA(−)-Akt1 or the
empty vector alone were previously established by our team
(35) and kept in our laboratory.
Cells were grown in Dulbecco's modified Eagle's medium (DMEM;
Hyclone/GE Healthcare Life Sciences, Logan, UT, USA) supplemented
with 10% fetal bovine serum (FBS) and penicillin-streptomycin (both
from Gibco Life Technologies, Carlsbad, CA, USA), and incubated at
37°C in a humidified incubator with 5% CO2. CVB3 Nancy
strain was obtained from the Shanghai Jiao Tong University School
of Medicine and stored at −80°C in our laboratory. CVB3 was
propagated in HeLa cells in DMEM supplemented with 2% FBS, and the
virus titer was routinely determined by plaque assay.
Virus infection
HeLa cells were infected with CVB3 at a multiplicity
of infection (MOI) of 10. Then, the cells were washed with PBS and
replenished with fresh DMEM containing 10% FBS. DMEM containing 2%
FBS was used for the sham group. Cells were harvested at different
times of infection for the next experiments. For inhibition
experiments, HeLa cells were pre-treated with the inhibitors
(dissolved in DMEM containing 10% FBS) for 2 h before viral
infection, and DMEM containing 10% FBS for the sham group. Then,
the cells were washed with PBS and infected with CVB3 for 1 h, and
fresh DMEM containing 2% FBS for the sham group.
Cell transfection
HeLa cells at 80% confluency were transfected with
pEGFP-LC3 or an empty vector (pEGFP-C3). Four milligrams of
expression plasmid combined with 10 µl Lipofectamine 2000
reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) was added
to the cells according to the manufacturer's instructions. At 5 h
post-transfection, the cells were refreshed with DMEM containing
10% FBS. Twenty-four hours later, the cells were seeded in 6-well
plates for the next experiments.
Immunofluorescence assay
HeLa cells transfected with plasmid pEGFP-LC3 or
pEGFP-C3 were seeded on cover glasses and exposed to CVB3 or not.
Cells were washed thrice with cold PBS. The different groups were
fixed in 4% paraformaldehyde for 10 min at room temperature,
permeabilized and then blocked with 5% bovine serum albumin in 0.3%
Triton X-100 (diluted in PBS) for 2 h. The samples were incubated
with anti-lysosomal-associated membrane protein 1 (LAMP-1) (1:100)
overnight at 4°C and then washed thrice with 0.3% Triton X-100
(diluted with deionized water) and incubated for 60 min with
HRP-conjugated goat anti-mouse IgG. After three washes, the cells
were stained with DAPI (1:1,000) for 5 min and washed again.
Afterwards, cover slips were mounted onto glass microscopic slides,
and the samples were observed under an Olympus microscope equipped
with a MetaMorph image acquisition system (DP2-BSW software) (both
from Olympus Corp., Tokyo, Japan).
Western blot analysis
HeLa cells exposed to various conditions were washed
twice with ice-cold PBS and then lysed with RIPA lysis buffer
(CWBIO) containing 0.1% phenylmethylsulfonyl fluoride (PMSF;
CWBIO). Afterwards, the lysates were sonicated and centrifuged at
13,000 rpm for 15 min at 4°C. The samples were subsequently boiled
and denatured. The protein concentration was determined by the
Enhanced BCA protein Assay kit (Beyotime Institute of
Biotechnology, Shanghai, China). Briefly, 30 µg of cell
protein samples was subjected to 10–12% SDS polyacrylamide gel
electrophoresis and transferred to 0.45-mm PVDF membranes (EMD
Millipore, Billerica, MA, USA). After blocking with 5% non-fat milk
for 3 h (room temperature), the membranes were incubated with the
primary antibody at 4°C overnight. After washing with PBST thrice,
the membranes were incubated with horseradish peroxidase-conjugated
secondary antibodies (CWBIO) for 60 min followed by washing.
Finally, protein bands were detected with X-ray film, and protein
expression was quantitated by densitometric analysis using NIH
Image J.
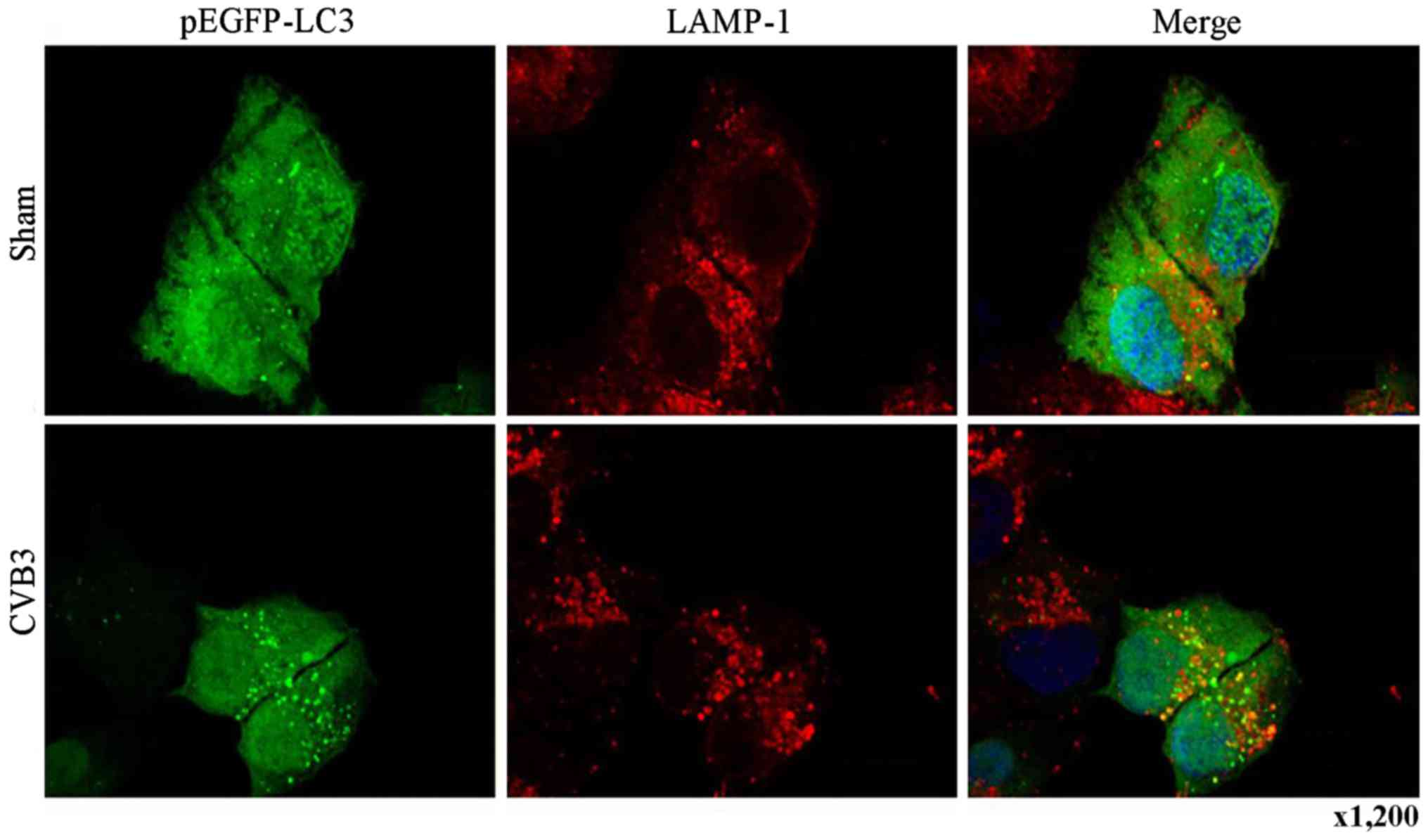
Confocal microscopy
To determine whether autophagosomes in CVB3-infected
HeLa cells fused with lysosomes, cells were transiently transfected
with the pEGFP-LC3 plasmid using Lipofectamine 2000 (Invitrogen
Life Technologies). After 24 h of transfection, the cells were
infected with CVB3 or not and then incubated with anti-LAMP-1
overnight (see details in 'Immunofluorescence assay'). We observed
the co-localization of LAMP-1 and GFP-LC3 under a Leica SP2 AOBS
confocal fluorescence microscope. Autophagosomes can be
distinguished from amphisomes or autolysosomes as follows: green
vesicles [GFP-LC3+ LAMP-1-negative (LAMP-1−)]
are autophagosomes; yellow vesicles [GFP-LC3+
LAMP-1-positive (LAMP-1+)] are amphisomes; and red
vesicles [GFP-LC3-negative (pEGFP-LC3−)
LAMP-1+] cannot be categorized solely on the basis of
LAMP-1 expression, as they may be endosomes, lysosomes, or
autolysosomes.
