Introduction
Sepsis, caused by severe infections, burns and
trauma, remains a leading cause of mortality worldwide. In the US,
the mortality rate due to sepsis is as high as 50% and is
increasing at a rate of 1.5% per year. Thus, approximately 200,000
individuals die from sepsis each year, and health care costs
associated with this exceed $16 billion annually (1,2).
In China, severe sepsis has a mortality rate of 40.7% during the
first 28 days, and health care costs are at 90,000 Yuan/patient
(3). Accordingly, further studies
are required in order to obtain a better understanding of the
pathophysiology of sepsis and available clinical treatment
options.
Lipopolysaccharide (LPS)-induced endotoxemia mimics
many features of septic shock (4), including the persistent activation
of systemic inflammatory responses, immune suppression, coagulation
disorders and mitochondrial damage (2). Macrophage mitochondrial structural
damage and consequent oxidative stress responses are associated
with apoptosis, energy metabolism fluctuations and systemic
inflammatory responses; these all contribute to septic shock and
multiple organ failure (5).
Macrophages are the first line of defense against
pathogens and are important regulators of innate and adaptive
immune responses (6). Macrophage
mitochondrial dysfunction is responsible for changes in energy
metabolism, the intrinsic apoptotic pathway, oxidative stress and
systemic inflammatory responses. The association between the degree
of macrophage mitochondrial dysfunction and deleterious outcomes
has been documented; inflammatory factors, free radicals, calcium
overload, adenosine triphosphate (ATP)-sensitive potassium channel
perturbations and respiratory chain complex damage are responsible
for the noted mitochondrial damage during endotoxemia (7). However, the upstream signaling that
mediates these molecular events leading to mitochondrial
dysfunction is poorly understood.
Interferon regulatory factor-1 (IRF-1) is an
important nuclear transcription factor with various biological
functions, including the promotion of systemic inflammatory
responses and apoptosis, the regulation of the development and
differentiation of immune cells, and the inhibition of cell
proliferation (8). IRF-1 was
first recognized for regulating type I interferon production, and
it has been indicated that IRF-1 can promote the transcription of
diverse inflammatory factors, such as interleukin (IL)-6, IL-12 and
IL-18 (9). IRF-1 has also been
shown to be involved in LPS-induced multiple organ damage and
death. IRF-1 can also promote the release of numerous inflammatory
factors, including the early inflammatory cytokines tumor necrosis
factor-α (TNF-α) and interferon-γ (IFN-γ), as well as the late
inflammatory danger signal, high mobility group box 1 (HMGB1)
(10,11). We have previously demonstrated
that IRF-1 inhibited the autophagy responses of immune cells, such
as macrophages and splenocytes (12,13). Since autophagy is important for
mitochondrial quality control and the maintenance of mitochondrial
homeostasis (14–16), we hypothesized that IRF-1
activation may disrupt mitochondrial homeostasis.
IRF-1 activation may also directly damage
mitochondria in both primary cells and cancer cell lines.
Mitochondrial membrane permeability increases and cytochrome
c is released in gastric cancer cells overexpressing IRF-1
(17). IRF-1 gene knockout can
alleviate LPS/D-GalN-induced hepatocellular mitochondrial damage
and reduce intrinsic apoptosis (18). However, whether IRF-1 participates
in LPS-induced macrophage mitochondrial damage and oxidative stress
remains unknown. We previously reported that IRF-1-mediated immune
cell apoptosis and autophagy plays an important role in LPS-induced
multiple organ failure and death (12,13). In this study, we report a novel
role for IRF-1 in LPS-induced oxidative stress responses and
mitochondrial structural damage in macrophages.
Materials and methods
Animals
IRF-1 knockout (KO; n=48) and matched C57BL/6J
wild-type (WT; n=48) (8–10 weeks of age, male, 25–30 g) mice were
purchased from the Jackson Laboratory (Bar Harbor, ME, USA).
Animals were maintained in a specific pathogen-free, laminar-flow
housing apparatus under controlled temperature, humidity and a 12 h
light/dark regimen. The Animal Care and Use Committee of the
Central South University approved all animal protocols. All
experiments were conducted in accordance with the National
Institutes of Health Guidelines for the Care and Use of Laboratory
Animals.
In vivo experimental design
The mice were randomly assigned to 4 groups (n=8 in
each) as follows: WT + phosphate-buffered saline (PBS), WT + LPS,
IRF-1 KO + PBS and IRF-1 KO + LPS groups. The mice in the LPS
groups were administered LPS (Escherichia coli 0111:B4)
(Sigma-Aldrich, St. Louis, MO, USA) (20 mg/kg, i.p.). The mice in
the PBS groups received treatment with sterilized PBS. At 16 h
after the administration of PBS or LPS, the mice were anesthetized
with chloral hydrate (400 mg/kg). Blood samples and peritoneal
macrophages were collected and the mice were sacrificed.
Isolation of peritoneal macrophages
Mouse abdomens were washed with 70% ethanol and a
lateral incision was made with scissors along the bottom midline of
the peritoneum. With forceps, abdominal skin was retracted to
expose the transparent peritoneal skin. Subsequently, 5 ml syringes
were attached to 20 G needles and 3 ml of cold RPMI-1640 cell
culture medium was injected into the peritoneal cavity of each
mouse. The peritoneal cavity was massaged and peritoneal fluid was
carefully aspirated and placed into 15 ml centrifuge tubes
(19). This was repeated for 3
treatments and the samples were centrifuged for 10 min at 300 × g.
The supernatant was discarded and the cell pellet was resuspended
in RPMI-1640 cell culture medium.
Cell culture and treatment
Murine monocyte/macrophage-like cells (RAW264.7;
106 cells; American Type Culture Collection, Manassas,
VA, USA) were cultured in RPMI-1640 cell culture medium,
supplemented with 10% FBS, 50 U/ml penicillin, and 50 µg/ml
streptomycin (Gibco, Grand Island, NY, USA) in 6 cm culture plates.
