Introduction
Acute myocardial infarction (MI) is a tremendous
threat to the life and health of the population. However,
accumulating evidence indicates that directly restoring the blood
supply through reperfusion following MI in fact aggravates the
damage. This additional damage, namely ischemia/reperfusion (I/R)
injury (1–3) is a hotspot of current research.
Recent studies have reported that the pathogenesis of myocardial
I/R injury is multifactorial, involving the dysfunction of energy
metabolism, robust reactive oxygen species (ROS) generation, cell
calcium overload, inflammatory responses and apoptosis (4–8).
Oxidative stress resulting from ROS may be a significant feature in
the development of cardiac injury induced by I/R (9); however, the underlying regulatory
mechanisms of cardiac oxidative stress resistance remain unclear.
Forkhead box O3a (FoxO3a) plays an important role in metabolism,
cell survival and resistance to oxidative stress in multiple cell
types (10). However, its role in
regulating tolerance to cardiac stress remains to be fully
elucidated. FoxO3a interacts with the promoter of the superoxide
diamutase 2 (SOD2) gene at a specific binding site in
vascular smooth muscle cells (11); however, whether it influences SOD2
expression in cardiomyocytes has not yet been reported, at leas to
the best of our knowledge.
The nucleosome is the basic structural unit of
eukaryotic chromatin, which is made up of histones and DNA. Histone
acetylation modification affects chromatin packaging and gene
expression (12). Transcriptional
activity is found in areas where the histone acetylation level is
higher, and transcriptional silencing in areas where the histone
acetylation level is usually lower (13). The acetylation of histone is a
reversible process, which is regulated by histone
acetyltransferases (HATs) and histone deacetylases (HDACs)
(14), and the dynamic balance
between them plays a very important role in eukaryotic gene
regulation (15). It has been
demonstrated that HDAC inhibitors play a protective role in
cardiovascular diseases, such as cardiac hypertrophy, inflammatory
responses and heart failure (16).
Trichostatin A (TSA), a histone deacetylase
inhibitor, is used as a promising anticancer agent (17,18). Our previous study demonstrated
that TSA attehnuated myocardial I/R injury (19); however, the underlying mechanisms
remain to be elucidated. It has been shown that FoxO3a attehnuates
myocardial injury by increasing resistance to oxidative stress in
mice (20) and that the
endogenous HDAC inhibitor, β-hydroxybutyrate, can increase histone
acetylation at the FoxO3a promoters in 293 cells (21). We therefore speculated that the
mechanisms through which TSA attenuates myocardial injury may be
associated with FoxO3a. In the present study, we demonstrate that
TSA attenuates myocardial injury by increasing resistance to
oxidative stress. We further investigated whether the mechanisms
involved in these effects are mediated through the FoxO3a signaling
pathway.
Materials and methods
Ethical approval of the study
protocol
The study was approved by the Ethics Committee of
Jilin University, and all experimental procedures conformed to the
guidelines for the Animal Care and Use Committee of Jilin
University, Changchun, China.
Animals and model of myocardial I/R in
rats
A total of 80 healthy male Wistar rats (weighing,
180–220 g) were purchased from the Animal Centre of Jilin
University (certificate number SCXK (JI) 2007-0003). In total 40
Wistar rats first were subjected to MI by ligating the left
anterior descending branch of the coronary artery and to 30 min of
ischemia, followed by 0, 6, 12 or 24 h reperfusion in order to
determine the effects at different time points on FoxO3a
expression; 10 rats were used at each time point. The other 40 rats
were randomly divided into 5 groups as follows: i) the
sham-operated (control), treated with saline (n=8); ii) the I/R
group, in which rats were subjected to I/R and treated with saline
(n=8); iii) the TSA 1 group, in which rats were subjected to I/R
and treated with 0.2 mg/kg (I/R+T1) (n=8); iv) the TSA 2 group, in
which rats were subjected to I/R and treated with 0.1 mg/kg
(I/R+T2) (n=8); and v) the TSA 3 group, in which rats were
subjected to I/R and treated with 0.05 mg/kg (I/R+T3) (n=8). The
rats in the 5 groups were injected with TSA or saline by
intraperitoneal administration once daily for 5 days. Standard rat
chow and water were available ad libitum. On day 5, the
animals in the 5 groups underwent 30 min left anterior descending
(LAD) coronary artery ligation and 24 h of reperfusion according to
the method previously described by Li et al (22). Briefly, the rats were anesthetized
by an intraperitoneal injection of 10% chloral hydrate (0.35 g/kg)
before undergoing a thoracotomy. After ensuring the appropriate
depth of anesthesia, the rats were intubated without incision using
a rodent ventilator. The thoracic cavity was opened and the heart
was rapidly exposed. The left anterior descending coronary artery
(LAD) was ligated by a 6-0 silk suture (2–3 mm below the left
auricle) and the chest was immediately closed. Successful I/R
injury was monitored by standard lead II electrocardiography (ECG).
The sham-operated rats only underwent the suture around the LAD,
but were not ligated. Following reperfusion, the myocardial infarct
size, serum myocardium enzyme activities and protein expression
were evaluated.
Cell culture
The H9c2 rat myocardial cell line was obtained from
the Cell Bank of Chinese Academy of Sciences (Shanghai, China). The
cells were cultured in a 5% CO2 humidified atmosphere at
37°C in 10% fetal bovine serum (FBS)-containing Dulbecco's modified
Eagle's medium (DMEM). The cells were seeded in 6-well-plates for
24 h and were then incubated with/without TSA (50 nmol/l) for 1 h
and then incubated with/without H2O2 (400
µM) for 2 h.
Chemicals and antibodies
TSA was purchased from TCI Development Company Ltd.
(Shanghai, China). Polyclonal rabbit antibody against catalase was
purchased from Abcam (ab76110; Cambridge, CA, USA). Polyclonal
rabbit antibodies to FoxO3a (12829) and SOD2 (13141) were purchased
from Cell Signaling Technology (Danvers, MA, USA). Glyceraldehyde
3-phosphate dehydrogenase (GAPDH; 60004-1-lg) antibody was from the
ProteinTech Group, Inc. (Chicago, IL, USA). Goat anti-rabbit IgG
horseradish peroxidase (HRP)-conjugated secondary antibody (A0208),
goat anti-mouse IgG HRP-labeled secondary antibody (A0216), the
cell counting kit-8 (CCK-8), ROS assay kit and mitochondrial
membrane potential detection kit were from the Beyotime Institute
of Biotechnology (Jiangsu, China). The EZ ChIP™ Chromatin
Immunoprecipitation kit was purchased from Millipore Corp.
(Billerica, MA, USA). Enhanced chemiluminescence (ECL) detection
reagents were from Thermo Fisher Scientific (Waltham, MA, USA).
Other chemical reagents were commercially available and
analytically pure.
