Introduction
Epithelial-mesenchymal transition (EMT) is a
fundamental process driving morphogenesis in most metazoans
conserved throughout evolution. Cells engaged in the EMT program
will undergo complex changes in intercellular junctions, adhesive
structures and apicobasal polarization (1,2).
The process of EMT is known to be involved in various physiological
and pathological states, including organ fibrosis (3,4),
cancer metastasis (5) and
resistance to chemotherapy (6).
Recently, increasing evidence has shed light on the transforming
growth factor-β1 (TGF-β1), which is a well-known inducer of EMT in
epithelial cells (7,8). It binds to its receptors (TβRI,
TβRII and TβRIII) and activates various transcription factors for
cadherin isoform switching (9).
TGF-β induces EMT primarily via a Smad-dependent mechanism. Smad2/3
is phosphorylated by Smad4 and then translocates to the nucleus,
where it regulates the transcription of the target genes
responsible for EMT (10,11). However, there are few studies
available on TGF-β-induced EMT through the activation of
non-canonical pathways (12). For
example, it is still not well characterized that the
Cdc42-interacting protein-4 (CIP4)/partitioning-defective protein 6
(Par6) pathway is responsible for TGF-β-induced EMT in NRK-52E
cells.
Par6 is part of the Par polarity complex that
localizes to the tight junction (TJ) and is comprised of 3 highly
conserved proteins, Par6, Par3 and atypical protein kinase C (aPKC)
(13). Previous studies have
shown that Par6, a regulator of epithelial cell polarity and TJ
assembly, is directly phosphorylated (at Ser345) and is activated
by TβRII in response to TGF-β, which is essential for TGF-β-induced
EMT and facilitates metastasis (13–15). CIP4 belongs to the
Bin/amphiphysin/Rvs (BAR) domain protein superfamily. Members of
this superfamily are noted for their involvement in membrane
remodeling processes that occur in various cellular pathways, such
as endocytosis, cytokinesis, T-tubule formation, cell migration and
neuromorphogenesis (16). Earlier
studies have demonstrated that CIP4 is capable of inducing
extracellular matrix (ECM) deposition and exacerbating progressive
fibrosis in chronic renal failure and is required for E-cadherin
endocytosis (16–19). The interaction between
Cdc42/Par6/aPKC and CIP4, as a downstream effector protein of
Cdc42, may act as a link between Cdc42 signaling and the regulation
of the actin cytoskeleton (17,20). However, the interaction between
CIP4 and Par6 in NRK-52E cells undergoing TGF-β-induced EMT remains
largely unknown.
In the present study, we demonstrated that the
expression of CIP4 was significantly increased in NRK-52E cells
stimulated with TGF-β1. Intriguingly, CIP4 lose-of-function with
small interfering RNA (siRNA) reversed TGF-β1-induced EMT in
NRK-52E cells. Moreover, we found that there was an interaction
between Par6 and CIP4 in TGF-β1-stimulated NRK-52E cells undergoing
EMT.
Materials and methods
Cell culture
Rat NRK-52E cells were obtained from the Chinese
Academy of Sciences (Institute of Shanghai Cell Biology and Chinese
Type Culture Collection, Shanghai, China), and maintained in
Dulbecco's modified Eagle's medium (DMEM; Invitrogen Life
Technologies, Carlsbad, CA, USA), supplemented with 10% fetal
bovine serum (FBS) (HyClone, Logan, UT, USA), 100 U/ml penicillin,
and 100 mg/ml streptomycin (Invitrogen Life Technologies) at 37°C
in a humidified, 5% CO2, 95% air atmosphere. The medium
was replenished every day. The cells were stimulated with TGF-β1 at
20 ng/ml.
Gene silencing by siRNA
For siRNA experiments, RNA primers complementary to
rat Par6 and CIP4 were designed and synthesized by the Invitrogen
Biotechnology Co., Ltd. (Shanghai, China). The NRK-52E cells were
transfected with the annealed RNA primer pair using Lipofectamine
2000 (Invitrogen Life Technologies) in accordance with the
instructions provided by the manufacturer. The cells transfected
with scrambled siRNA served as controls. Five hours after
transfection, the cells were incubated with 20 ng/ml of TGF-β1 for
72 h in order to observe the effects of Par6 and CIP4 silencing.
The siRNA used had the following primers: Par6 forward,
5′-CUCACUGAGUGCGACAAGGUCUUCA-3′ and reverse,
5′-AGAAGAGGUUGUCGCUCACACACUG-3′; CIP4 forward,
5′-GCUAACAGUCGACUGUGCUAGUGU-3′ and reverse,
5′-UCGACAACGAGAGUGGACUCUAGC-3′; scramble forward,
5′-GACCAGUCGCUAUCACACAUGUCA-3′ and reverse,
5′-AGAUGACGAGUCUUACGCACUCGU-3′.
Reverse transcription-polymerase chain
reaction (RT-PCR)
RNA extraction was performed using TRIzol reagent
according to the manufacturer's instructions (Invitrogen Life
Technologies). RNA integrity was verified by agarose gel
electrophoresis. The synthesis of cDNA was performed by reverse
transcription reactions with 2 μg of total RNA using moloney
murine leukemia virus reverse transcriptase (Invitrogen Life
Technologies) with oligo(dT) (15) primers (Fermentas, Pittsburgh, PA,
USA) as described by the manufacturer. The first-strand cDNA served
as the template for the regular PCR performed using a DNA engine
(ABI 7300; Applied Biosystems, Foster City, CA, USA).
Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as an internal
control was used to normalize the data to determine the relative
expression of the target genes. The reaction conditions were set
according to the kit instructions. The PCR primers used in this
study were as follows: Par6 forward, 5′-GCACGCAGAATGATGACGATT-3′
and reverse, 5′-GTCGTGCTACGATCGTAGTA-3′; CIP4 forward,
5′-ACACGGAGTTTGATGAGGAT-3′ and reverse, 5′-ATGGTGGAACGATGGTAGAA-3′;
GAPDH forward, 5′-GGATTTGGTCGTATTGGG-3′ and reverse,
5′-GGAAGATGGTGATGGGATT-3′.