The cells were stimulated with LPS at various concentrations (0,
10, 100, 500 and 1,000 ng/ml) for different periods of time (0, 1,
2, 4, 8 and 16 h).
Stable transfection of RAW264.7 cells for
overexpression or knockdown of IRF-1
Lentiviral constructs expressing shRNA directed
against mouse IRF-1 mRNA were custom-manufactured (Hanyin,
Shanghai, China). shRNA sequences were as follows: IRF-1 shRNA,
5′-GCACTAAATGAGTCCTATTCC-3′, and non-specific shRNA,
5′-TTCTCCGAACGTGTCACGT-3′. The lentiviral constructs expressing
mouse IRF-1 were also manufactured by the same vendor (Hanyin).
IRF-1 cDNA was obtained from a commercially available source
(National Center for Biotechnology Information, Bethesda, MD, USA).
All lentiviral vectors were purified to a titer of 1×109
TU/ml. The constructs were transfected into the cells at a
multiplicity of infection (MOI) of 20:1 using 5 µg/ml
Polybrene in RPMI-1640 cell culture medium. At 4 h after
transfection, the medium was exchanged, and the cells were cultured
for an additional 48 h. The effects of IRF-1 knockdown and
overexpression were measured by western blot analysis.
Nuclear protein extraction and western
blot analysis
The collected cells were centrifuged at 300 × g for
5 min at 4°C The pellets were then resuspended on ice with
cytoplasmic extraction reagent (Vazyme, Nanjing, China) combined
with protease inhibitor and mixed for 10 min, and centrifuged at
16,000 × g for 5 min at 4°C to obtain protein. For nuclear protein
isolation, the cell pellet was lysed with nuclear extraction
reagent (Vazyme) plus protease inhibitor mix.
Protein concentrations were determined by a BCA
protein assay kit (Beyotime, Haimen, China), and 40 mg protein per
sample were mixed with sample loading buffer and boiled at 96°C for
5 min. The samples were then separated using sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred onto polyvinylidene fluoride (PVDF) membranes
(Millipore, Billerica, MA, USA). After blocking with 5% non-fat
milk in 10 mM Tris-buffered saline (pH 7.5) with 0.1% Tween-20
(TBS-T) for 1 h at room temperature, the membranes were incubated
overnight at 4°C with primary antibodies against rabbit monoclonal
antibody of IRF-1 (1:1,000; Cat. no. 8478) and Histone H3 (1:2,000;
Cat. no. 4499) (Cell Signaling Technology, Beverly, MA, USA). After
3 washes in TBS-T, the membranes were incubated with secondary
antibody conjugated with horseradish peroxidase (HRP) at room
temperature for 1 h. After 3 washes, the blots were developed with
electrochemiluminescence (ECL) (Beyotime) and visualized with a
ChemiDoc MP imaging system (Bio-Rad Laboratories, Berkeley, CA,
USA). Band density was quantified using ImageJ software version
1.49 (National Institutes of Health, Bethesda, MD, USA).
Detection of mitochondrial transmembrane
potential and reactive oxygen species (ROS)
Mitochondrial transmembrane potential was assessed
using the sensitive fluorescent probe,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-benzimidazolylcarbocy-anine
iodide (JC-1) (Invitrogen, Carlsbad, CA, USA). Red emission from
the dye is attributed to a potential-dependent aggregation of JC-1
in the mitochondria. Green fluorescence reflects the monomeric form
of JC-1, appearing in the cytoplasm after mitochondrial membrane
depolarization. The cells were seeded at 3×105
cells/well into 6-well plates and incubated overnight at 37°C in a
carbon dioxide incubator. The cells were then washed with PBS and
trypsinized, resuspended in 2 mM of JC-1 at 37°C for 30 min. The
cells were washed 3 times with PBS, resuspended in 500 µl
PBS, and then analyzed immediately with a BD FACSCanto II flow
cytometer (BD Biosciences, San Jose, CA, USA).
The collected cells were resuspended in 5 µM
2,7-dichlorofluorescin diacetate (DCFH-DA) (Sigma-Aldrich, St.
Louis, MO, USA) staining solution at 37°C for 25 min. Cell
suspensions were mixed by inversion every 5 min, making full
contact with the probe. The cells were then washed 3 times with PBS
to remove DCFH-DA that did not enter the cell. Fluorescence
intensity was measured by flow cytometry, as previously described
(20).
Transmission electron microscopy
(TEM)
The cells were fixed in cold 2.5% glutaraldehyde in
PBS and post-fixed in 1% osmium tetroxide with 0.1% potassium
ferricyanide, dehydrated through a graded series of ethanol
(30–90%) and embedded in Epon. The Epon blocks were then sectioned.
Ultrathin sections (65 nm) were stained with 2% uranyl acetate and
Reynolds lead citrate. The sections were examined under a H-7500
TEM (Hitachi, Tokyo, Japan) by an electron microscopy specialist
from the Electron Microscopy Laboratory of Central South
University.
Biochemical analysis of intracellular
ATP, superoxide dismutase (SOD) and malondialdehyde (MDA)
The cells were seeded at 3×105 cells/well
into 6-well plates and incubated overnight at 37°C in a carbon
dioxide incubator and stimulated with LPS (500 ng/ml, 16 h). The
cells were then washed with PBS and trypsinized, resuspended in PBS
and lysed on ice by sonication (Sonics Inc., Qingdao, China) at
100% amplitude (10×, 30 sec each with 30 sec intervals).
Biochemical analysis of MDA, SOD and ATP was carried out using
respective kits (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China) following the manufacture's instructions.
Purification of circulating DNA and
quantitative PCR (qPCR)
Circulating DNA was isolated from plasma with an
EZNA circulating DNA kit (Omega Bio-Tek, Norcross, GA, USA)
following manufacturer's instructions. The concentrations of DNA
were assessed using a NanoDrop 2000 spectrophotometer (Thermo
Fisher Scientific, Waltham, MA, USA). Ratios of A260/A280 ranged
from 1.9–2.1.