Cell viability
The viability of the H9c2 cells (in 96-well plates)
was measured by CCK-8 assay according to the manufacturer's
instructions. Briefly, the cells were exposed to
H2O2 at various concentrations for different
durations. CCK-8 solution (10 µl) was added followed by
further incubation for 2 h and the absorbance were measured at 450
nm using a microplate reader (Infinite M200 PRO; Tecan Corp.,
Mannedorf, Switzerland).
Measurement of myocardial infarct size
(MIS)
Following 0, 6, 12 and 24 h of reperfusion, the
animals were euthanized and the hearts were rapidly removed. The
MIS size was assessed by the 2,3,5-triphenyltetrazolium chloride
(TTC) method as previously described (23). Briefly, the excised hearts were
washed using 0.9% saline. The right ventricular tissues were
removed and left ventricles were frozen and stored at −20°C for 5
min and then were sliced into 1-mm-thick sections, giving 5 slices
of equal thickness along the long axis from the apex to the base.
The slices were incubated for 15–30 min in TTC at pH 7.4 at 37°C to
identify the viable myocardium stained red, while the infarct
tissue remained white. The slices were fixed in 10% formaldehyde
solution and photographed with a digital camera. Areas stained in
red and white were measured by a BI-2000 Medical Image Analysis
System (Chengdu TME Technology, Chengdu, China). The infarct size
was calculated using the following equation: %Infarct volume =
infarct volume/total left ventricle volume ×100%.
Measurement of myocardial enzyme
[creatine kinase (CK), lactate dehydrogenase (LDH) and aspartate
aminotransferase (AST)] activities in serum
At the end of the experiment, blood samples were
obtained from the abdominal aorta, incubated at 37°C for 30 min and
then centrifuged at 3,000 rpm for 10 min. The supernatant from each
group was collected and frozen at −80°C for further analysis. The
activities of CK, LDH and AST were measured using CK, LDH and AST
kits (Jiancheng Bioengineering Institute, Nanjing, China) with an
automatic biochemistry analyzer (EOS880; Hospitex Diagnostics,
Italy) according to the manufacturer's instructions.
Measurement of malondialdehyde (MDA)
levels and SOD activities
At the end of the experiment, the myocardial tissue
was homogenated for 5 min in the ice and then centrifuged at 2,000
rpm for 5 min. The supernatant from each group was collected for
measuring the levels of MDA and the activities of SOD using
respective kits (Jiancheng Bioengineering Institute).
Western blot analysis
Proteins were separated by 5–18% sodium
dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto polyvinylidene fluoride membranes. The membranes
were blocked with 5% skimmed dry milk for 2 h at room temperature
and washed 3 times in Tris-buffered saline with Tween (TBST); the
membranes were then incubated with the primary antibodies to
FoxO3a, SOD2, catalase and GAPDH overnight at 4°C. After washing
with TBST again, HRP-labeled secondary antibodies goat anti-rabbit
(1:1,000) or goat anti-mouse (1:2,000) were added followed by
incubation for 1.5 h at room temperature. Immunoblots were
developed by ECL detection kits and the bands were quantified by
Quantity One software.
Immunofluorescence staining
The cultured H9c2 cells were fixed with 4%
paraformaldehyde for 30 min and then washed 3 times in
phosphate-buffred saline (PBS). Blocking solutions (0.2% Triton
X-100 and sheep serum in PBS) were added to the fixed cells
followed by incubation for 30 min at 37°C. The cells were then
incubated with primary antibody against FoxO3a overnight at 4°C.
After washing with PBS, the appropriate secondary antibody was
added followed by incubation for 1 h in the dark at room
temperature. The nuclei were then stained with Hoechst 33342 for 20
min. After washing with PBS, immunofluorescence was examined under
a fluorescence microscope.
Measurement of ROS production
Intracellular ROS of H9c2 was measured by
2′7′-dichlorodihydrofluorescein diacetate (DCFH-DA) as a
fluorescent probe according to the manufacturer's instructions.
Briefly, H9c2 cells were loaded with DCFH-DA (10 µM) for 30
min at 37°C and washed 3 times with serum-free medium. The
fluorescence image was observed under a fluorescence
microscope.
Detection of mitochondrial membrane
potential (Δψm)
The dye, JC-1, was used as a fluorescent probe to
detect Δψm as previously described (24). CCCP (10 mM) provided in the kit
was added to the cell culture medium at 1:1,000 for 20 min at room
temperature, then the H9c2 cells were incubated with JC-1 dye (10
µM) for 30 min at 37°C and washed twice with JC-1 dyeing
buffer (1X). The red and green fluorescence were observed under a
fluorescence microscope.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation assay (25) was performed in accordance with the
manufacturer's instructions with slight modifications. In brief,
protein/DNA complexes of H9c2 cells (5×106) were
cross-linked with formaldehyde and were sonicated to 200–1000 bp of
DNA fragments. The crosslinked protein/DNA was then immunoselected
with specific antibodies and the protein/DNA complexes were then
eluted. Protein-DNA crosslinks were reversed during incubation at
65°C and DNA was purified to subject to quantitative PCR using the
following primers: 5′-AAAGGCATCCCAAGGTAT-3′ (forward) and 5′-CCA
GCTCTACAGCTCGTAC-3′ (reverse).
Statistical analyses
Data are shown as the means ± SD. Analysis of
variance (ANOVA) was used to compare quantitative variables between
more than 2 groups. Intergroup differences were assessed by the
Chi-square test and Student's t-test for quantitative variables. A
p-value <0.05 (two-sided) was considered statistically
significant. Data were analyzed with GraphPad Prism 5.0
software.