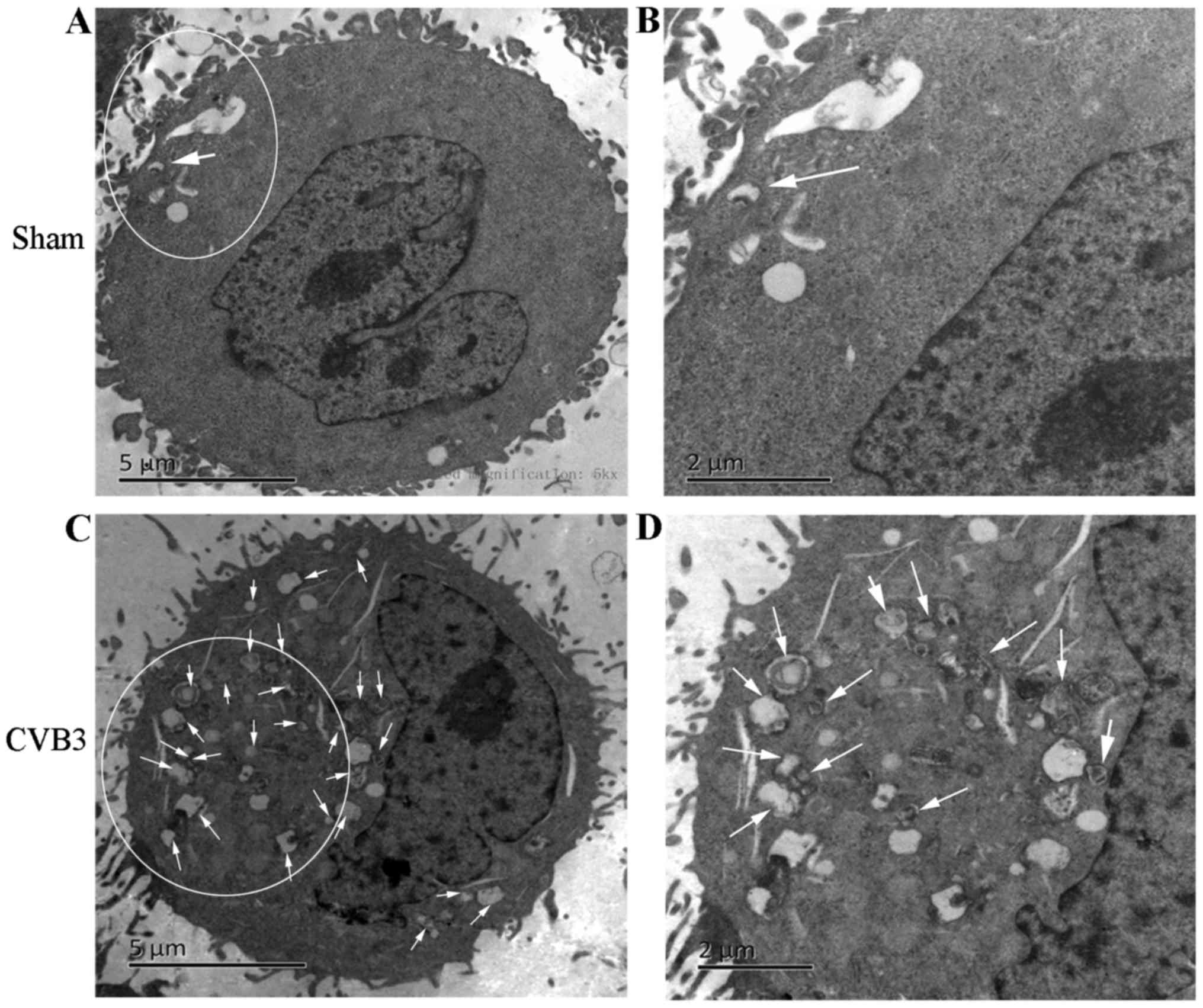
Transmission electron microscopy
(TEM)
For ultrastructural analysis, HeLa cells were mock
infected or infected with CVB3 at MOI of 10 for 1 h. Cells were
collected at 24 hours post-inoculation (h.p.i.) and centrifuged at
1,200 rpm for 10 min at room temperature. Cells were then fixed in
2.5% glutaraldehyde in PBS for 10 min at room temperature.
Following three washes in PBS, the cells were post-fixed in 2%
osmium tetroxide for 2 h. Then, the cells were dehydrated in a
graded series of acetone washes successively. Then, the cells were
soaked in epoxy resin mixed with an equal volume of pure acetone
and embedded in Eponate 12 resin for 24 h. Finally, the dyed slices
were observed, and images were obtained via TEM (Hitachi H-7500;
Hitachi, Ltd., Tokyo, Japan). Approximately 15 cells were counted.
The number of double-membrane vesicles for each cell was examined.
Autophagosomes were defined as double-membrane vacuoles measuring
0.2–0.5 µm.
Semi-quantitative PCR
HeLa cells either untreated or treated with
different inhibitors were harvested at 24 h.p.i. mRNA was extracted
following the method of the RNA extraction kit (Omega Bio-Tek,
Inc., Norcross, GA, USA). Next, mRNA was reverse transcribed into
cDNA following the RevertAid First Strand cDNA Synthesis kit
(Thermo Fisher Scientific, Waltham, MA, USA). Finally, PCR
amplification was performed. The primer sequences and PCR
conditions are provided in Tables
I and II [refer to Li et
al (36)].
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Primer
sequences |
|---|
| CVB3 | F:
5′-TGGTGGGCTATGGAGTATGG-3′ |
| R:
5′-CACTGGATGGGGTGTTGTCT-3′ |
| β-actin | F:
5′-CTAAGGCCAACCGTGAAAAGATGAC-3′ |
| R:
5′-TGGGTACATGGTGGTGCCACCAGAC-3′ |
| Table IIPCR conditions. |
Table II
PCR conditions.
| Genes | Conditions |
|---|
| CVB3 | 95°C for 2 min, 30
sec; 94°C for 40 sec; 59°C for 40 sec, 72°C for 40 sec, 35 cycles;
72°C for 5 min |
Drug treatment
To identify the autophagic response in HeLa cells
infected by CVB3, rapamycin (100 mM dissolved in DMEM containing
10% FBS) served as a positive control and 24 h later, the cells
were harvested for western blot analysis. Chloroquine phosphate
(inhibition of lysosome function, 40 µM) was used to assess
autophagic flux caused by CVB3 infection, and rapamycin (10 nM
dissolved in DMEM containing 10% FBS) was used to inhibit the
function of mTOR; 2 h later, the cells were washed thrice and
infected with CVB3. ZSTK474 (50 µM) was used at a
concentration of 50 µM to inhibit PI3K and 2 h later, the
cells were washed thrice and infected or not with CVB3. To suppress
the overexpression of Akt1, MK2206 (inhibitor of Akt, 50 µM,
dissolved in DMEM containing 10% FBS) was used to pre-treat the
pcDNA3.1-myc-HisA(−)-Akt1-HeLa cells. After 2 h, the cells were
infected and harvested at 24 h.p.i. for western blot analysis.
Statistical analysis
Two-way analysis of variance with multiple
comparisons and paired Student's t-tests were performed. Data are
presented as the means ± standard error (SE). A value of P<0.05
was considered to indicate a statistically significant
difference.
Results
CVB3 induces an autophagic response at
different times during CVB3 infection in HeLa cells
During the process of autophagosome formation, the
conversion of cytosolic LC3-I into the lipidated form LC3-II is
pivotal. Thus, the processing from LC3-I to LC3-II represents the
appearance of autophagosomes or autophagic response. The
membrane-bound form of LC3 (LC3-II) is considered as the most
appropriate marker of autophagosomes in mammalian cells. The
protein p62 is considered as a marker for autophagy-mediated
protein degradation or autophagic flux (37); it functions as an autophagy
receptor targeting ubiquitinated proteins to autophagosomes for
degradation. To characterize the autophagic reaction and the change
of viral replication during CVB3 infection, HeLa cells were
infected with CVB3. Cell lysates were collected at 1, 3, 5, 7, 9,
12 and 24 h.p.i. Mock-infected HeLa cells served as the sham
control. We examined LC3, p62, p-mTOR and the viral capsid protein
VP1 by western blot analysis at different times during CVB3
infection (Fig. 1). The
LC3-II/LC3-I ratio increased at 1 h.p.i. and reached its peak at 5
h.p.i. (p<0.05). p62 showed a decrease at 1, 3, 5, 7 and 9
h.p.i. (p<0.05). No changes were noted at 12 and 24 h.p.i.
compared with the sham group. p-mTOR exhibited a decrease at 5, 7,
and 9 h.p.i (p<0.05), which was more obvious at 12 and 24 h.p.i.
We detected obvious VP1 expression 5 h.p.i. (p<0.05) which
increased as the infected time increased (p<0.01).