Relative mitochondrial DNA was measured by qPCR
using SYBR-Green PCR Master Mix (GeneCopoeia, Guangzhou, China).
Reactions were carried out in a 20 µl final volume,
containing 0.2 µM of each forward and reverse primer, 20 ng
DNA sample, and 10 µl SYBR-Green PCR Master Mix.
Amplification was performed with an Applied Biosystems 7300
Real-Time PCR machine (Life Technologies, Carlsbad, CA, USA) under
the thermal profile of 95°C for 10 min followed by 40 cycles at
95°C for 15 sec, 60°C for 20 sec and 72°C for 20 sec. Samples that
produced no PCR products after 40 cycles were considered
'undetectable'. The threshold cycle (Ct) was obtained from
triplicate samples and averaged.
mtDNA copies were calculated based on the 'ΔΔCt'
method, using the equation R (ratio) = 2−ΔΔCt,
cytochrome c oxidase 1 (mtCOI) DNA standardized by the
housekeeping gene, 18s RNA (encoding 18S ribosomal RNA). The
following primers were used: mtCOI forward,
5′-GCCCCAGATATAGCATTCCC-3′; and reverse, 5′-GTTCATCCTGTTCCTGCTCC-3′
and 18S RNA forward, 5′-TAGAGGGACAAGTGGCGTTC-3′; and reverse,
5′-CGCTGAGCCAGTCAGTGT-3′ (21).
Sequence data were analyzed using Blast nucleic acid database
searches from the National Centre for Biotechnology Information,
and had no significant homology with DNA found in any bacterial
species.
Statistical analyses
All data are expressed as the means ± SEM.
Significant differences within groups were analyzed with repeated
measures ANOVA followed by an LSD test and significant differences
between groups were assessed with one-way ANOVA (a value of
p<0.05 was considered to indicate a statistically significant
difference). All calculations and statistical analyses were
performed using SPSS software for Windows (version 17.0).
Results
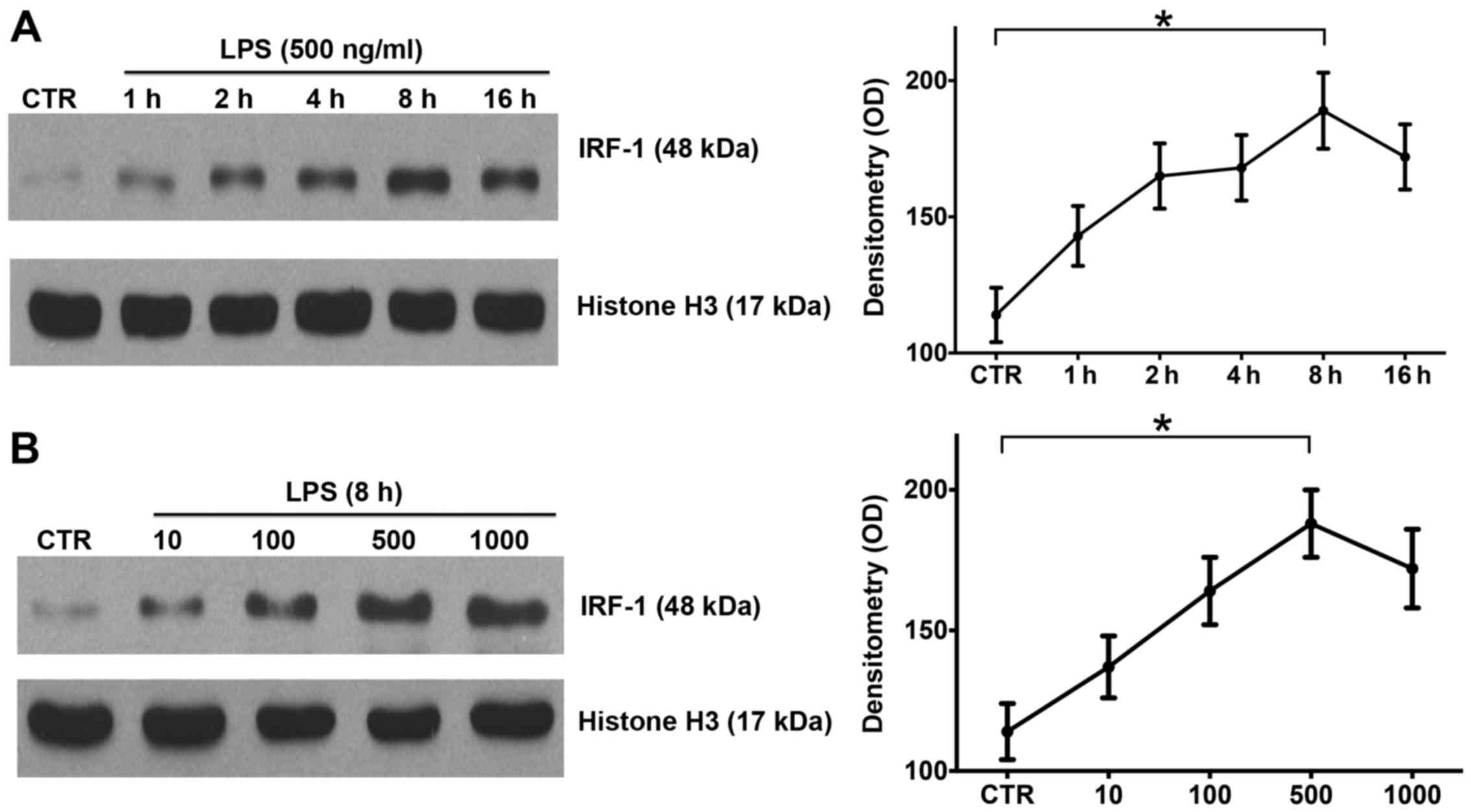
LPS induces IRF-1 activation in a time-
and dose-dependent manner in RAW264.7 cells
IRF-1 is a key regulator of immunity and plays an
important role in the progression of endotoxemia (9). In order to investigate the
expression pattern of IRF-1, the RAW264.7 cells were stimulated
with LPS at various concentrations and durations as described in
the Materials and methods. Western blot analysis confirmed that
IRF-1 nuclear protein peaked at 8 h after LPS stimulation (Fig. 1A), and LPS induced the peak
activation of nuclear IRF-1 at 500 ng/ml (Fig. 1B). Thus, LPS induced IRF-1
activation in the RAW264.7 cells in a time- and dose-dependent
manner.