Results
Oxidative stress induces myocardial
injury
In order to observe the effects of oxidative stress
on H9c2 rat myocardial cells, H2O2 was used
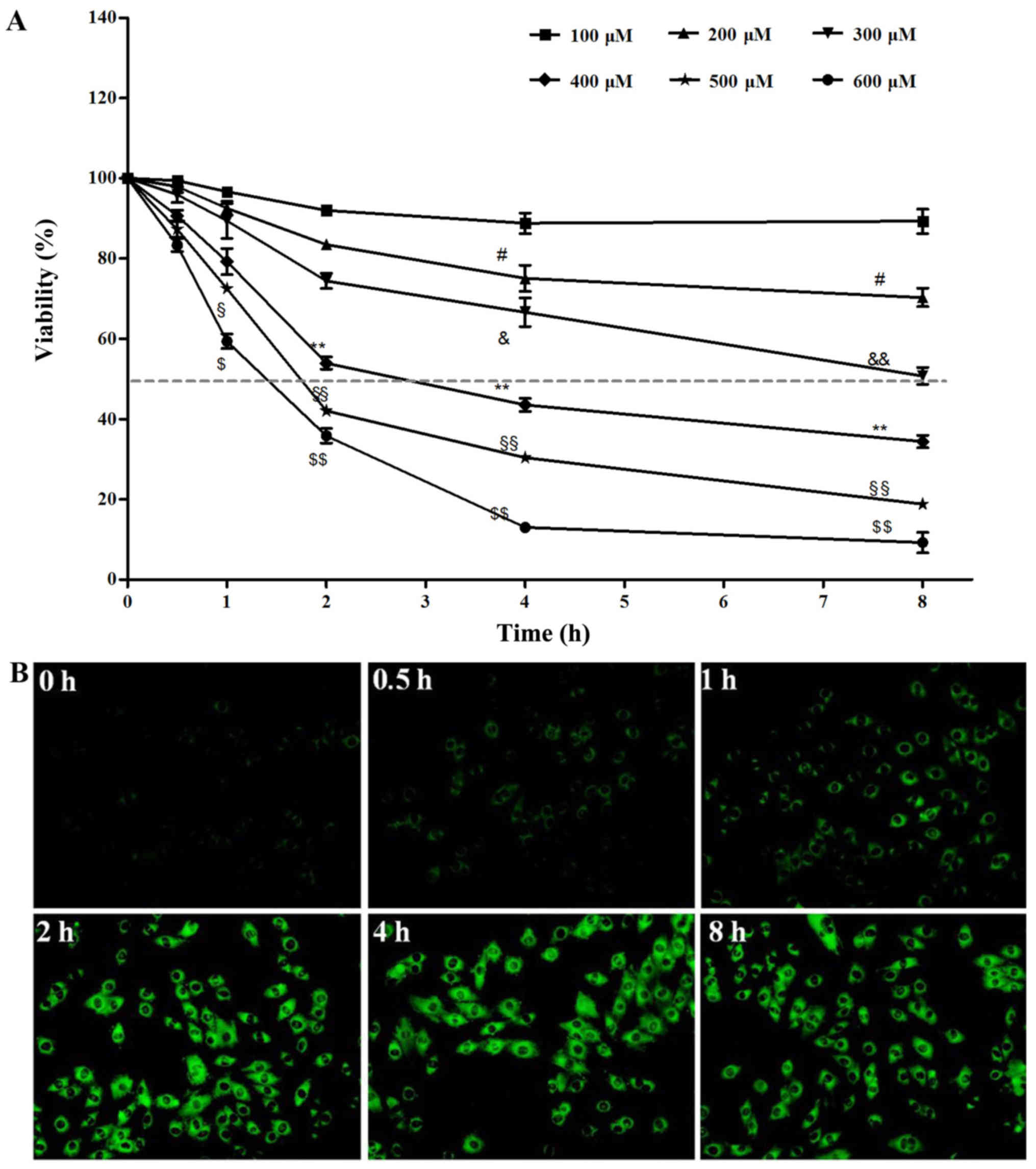
as an inducer. The results from CCK-8 assay revealed that cell
viability decreased following exposure to
H2O2 in a dose- and time-dependent manner.
The cell survival rate at H2O2 400 µM
for 2 h was 53.96±5.6% (Fig. 1A),
compared with the control group, and the cell viability was
significantly decreased (p<0.01). Subsequently, intracellular
ROS levels were detected using the DCFH-DA probe; the strength of
the green fluorescence directly reflects intracellular ROS. The
levels of ROS increased gradually following exposure to
H2O2 compared with the untreated group, and
the fluorescence intensity was significantly increased at 2 h
(Fig. 1B).
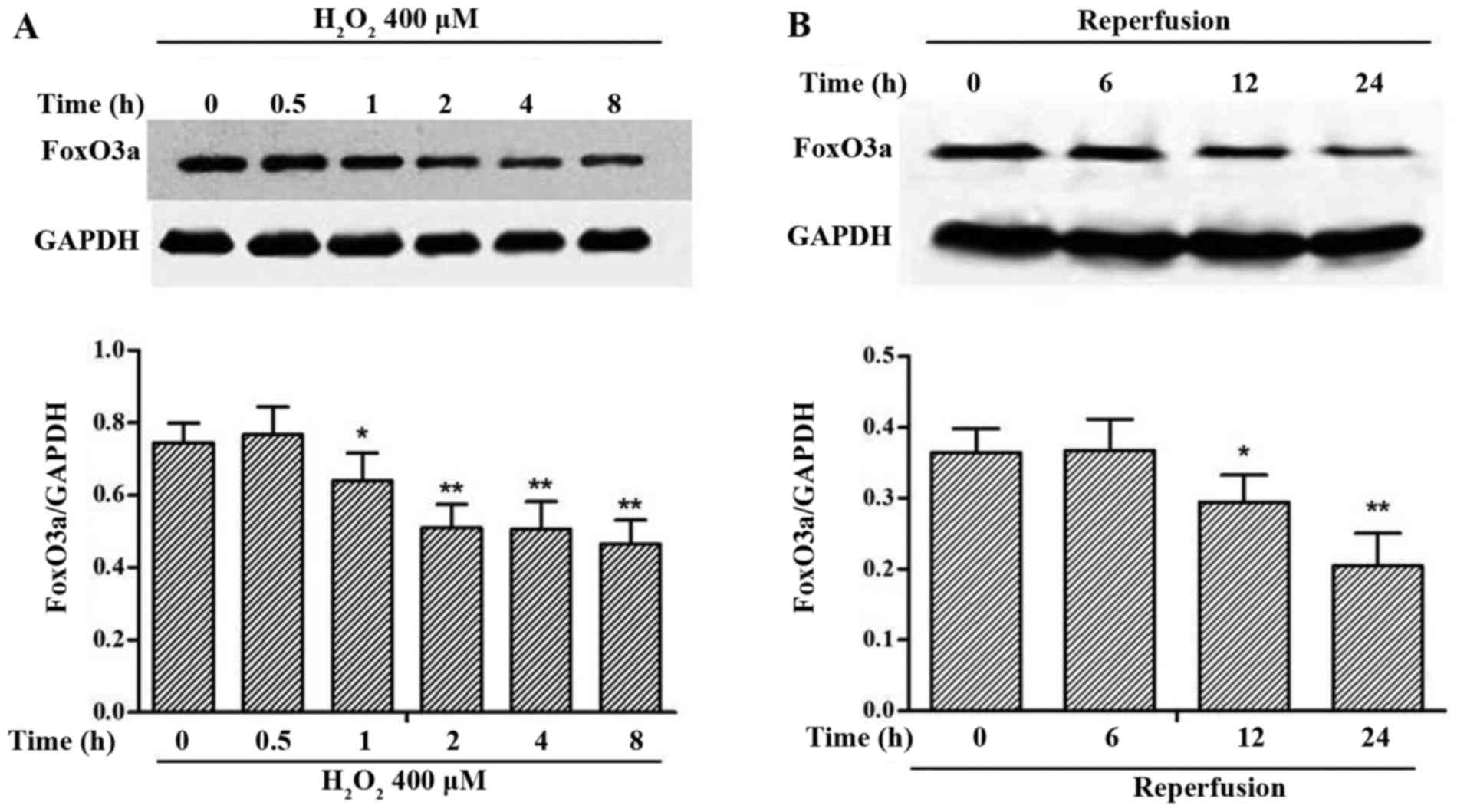
FoxO3a is downregulated in oxidative
stress-induced myocardial injury
To investigate the role of FoxO3a under myocardial
oxidative stress conditions, we performed the experiments in
vitro and in vivo. We examined the expression of FoxO3a
in the H9c2 cells exposed to H2O2 at 400
µM for different periods of time. A significant
downregulation in FoxO3a expression was observed following exposure
of the cells to H2O2 at 400 µM for 2 h
(Fig. 2A). We also detected the
expression of FoxO3a in rats subjected to MI and 30 min of
ischemia, followed by 0, 6, 12 or 24 h of reperfusion. The
expression of FoxO3a was also decreased in the myocardial tissue in
the rats with I/R injury (Fig.
