Introduction
Stroke is a neural degenerative disease with high
rates of mortality and disability, which is characterized by
ischemic-hypoxic (IH) injury in neurons (1). Strokes are classified as ischemic
and hemorrhagic, with ~80% of cases being ischemic (2). Ischemic stroke is induced by a
sudden reduction of blood circulation, resulting in the cutoff of
oxygen to the brain and corresponding loss of neurological
function. Ischemic stroke can lead to permanent and transient IH
injuries, depending on crucial artery reperfusion within a limited
period of time, with permanent IH dominant in clinic cases. The
pathological processes of IH injury include hypoxia response,
postischemic inflammatory response, cell apoptosis and peri-infarct
depolarization (3–5).
Although stroke has attracted widespread attention
clinically, effectiveness of treatment is finite, as the mechanism
underlying IH injury remains to be fully elucidated. Previous
studies have revealed that autophagy is important in inflammation,
nutrient deprivation and the processes of IH. For example, studies
have indicated that autophagy is involved in IH injury affecting
neuronal survival following chronic and moderate hypoxia (6). However, considering the existence of
cell death during autophagic processes, how autophagy mediates
neuronal survival following IH injury remains a topic of debate,
particularly in permanent IH injury (7–11).
Autophagy involves a cascade of morphological and chemical
processes, with the involvement of several apoptotic proteins and
signaling transduction pathways, including Beclin 1 and
autophagy-related (Atg) proteins, and the Notch pathway. Previous
studies have shown that the Wnt signaling pathway is also involved
in autophagy (12–15), despite the Wnt signaling pathway
being involved primarily in the regulation of cell proliferation
and differentiation during development (16,17). In addition, previous studies have
shown that the Wnt signaling pathway is likely to be important in
the pathogenesis of neurodegenerative diseases, including IH, as
cytokines in the Wnt pathway can improve the microenvironment of
neuronal repair following post-ischemic injury (18). However, how the Wnt signaling
pathway regulates autophagy in permanent neuronal IH injury remains
to be fully elucidated, particularly the role of autophagic flux in
regulation.
In the present study, PC12 cells were used to
establish an oxygen-glucose deprivation (OGD) model to imitate
permanent IH injury. Following the induction of autophagy in the IH
model, the expression levels of hypoxia-inducible factor-α (HIF-α),
a key regulator in hypoxia, and cyclooxygenase 2 (COX2), an
inflammatory indicator, were investigated. The apoptotic rates of
cells and levels of lactate dehydrogenase (LDH) were also analyzed
(19), and the role of autophagy
during IH injury and the regulatory mechanism of the Wnt pathway in
autophagy were discussed. The data may provide novel insight for
treatment strategies following stroke.
Materials and methods
Cell culture and the model of OGD
Cell culture
As a neuronal alternative, PC12 cells, obtained from
the Institute of Cell Biology, Chinese Academy of Sciences
(Shanghai, China) were used in the present study as a neuronal
model. Following thawing, the PC12 cells on collagen-coated glass
were cultured in normal medium containing Dulbecco's modified
Eagle's medium (cat. no. 12800-017; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 5% fetal bovine serum (FBS;
Zhejiang Tiangeng Biotechnology, Zhejiang, China), 5%
heat-inactivated horse serum (cat. no. 26050070; Thermo Fisher
Scientific, Inc.), 50 U/ml penicillin and 50 µg/ml
streptomycin. The cells were cultured at 37°C with 5%
CO2 for 2 days, following which the cells were harvested
in the log phase with neuron-like morphology and at 70–80% cell
confluence.
OGD treatment
Initially, Biocoat plates were precoated with
poly-D-lysine (BD Biosciences, Bedford, MA, USA), and the PC12
cells were plated at the density of 1×104 cells/well in
the 24-well culture plate. The PC12 cells were grown for 12 h in
normal medium (DMEM with 5% heated horse serum and 5% FBS) at 37°C
with 5% CO2. For OGD treatment, the cells were rinsed
twice in DMEM without glucose (cat. no. 11966-025; Thermo Fisher
Scientific, Inc.), following which they were transferred into
modular incubator chambers (Billups-Rothenberg, Del Mar, CA, USA)
with OGD medium (glucose-free DMEM with 2% horse serum and 1% FBS).