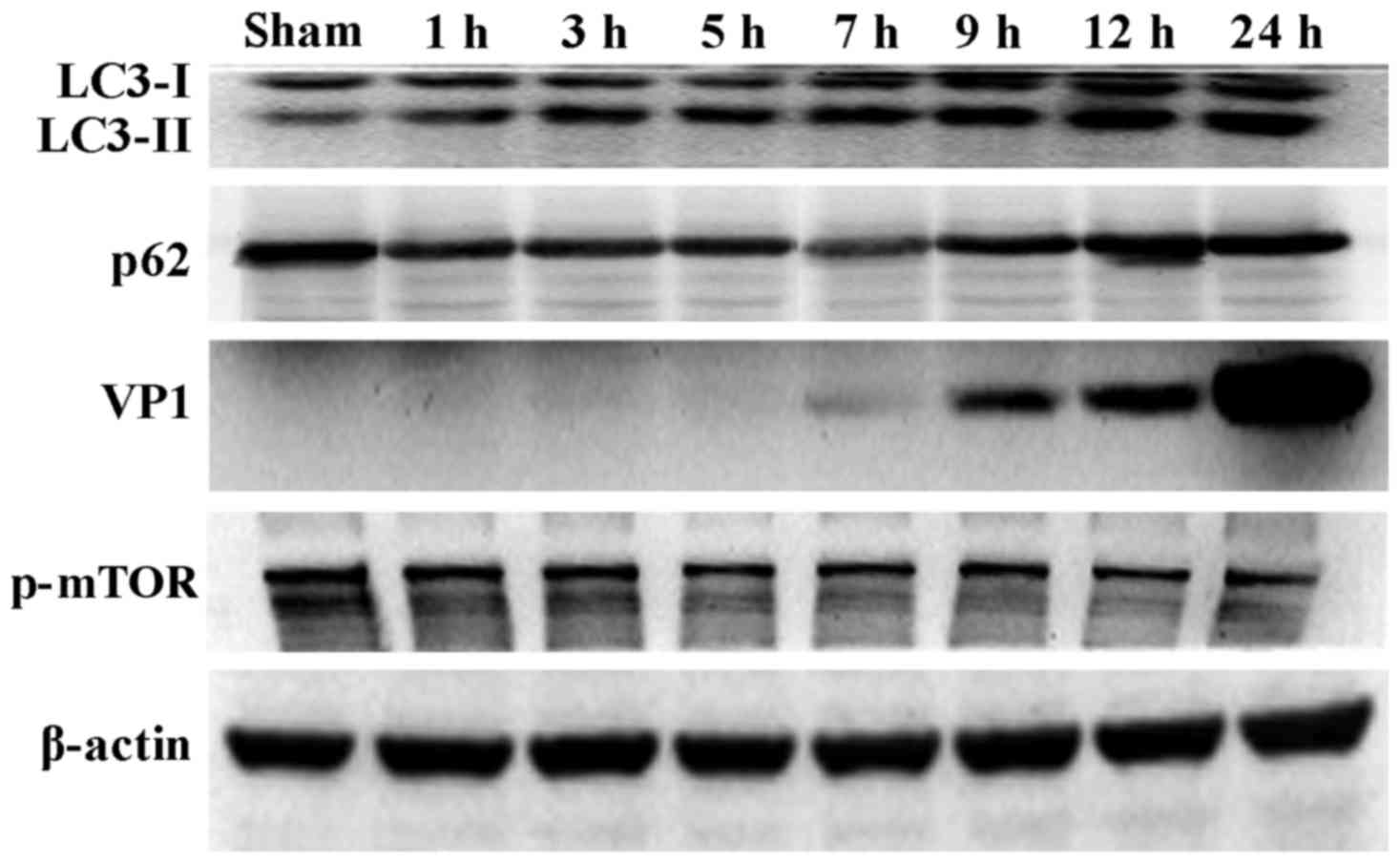 | Figure 1CVB3 induces an autophagic response
at different times during CVB3 infection in HeLa cells. HeLa cells
were infected with CVB3, cell lysates were collected at 1, 3, 5, 7,
9, 12 and 24 h.p.i., and mock-infected HeLa cells served as the
sham control. LC3, p62, p-mTOR and viral capsid protein VP1 were
examined by western blot analysis, and these samples were
immunoblotted with an antibody to β-actin to illustrate equal
protein loading. Compared with the sham group, the LC3-II/LC3-I
ratio increased at 1 h.p.i. then peaked at 5 h.p.i. (p<0.05).
p62 exhibited a decrease at 1, 3, 5, 7 and 9 h.p.i. (p<0.01). No
changes were noted at 12 and 24 h.p.i. compared with the sham
group. p-mTOR showed a decrease after 5 h.p.i. (p<0.05), which
was more obvious at 12 and 24 h.p.i. VP1 increased as the time of
infection increased. *P<0.05, compared with the sham
group; **p<0.01, compared with the sham group. CVB3,
coxsackievirus B3; h.p.i., hours post-inoculation; LC3, light chain
3; mTOR, mammalian target of rapamycin. |
Confirmation of autophagy activation in
HeLa cells at 24 h.p.i
Given that LC3 and p-mTOR was altered during CVB3
infection, to further characterize the effects of the mTOR pathway
during CVB3 infection, we selected 24 h.p.i. as our research time
point to conduct our next study.
Primarily, we identified the autophagic response in
HeLa cells infected by CVB3. We determined the activation of
autophagy using three criteria. First, we analyzed the lipidated
form of the microtubule-associated protein 1 LC3 in the
CVB3-infected HeLa cells and mock-infected cells, and rapamycin
(100 µM) served as a positive control (Fig. 2). The results revealed that the
total amount of LC3 (LC3-I, LC3-II) and the ratio of LC3-II/LC3-I
were markedly increased in the infected cells at 24 h.p.i. compared
with the mock-infected cells (p<0.05). The expression of p-mTOR
was decreased (p<0.05). Second, we found that double-membrane
autophagosomes, which are prominent features of autophagy (Fig. 3), were increased in the
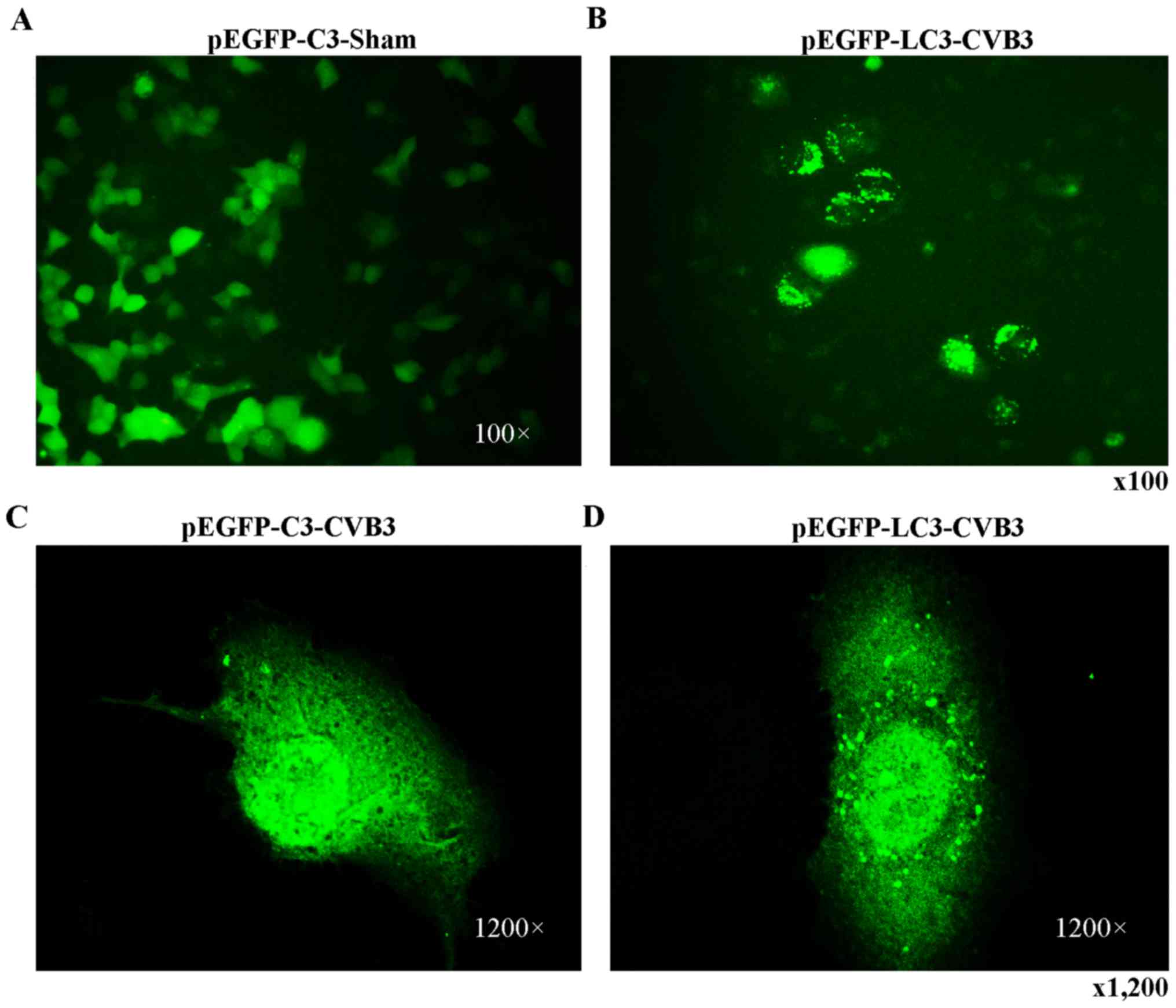
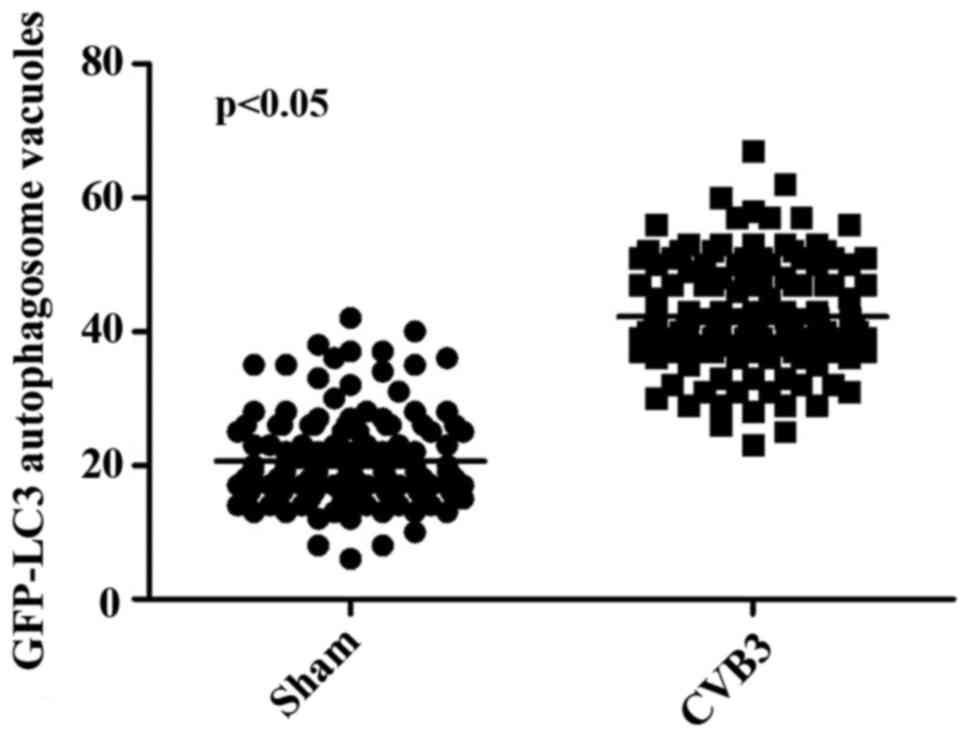
CVB3-infected HeLa cells. Finally, we observed that LC3 puncta
formation was increased following CVB3 infection (Fig. 4). We counted 50 transduced
cells/well for quantification of cells with high levels of GFP-LC3
autophagosome vacuoles, with 3 wells/group (represented as the mean
± SEM). Quantification of >30 punctate/cell was defined as a
high level of GFP-LC3 autophagosome vacuoles. We observed a
statistically significant increase in LC3 puncta formation within
the infected pEGFP-LC3-HeLa cells at 24 h.p.i. compared with that
in the mock-infected HeLa cells or the CVB3-infected pEGFP-C3-HeLa
cells (Fig. 5, p<0.05). To
assess the effects of CVB3 on autophagosome fusion with
endosomes/lysosomes, we observed the distribution of LAMP-1, a
protein found in endosomes and lysosomes. Autophagosomes can be
distinguished from amphisomes or autolysosomes as follows: green
vesicles (pEGFP-LC3+/LAMP-1−) are
autophagosomes; yellow vesicles
(pEGFP-LC3+/LAMP-1+) are amphisomes; and red
vesicles (pEGFP-LC3−/LAMP-1+) cannot be
categorized solely on the basis of LAMP-1 expression, as they may
be endosomes, lysosomes, or autolysosomes (pEGFP-LC3-II is degraded
in autolysosomes), In addition, GFP fluorescence is quenched in an
acidic environment (38). In our
research, we observed yellow dots, which suggested the
co-localization of GFP-LC3 and LAMP-1 (Fig. 6).