2B).
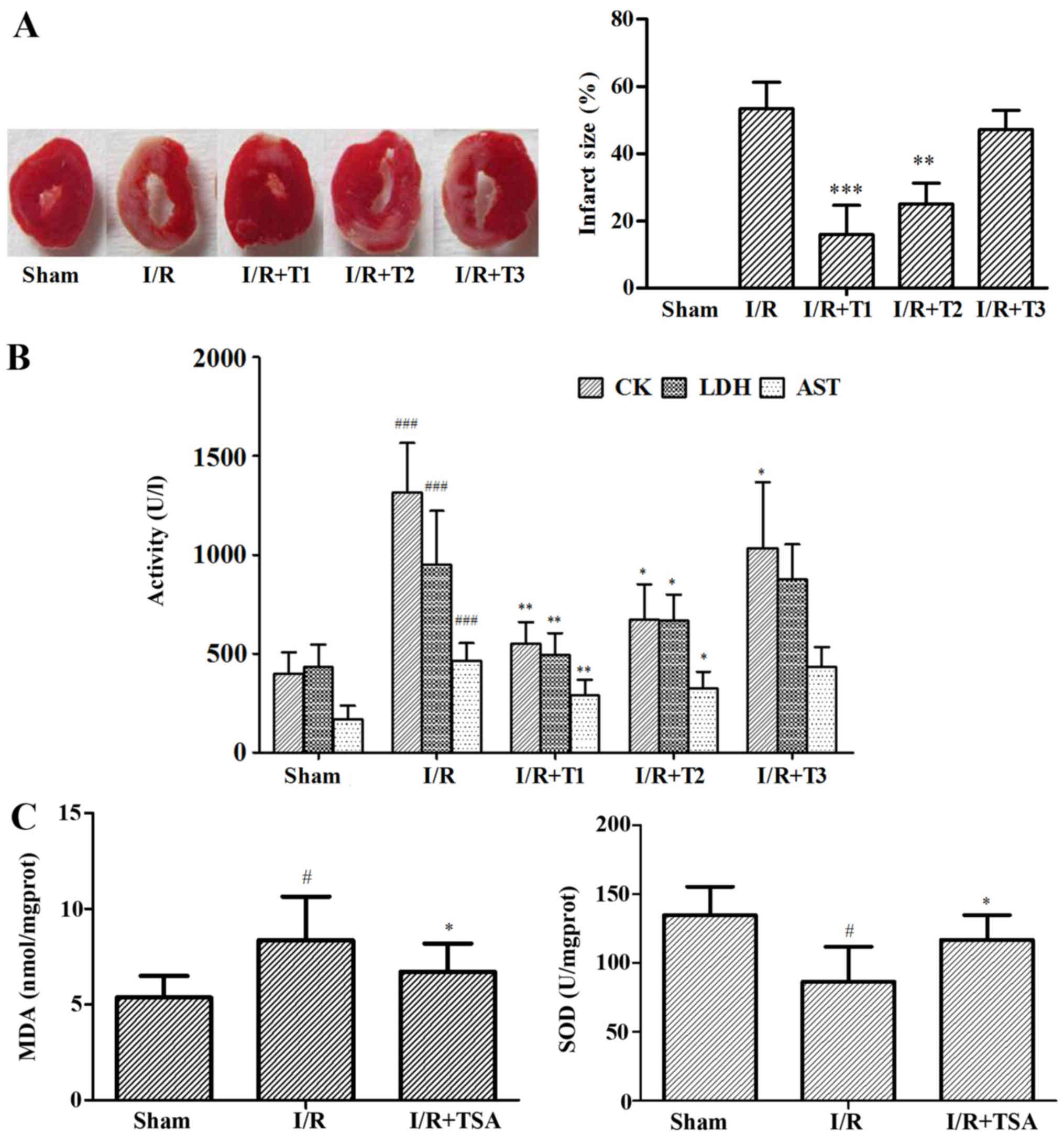
TSA attenuates myocardial injury mediated
by oxidative stress
To examine the effects of TSA on myocardial injury
mediated by oxidative stress, we performed the experiments in
vitro and in vivo. We measured the MIS by the TTC
method. Compared with the I/R group, the MIS was significantly
reduced in the TSA 0.2 mg/kg (T1)- and 0.1 mg/kg (T2)-treated
groups, from 53.36 to 15.89% (p<0.01) and 20.06% (p<0.001)
respectively; however, the MIS between the I/R and TSA 0.05 mg/kg
(T3)-treated groups was not significantly altered (Fig. 3A). Subsequently, in order to
evaluate the extent of myocardial injury in the rats, we measured
the activities of CK, LDH and AST in serum after 24 h of
reperfusion. In accordance with MIS, TSA significantly decreased
the activities of CK, LDH and AST in serum (Fig. 3B). In addition, the MDA level was
decreased and the SOD activity was increased in the TSA-treated
groups compared with those in the sham-operated group (Fig. 3C).
In order to investigate whether TSA reduces the ROS
levels following exposure of the H9c2 cells to
H2O2, intracellular ROS production was
detected by the DCFH-DA probe. Compared with the
H2O2-exposed group, treatment with TSA
reduced the intracellular ROS level (Fig. 3D). It has been reported that
oxidative stress induces mitochondrial dysfunction (26). Thus, in this study, to investigate
the protective effect of TSA against oxidative stress-induced
mitochondrial damage, Δψm in the H9c2 cells was assessed
using the JC-1 probe. The Δψm of the H9c2 cells declined
markedly following exposure to H2O2, whereas
treatment with TSA increased the Δψm in the H9c2 cells
(Fig. 3E).