The chambers were perfused with 95% N2 and 5%
CO2 for 30 min and 3 l/min speed at room temperature.
Finally, the chambers were sealed and cultured at 37°C for
designated durations (0.5, 1, 6, 12 and 24 h), respectively.
Cell grouping
In order to understand the effect of time on IH
injury and autophagy, the cells were divided into a control group,
in which cells were cultured in normal medium under normoxia, and
OGD groups, in which cells were cultured in OGD medium for 0.5 h
(OGD+0.5 h), 2 h (OGD+2 h), 6 h (OGD+6 h), 12 h (OGD+12 h) and 24 h
(OGD+24 h). The 0.5, 2 and 6 h time-points represented the
condition of transient IH, whereas the 12 and 24 h time-points
represented permanent IH. In the present study, autophagy was
examined at the various time-points under OGD conditions and
following chemical intervention (20), including exposure to rapamycin
(RAP; autophagy agonist), 3-methyladenine (3-MA; autophagy
antagonist) and MHY1485 (MHY; lysosome antagonist). In these
intervention groups, the cells were first cultured in normal medium
containing the respective intervention agents at 37°C for 2 h,
followed by continuous culture under OGD conditions for 6 h. The
intervention agents and their doses were as follows: 10 µM
rapamycin (RAP+OGD), 10 mM 3-MA (3-MA+OGD) and 2 µM MHY
(MHY+OGD).
Assays for neuronal injury
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The cells were seeded at a density of
1×l04 cells/ml in 96-well culture plates. After 24 h,
the cells were treated with the OGD treatment. Control wells
consisted of cells incubated with medium only. Following the
different durations of treatment, the cells were incubated with 20
µl MTT (5 mg/ml; Sigma; Merck Millipore, Darmstadt,
Germany). After 4 h at 37°C, the supernatant was removed and 150
µl dimethyl sulfoxide (DMSO) was added. The blue crystals
were dissolved in DMSO and the optical density (OD) was detected at
a wave-length of 570 nm using a 96-well multiscanner autoreader
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). The following
formula was used: Cell proliferation inhibition (%) = [1 − (OD of
experimental sample/OD of control)] × 100.
LDH analysis for necrosis
The cells were seeded at 1×106 cells in
6-well Biocoat plates. As necrotic cells can release LDH, the
quantity of LDH can be used to evaluate the condition of cell
death. Following the collection of culture medium and
centrifugation at the speed in 250 × g for 10min at 4°C, the
supernatant was obtained. Using the LDH assay kit (Sigma; Merck
Millipore), the supernatant was incubated with 100 µl
substrate buffer/well for 30 min at 37°C. Following centrifugation
at the speed in 250 × g for 10 min at room temperature, cellular
debris was removed, and LDH was assessed at 490 nm excitation. The
LDH value was calculated according to the following formula: LDH =
(sample OD value − control OD value)/(standard OD value − blank
control OD) × standard substance concentration × 1,000 U/l. The
percentage LDH was calculated to reflect the condition of cell
death. The percentage LDH was calculated as: sample LDH
value/control LDH value.
Western blot analysis
The proteins were extracted from the PC12 cells
using RIPA buffer, containing 20 mmol/l Tris-HCl (pH 7.5), 150
mmol/l NaCl, 1 mmol/LEDTA, 1% Triton X-100 (cat. no. T0694; Sangon
Biotech Co., Ltd., Beijing, China) and 1 mmol/l PMSF. Protein
concentrations were determined by a reducing agent-compatible BCA
assays kit (Pierce, Nepean, ON, Canada). The proteins (30
µg) were separated on 10 or 15% sodium dodecyl
sulfate-polyacryl-amide gel electrophoresis (SDS-PAGE) gels and
transferred onto a polyvinylidene difluoride membrane (EMD
Millipore, Billerica, MA, USA). Following blocking with 5% skim
milk in Tris-buffered saline, the membrane was incubated with
primary antibodies at 4°C overnight. The following primary
antibodies were used: Rabbit polyclonal anti-microtubule-associated
protein 1A/1B-light chain 3 (MAP1LC3A; 1:1,500; cat. no. ab52768),
mouse monoclonal anti-HIF-α (1:1,500, cat. no. ab16066) (both from
Abcam, Cambridge, UK), mouse monoclonal anti protein-COX2 (1:1,000;
cat. no. sc166475), goat polyclonal anti-Beclin 1 (1:1,000; cat.