Immunoprecipitation (IP) and
immunoblotting (IB)
Cell lysates were prepared as previously described
(21). The lysates were then
incubated with the indicated antibodies for 1 h at 4°C. The immune
complexes were precipitated with protein A/G agarose (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) for 1 h at 4°C, washed
extensively with lysis buffer, resolved in 4–20% gradient SDS-PAGE,
and analyzed by immunoblotting. All immunoblots were developed by
enhanced chemiluminesence (Amersham Pharmacia Biotech, Inc.,
Piscataway, NJ, USA).
Western blot analysis
The NRK-52E cells were homogenized and extracted in
NP-40 buffer, followed by 5–10 min boiling and centrifugation to
obtain the supernatant. Samples containing 50 μg of protein
were separated on a 10% SDS-PAGE gel and transferred onto
nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, CA,
USA). After saturation with 5% (w/v) non-fat dry milk in TBS and
0.1% (w/v) Tween-20 (TBST), the membranes were incubated with the
following antibodies: E-cadherin (sc-71008; Santa Cruz
Biotechnology, Inc.), α-SMA (SAB5500002; Sigma-Aldrich, St. Louis,
MO, USA), rCIP4 (ab72220; Abcam, Cambridge, MA, USA), Par6
(sc-365323) and p-Par6 (both from Santa Cruz Biotechnoogy, Inc.),
at dilutions ranging from 1:500 to 1:2,000 at 4°C overnight.
Following 3 washes with TBST, the membranes were incubated with
secondary immunoglobulins (Igs) conjugated to IRDye 800CW Infrared
Dye (LI-COR Biotechnology, Lincoln, NE, USA), including donkey
anti-goat IgG and donkey anti-mouse IgG at a dilution of
1:10,000–1:20,000. Following 1 h of incubation at 37°C, the
membranes were washed 3 times with TBST. The blots were visualized
by the Odyssey Infrared Imaging System (LI-COR Biotechnology).
Signals were densitometrically assessed (Odyssey Application
software version 3.0) and normalized to the GAPDH or β-actin
signals to correct for unequal loading using the mouse monoclonal
anti-GAPDH (MB001H) or anti-β-actin antibody (BS6007MH) (both from
Bioworld Technology, Inc., St. Louis Park, MN, USA),
respectively.
Confocal fluorescence microscopy
For immunocytochemical analysis, the NRK-52E cells
were cultured on sterile glass cover-slips in 6-well plates.
Thereafter, the cells were treated and fixed with iced acetone for
10 min, incubated with 0.1% Triton X-100 for 10 min in order to
induce membrane rupture, and incubated with 1% BSA at 37°C for an
additional 30 min for blocking. The cells were then incubated with
anti-E-cadherin (sc-71008; Santa Cruz Biotechnology, Inc.),
anti-α-SMA (55135-1-AP; Proteintech Group, Inc., St. Pearl, IL,
USA), anti-Par6 (sc-365323; Santa Cruz Biotechnology, Inc.) and
anti-CIP4 (ab72220; Abcam) antibodies (1:100) overnight at 4°C,
followed by incubation with FITC-goat anti-rabbit IgG (SA00003-2;
Proteintech Group, Inc.) at 37°C for 1 h. Thereafter, the nuclei
were stained with DAPI (Sigma-Aldrich) for 5 min and analyzed using
a laser confocal scanning microscope (Leica, Heidelberg, Germany).
The imaging using the laser confocal scanning microscopy for the
cell culture is better than that of total internal reflection
fluorescence (TIRF) microscopy (22) that can only exploit a limited
region adjacent to the substrate.
Statistical analysis
The data from these experiments are reported as the
means ± standard error of the mean (SEM) for each group. All
statistical analyses were performed by using GraphPad Prism version
5.0 software (GraphPad Software, La Jolla, CA, USA). Inter-group
differences were analyzed by one-way analysis of variance (ANOVA),
followed by Tukey's multiple comparison test as a post-test to
compare the group means with an overall value of P<0.05.
Differences with a P-value <0.05 were considered statistically
significant.