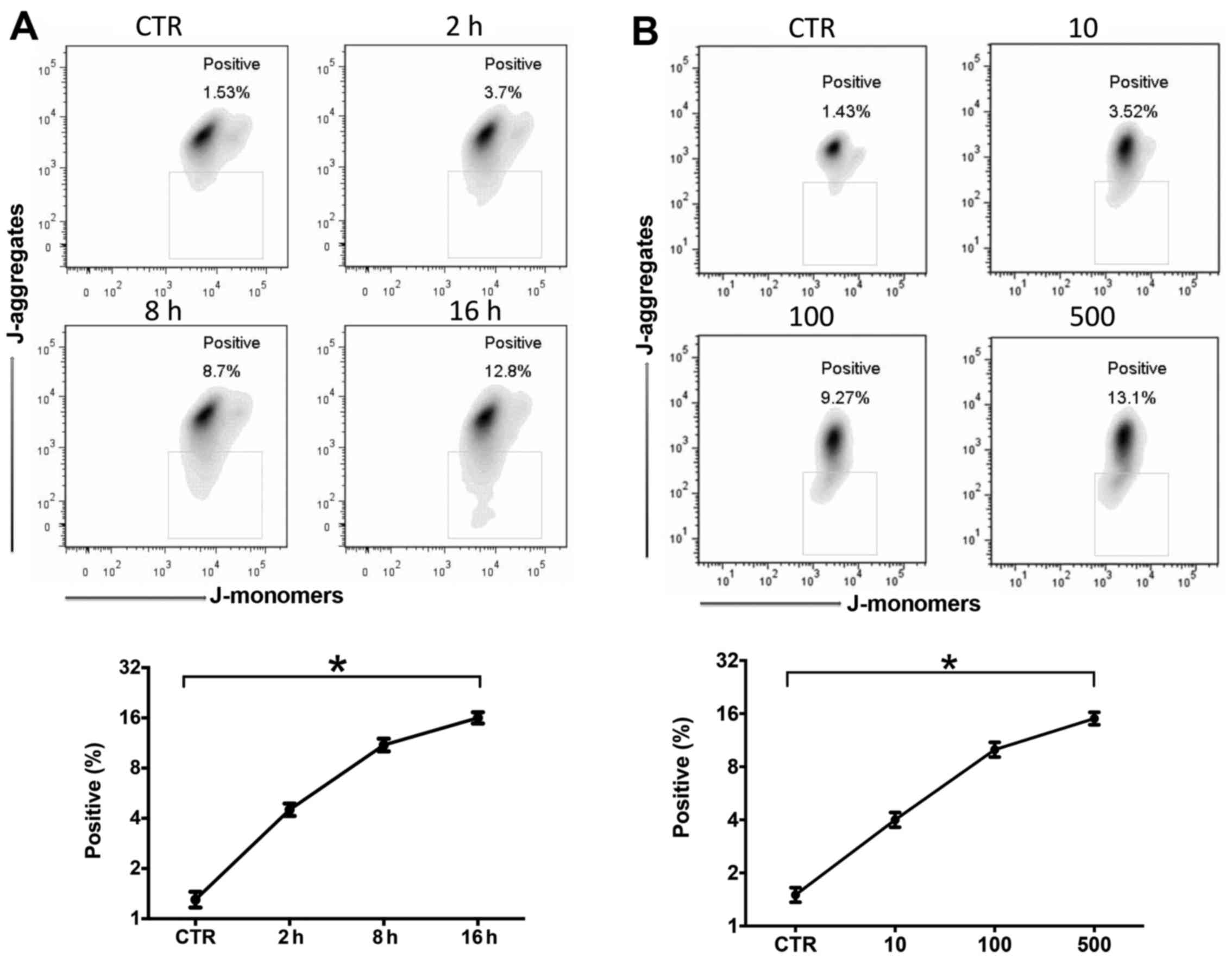
LPS induces mitochondrial damage and ROS
production was time- and dose-dependent
It has been clearly described that mitochondrial
damage and ROS production are very important in the pathophysiology
of sepsis (22). In this study,
we stimulated the RAW264.7 cells with LPS (10–500 ng/ml) for 2–16
h. Mitochondrial membrane depolarization and ROS production were
measured by JC-1 or DCFH-DA flow cytometry. Mitochondrial
depolarization increased after LPS stimulation in a time- (Fig. 2A) and dose-dependent (Fig. 2B) manner. LPS also induced ROS
production in a time- (Fig. 2C)
and dose-dependent (Fig. 2D)
manner.
LPS induces ATP depletion, SOD
consumption and MDA accumulation in RAW264.7 cells
As previously indicated, LPS-induced oxidative
stress responses are associated with apoptosis, energy metabolism
fluctuations and systemic inflammation responses, all of which
contribute to septic shock and multiple organ failure (5). In this study, we found that LPS
stimulation induced ATP depletion (Fig. 3A), SOD consumption (Fig. 3B) and MDA accumulation (Fig. 3C) in the RAW264.7 cells in both a
time- and dose-dependent manner.