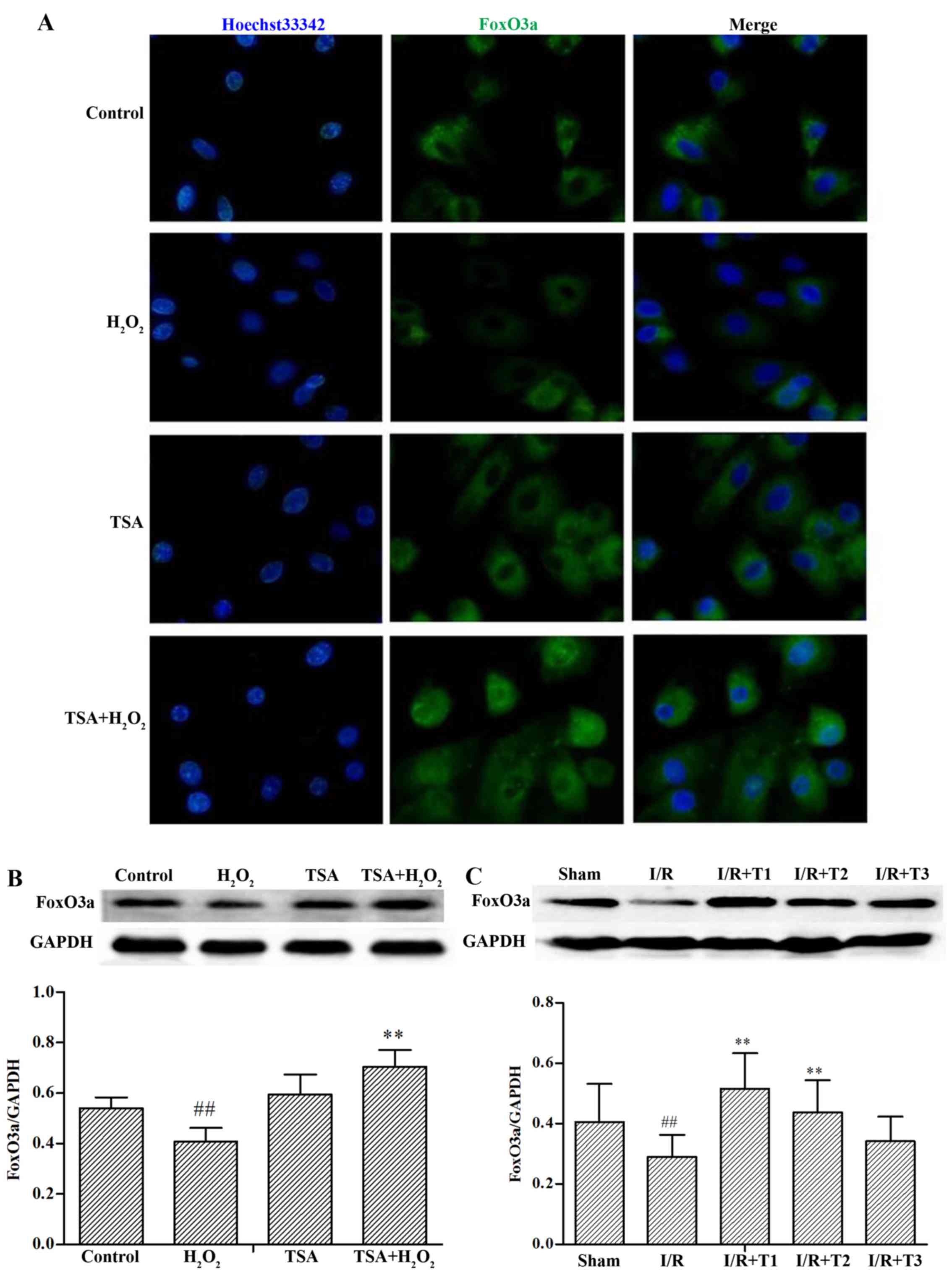
Effect of TSA on the expression of
FoxO3a
FoxO3a is an important regulator of the resistance
to oxidative stress and the downregulation of FoxO3a increases cell
death in response to oxidative stress in human chondrocytes
(27). Resveratrol, a potent
antioxidant, has previously been demonstrated to protect
photoreceptor cells from apoptosis by upregulating the FoxO family
in experimental retinal detachment (28). Our experimental results revealed
that the expression of FoxO3a in the H9c2 cells was markedly
reduced in the presence of H2O2 compared with
that of the control group (p<0.01). However, TSA elicited a
significant increase in FoxO3a expression compared with the
H2O2 alone-treated group (p<0.01)
(Fig. 4A and B). In line with the
in vitro experimental results, TSA increased the expression
of FoxO3a following 24 h of reperfusion in rats (Fig. 4C; p<0.01 vs. sham-operated
group; p<0.01 vs. I/R group).
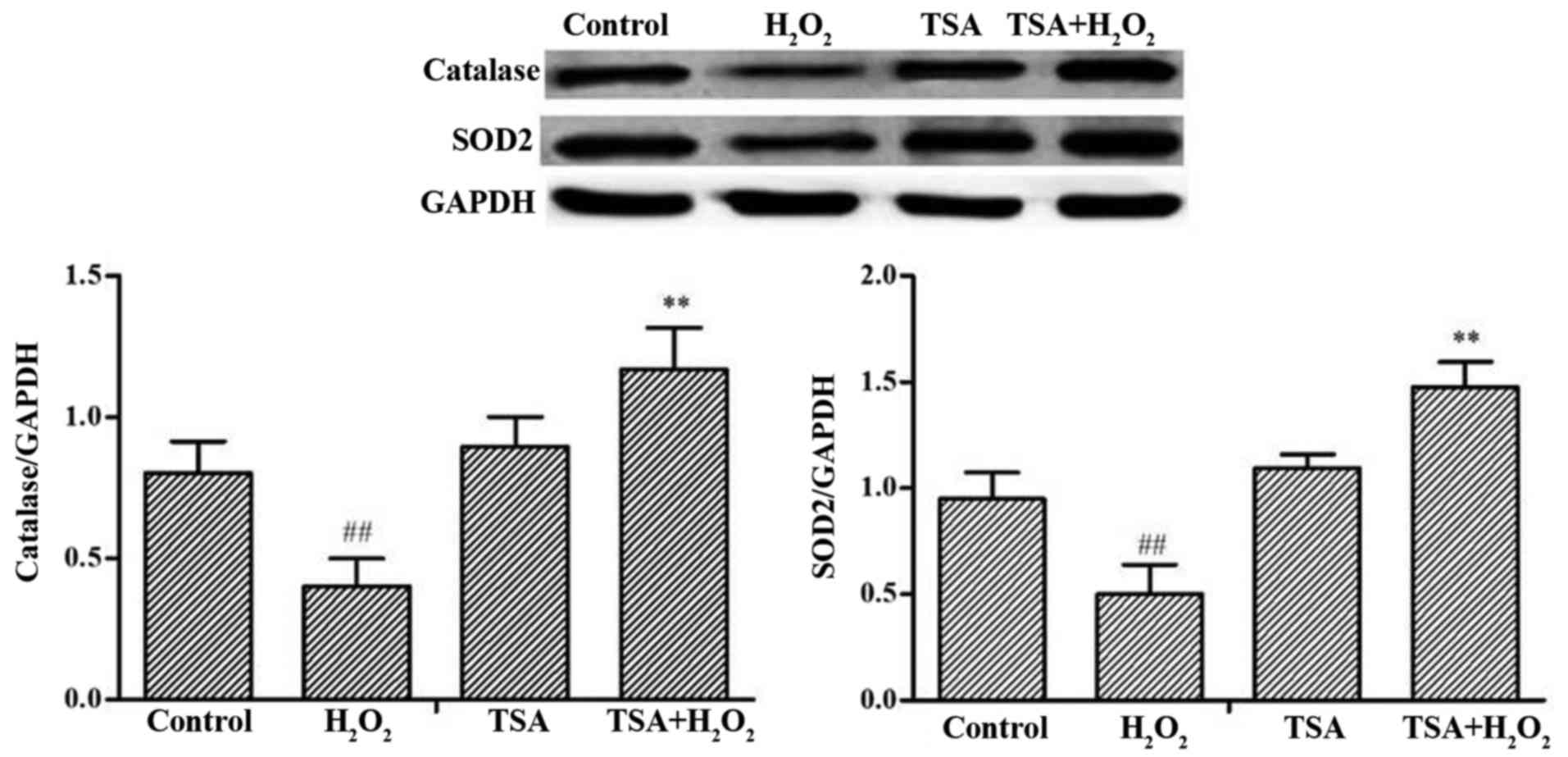
Effect of TSA on SOD2 and catalase
levels
Mitochondrial SOD and catalase as endogenous enzymes
are known to be the target proteins of FoxO3a that contributes to
their protective effects against oxidative stress (29,30). The levels of SOD2 and catalase in
the H9c2 cells markedly decreased following exposure to
H2O2, whereas treatment with TSA elicited a
significant increase in SOD2 and catalase levels in the H9c2 cells
(p<0.01) compared with cells exposed to
H2O2 alone (Fig. 5).