no. sc10086) (both from Santa Cruz Biotechnology, Inc., Dallas, TX,
USA), mouse monoclonal anti-sequestosome (SQSTM1/p62; 1:1,000; cat.
no. ab56416), rabbit polyclonal anti-Wnt1 (1:1,000; cat. no.
ab15251), rabbit polyclonal anti-Wnt3a (1:1,500; cat. no. ab19925),
rabbit polyclonal anti-β-catenin (1:1,000; cat. no. ab6302) (all
from Abcam), mouse polyclonal anti-dishevelled segment polarity
protein 2 (Dvl2; 1:800; cat. no. 12037-1-AP; ProteinTech Group,
Inc., Chicago, IL, USA), mouse mono-clonal anti-cyclin D1 (A-12;
1:1,000; cat. no. sc8396; Santa Cruz Biotechnology, Inc.), rabbit
polyclonal anti-C-myc (1:800; cat. no. 10828-1-AP; ProteinTech
Group, Inc.). Following washing, secondary antibodies conjugated
with horseradish peroxidase were added. Anti-rabbit (1:4,000; cat.
no. ab6721), anti-mouse (1:4,000; cat. no. ab6728) or anti-goat
IgGs (1:4,000; cat. no. ab6741) (all from Abcam) were used for 1 h
at room temperature. Visualization of the immunoreactive bands was
visualized with an enhanced chemoluminescence detection kit
(Amersham; GE Healthcare Life Sciences, Piscataway, NJ, USA).
β-actin (1:6,000; cat. no. a2228; Sigma-Aldrich; Merck Millipore)
was used as an internal reference, and quantification of the
intensities (target protein/β-actin) of the immunoreactive bands
was performed using ImageJ 10.0 software (National Institutes of
Health, Bethesda, MA, USA).
Immunocytochemistry
The cells were fixed with 4% paraformaldehyde in
phosphate buffer, and rinsed with 0.01 M phosphate buffer,
following which nonspecific antigens were blocked with 10% normal
goat serum with 0.3% Triton X-100 and 1% BSA in 0.01 M phosphate
buffer for 30 min at room temperature. The cells were incubated
with primary antibodies at 4°C overnight. Following rinsing with
phosphate-buffered saline (PBS) three times, each time for 10 min,
the cells were incubated with secondary antibodies for 3 h at room
temperature. The primary antibodies were as follows: Rabbit
anti-microtubule-associated LC3 (1:200; cat. no. sc28266; Santa
Cruz Biotechnology, Inc.) polyclonal antibody, mouse anti-HIF-α
(1:200; cat. no. ab16066; Abcam) monoclonal antibody, and mouse
anti-COX2 (1:200; cat. no. sc166475; Santa Cruz Biotechnology,
Inc.) monoclonal antibody. The secondary antibodies used were Alexa
Fluor 488-conjugated anti-rabbit IgG (1:600; cat. no. a11034;
Invitrogen; Thermo Fisher Scientific, Inc.) and Alexa Fluor
568-conjugated anti-mouse IgG (1:300; cat. no. a10037; Invitrogen;
Thermo Fisher Scientific, Inc.). The cells were coverslipped with
65% glycerol in 0.01 M phosphate buffer with DAPI (1:2,000) for
counter-staining. Finally, images of the cells were captured with
an epifluorescence microscope (BX61; Olympus Corp., Tokyo, Japan)
under rhodamine, fluorescein isothiocyanate or ultraviolet
excitation. Images of high quality sections were captured under a
laser confocal microscope (FV1000; Olympus Corp.).