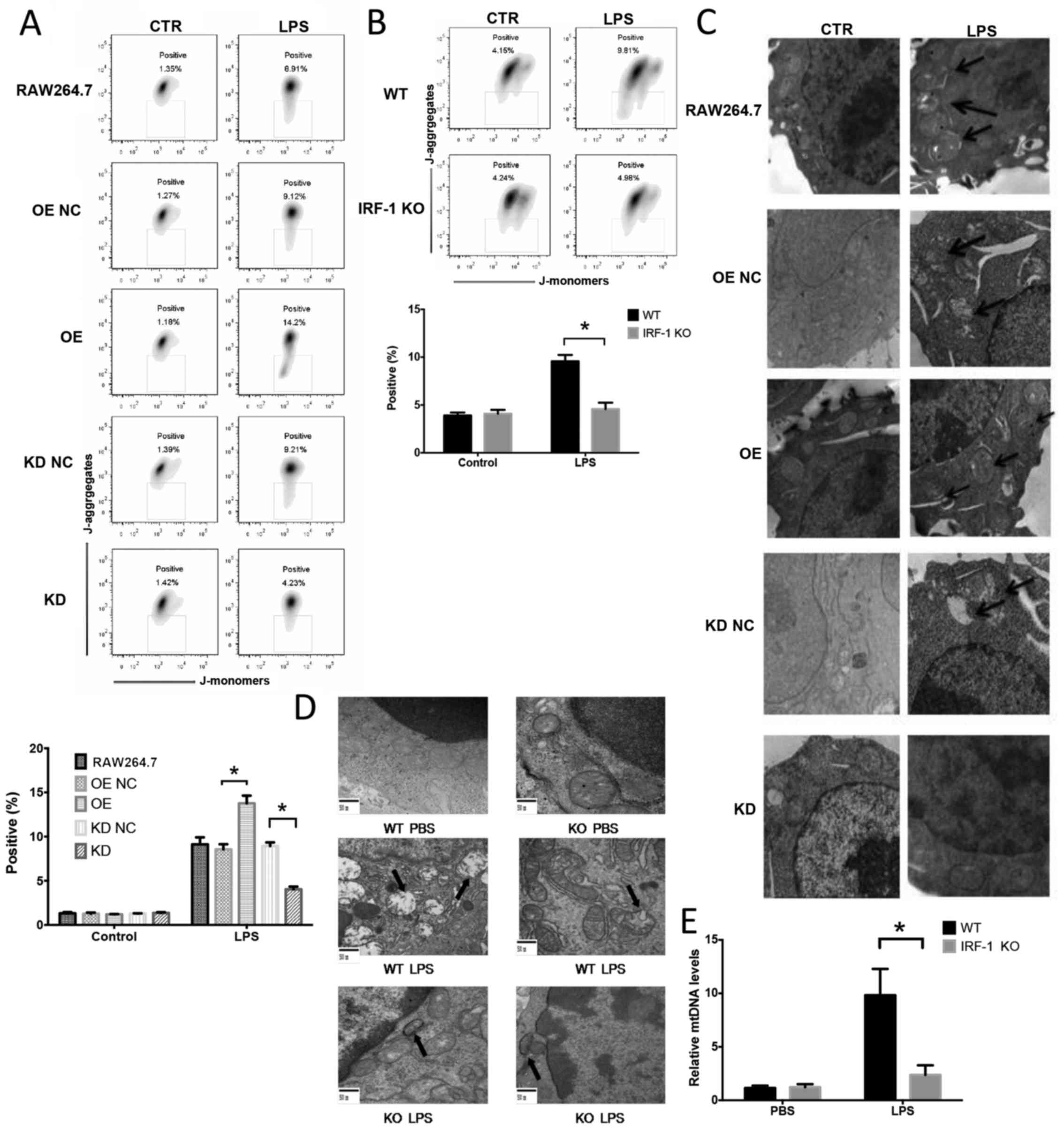
IRF-1 leads to mitochondrial damage both
in vivo and in vitro
To determine the role of IRF-1 in LPS-induced
mitochondrial damage and ROS production in macrophages, the effects
of the stable overexpression or knockdown of IRF-1 in murine
RAW264.7 macrophage cells were assessed by western blot analysis
(data not shown). IRF-1 KO and WT mice were administered LPS (20
mg/kg) and peritoneal macrophages were evaluated for mitochondrial
depolarization by flow cytometry, mitochondrial structure damage by
TEM and plasma mitochondrial DNA by qPCR. Mitochondrial
depolarization was exacerbated in the cells overexpressing IRF-1
(Fig. 4A) and was attenuated in
the cells in which IRF-1 was knocked down (Fig. 4A). Following the LPS injection,
peritoneal macrophages from the IRF-1 KO mice demonstrated
decreased mitochondrial depolarization (Fig. 4B). In addition, mitochondrial
structural damage was exacerbated in IRF-1-overexpressing cells
(Fig. 4C), and the damage was
attenuated in the cells in which IRF-1 was knocked down (Fig. 4C). The LPS-induced mitochondrial
structural damage in the peritoneal macrophages was also diminished
in the IRF-1 KO mice (Fig. 4D).
Furthermore, IRF-1 KO significantly reduced circulating
mitochondrial DNA release in vivo (Fig. 4E).