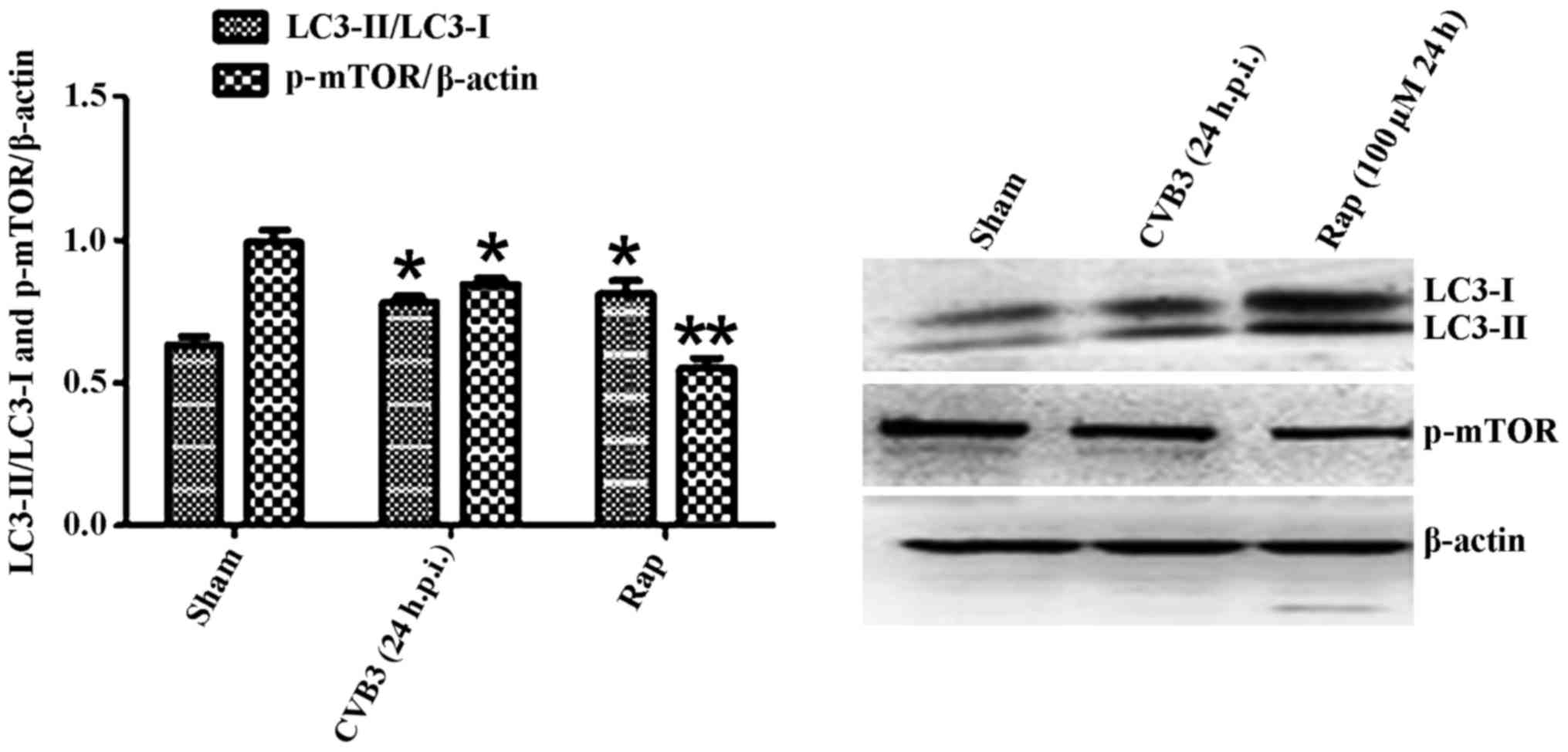 | Figure 2LC3 and p-mTOR-are altered in HeLa
cells at 24 h.p.i. HeLa cells were infected with CVB3. DMEM
containing 2% FBS served as the sham control, and rapamycin (Rap)
(100 µM) served as a positive control. At 24 h.p.i. cells
were collected for western blot analysis. Compared with the sham
group, the total amount of LC3 (LC3-I, LC3-II) and the ratio of
LC3-II/LC3-I were markedly increased in the infected cells at 24
h.p.i. (p<0.05). The expression of p-mTOR was downregulated
(p<0.05). *p<0.05, compared with the sham group;
**p<0.01, compared with the sham group. LC3, light
chain 3; mTOR, mammalian target of rapamycin; h.p.i., hours
post-inoculation; CVB3, coxsackievirus B3; DMEM, Dulbecco's
modified Eagle's medium; FBS, fetal bovine serum. |
Our findings revealed that CVB3 triggered auto
phagic response in HeLa cells, in accordance with previous studies
(19,20).
Rapamycin aggravates the autophagic
reaction induced by CVB3 infection
To further elucidate the impact of mTOR during the
course of CVB3 infection, HeLa cells were treated with rapamycin
(10 nM dissolved in DMEM containing 10% FBS) for 2 h. Then, the
cells were washed thrice and infected with CVB3 (rapamycin group).
In the CVB3 group, HeLa cells were incubated with DMEM containing
10% FBS for 2 h and then infected with CVB3. The control group was
incubated with DMEM containing 10% FBS. Two hours later, the medium
was refreshed with DMEM containing 2% FBS. At 24 h.p.i., the cells
were collected for western blot analysis (Fig. 7). The LC3-II/LC3-I ratio was
increased in the CVB3 group and the rapamycin group (p<0.01),
and p-mTOR was decreased in the CVB3 group (p<0.05) and
rapamycin group (p<0.01), compared with the control group. When
comparing the virus-infected condition with the rapamycin +
virus-infected condition, we found that pre-treatment of the cells
with rapamycin aggravated the autophagy induced by CVB3 infection
(p<0.05). Furthermore, we used chloroquine phosphate (inhibition
of lysosome function, 40 µM, 2 h before infection) to assess
autophagic flux caused by CVB3 infection (CQ group) and monitored
p62. The LC3-II/LC3-I ratio and p62 were increased more obviously
in the CQ group compared with the CVB3 group (p<0.05).