Results
TGF-β1-induced EMT in NRK-52E cells
To assess TGF-β-induced EMT, we used rat NRK-52E
cells as an in vitro model system for assessing EMT. The
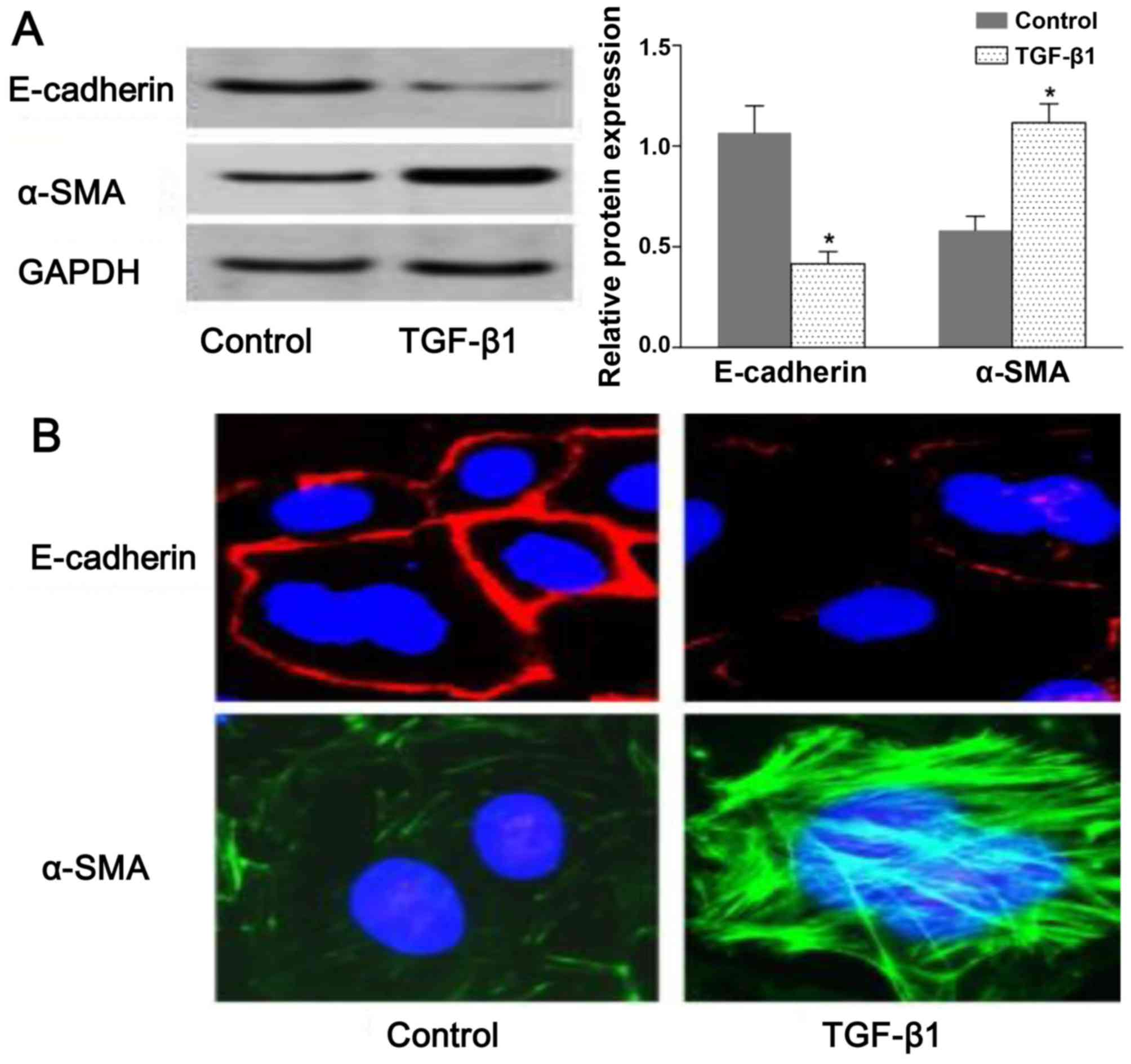
resutls of western blot analysis demonstrated that the expression
of E-cadherin and α-SMA was decreased and increased, respectively,
in the TGF-β1-stimulated group compared with the untreated group
(Fig. 1A). Consistent with the
results of western blot analysis, we observed a change in
E-cadherin and α-SMA distribution in the NRK-52E cells following
exposure to TGF-β by confocal fluorescence microscopy. The
morphologic observations of the NRK-52E cells revealed a continuous
distribution of E-cadherin near the perimeter of the control cells.
On the contrary, a discontinuous distribution of E-cadherin was
observed in the TGF-β1-stimulated cells (Fig. 1B). Furthermore, α-SMA was present
exclusively in the cytosol of the TGF-β1-stimulated cells, and
little endogenous expression in the control cells was observed
(Fig. 1B). The results shown in