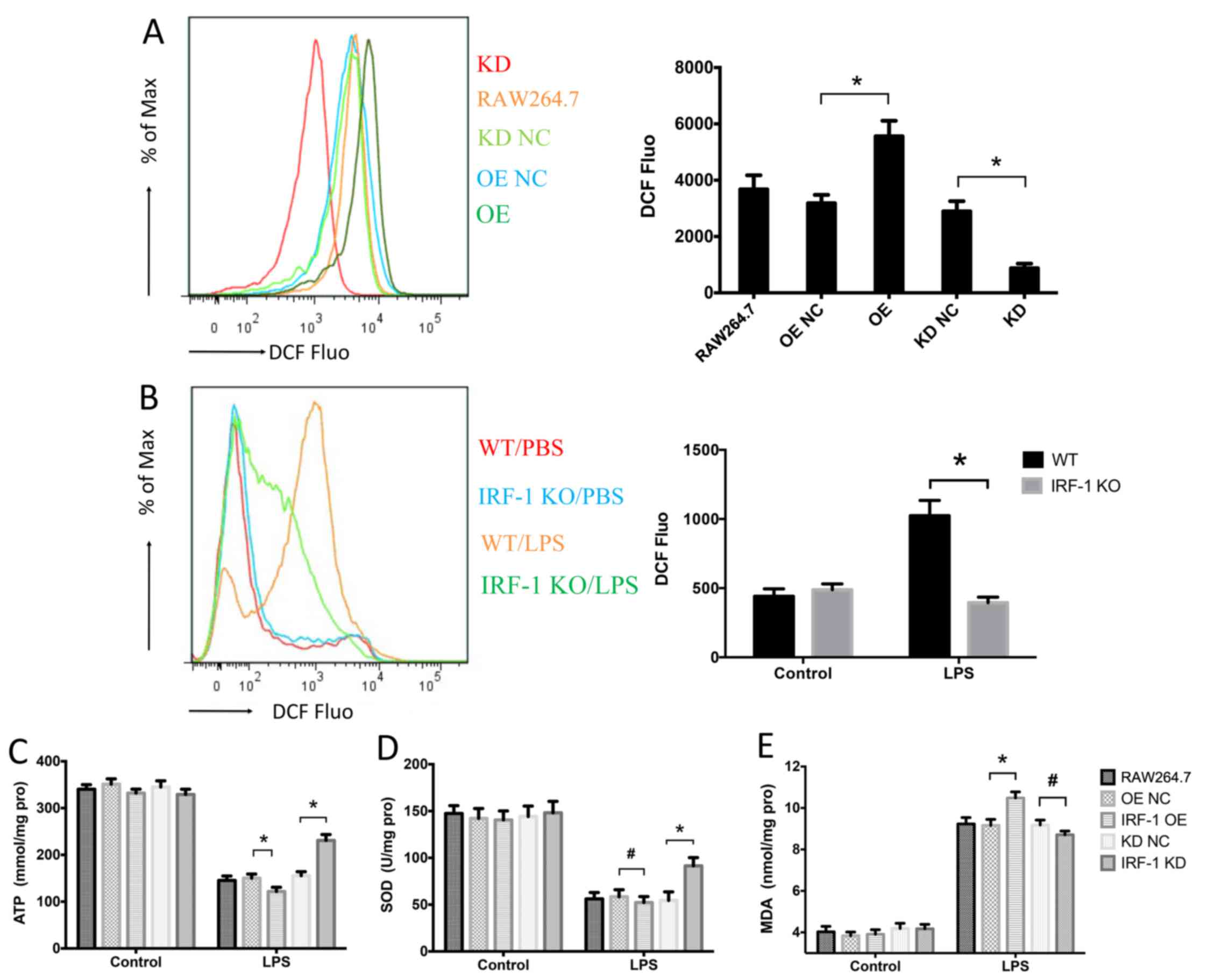
IRF-1 leads to oxidative stress in
macrophages
Since oxidative stress is a crucial component of
sepsis (22), we assessed the
role of IRF-1 in LPS-induced oxidative stress by measuring the
levels of ROS, ATP, SOD and MDA. ROS production was increased in
the IRF-1-overexpressing cells and was reduced in the cells which
IRF-1 was knocked down (Fig. 5A).
In addition, LPS-induced ROS production in the peritoneal
macrophages was significantly decreased in the IRF-1 KO mice
(Fig. 5B). IRF-1 overexpression
further enhanced ATP depletion, while IRF-1 knockdown attenuated
ATP depletion (Fig. 5C).
Furthermore, although SOD consumption was reduced in the cells in
which IRF-1 was knocked down, there was no apparent difference in
the IRF-1-overexpressing cells (Fig.
5D). By contrast, IRF-1 overexpression increased the
LPS-induced MDA accumulation; however, the difference in the cells
in which IRF-1 was knocked down was not significant (Fig. 5E).
Discussion
Despite improvements in antibiotic therapies and
critical care techniques, sepsis remains a fatal syndrome due to
the lack of effective and targeted interventions. The
life-threatening effects of sepsis involve excessive formation of
inflammatory cytokines, ROS and reactive nitrogen species (RNS),
which lead to oxidative stress responses, mitochondrial dysfunction
and organ failure (2,23). Since the mitochondria regulate
vital cellular functions, including energy production, ROS and RNS
generation, intrinsic apoptosis, stress and metabolic signals
transduction, they are important to the pathogenesis of sepsis
(24–26). Thus, attenuating mitochondrial
damage and oxidative stress responses may be promising therapeutic
targets for the treatment of sepsis.
Mitochondrial structural damage and dysfunction has
been recognized as an important molecular pathology in sepsis and
is linked to the severity of organ dysfunction and outcome of
sepsis (27). Peripheral blood
mononuclear cells in septic patients have significant mitochondrial
damage, and this is associated with the abundance of inflammatory
cytokines. For patients with severe sepsis, mitochondrial
functional deficiency is associated with clinical outcomes
(28). Macrophage mitochondrial
damage is involved in the pathogenesis of sepsis through several
mechanisms (5). Specifically,
mitochondrial damage-induced intrinsic apoptotic pathways cause
macrophage apoptosis, leading to immunosuppression (29). In addition, damaged mitochondria
release mtDNA, which join forces with ROS, and also activates NALP3
IL-1 and IL-18, leading to caspase-1 processing and maturation in
macrophages (30,31). Thus, the upstream signaling that
mediates macrophage mitochondrial dysfunction may be a potential
therapeutic target.
Mitochondria are membrane-bound organelles that
maintain cellular energy production through oxidative
phosphorylation. As the mitochondria are prone to damage during
stress, mitochondrial dynamics are tightly controlled to maintain
homeostasis (32–34). Autophagy, through the
identification and removal of damaged mitochondria, is an important
mechanism of mitochondrial quality control to maintain homeostasis.
Defects in autophagy lead to the delayed removal and heightened
accumulation of damaged mitochondria in the cytoplasm, which then
triggers oxidative stress, inflammation, apoptosis and other
reactions (32,33). The presence of IRF-1 has been
shown to be associated with less autophagy in both splenocytes and
macrophages (12,13).