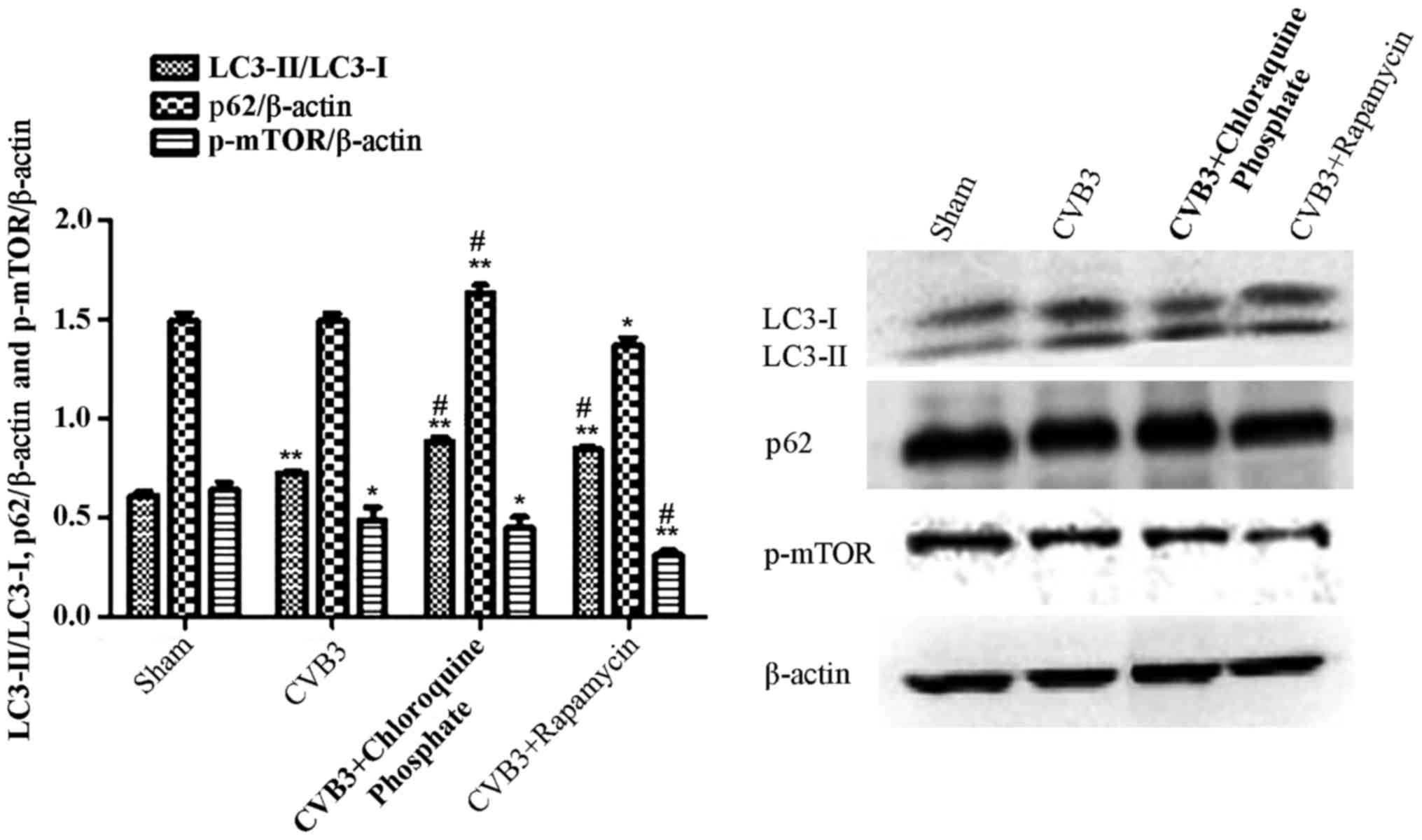 | Figure 7Rapamycin aggravates the autophagic
reaction induced by CVB3 infection. HeLa cells were treated with 10
nM rapamycin and chloroquine phosphate (CQ group) for 2 h. The
cells were washed thrice and infected with CVB3. In the CVB3 group,
HeLa cells were incubated with DMEM containing 10% FBS for 2 h and
infected with CVB3. The sham group was incubated with DMEM
containing 10% FBS. Two hours later, the medium was refreshed with
DMEM containing 2% FBS. At 24 h.p.i., cells were collected for
western blot analysis. The LC3-II/LC3-I ratio was increased in the
CVB3 and rapamycin group (p<0.01), and p-mTOR was decreased in
the CVB3 group (p<0.05) and rapamycin group (p<0.01) compared
with the sham group. The changes in the LC3-II/LC3-I ratio and
p-mTOR were more obvious in the Rap group compared with the CVB3
group (p<0.05). The LC3-II/LC3-I ratio and p62 was increased in
the CQ group compared with the CVB3 group (p<0.05).
*p<0.05, compared with the sham group;
**p<0.01, compared with the sham group;
#p<0.05, compared with the CVB3 group. CVB3,
coxsackievirus B3; DMEM, Dulbecco's modified Eagle's medium; FBS,
fetal bovine serum; h.p.i., hours post-inoculation; LC3, light
chain 3; mTOR, mammalian target of rapamycin. |
ZSTK474 alleviates the autophagic
response caused by CVB3 infection in HeLa cells
To further elucidate whether PI3K is involved in
CVB3-induced autophagy, we used ZSTK474, a novel PI3KC1 inhibitor,
to assess the effects of PI3KC1 on autophagy during CVB3 infection.
HeLa cells were pre-treated with ZSTK474 (50 µM, 2 h), and
the cells were infected with CVB3 or not. Cell lysates were
collected at 24 h.p.i. The expression levels of LC3 and
phosphorylated Akt1 (p-Akt1) were determined by western blot
analysis (Fig. 8). The results
showed that p-Akt1 was decreased in the CVB3 infection group
(p<0.05) and the ZSTK474 treatment group (p<0.01) accompanied
by a significant increase in the LC3-II/LC3-I ratio (p<0.01).
The LC3-II/LC3-I ratio in the CVB3-infected + ZSTK474 group was
reduced compared with that in the CVB3-infected group
(p<0.05).
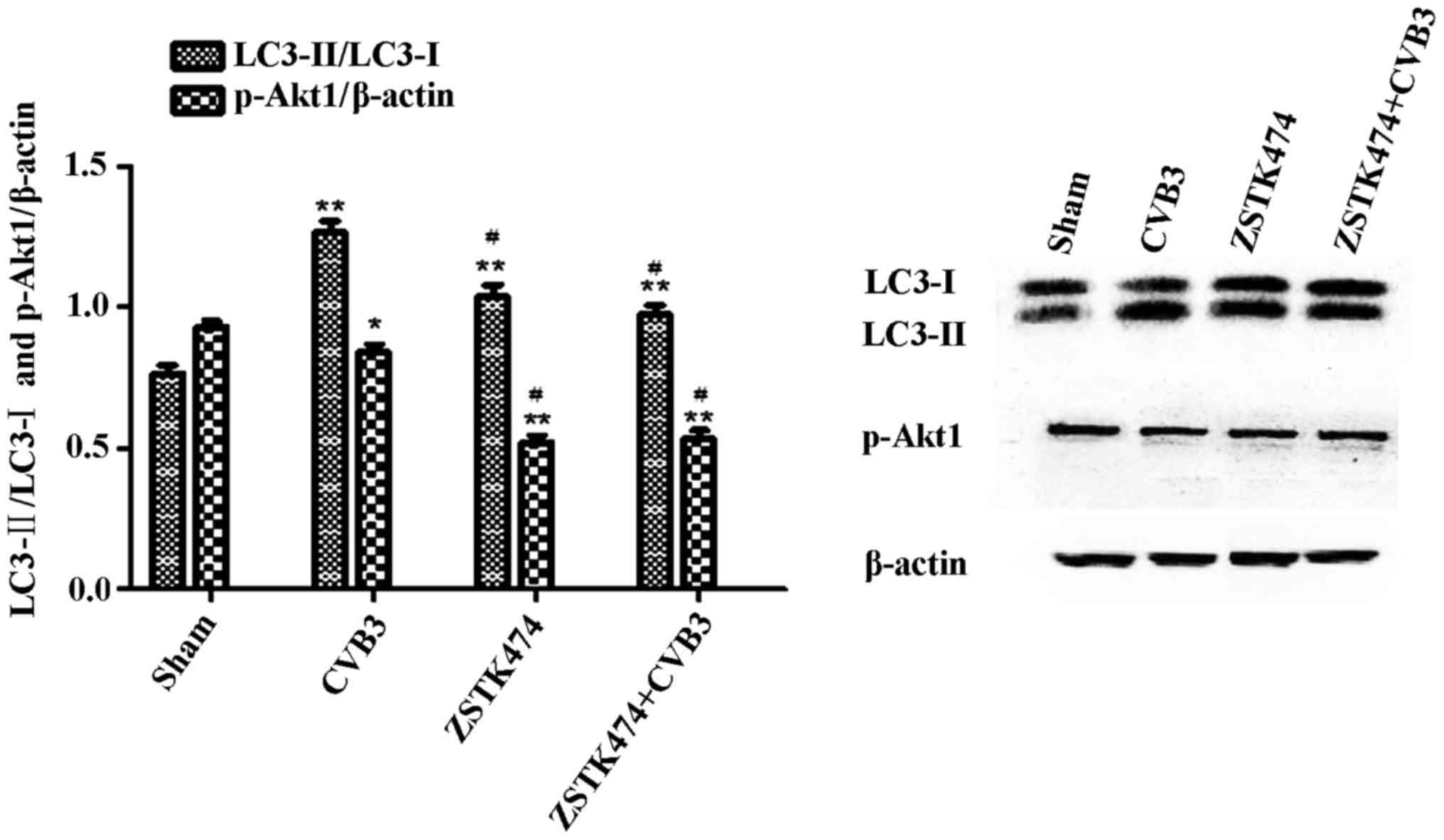 | Figure 8Inhibition of PI3K with ZSTK474
alleviates the autophagic reaction caused by CVB3 infection. HeLa
cells were pre-treated with ZSTK474 (50 µM, 2 h) and then
infected with CVB3 (ZSTK4747 + CVB3 group) or not (CVB3 group).