Measurements and statistical
tests
In order to understand the levels of cell apoptosis
and cell death, the levels of LDH and the apoptotic rates of the
cells were quantified: i) Autophagic rate (%) = LC3-positive
apoptotic cells/total cells; ii) levels of LDH were determined to
evaluate the presence of necrosis as LDH (%) = sample LDH
value/control LDH value; iii) using immunochemistry, fluorescent
intensity was used for measuring expression levels of proteins,
including COX2 and HIF-α, as fluorescent intensity = Σ positive
cells IOD/Σ cells.
Statistical analysis
All data were analyzed in a double-blinded manner.
Data are shown as the mean ± standard deviation. Comparisons were
made among various groups using one-way analysis of variance and
were analyzed using the SPSS statistical package (SPSS 13.0; IBM
SPSS, Armonk, NY, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
OGD and autophagic flux
In the present study, PC12 cell autophagy and
autophagic flux were investigated following OGD treatment. LC3 was
used to label the autophagosome in autophagic cells. In the control
group, it was difficult to identify LC3-positive puncta in the PC12
cells (Fig. 1A), however, in OGD
groups between 0.5 and 24 h, dense LC3-positive puncta were found
in the cytoplasm surrounding the nucleus. The expression levels of
proteins LC3-II, LC3-I, Beclin 1 and p62, which reflect autophagic
flux, were examined using western blot analysis (Fig. 1B–F). In the presence of autophagy,
LC3-I in the cytoplasm is converted to LC3-II, which moves to the
membrane of autophagosomes (21)
(Fig. 1B). In the present study,
the levels of LC3-II/I in the OGD groups were significantly
increased in a dose-dependent manner (P<0.05), compared with
those in the control group (Fig. 1B
and D). The protein expression levels of p62 and Beclin 1 were
also examined by western blot analysis. Compared with the control,
the expression of p62 was decreased, however, the expression of
Beclin 1 was increased and showed a similar trend in expression