In this study, we reported that LPS promoted IRF-1
activation in macrophages in both a time-and dose-dependent manner.
IRF-1 overexpression can lead to mitochondrial structural damage
and oxidative stress responses, whereas IRF-1 knockdown or knockout
is associated with less mitochondrial damage and oxidative stress,
which may be explained by decreased autophagic responses and
increased mitochondrial damage triggering intrinsic macrophage
apoptosis.
The mechanisms underlying macrophage mitochondrial
damage in during sepsis involve defects in energy production,
apoptosis and the induction of inflammation (5). Increased mitochondrial ROS during
sepsis can inhibit oxidative phosphorylation and ATP generation,
which unbalance energy metabolism and cause systemic inflammation
and multiple organ failure (22,35). ROS can subsequently impair
mitochondrial structures and halt mitochondrial biogenesis
(36–38). These events form a feedback loop,
such that targeting IRF-1 may inhibit macrophage mitochondrial
damage and ROS production, alleviate oxidative stress, and in turn
improve endotoxemia outcomes.
A recent study implicated the mitochondria in the
regulation of inflammation and mitochondrial derived
danger-associated molecular patterns (DAMPs), including ROS, mtDNA,
N-formyl peptides and cytochrome c (39). During sepsis, damaged
mitochondrial release mitochondrial DNA, a type of DAMP, through
the TLR9 receptor, activating the MAPK signal transduction pathway.
Following the tail vein injection of LPS, mitochondrial fragments
containing mtDNA can be found to directly cause systemic
inflammatory responses, acute lung injury, and neutrophil
infiltration in the liver and kidney of the mice (39,40). In this study, we confirmed that
circulating mtDNA release was decreased in IRF-1 KO mice; however,
the role of mtDNA in IRF-1-induced systemic inflammatory responses
remains unclear. Although the hallmarks of mitochondrial
dysfunction, including oxidative stress and impaired ATP
production, are evident in macrophages during endotoxemia, whether
these processes cause or are a result of systemic inflammation is
unknown. To study mitochondrial function in the setting of
overwhelming systemic inflammation during endotoxemia may elucidate
these specifics. We confirmed that IRF-1 is important to
LPS-induced mitochondrial structural damage and oxidative stress
response in macrophages and suggest that IRF-1 may be a potential
therapeutic target in the treatment of endotoxemia.
Abbreviations:
|
LPS
|
lipopolysaccharide
|
|
IRF-1
|
interferon regulatory factor-1
|
|
mtDNA
|
mitochondrial DNA
|
|
WT
|
wild-type
|
|
KO
|
knockout
|
|
KD
|
knockdown
|
|
PBS
|
phosphate-buffered saline
|
|
IP
|
intraperitoneal
|
|
ROS
|
reactive oxygen species
|
|
SOD
|
superoxide dismutase
|
|
MDA
|
malondialdehyde
|
|
ATP
|
adenosine triphosphate
|
|
TEM
|
transmission electron microscope
|
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81401631) and the Health and
Family Planning Commission of Hunan Province (no. 14JJ7010).
References
|
1
|
Aneja R and Fink MP: Promising therapeutic
agents for sepsis. Trends Microbiol. 15:31–37. 2007. View Article : Google Scholar
|
|
2
|
Stearns-Kurosawa DJ, Osuchowski MF,
Valentine C, Kurosawa S and Remick DG: The pathogenesis of sepsis.
Annu Rev Pathol. 6:19–48. 2011. View Article : Google Scholar
|
|
3
|
Cheng B, Xie G, Yao S, Wu X, Guo Q, Gu M,
Fang Q, Xu Q, Wang D, Jin Y, et al: Epidemiology of severe sepsis
in critically ill surgical patients in ten university hospitals in
China. Crit Care Med. 35:2538–2546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park BS, Song DH, Kim HM, Choi BS, Lee H
and Lee JO: The structural basis of lipopolysaccharide recognition
by the TLR4-MD-2 complex. Nature. 458:1191–1195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singer M: The role of mitochondrial
dysfunction in sepsis-induced multi-organ failure. Virulence.
5:66–72. 2014. View Article : Google Scholar :
|
|
6
|
Mège JL, Mehraj V and Capo C: Macrophage
polarization and bacterial infections. Curr Opin Infect Dis.
24:230–234. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garrabou G, Morén C, López S, Tobías E,
Cardellach F, Miró O and Casademont J: The effects of sepsis on
mitochondria. J Infect Dis. 205:392–400. 2012. View Article : Google Scholar
|
|
8
|
Chen W and Royer WE Jr: Structural
insights into interferon regulatory factor activation. Cell Signal.
22:883–887. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tamura T, Yanai H, Savitsky D and
Taniguchi T: The IRF family transcription factors in immunity and
oncogenesis. Annu Rev Immunol. 26:535–584. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pan PH, Cardinal J, Li ML, Hu CP and Tsung
A: Interferon regulatory factor-1 mediates the release of high
mobility group box-1 in endotoxemia in mice. Chin Med J (Engl).
126:918–924. 2013.
|
|
11
|
Senaldi G, Shaklee CL, Guo J, Martin L,
Boone T, Mak TW and Ulich TR: Protection against the mortality
associated with disease models mediated by TNF and IFN-gamma in
mice lacking IFN regulatory factor-1. J Immunol. 163:6820–6826.
1999.PubMed/NCBI
|
|
12
|
Zhang L, Cardinal JS, Bahar R, Evankovich
J, Huang H, Nace G, Billiar TR, Rosengart MR, Pan P and Tsung A:
Interferon regulatory factor-1 regulates the autophagic response in
LPS-stimulated macrophages through nitric oxide. Mol Med.