Fig. 1 suggested that the NRK-52E
cells underwent EMT following exposure to TGF-β1.
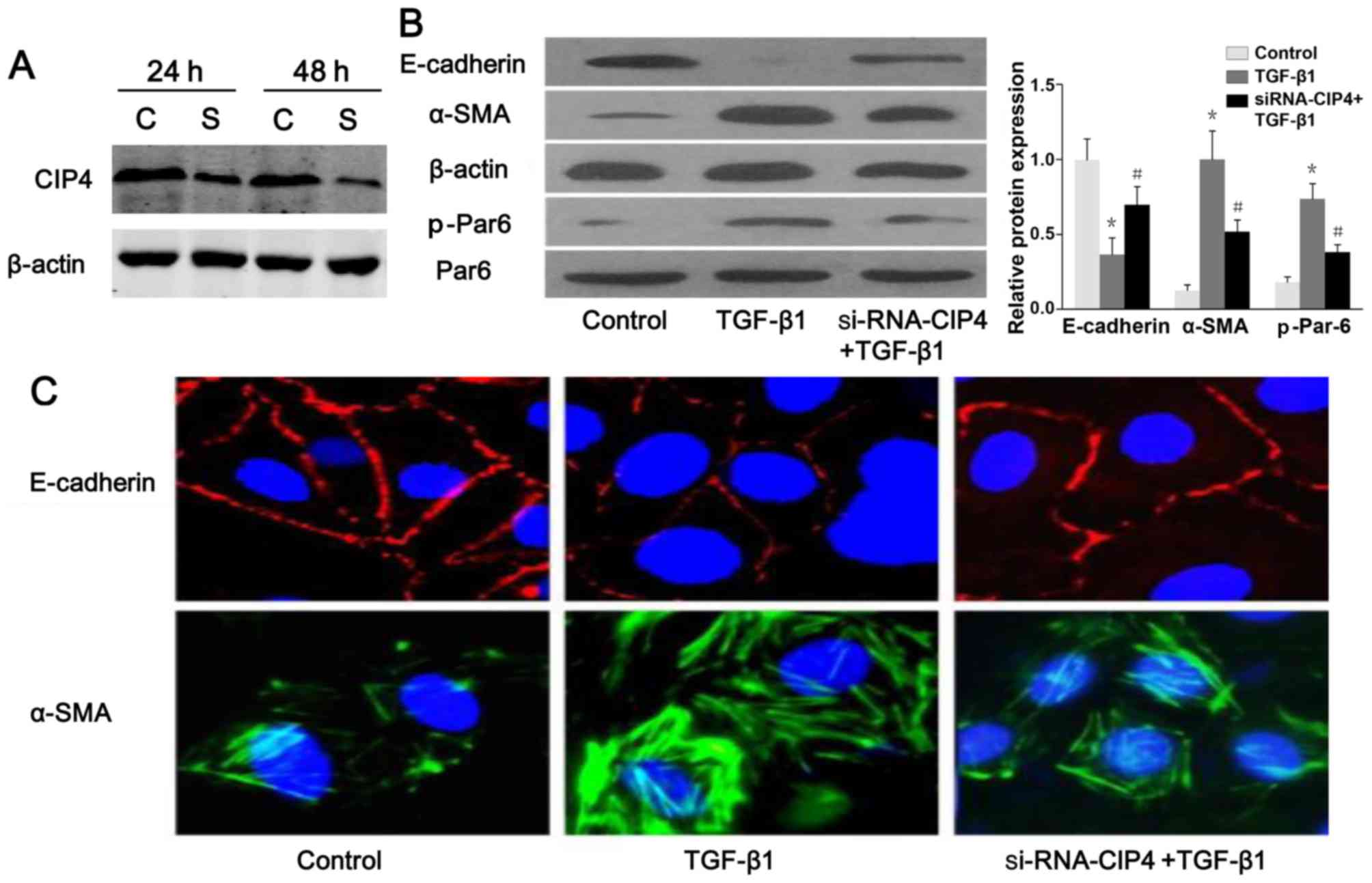
TGF-β1 regulates the expression of CIP4
in rat NRK-52E cells
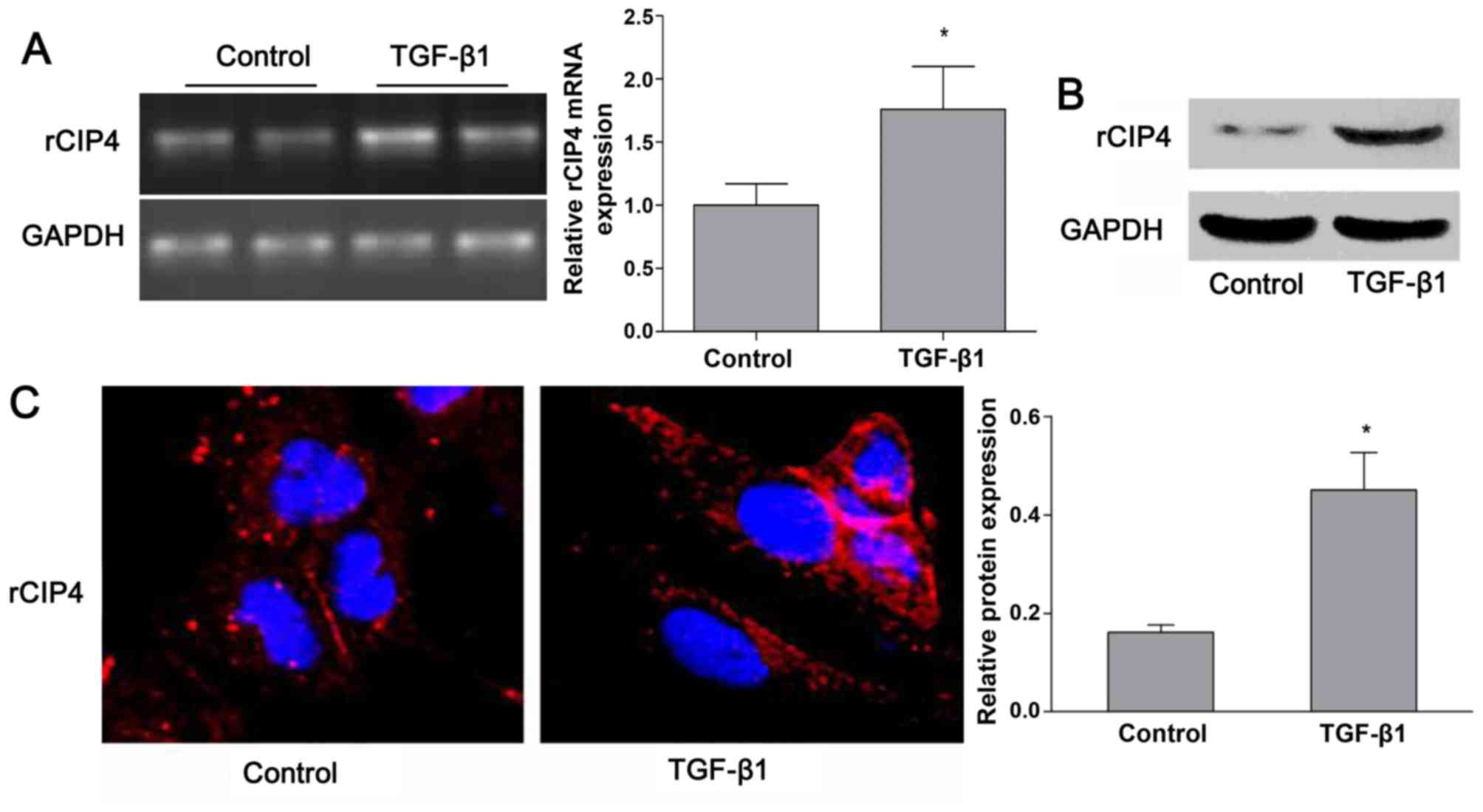
Based on the previous findings shown in the
5/6-nephrectomy rat model (23),
the expression of rat CIP4 (rCIP4) in the NRK-52E cells stimulated
with TGF-β1 in vitro was demonstrated. Following stimulation
with TGF-β1 (20 ng/ml) for 72 h, the mRNA and protein levels of
rCIP4 in NRK-52E cells were upregulated as compared to those of the
control group (Fig. 2A and B).
Confocal fluorescence microscopy revealed that rCIP4 exhibited a
punctate localization throughout the cytosol, with the highest
levels localized in the perinuclear region of the NRK-52E cells.
Following exposure to TGF-β1, the rCIP4 levels increased, and rCIP4
was recruited into cluster formations located adjacent to the cell
periphery. Gradually, these clusters were observed to be
redistributed into the cytoplasm (Fig. 2C).