Mock-infected cells served as the sham control. Cell lysates were
collected at 24 h.p.i., and the expression of LC3 and p-Akt1 was
determined by western blot analysis. Compared with the sham group,
p-Akt1 was decreased in the CVB3 group (p<0.05) and the ZSTK474
group (p<0.01). The LC3-II/LC3-I ratio was increased
(p<0.01). The LC3-II/LC3-I ratio was increased in the CVB3 group
compared with the ZSTK474 + CVB3 group (p<0.05).
*p<0.05, compared with the sham group;
**p<0.01, compared with the sham group;
#p<0.05, compared with the CVB3 group. PI3K,
phosphatidylinositol 3-kinase; CVB3, coxsackievirus B3; h.p.i.,
hours post-inoculation; LC3, light chain 3; p-Akt1, phosphorylated
Akt1. |
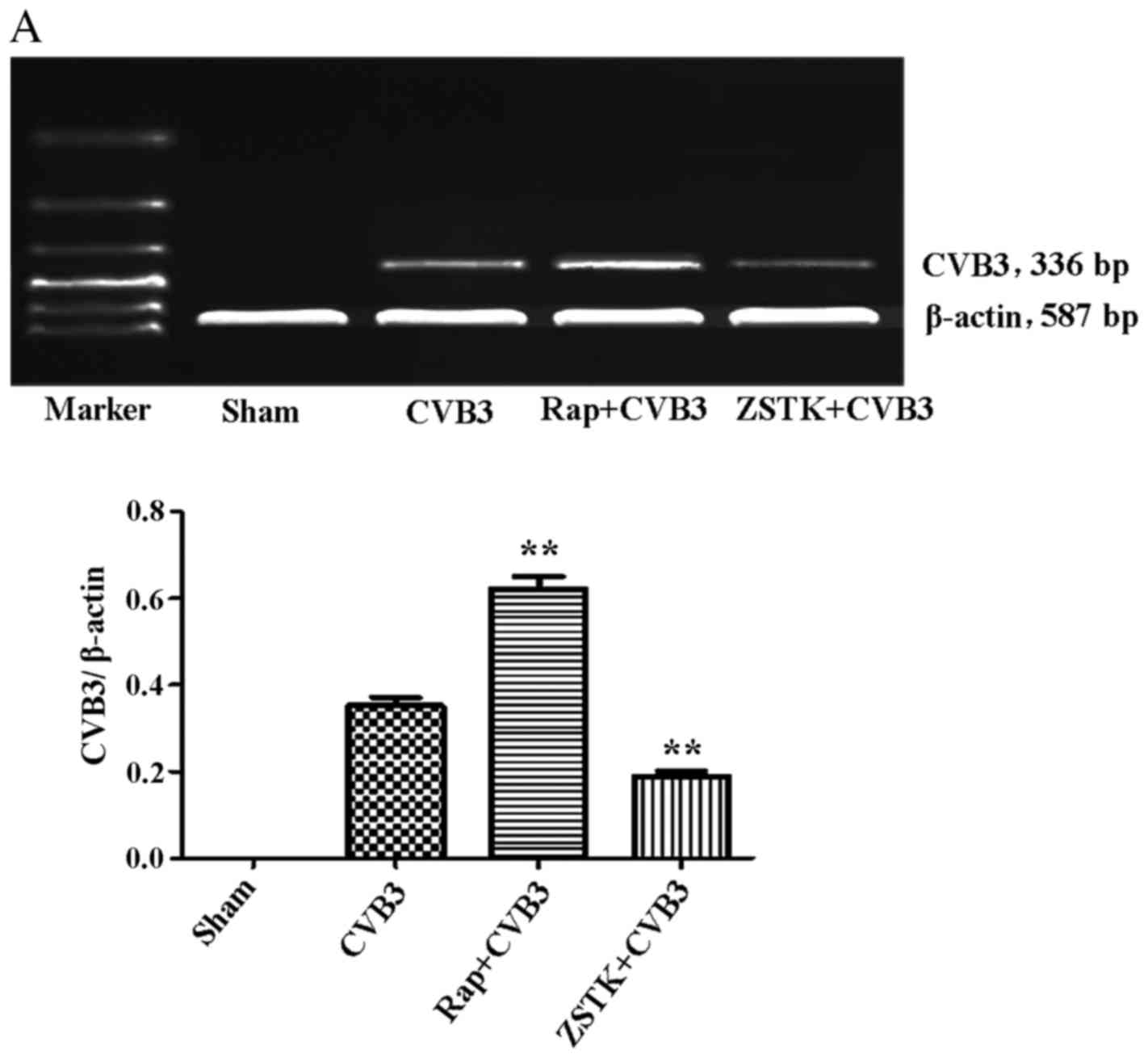
Rapamycin and ZSTK474 affect viral
replication
To further characterize the change in viral
replication when this pathway was blocked, HeLa cells pre-treated
with rapamycin (10 nM) and ZSTK474 (50 µM) were infected
with CVB3, and we examined mRNA (Fig.
9A) and viral capsid protein VP1 (Fig. 9B) by semi-quantitative PCR and
western blot analysis, respectively. ZSTK474 inhibited viral mRNA
synthesis and blocked viral protein VP1 expression caused by CVB3
infection (p<0.01), whereas rapamycin stimulated viral mRNA
synthesis (p<0.01). However, the viral protein VP1 did not
mirror the mRNA changes in the rapamycin group.
Inhibition of Akt1 aggravates the
autophagic response caused by CVB3 infection in Akt1-overexpressing
cells
To clarify whether the changes in Akt1 affect the
autophagic reaction in CVB3-induced autophagy, we examined the
autophagy activation in cell lines containing the plasmid
pcDNA3.1-myc-HisA(−)-Akt1 or the empty vector alone. The
establishment of stable cell lines overexpressing Akt1 was
described in our previous study (35). The cell lines overexpressing Akt1
or the containing empty vector alone were infected with CVB3. The
cells were collected for western blot analysis at 24 h.p.i. To
suppress the overexpression of Akt1, we used MK2206 (inhibitor of
Akt, 50 µM, dissolved in DMEM containing 10% FBS) to
pre-treat the pcDNA3.1-myc-HisA(−)-Akt1-HeLa cells. Two hours
later, the cells were infected and harvested at 24 h.p.i. LC3 and
p-Akt1 were determined by western blot analysis (Fig. 10), with mock-infected cells
serving as the sham control. In the Akt1-overexpressing cell line,
CVB3 infection increased the LC3-II/LC3-I ratio (p<0.05).
Moreover, the LC3-II/LC3-I ratio increased to greater levels when
the infected cells were pre-treated with MK2206. In addition, under
uninfected conditions, the LC3-II/LC3-I ratio was higher in the
empty vector cells compared with that in the Akt1-overexpressing
cells (p<0.05).
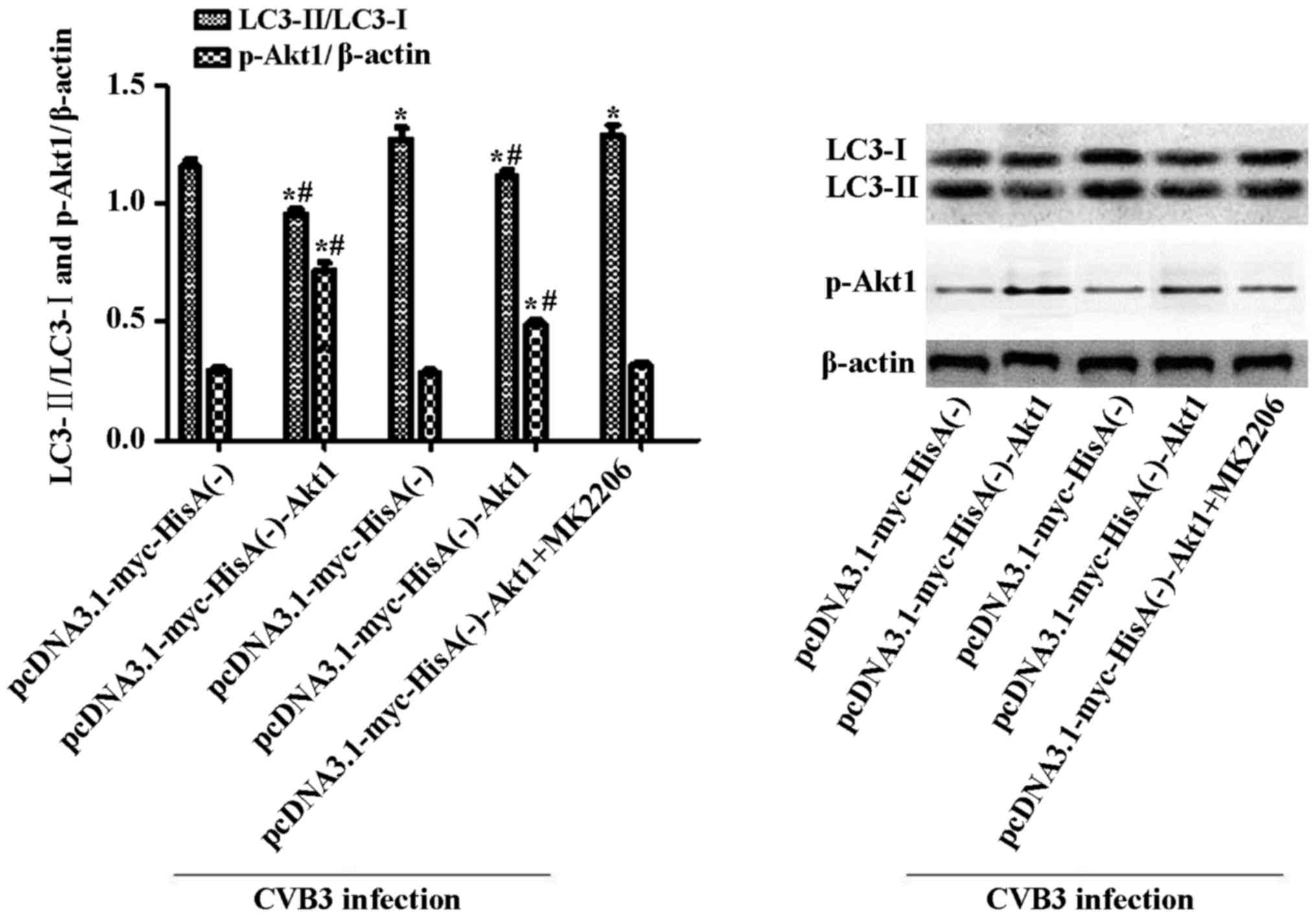 | Figure 10Inhibition of Akt1 aggravates the
autophagic response caused by CVB3 infection in Akt1-overexpressing
cells. Cells overexpressing Akt1 or harboring empty vector alone
were infected with CVB3 or not. Cells were collected for western
blot analysis at 24 h.p.i. To suppress the overexpression of Akt1,
MK2206 (50 µM) was used to pre-treat the
pcDNA3.1-myc-HisA(−)-Akt1 HeLa cells. Two hours later, the cells
were infected and harvested at 24 h.p.i., and mock-infected cells
served as the sham control. In Akt1-overexpressing cell lines, CVB3
infection increased the LC3-II/LC3-I ratio (p<0.05). The ratio
was more significantly increased when the infected cells were
pre-treated with MK2206 (p<0.05). Under uninfected conditions,
the LC3-II/LC3-I ratio was higher in Akt1-overexpressing cells
compared with the empty vector cells (p<0.05).