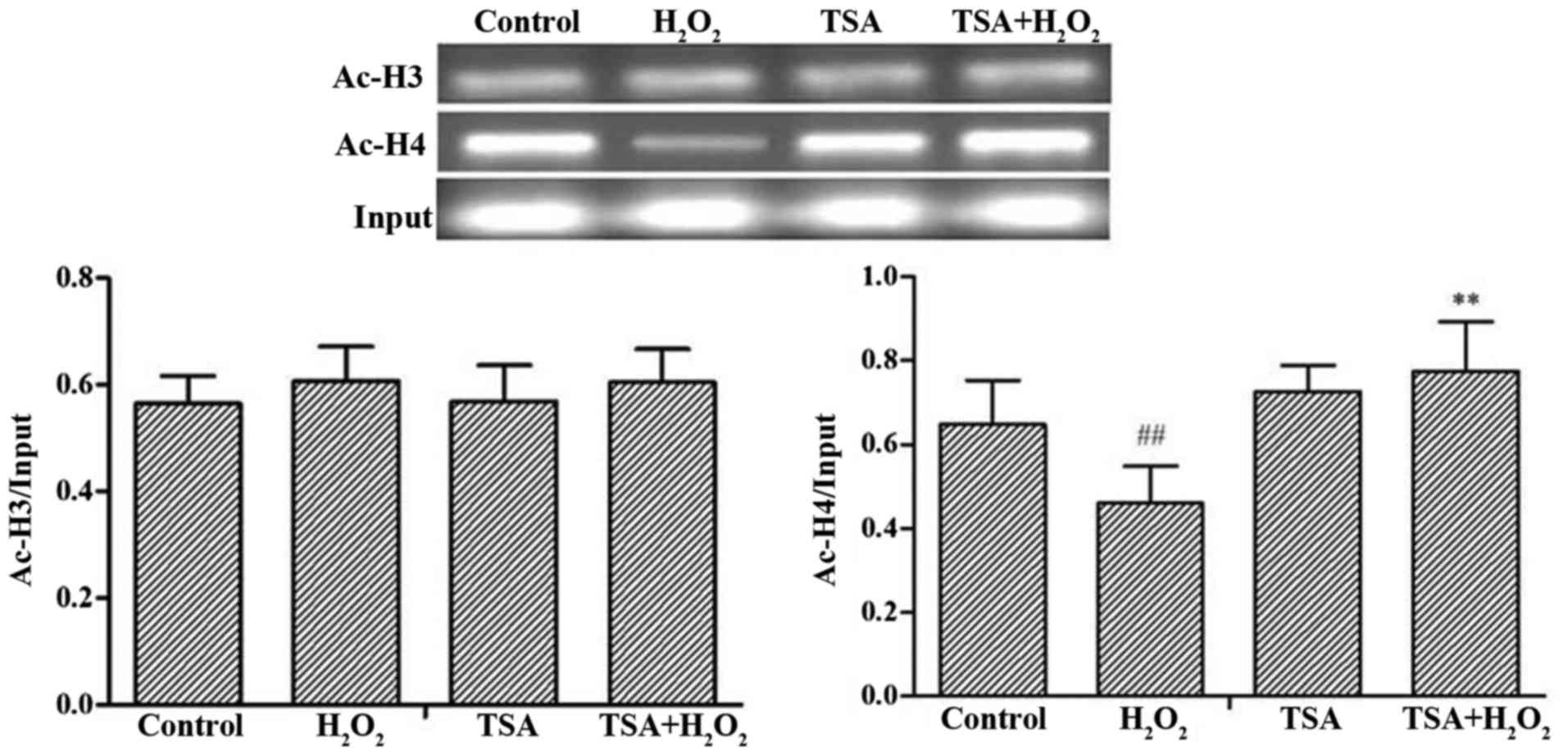
Effects of TSA on the acetylation levels
of histone H3 and H4 in the promoter region of FoxO3a
To investigate whether TSA affects the levels of
histone acetylation of the FoxO3a gene promoter and then FoxO3a
gene expression, we performed in vitro experiments to
observe the changes in the H3 and H4 acetylation levels of FoxO3a.
The H4 acetylation of the FoxO3a promoter region was markedly
decreased in the H2O2-exposed cells compared
with that of the control group (p<0.01), whereas TSA elicited a
significant increase in H4 acetylation of the FoxO3a promoter
region compared with that of the H2O2 group.
The level of H3 acetylation of FoxO3a was not significantly altered
(Fig. 6). The above-mentioned
results thus indicate that TSA alters the H4 acetylation of the
FoxO3a promoter region, thus affecting the expression of
FoxO3a.
Discussion
The present study demonstrated that TSA attenuates
myocardial injury mediated by oxidative stress in vivo and
in vitro. This attenuation may be associated with the
increased histone acetylation in the FoxO3a promoter region and the
upregulation of the expression of FoxO3a, as well as the target
proteins (SOD2 and catalase), which regulate the expression of ROS
scavengers in response to ROS.
Oxidative stress is an important pathophysiological
mechanism in myocardial injury (31). Oxidative stress means that the
body suffers various harmful stimulations, disrupting the balance
between the generation and clearance of ROS; the overproduction of
ROS as byproducts by the mitochondria results in myocardial injury
(32,33). In this study, exposure to
H2O2 at various concentrations for different
periods of time in the H9c2 cells decreased cell viability in a
dose- and time-dependent manner (Fig.