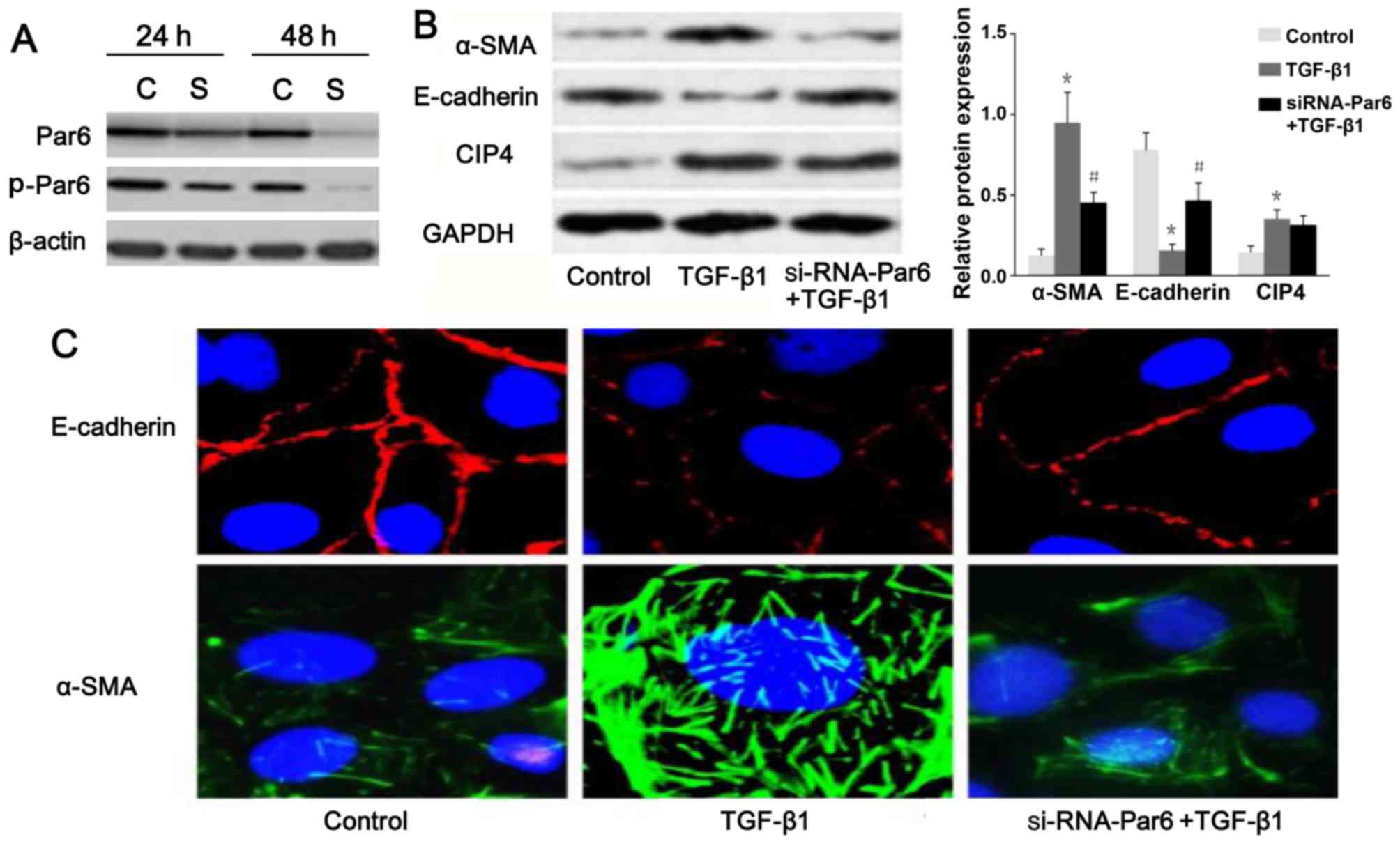
TGF-β1 upregulates the expression of
p-Par6 in rat NRK-52E cells
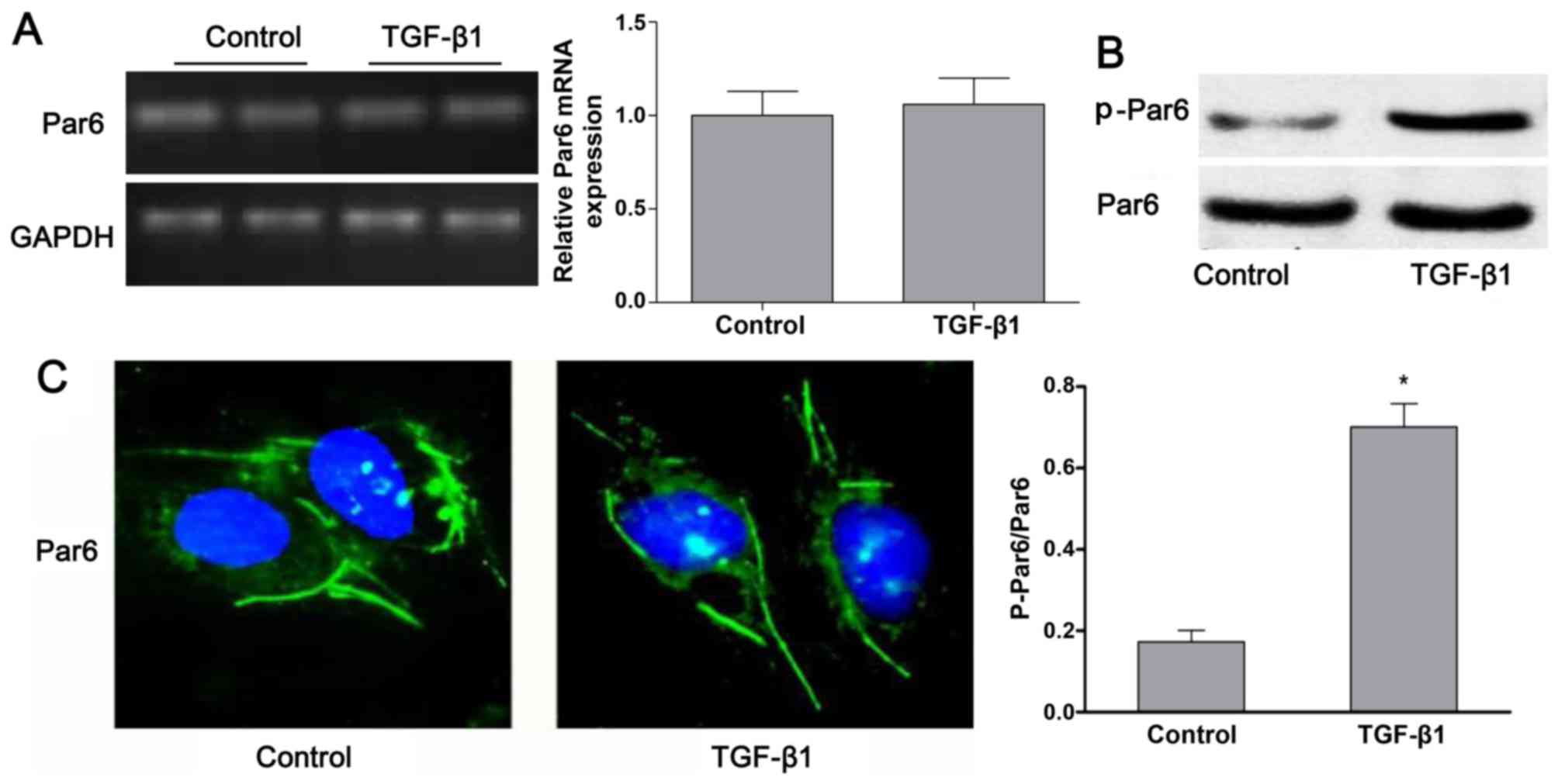
As shown in Fig.
3B, the protein expression of p-Par6 was increased by TGF-β1
stimulation as compared to the control group. There was no obvious
difference in the mRNA and protein expression of Par6 between the
control cells and the cells exposed to TGF-β1; this was confirmed
by RT-PCR, western blot analyses and confocal fluorescence
microscopy (Fig. 3A–C).
Stimulation with TGF-β1 enhances the
interaction between Par6 and CIP4
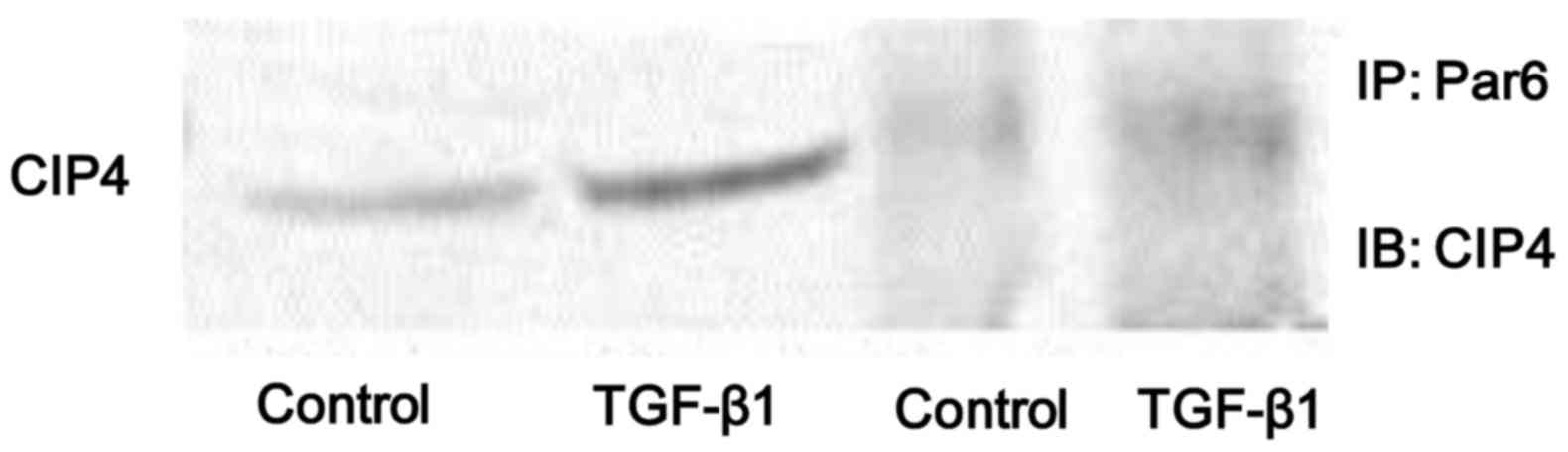
In the present study, we found that the protein
expression of both p-Par6 and CIP4 was significantly increased in
the NRK-52E cells stimulated with TGF-β1 in vitro. Thus, in
order to examine whether the interaction between Par6 and CIP4 is
dependent on TGF-β1, the cell lysates were prepared from
TGF-β1-stimulated cells and immunoprecipitated with anti-Par6
antibody. The results indicated that the binding capacity between
Par6 and CIP6 was weak in the control cells, but was increased in
the NRK-52E cells stimulated with TGF-β1 (Fig. 4). Therefore, our data suggest that
the interaction between Par6 and CIP6 may play a vital role in
TGF-β1-induced EMT.
Silencing of Par6 inhibits TGF-β1-induced
EMT
To inhibit the function of Par6, the NRK-52E cells
were transfected with siRNA. The results of western blot analysis
demonstrated that the expression of p-Par6 was markedly inhibited
by transfection with siRNA-Par6. Moreover, the protein expression
of p-Par6 was decreased with increasing treatment durations
(Fig. 5A). Therefore, our data
suggested that the siRNA experiments were successfully performed.