18:201–208. 2012. View Article : Google Scholar :
|
|
13
|
Zhang L, Cardinal JS, Pan P, Rosborough
BR, Chang Y, Yan W, Huang H, Billiar TR, Rosengart MR and Tsung A:
Splenocyte apoptosis and autophagy is mediated by interferon
regulatory factor 1 during murine endotoxemia. Shock. 37:511–517.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen F, Chen B, Xiao FQ, Wu YT, Wang RH,
Sun ZW, Fu GS, Mou Y, Tao W, Hu XS, et al: Autophagy protects
against senescence and apoptosis via the RAS-mitochondria in
high-glucose-induced endothelial cells. Cell Physiol Biochem.
33:1058–1074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen S, Huang J, Zeng Q, Jia Y and Wang J:
Effect of autophagy and mitochondrial coenzyme Q on exocrine
function of pancreas in rats with acute sepsis. Zhonghua Wei Zhong
Bing Ji Jiu Yi Xue. 27:86–91. 2015.In Chinese. PubMed/NCBI
|
|
16
|
Su Y, Qu Y, Zhao F, Li H, Mu D and Li X:
Regulation of autophagy by the nuclear factor κB signaling pathway
in the hippocampus of rats with sepsis. J Neuroinflammation.
12:1162015. View Article : Google Scholar
|
|
17
|
Gao J, Senthil M, Ren B, Yan J, Xing Q, Yu
J, Zhang L and Yim JH: IRF-1 transcriptionally upregulates PUMA,
which mediates the mitochondrial apoptotic pathway in IRF-1-induced
apoptosis in cancer cells. Cell Death Differ. 17:699–709. 2010.
View Article : Google Scholar
|
|
18
|
Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY,
Park YS, Kwon Y, Choi KH, Jo I, Park SI, et al: The role of
STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial
transmembrane potential during hepatic cell death induced by
LPS/d-GalN. J Mol Biol. 369:967–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Layoun A, Samba M and Santos MM: Isolation
of murine peritoneal macrophages to carry out gene expression
analysis upon Toll-like receptors stimulation. J Vis Exp.
98:e527492015.
|
|
20
|
Carchman EH, Rao J, Loughran PA, Rosengart
MR and Zuckerbraun BS: Heme oxygenase-1-mediated autophagy protects
against hepatocyte cell death and hepatic injury from
infection/sepsis in mice. Hepatology. 53:2053–2062. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng G, Lyu J, Liu S, Huang J, Liu C,
Xiang D, Xie M and Zeng Q: Silencing of uncoupling protein 2 by
small interfering RNA aggravates mitochondrial dysfunction in
cardiomyocytes under septic conditions. Int J Mol Med.
35:1525–1536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Galley HF: Oxidative stress and
mitochondrial dysfunction in sepsis. Br J Anaesth. 107:57–64. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Víctor VM, Espulgues JV, Hernández-Mijares
A and Rocha M: Oxidative stress and mitochondrial dysfunction in
sepsis: A potential therapy with mitochondria-targeted
antioxidants. Infect Disord Drug Targets. 9:376–389. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arulkumaran N, Deutschman CS, Pinsky MR,
Zuckerbraun B, Schumacker PT, Gomez H, Gomez A, Murray P and Kellum
JA; ADQI XIV Workgroup: Mitochondrial function in sepsis. Shock.
45:271–281. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Galluzzi L, Kepp O and Kroemer G:
Mitochondria: Master regulators of danger signalling. Nat Rev Mol
Cell Biol. 13:780–788. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tait SW and Green DR: Mitochondria and
cell signalling. J Cell Sci. 125:807–815. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Azevedo LC: Mitochondrial dysfunction
during sepsis. Endocr Metab Immune. Disord Drug Targets.
10:214–223. 2010.
|
|
28
|
Brealey D, Brand M, Hargreaves I, Heales
S, Land J, Smolenski R, Davies NA, Cooper CE and Singer M:
Association between mitochondrial dysfunction and severity and
outcome of septic shock. Lancet. 360:219–223. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chung CS, Song GY, Lomas J, Simms HH,
Chaudry IH and Ayala A: Inhibition of Fas/Fas ligand signaling
improves septic survival: Differential effects on macrophage
apoptotic and functional capacity. J Leukoc Biol. 74:344–351. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakahira K, Haspel JA, Rathinam VA, Lee
SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim
HP, et al: Autophagy proteins regulate innate immune responses by
inhibiting the release of mitochondrial DNA mediated by the NALP3
inflammasome. Nat Immunol. 12:222–230. 2011. View Article : Google Scholar
|
|
31
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar
|
|
32
|
Green DR, Galluzzi L and Kroemer G:
Mitochondria and the autophagy-inflammation-cell death axis in
organismal aging. Science. 333:1109–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Youle RJ and Narendra DP: Mechanisms of
mitophagy. Nat Rev Mol Cell Biol. 12:9–14. 2011. View Article : Google Scholar
|
|
35
|
Andrades M, Ritter C, de Oliveira MR,
Streck EL, Fonseca Moreira JC and Dal-Pizzol F: Antioxidant
treatment reverses organ failure in rat model of sepsis: Role of
antioxidant enzymes imbalance, neutrophil infiltration, and
oxidative stress. J Surg Res. 167:e307–e313. 2011. View Article : Google Scholar
|
|
36
|
Shokolenko I, Venediktova N, Bochkareva A,
Wilson GL and Alexeyev MF: Oxidative stress induces degradation of
mitochondrial DNA. Nucleic Acids Res. 37:2539–2548. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and mitochondrial DNA damage in heart failure.
Circ J. 72(Suppl A): A31–A37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Q, Itagaki K and Hauser CJ:
Mitochondrial DNA is released by shock and activates neutrophils
via p38 map kinase. Shock. 34:55–59. 2010. View Article : Google Scholar
|
|
39
|
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal
T, Junger W, Brohi K, Itagaki K and Hauser CJ: Circulating
mitochondrial DAMPs cause inflammatory responses to injury. Nature.
464:104–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang Z, Shi L, Song L, Ephrem E, Petri M
and Sullivan KE: Interferon regulatory factor 1 marks activated
genes and can induce target gene expression in systemic lupus
erythematosus. Arthritis Rheumatol. 67:785–796. 2015. View Article : Google Scholar :
|