*p<0.05, compared with the pcDNA3.1-myc-HisA(−)
group; #p<0.05, compared with the
pcDNA3.1-myc-HisA(−)-Akt1+CVB3 group. CVB3, coxsackievirus B3;
h.p.i., hours post-inoculation; LC3, light chain 3. |
Discussion
Autophagy is a significant cellular catabolic
process in which long-lived proteins and damaged organelles are
degraded in lysosomes. Autophagy also serves as an important
function in innate host defense by eliminating intracellular
pathogens (39,40). Autophagy begins with the
generation of phagophores, which elongate and self-fuse to form
double-membrane vesicles, known as autophagosomes (41). The formation of phagophores and
autophagosomes is meditated by autophagy-related genes (ATGs).
Microtubule-associated protein LC3, the mammalian homolog of yeast
ATG8, is one well-studied marker of the presence of autophagic
membranes (42). LC3-I localizes
to the cytoplasm when autophagy occurs. LC3-I is conjugated with
phosphatidylethanolamine (PE) to form lipidated LC3-PE, also known
as LC3-II. The p62 protein interacts with LC3-II to target cargo to
the autophagosomes for degradation. Autophagosomes fuse with
endosomes to generate amphisomes, which obtain vacuolar-ATPase and
become acidic. Subsequently, amphisomes fuse with incoming
lysosomes to form autolysosomes. The cargo in the lumina of
autolysosomes is then degraded by lysosomal proteases. The p62
protein can also be used to indicate the level of autophagic flux,
as this protein is degraded along with LC3-II during complete
autophagy (43,44). However, during CVB3 infection,
viral protease 2Apro can also account for p62 cleavage
(45).
CVB3 infection induces autophagic reactions in
vivo and in vitro, but the exact mechanism is unclear.
The PI3K/Akt/mTOR pathway plays a vital role in the regulation of
autophagy (46). Class I PI3K is
the traditional upstream activator of mTOR. As the major upstream
modulator, the PI3K pathway regulates autophagy by phosphorylating
Akt. Activation of Akt activates mTORC1 by phosphorylation of
tuberous sclerosis complex (TSC)2, causing the inhibition of the
GTPase-activating protein (GAP) domain of TSC2 leading to the
activation of mTORC1 and inhibition of autophagy (47).
Our research demonstrated that CVB3 induces
autophagosome formation in HeLa cells, including an increase in the
LC3-II/LC3-I ratio, LC3 punctate dot formation as demonstrated by
immunofluorescence and autophagosome-like vesicle formation as
detected via TEM. These data suggested a successful induction of
autophagy caused by CVB3. Previous in vitro studies
suggested that CVB3 could inhibit the degradation of virus-induced
autophagosomes by blocking their fusion with lysosomes (48). In our time point experiment of
CVB3 infection, autophagy was rapidly activated after viral
infection and reached its peak at 5 h.p.i. accompanied by an
obvious decrease of p62. Then, p62 increased as the time of
infection increased. We did not detect an obvious expression of VP1
until 7 h.p.i. We then hypothesized that autophagy may play a
protective role at the beginning of virus infection given that
activated autophagy was accompanied by a degradation in p62. This
may explain why we were unable to detect VP1 until 7 h.p.i. Then,
the increase in autophagosomes affected the function of lysosomes,
resulting in accumulation of autophagosomes, as demonstrated by the
increase in p62. However, we did not observe a continuous increase
in p62 as the infected time increased, which is inconsistent with
previous findings (21). We
believe that different cell types and different infection times
accounted for this discrepancy. Moreover, during CVB3 infection,
viral protease 2Apro is responsible for p62 cleavage
(45). Thus, p62 expression in
the present study may not accurately represent the level of p62
caused directly by CVB3 infection. Our findings simply imply a
correlation between autophagy caused by CVB3 infection and protein
degradation, and the details require further study.
The autophagic process may be subverted by viruses
to provide a surface for RNA replication as the time of infection
increases. At 24 h.p.i., modulation of the autophagic pathway
(using rapamycin or ZSTK474) to enhance or reduce autophagy
resulted in an increase and a decrease in CVB3 mRNA replication,
respectively. It appears that the virus can subvert components of
the autophagic machinery for their own benefit. To our surprise,
the increased autophagy caused by rapamycin did not increase VP1
expression. We hypothesized that the inactivation of mTORC1 by
rapamycin would inhibit cap-dependent mRNA translation. We observed
a co-localization of LC3-positive puncta with LAMP-1 by confocal
microscopy at 24 h.p.i., suggesting that CVB3 infection may induce
autophagosomes to fuse with lysosomes/endosomes to form
autolysosomes/amphisomes. Autophagosomes primarily fuse with
lysosomes during CVB3 infection, excluding a small proportion of
the autophagosomes that fuse with endosomes to form amphisomes
(49). We inhibited lysosome
function using chloroquine phosphate and monitored the levels of
p62 and LC3 protein. After blockade of lysosome function, the
LC3-II/LC3-I ratio and p62 protein levels increased significantly.
These data revealed that at 24 h.p.i., autophagy may partly retain
the function of promoting protein degradation (VP1) by lysosomes.
Hence, the high expression of CVB3 mRNA did not contribute to the
VP1 synthesis in cells treated with rapamycin.
In our research, we found that p-mTOR was altered
during the first 3 h and did not keep pace with the change in LC3.
The MEK/ERK pathway and the PI3K/Akt pathway cross-inhibit each
other (50). Xin et al
claimed that CVB3-induced autophagosome accumulation occurs via the
ERK pathway (33). We
hypothesized that autophagy caused by CVB3 infection was an outcome
of multiple factors, including activation or inactivation of
multiple signaling pathways. The balance or crosstalk among them
may account for the difference. p-mTOR and p-Akt1 decreased at 24
h.p.i., and pre-treatment of virus-infected cells with ZSTK474
alleviated the LC3-II/LC3-I ratio caused by CVB3 infection.
Inhibition of mTOR and Akt1 with rapamycin or MK2206 before virus
infection promoted autophagy induced by CVB3 infection. We
hypothesized that the incomplete blockade may explain this result.
Additionally, rapamycin may have aggravated CVB3 mRNA replication
in our study. Does the increased virus replication subsequently
prompt autophagy? This question warrants further study. In
addition, we found a significant phenomenon under uninfected
conditions; the LC3-II/LC3-I ratio was increased in
Akt1-overexpressing cells when compared with that in the empty
vector cells. This finding suggests that the overexpression of Akt1
may inhibit basal autophagy in HeLa cells. Our previous research
demonstrated that Akt1 overexpression promotes CVB3-induced
apoptosis in HeLa cells, thus Akt1 overexpression may activate the
crosstalk of autophagy and apoptosis, change the cell status or
activate other signaling pathways during viral infection, thus
explaining the differences between the two cell lines.
In summary, our study demonstrated that HeLa cells
respond to CVB3 infection by increasing LC3 expression and
autophagosome formation in vitro. We demonstrated that the
inhibition of the PI3K/Akt/mTOR signaling pathway could affect the
autophagic reaction induced by CVB3 infection, resulting in changes
in viral infection. These findings may provide a better
understanding of VMC pathogenesis.
Acknowledgments
We acknowledge the Institute of Oncology, Central
South University that provided the HeLa cells, and the Center
Laboratory at the Third Xiangya Hospital of the Central South
University that provided the experimental equipment and technical
guidance necessary to complete our study. This study was funded by
the National Natural Science Foundation of China (grant no.
81570346).
References
|
1
|
Yajima T and Knowlton KU: Viral
myocarditis: From the perspective of the virus. Circulation.