As shown in Fig. 5B and C, as the
E-cadherin protein levels increased, a marked decrease in α-SMA
expression was clearly evident following transfection of the
TGF-β1-stimulated cells with Par6-siRNA. The NRK-52E cells
transfected with Par6-siRNA, however, demonstrated resistance to
TGF-β1-induced EMT.
CIP4 is involved in TGF-β1-induced EMT
and the regulation of Par6 expression
The results of western blot analysis and confocal
fluorescence microscopy indicated a reduced protein expression and
a discontinuous distribution of E-cadherin near the perimeter of
TGF-β1-stimulated cells (Fig.
6A–C). Furthermore, α-SMA was present exclusively in the
cytosol of TGF-β1-stimulated cells, and little endogenous
expression was observed in the control cells (Fig. 6B and C), suggesting that these
cells underwent EMT in response to TGF-β1. Following treatment with
CIP4-siRNA, E-cadherin re-localized to regions surrounding the
cellular junctions, and a reduction in α-SMA expression was
observed compared with the TGF-β1-stimulated cells (Fig. 6B and C). Intriguingly, the protein
level of p-Par6 in the TGF-β1-stimulated cells transfected with
CIP4-siRNA was downregulated as compared to that of the
TGF-β1-treated group (Fig. 6B).
Therefore, our data suggest that CIP4 is involved in TGF-β1-induced
EMT, and the underlying mechanisms are mediated, at least in part,
through the upregulation of the expression of p-Par6.
Discussion
TGF-β1 promotes the development of renal tubule
interstitial fibrosis through EMT. In this study, the upregulation
of rCIP4 and p-Par6 was demonstrated in NRK-52E cells stimulated
with TGF-β1. Conversely, CIP4 or Par6 loss of function by siRNA
reversed TGF-β1-induced EMT by increasing E-cadherin (an epithelial
marker) and reducing α-SMA (a mesenchymal marker) expression. These
findings indicated that CIP4/Par6 was involved in the
TGF-β1-induced EMT processes, and subsequently in renal
fibrosis.
In epithelial tissue, CIP4 has been identified to
regulate the early E-cadherin trafficking/endocytosis (17), which is the key process of EMT in
renal tubular epithelial cell (16,24). In 5/6-nephrectomized rats, the
expression of rCIP4 has been shown to be increased in the renal
tubules, and the overexpression of hCIP4 has been demonstrated to
promote the development of EMT in HK-2 cells exposed to TGF-β1
(16). Moreover, Par6, a
conserved polarity protein, regulates cell polarization processes.
However, increasing evidence also suggests that Par6 is
phosphorylated to facilitate EMT (25). Given the recent important roles
reported for Par6 in the generation and progression of various
types of cancer (26,27), Par6 phosphorylation may be central
to various extrinsic cues that can lead to the EMT (28). It has also been revealed that Par6
can have different functions depending on the interacting partners
(28). Furthermore, in
cell-culture systems, E-cadherin endocytosis was identified as a
model of EMT (25,26), and CIP4 acts as a link between
Cdc42/Par6/aPKC and the early endocytic machinery to regulate
E-cadherin endocytosis in epithelial cells. Taken together, our
data suggested that CIP4/Par6 have a close interaction in the
process of EMT induced by TGF-β1.
To more accurately determine the location of CIP4
and Par6, we provided superior assessment of the role of CIP4 and
Par6 location in NRK-52E cells. Immunofluorescence laser scanning
confocal microscopy showed that CIP4 exhibited punctate
localization throughout the cytosol. Following stimulation with
TGF-β1, CIP4 was increased and formed visible clusters adjacent to
the cell periphery that gradually redistributed into the cytoplasm.
These findings suggest that the upregulation of CIP4 plays an
important role in the occurrence of EMT in NRK-52E cells. However,
the cluster distribution of Par6 has no obvious difference in cell
periphery. Interestingly, stimulation with TGF-β1 led to a closer
interaction between CIP4 and Par6 in NRK-52E cells. Our results
were measured by co-immunoprecipitation analysis, which showed that
the binding capacity of CIP4 and Par6 was increased in
TGF-β1-stimulated NRK-52E cells. CIP4 loss of function by siRNA
reversed the increase in p-Par6 protein expression in
TGF-β1-stimulated NRK-52E cells. A similar result was observed by
decreasing of CIP4 protein expression due to Par6 loss of function
by siRNA. These results suggested that there was an interaction
between CIP4 and Par6, and that Par6 phosphorylation may be
regulated by CIP4.
In conclusion, our data demonstrate that CIP4 and
Par6 play an important role in the occurrence of EMT in
TGF-β1-stimulated NRK-52E cells. The underlying mechanisms are
mediated, at least in part, through the upregulation of the
expression of CIP4 and p-Par6.
Acknowledgments
We would like to acknowledge the support by Natural
Science Foundation of Shanghai (13ZR1407200).
References
|
1
|
Baum B, Settleman J and Quinlan MP:
Transitions between epithelial and mesenchymal states in
development and disease. Semin Cell Dev Biol. 19:294–308. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chua KN, Poon KL, Lim J, Sim WJ, Huang RY
and Thiery JP: Target cell movement in tumor and cardiovascular
diseases based on the epithelial-mesenchymal transition concept.
Adv Drug Deliv Rev. 63:558–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Simic P, Williams EO, Bell EL, Gong JJ,
Bonkowski M and Guarente L: SIRT1 suppresses the
epithelial-to-mesenchymal transition in cancer metastasis and organ
fibrosis. Cell Reports. 3:1175–1186. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guarino M, Tosoni A and Nebuloni M: Direct
contribution of epithelium to organ fibrosis:
epithelial-mesenchymal transition. Hum Pathol. 40:1365–1376. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao D, Besser AH, Wander SA, Sun J, Zhou
W, Wang B, Ince T, Durante MA, Guo W, Mills G, et al: Cytoplasmic
p27 promotes epithelial-mesenchymal transition and tumor metastasis
via STAT3-mediated Twist1 upregulation. Oncogene. 43:5447–5459.