1A). Intracellular ROS levels in the H9c2 cells exposed to
H2O2 for different periods of time increased
gradually (Fig. 1B). This may be
associated with the downregulated expression of FoxO3a (Fig. 2). Depending on the different
environment and cell types, FoxO3a plays an important role in
lifespan extension, energy metabolism and resistance to oxidative
stress (34,35). FoxO3a also acts as the potential
target for the treatment of several types of diseases (10). It has been demonstrated that
FoxO3a plays an important role in maintaining cardiac function and
antagonizing oxidative stress responses (36). Increasing the expression of FoxO3a
reduces ROS and promotes cardiomyocyte survival (20). FoxO3a also upregulates the
expression of ROS scavengers to clear ROS (30,37). Mitochondria are considered to be
important cellular organelles as the source of ROS (38), and Δψm is an important
index maintaining mitochondrial function (39). A decrease in the Δψm
results in mitochondrial dysfunction, causing an increase in ROS
generation. MDA, which is the final product of lipid peroxidation,
can reflect the production of oxygen free radicals; the content can
not only reflect the formation of oxygen free radicals in
vivo, it can also reflect the severity of tissue damage caused
by oxygen free radicals. SOD, which is the specific scavenger, can
reduce free radical damage to myocardial cells. SOD plays an
important role in the free radical scavenging system. The present
study revealed that the downregulation of FoxO3a in H9c2 cells was
associated with the H2O2-induced increase in
intracellular ROS levels, and reduced Δψm, and SOD2 and
catalase levels. The opposite was observed following treatment with
TSA (Figs. 3D and E, 4A and B and 5). Consistent with the experimental
results obtained in vitro, TSA significantly reduced the MIS
(Fig. 3A) and decreased the
activities of serum CK, LDH and AST (Fig. 3B) in the rats. Moreover, TSA
decreased the MDA level and increased SOD activity in the rats
(Fig. 3C). These data suggest
that TSA attenuates myocardial I/R-induced oxidative stress-related
damage and upregulates the expression of FoxO3a, which is a
possible mechanism ensuring the protective effects of TSA against
I/R-induced myocardial injury (Fig.
4C).
Histone acetylation plays a pivotal role in the
epigenetic modulation of gene expression and maintains a dynamic
balance by affecting the activities of histone acetyltransferases
and histone deacetylases (40).
In most cases, the acetylation of transcription factors results in
increasing their transactivation functions, principally by
enhancing their DNA binding ability (41) and it has been shown that p300, an
acetyltransferase, enhances the transcriptional activity of FoxO3a
(42), reminding us that histone
acetylation participates in FoxO3a regulation. Similar to a
previous study showing that the acetylation level of the FoxO3a
promoter region can be upgraded by HDAC inhibitors (21), we demonstrated that TSA reduced
oxidative stress-induced myocardial injury by increasing the level
of H4 acetylation of the FoxO3a promoter region, thereby enhancing
the expression of FoxO3a (Fig.
6).
In conclusion, our data indicate that treatment with
TSA attenuates oxidative stress-mediated myocardial damage by
regulating the level of histone H4 acetylation in the promoter
region of FoxO3a, and upregulating the expression of FoxO3a, SOD2
and catalase. Our findings provide a novel therapeutic strategy for
TSA acting as a mediator of resistance to oxidative stress for
myocardial protection.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant no. 81570253).
Glossary
Abbreviations
Abbreviations:
|
TSA
|
trichostatin A
|
|
I/R
|
ischemia/reperfusion
|
|
MI
|
myocardial infarction
|
|
TTC
|
2,3,5-triphenyltetrazolium
chloride
|
|
AST
|
aspartate aminotransferase
|
|
CK
|
creatine kinase
|
|
LDH
|
lactate dehydrogenase
|
|
ROS
|
reactive oxygen species
|
|
Δψm
|
mitochondrial membrane potential
|
|
SOD2
|
superoxide dismutase 2
|
References
|
1
|
Frank A, Bonney M, Bonney S, Weitzel L,
Koeppen M and Eckle T: Myocardial ischemia reperfusion injury: From
basic science to clinical bedside. Semin Cardiothorac Vasc Anesth.
16:123–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cao J, Xie H, Sun Y, Zhu J, Ying M, Qiao
S, Shao Q, Wu H and Wang C: Sevoflurane post-conditioning reduces
rat myocardial ischemia reperfusion injury through an increase in
NOS and a decrease in phopshorylated NHE1 levels. Int J Mol Med.
36:1529–1537. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jennings RB, Sommers HM, Smyth GA, Flack
HA and Linn H: Myocardial necrosis induced by temporary occlusion
of a coronary artery in the dog. Arch Pathol. 70:68–78.
1960.PubMed/NCBI
|
|
4
|
Matsumoto T, Miura T, Miki T, Nishino Y,
Nakamura Y and Shimamoto K: Does enhanced expression of the
Na+-Ca2+ exchanger increase myocardial
vulnerability to ischemia/reperfusion injury in rabbit hearts? Mol
Cell Biochem. 248:141–147. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang M, Baker L, Tsai BM, Meldrum KK and
Meldrum DR: Sex differences in the myocardial inflammatory response
to ischemia-reperfusion injury. Am J Physiol Endocrinol Metab.
288:E321–E326. 2005. View Article : Google Scholar
|
|
6
|
Qin C, Yap S and Woodman OL: Antioxidants
in the prevention of myocardial ischemia/reperfusion injury. Expert
Rev Clin Pharmacol. 2:673–695. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sanada S, Komuro I and Kitakaze M:
Pathophysiology of myocardial reperfusion injury: Preconditioning,
postconditioning, and translational aspects of protective measures.
Am J Physiol Heart Circ Physiol. 301:H1723–H1741. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guo J, Zheng D, Li HR, Zhang AD and Li ZC:
Anti-apoptotic potency of TNFR:Fc gene in
ischemia/reperfusion-induced myocardial cell injury. Inflammation.
38:664–671. 2015. View Article : Google Scholar
|
|
9
|
Zhou T, Chuang CC and Zuo L: Molecular
characterization of reactive oxygen species in myocardial
ischemia-reperfusion injury. BioMed Res Int. 2015:8649462015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nho RS and Hergert P: FoxO3a and disease
progression. World J Biol Chem. 5:346–354. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li M, Chiu JF, Mossman BT and Fukagawa NK:
Downregulation of manganese-superoxide dismutase through
phosphorylation of FOXO3a by Akt in explanted vascular smooth
muscle cells from old rats. J Biol Chem. 281:40429–40439. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dell'Aversana C, Lepore I and Altucci L:
HDAC modulation and cell death in the clinic. Exp Cell Res.
318:1229–1244. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Grunstein M: Histone acetylation in
chromatin structure and transcription. Nature. 389:349–352. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shats I, Gatza ML, Liu B, Angus SP, You L
and Nevins JR: FOXO transcription factors control E2F1
transcriptional specificity and apoptotic function. Cancer Res.
73:6056–6067. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Icardi L, De Bosscher K and Tavernier J:
The HAT/HDAC interplay: Multilevel control of STAT signaling.