119:2615–2624. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yajima T: Viral myocarditis: Potential
defense mechanisms within the cardiomyocyte against virus
infection. Future Microbiol. 6:551–566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bowles NE, Ni J, Kearney DL, Pauschinger
M, Schultheiss HP, McCarthy R, Hare J, Bricker JT, Bowles KR and
Towbin JA: Detection of viruses in myocardial tissues by polymerase
chain reaction: Evidence of adenovirus as a common cause of
myocarditis in children and adults. J Am Coll Cardiol. 42:466–472.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esfandiarei M and McManus BM: Molecular
biology and pathogenesis of viral myocarditis. Annu Rev Pathol.
3:127–155. 2008. View Article : Google Scholar
|
|
5
|
Sin J, Mangale V, Thienphrapa W, Gottlieb
RA and Feuer R: Recent progress in understanding coxsackievirus
replication, dissemination, and pathogenesis. Virology.
484:288–304. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liao YH, Xia N, Zhou SF, Tang TT, Yan XX,
Lv BJ, Nie SF, Wang J, Iwakura Y, Xiao H, et al: Interleukin-17A
contributes to myocardial ischemia/reperfusion injury by regulating
cardiomyocyte apoptosis and neutrophil infiltration. J Am Coll
Cardiol. 59:420–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garmaroudi FS, Marchant D, Hendry R, Luo
H, Yang D, Ye X, Shi J and McManus BM: Coxsackievirus B3
replication and pathogenesis. Future Microbiol. 10:629–653. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen Z, Yang L, Liu Y, Tang A, Li X, Zhang
J and Yang Z: LY294002 and Rapamycin promote coxsackievirus-induced
cytopathic effect and apoptosis via inhibition of PI3K/AKT/mTOR
signaling pathway. Mol Cell Biochem. 385:169–177. 2014. View Article : Google Scholar
|
|
9
|
He B: Viruses, endoplasmic reticulum
stress, and interferon responses. Cell Death Differ. 13:393–403.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zha X, Yue Y, Dong N and Xiong S:
Endoplasmic reticulum stress aggravates viral myocarditis by
raising inflammation through the IRE1-associated NF-κB pathway. Can
J Cardiol. 31:1032–1040. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein
DA and Yang D: Coxsackievirus B3 infection activates the unfolded
protein response and induces apoptosis through downregulation of
p58IPK and activation of CHOP and SREBP1. J Virol. 84:8446–8459.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang C and Jung JU: Autophagy genes as
tumor suppressors. Curr Opin Cell Biol. 22:226–233. 2010.
View Article : Google Scholar :
|
|
13
|
Sarkar S and Rubinsztein DC: Huntington's
disease: Degradation of mutant huntingtin by autophagy. FEBS J.
275:4263–4270. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cadwell K, Stappenbeck TS and Virgin HW:
Role of autophagy and autophagy genes in inflammatory bowel
disease. Curr Top Microbiol Immunol. 335:141–167. 2009.PubMed/NCBI
|
|
15
|
Lerena MC, Vázquez CL and Colombo MI:
Bacterial pathogens and the autophagic response. Cell Microbiol.
12:10–18. 2010. View Article : Google Scholar
|
|
16
|
Tal MC and Iwasaki A: Autophagy and innate
recognition systems. Curr Top Microbiol Immunol. 335:107–121.
2009.PubMed/NCBI
|
|
17
|
Wong J, Zhang J, Si X, Gao G, Mao I,
McManus BM and Luo H: Autophagosome supports coxsackievirus B3
replication in host cells. J Virol. 82:9143–9153. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubli DA and Gustafsson AB: Cardiomyocyte
health: Adapting to metabolic changes through autophagy. Trends
Endocrinol Metab. 25:156–164. 2014. View Article : Google Scholar :
|
|
19
|
Robinson SM, Tsueng G, Sin J, Mangale V,
Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, et
al: Coxsackievirus B exits the host cell in shed microvesicles
displaying autophagosomal markers. PLoS Pathog. 10:e10040452014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhai X, Bai B, Yu B, Wang T, Wang H, Wang
Y, Li H, Tong L, Wang Y, Zhang F, et al: Coxsackievirus B3 induces
autophagic response in cardiac myocytes in vivo. Biochemistry
(Mosc). 80:1001–1009. 2015. View Article : Google Scholar
|
|
21
|
Kemball CC, Alirezaei M, Flynn CT, Wood
MR, Harkins S, Kiosses WB and Whitton JL: Coxsackievirus infection
induces autophagy-like vesicles and megaphagosomes in pancreatic
acinar cells in vivo. J Virol. 84:12110–12124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alirezaei M, Flynn CT, Wood MR and Whitton
JL: Pancreatic acinar cell-specific autophagy disruption reduces
coxsackievirus replication and pathogenesis in vivo. Cell Host
Microbe. 11:298–305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xin L, Xiao Z, Ma X, He F, Yao H and Liu
Z: Coxsackievirus B3 induces crosstalk between autophagy and
apoptosis to benefit its release after replicating in
autophagosomes through a mechanism involving caspase cleavage of
autophagy-related proteins. Infect Genet Evol. 26:95–102. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Luo H and McManus BM: Is autophagy an
avenue to modulate coxsackievirus replication and pathogenesis?
Future Microbiol. 7:921–924. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mohankumar V, Dhanushkodi NR and Raju R:
Sindbis virus replication, is insensitive to rapamycin and torin1,
and suppresses Akt/mTOR pathway late during infection in HEK cells.
Biochem Biophys Res Commun. 406:262–267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma J, Sun Q, Mi R and Zhang H: Avian
influenza A virus H5N1 causes autophagy-mediated cell death through
suppression of mTOR signaling. J Genet Genomics. 38:533–537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shelly S, Lukinova N, Bambina S, Berman A
and Cherry S: Autophagy is an essential component of Drosophila
immunity against vesicular stomatitis virus. Immunity. 30:588–598.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang H, Kang R, Wang J, Luo G, Yang W and
Zhao Z: Hepatitis C virus inhibits AKT-tuberous sclerosis complex
(TSC), the mechanistic target of rapamycin (MTOR) pathway, through
endoplasmic reticulum stress to induce autophagy. Autophagy.
9:175–195. 2013. View Article : Google Scholar :
|
|
29
|
Hu B, Zhang Y, Jia L, Wu H, Fan C, Sun Y,
Ye C, Liao M and Zhou J: Binding of the pathogen receptor HSP90AA1
to avibirnavirus VP2 induces autophagy by inactivating the AKT-MTOR
pathway. Autophagy. 11:503–515. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Vlahakis A and Powers T: A role for TOR
complex 2 signaling in promoting autophagy. Autophagy.
10:2085–2086. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bernard M, Dieudé M, Yang B, Hamelin K,
Underwood K and Hébert MJ: Autophagy fosters myofibroblast
differentiation through MTORC2 activation and downstream
upregulation of CTGF. Autophagy. 10:2193–2207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xin L, Ma X, Xiao Z, Yao H and Liu Z:
Coxsackievirus B3 induces autophagy in HeLa cells via the
AMPK/MEK/ERK and Ras/Raf/MEK/ERK signaling pathways. Infect Genet
Evol. 36:46–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li X, Li Z, Zhou W, Xing X, Huang L, Tian
L, Chen J, Chen C, Ma X and Yang Z: Overexpression of 4EBP1,
p70S6K, Akt1 or Akt2 differentially promotes Coxsackievirus
B3-induced apoptosis in HeLa cells. Cell Death Dis. 4:e803–e809.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li X, Zhang J, Chen Z, Yang L, Xing X, Ma
X and Yang Z: Both PI3K- and mTOR-signaling pathways take part in
CVB3-induced apoptosis of Hela cells. DNA Cell Biol. 32:359–370.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Klionsky DJ, Abeliovich H, Agostinis P,
Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA,
Ballabio A, et al: Guidelines for the use and interpretation of
assays for monitoring autophagy in higher eukaryotes. Autophagy.
4:151–175. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar
|
|
39
|
Gutierrez MG, Master SS, Singh SB, Taylor
GA, Colombo MI and Deretic V: Autophagy is a defense mechanism
inhibiting BCG and Mycobacterium tuberculosis survival in infected
macrophages. Cell. 119:753–766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Eskelinen EL: Maturation of autophagic
vacuoles in Mammalian cells. Autophagy. 1:1–10. 2005. View Article : Google Scholar
|
|
43
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lai JK, Sam IC and Chan YF: The autophagic
machinery in enterovirus infection. Viruses. 8:E322016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shi J, Wong J, Piesik P, Fung G, Zhang J,
Jagdeo J, Li X, Jan E and Luo H: Cleavage of sequestosome 1/62 by
an enteroviral protease results in disrupted selective autophagy
and impaired NFKB signaling. Autophagy. 9:1591–1603. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yao H and Han X and Han X: The
cardioprotection of the insulin-mediated PI3K/Akt/mTOR signaling
pathway. Am J Cardiovasc Drugs. 14:433–442. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Taylor MP and Jackson WT: Viruses and
arrested autophagosome development. Autophagy. 5:870–871. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shi X, Chen Z, Tang S, Wu F, Xiong S and
Dong C: Coxsackievirus B3 infection induces autophagic flux, and
autophagosomes are critical for efficient viral replication. Arch
Virol. 161:2197–2205. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hoeflich KP, O'Brien C, Boyd Z, Cavet G,
Guerrero S, Jung K, Januario T, Savage H, Punnoose E, Truong T, et
al: In vivo antitumor activity of MEK and phosphatidylinositol
3-kinase inhibitors in basal-like breast cancer models. Clin Cancer
Res. 15:4649–4664. 2009. View Article : Google Scholar : PubMed/NCBI
|