2015. View Article : Google Scholar
|
|
6
|
Ma J, Fang B, Zeng F, Ma C, Pang H, Cheng
L, Shi Y, Wang H, Yin B, Xia J, et al: Down-regulation of miR-223
reverses epithelial-mesenchymal transition in gemcitabine-resistant
pancreatic cancer cells. Oncotarget. 6:1740–1749. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chung H, Choi HS, Seo EK, Kang DH and Oh
ES: Baicalin and baicalein inhibit transforming growth
factor-β1-mediated epithelial-mesenchymal transition in human
breast epithelial cells. Biochem Biophys Res Commun. 458:707–713.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Y, Liu N, Su X, Zhou G, Sun G, Du F,
Bian X and Wang B: Epigallocatechin-3-gallate attenuates
transforming growth factor-beta1 induced epithelial-mesenchymal
transition via Nrf2 regulation in renal tubular epithelial cells.
Biomed Pharmacother. 70:260–267. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Piek E, Moustakas A, Kurisaki A, Heldin CH
and ten Dijke P: TGF-(beta) type I receptor/ALK-5 and Smad proteins
mediate epithelial to mesenchymal transdifferentiation in NMuMG
breast epithelial cells. J Cell Sci. 112:4557–4568. 1999.PubMed/NCBI
|
|
10
|
Bae E, Kim SJ, Hong S, Liu F and Ooshima
A: Smad3 linker phosphorylation attenuates Smad3 transcriptional
activity and TGF-β1/Smad3-induced epithelial-mesenchymal transition
in renal epithelial cells. Biochem Biophys Res Commun. 427:593–599.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Meng XM, Huang XR, Chung AC, Qin W, Shao
X, Igarashi P, Ju W, Bottinger EP and Lan HY: Smad2 protects
against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol.
21:1477–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar :
|
|
13
|
Avery-Cooper G, Doerr M, Gilbert RW,
Youssef M, Richard A, Huether P and Viloria-Petit AM: Par6 is an
essential mediator of apoptotic response to transforming growth
factor beta in NMuMG immortalized mammary cells. Cancer Cell Int.
14:192014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozdamar B, Bose R, Barrios-Rodiles M, Wang
HR, Zhang Y and Wrana JL: Regulation of the polarity protein Par6
by TGFbeta receptors controls epithelial cell plasticity. Science.
307:1603–1609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Viloria-Petit AM, David L, Jia JY, Erdemir
T, Bane AL, Pinnaduwage D, Roncari L, Narimatsu M, Bose R, Moffat
J, et al: A role for the TGFbeta-Par6 polarity pathway in breast
cancer progression. Proc Natl Acad Sci USA. 106:14028–14033. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bai S, Zeng R, Zhou Q, Liao W, Zhang Y, Xu
C, Han M, Pei G, Liu L, Liu X, et al: Cdc42-interacting protein-4
promotes TGF-B1-induced epithelial-mesenchymal transition and
extracellular matrix deposition in renal proximal tubular
epithelial cells. Int J Biol Sci. 8:859–869. 2012. View Article : Google Scholar :
|
|
17
|
Leibfried A, Fricke R, Morgan MJ, Bogdan S
and Bellaiche Y: Drosophila Cip4 and WASp define a branch of the
Cdc42-Par6-aPKC pathway regulating E-cadherin endocytosis. Curr
Biol. 18:1639–1648. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wirtz-Peitz F and Zallen JA: Junctional
trafficking and epithelial morphogenesis. Curr Opin Genet Dev.
19:350–356. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lu H and Bilder D: Endocytic control of
epithelial polarity and proliferation in Drosophila. Nat Cell Biol.
7:1232–1239. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aspenström P: A Cdc42 target protein with
homology to the non-kinase domain of FER has a potential role in
regulating the actin cytoskeleton. Curr Biol. 7:479–487. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu J, Kimura A, Baumann CA and Saltiel
AR: APS facilitates c-Cbl tyrosine phosphorylation and GLUT4
translocation in response to insulin in 3T3-L1 adipocytes. Mol Cell
Biol. 22:3599–3609. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang D, He C, Stoykovich MP and Schwartz
DK: Nanoscale topography influences polymer surface diffusion. ACS
Nano. 9:1656–1664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu C, Zhou Q, Liu L, Liu P, Pei G, Zeng R,
Han M and Xu G: Cdc42-interacting protein 4 represses E-Cadherin
expression by promoting β-Catenin translocation to the nucleus in
murine renal tubular epithelial cells. Int J Mol Sci.
16:19170–19183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zheng G, Lyons JG, Tan TK, Wang Y, Hsu TT,
Min D, Succar L, Rangan GK, Hu M, Henderson BR, et al: Disruption
of E-cadherin by matrix metalloproteinase directly mediates
epithelial-mesenchymal transition downstream of transforming growth
factor-beta1 in renal tubular epithelial cells. Am J Pathol.
175:580–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gunaratne A and Di Guglielmo GM: Par6 is
phosphorylated by aPKC to facilitate EMT. Cell Adhes Migr.
7:357–361. 2013. View Article : Google Scholar
|
|
26
|
Nolan ME, Aranda V, Lee S, Lakshmi B, Basu
S, Allred DC and Muthuswamy SK: The polarity protein Par6 induces
cell proliferation and is overexpressed in breast cancer. Cancer
Res. 68:8201–8209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ruan L, Shen Y, Lu Z, Shang D, Zhao Z, Lu
Y, Wu Y, Zhang Y, Tu Z and Liu H: Roles of partitioning-defective
protein 6 (Par6) and its complexes in the proliferation, migration
and invasion of cancer cells. Clin Exp Pharmacol Physiol.
44:909–913. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gunaratne A, Thai BL and Di Guglielmo GM:
Atypical protein kinase C phosphorylates Par6 and facilitates
transforming growth factor β-induced epithelial-to-mesenchymal
transition. Mol Cell Biol. 33:874–886. 2013. View Article : Google Scholar :
|