Cytokine Growth Factor Rev. 23:283–291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cardinale JP, Sriramula S, Pariaut R,
Guggilam A, Mariappan N, Elks CM and Francis J: HDAC inhibition
attenuates inflammatory, hypertrophic, and hypertensive responses
in spontaneously hypertensive rats. Hypertension. 56:437–444. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Usami M, Kishimoto K, Ohata A, Miyoshi M,
Aoyama M, Fueda Y and Kotani J: Butyrate and trichostatin A
attenuate nuclear factor kappaB activation and tumor necrosis
factor alpha secretion and increase prostaglandin E2 secretion in
human peripheral blood mononuclear cells. Nutr Res. 28:321–328.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lu JC, Chang YT, Wang CT, Lin YC, Lin CK
and Wu ZS: Trichostatin A modulates thiazolidinedione-mediated
suppression of tumor necrosis factor α-induced lipolysis in 3T3-L1
adipocytes. PLoS One. 8:e715172013. View Article : Google Scholar
|
|
19
|
Yu L, Lu M, Wang P and Chen X:
Trichostatin A ameliorates myocardial ischemia/reperfusion injury
through inhibition of endoplasmic reticulum stress-induced
apoptosis. Arch Med Res. 43:190–196. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sengupta A, Molkentin JD, Paik JH, DePinho
RA and Yutzey KE: FoxO transcription factors promote cardiomyocyte
survival upon induction of oxidative stress. J Biol Chem.
286:7468–7478. 2011. View Article : Google Scholar :
|
|
21
|
Shimazu T, Hirschey MD, Newman J, He W,
Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD,
et al: Suppression of oxidative stress by β-hydroxybutyrate, an
endogenous histone deacetylase inhibitor. Science. 339:211–214.
2013. View Article : Google Scholar
|
|
22
|
Li H, Liu Z, Wang J, Wong GT, Cheung CW,
Zhang L, Chen C, Xia Z and Irwin MG: Susceptibility to myocardial
ischemia reperfusion injury at early stage of type 1 diabetes in
rats. Cardiovasc Diabetol. 12:1332013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bollini S, Cheung KK, Riegler J, Dong X,
Smart N, Ghionzoli M, Loukogeorgakis SP, Maghsoudlou P, Dubé KN,
Riley PR, et al: Amniotic fluid stem cells are cardioprotective
following acute myocardial infarction. Stem Cells Dev.
20:1985–1994. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rukoyatkina N, Mindukshev I, Walter U and
Gambaryan S: Dual role of the p38 MAPK/cPLA2 pathway in the
regulation of platelet apoptosis induced by ABT-737 and strong
platelet agonists. Cell Death Dis. 4:e9312013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nechipurenko IV and Broihier HT: FoxO
limits microtubule stability and is itself negatively regulated by
microtubule disruption. J Cell Biol. 196:345–362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Torres-Gonzalez M, Gawlowski T, Kocalis H,
Scott BT and Dillmann WH: Mitochondrial 8-oxoguanine glycosylase
decreases mitochondrial fragmentation and improves mitochondrial
function in H9C2 cells under oxidative stress conditions. Am J
Physiol Cell Physiol. 306:C221–C229. 2014. View Article : Google Scholar :
|
|
27
|
Akasaki Y, Alvarez-Garcia O, Saito M,
Caramés B, Iwamoto Y and Lotz MK: FoxO transcription factors
support oxidative stress resistance in human chondrocytes.
Arthritis Rheumatol. 66:3349–3358. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Huang W, Li G, Qiu J, Gonzalez P and
Challa P: Protective effects of resveratrol in experimental retinal
detachment. PLoS One. 8:e757352013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lijnen PJ, van Pelt JF and Fagard RH:
Downregulation of manganese superoxide dismutase by angiotensin II
in cardiac fibroblasts of rats: Association with oxidative stress
in myocardium. Am J Hypertens. 23:1128–1135. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tan WQ, Wang K, Lv DY and Li PF: Foxo3a
inhibits cardiomyocyte hypertrophy through transactivating
catalase. J Biol Chem. 283:29730–29739. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Adluri RS, Thirunavukkarasu M, Zhan L,
Maulik N, Svennevig K, Bagchi M and Maulik G: Cardioprotective
efficacy of a novel antioxidant mix VitaePro against ex vivo
myocardial ischemia-reperfusion injury. Cell Biochem Biophys.
67:281–286. 2013. View Article : Google Scholar
|
|
32
|
Chen Q, Moghaddas S, Hoppel CL and
Lesnefsky EJ: Reversible blockade of electron transport during
ischemia protects mitochondria and decreases myocardial injury
following reperfusion. J Pharmacol Exp Ther. 319:1405–1412. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ji L, Fu F, Zhang L, Liu W, Cai X, Zhang
L, Zheng Q, Zhang H and Gao F: Insulin attenuates myocardial
ischemia/reperfusion injury via reducing oxidative/nitrative
stress. Am J Physiol Endocrinol Metab. 298:E871–E880. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Salih DA and Brunet A: FoxO transcription
factors in the maintenance of cellular homeostasis during aging.
Curr Opin Cell Biol. 20:126–136. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeng Y, Cheng L, Chen H, Cao H, Hauser ER,
Liu Y, Xiao Z, Tan Q, Tian XL and Vaupel JW: Effects of FOXO
genotypes on longevity: A biodemographic analysis. J Gerontol A
Biol Sci Med Sci. 65:1285–1299. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ronnebaum SM and Patterson C: The FoxO
family in cardiac function and dysfunction. Annu Rev Physiol.
72:81–94. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kops GJ, Dansen TB, Polderman PE, Saarloos
I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH and Burgering
BM: Forkhead transcription factor FOXO3a protects quiescent cells
from oxidative stress. Nature. 419:316–321. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Paradies G, Petrosillo G, Paradies V and
Ruggiero FM: Mitochondrial dysfunction in brain aging: Role of
oxidative stress and cardiolipin. Neurochem Int. 58:447–457. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang L, Lu K, Hao H, Li X, Wang J, Wang K,
Wang J, Yan Z, Zhang S, Du Y, et al: Decreased autophagy in rat
heart induced by anti-β1-adrenergic receptor autoantibodies
contributes to the decline in mitochondrial membrane potential.
PLoS One. 8:e812962013. View Article : Google Scholar
|
|
40
|
Peserico A and Simone C: Physical and
functional HAT/HDAC interplay regulates protein acetylation
balance. J Biomed Biotechnol. 2011:3718322011. View Article : Google Scholar
|
|
41
|
Chen H, Tini M and Evans RM: HATs on and
beyond chromatin. Curr Opin Cell Biol. 13:218–224. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Motta MC, Divecha N, Lemieux M, Kamel C,
Chen D, Gu W, Bultsma Y, McBurney M and Guarente L: Mammalian SIRT1
represses forkhead transcription factors. Cell. 116:551–563. 2004.
View Article : Google Scholar : PubMed/NCBI
|