with LC3 in the OGD groups, with time-dependency (Fig. 1B, C, E and F).
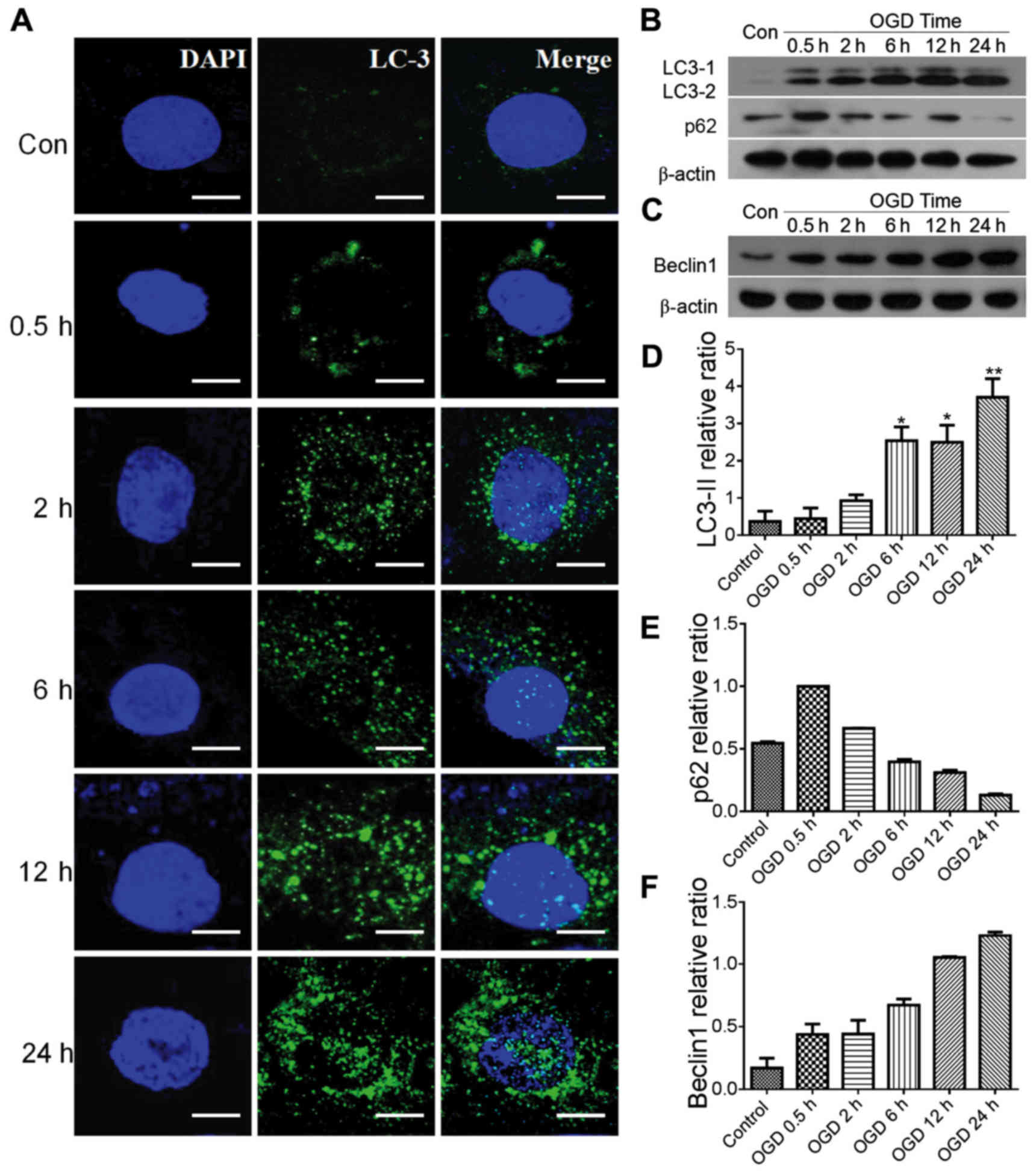 | Figure 1Autophagy in PC12 cells following IH.
(A) Autophagosomes (green) in PC12 cells at various OGD treatment
times (LC3 immunolabeling). IH induced autophagy in the PC12 cells.
The number of autophagosomes increased with OGD in a time-dependent
manner. Scale bar, 30 µm. Expression of (B) LC3-II, p62 and
(C) Beclin 1, determined using western blot analysis. The
expression levels of LC3-II and Beclin 1 increased in a
time-dependent manner, and the expression of p62 decreased in
time-dependent manner. Grayscale ratios of LC3-II, p62 and Beclin 1
(target protein/β-actin) are shown in histograms. (D) Expression of
LC3-II increased significantly from OGD 6 h in a time-dependent
manner, (E) expression of p62 decreased from OGD 0.5 h in a
time-dependent manner (F) Expression of Beclin 1 was similar to
that of LC3-II. OGD groups differed significantly from the control
with time-dependency. *P<0.05 and **P<0.01 vs.
control group. Data are expressed as the mean ± standard error of
the mean of four independent experiments. IH, ischemia-hypoxia;
OGD, oxygen-glucose deprivation; LC3, microtubule-associated
protein 1A/1B-light chain 3; Con, control. |
Autophagic intervention and function
during IH
Using western blot analysis and immunochemistry, the
present study revealed that the activation of autophagy was
controlled under an autophagic flux during IH. Subsequent
investigations were performed to examine the neural protective role
of autophagy during IH. This was examined using an agonist and
antagonist of autophagy, RAP and 3-MA, respectively, which were
added to the OGD group (OGD+6 h). The neural protection of
autophagy was evaluated through the expression levels of
inflammatory and stress response factors, HIF-α and COX2. As OGD
treatment for 6 h is considered the beginning of chronic IH injury,
it shows marked injury. Therefore, in the present study, OGD for 6
h was used for the intervention experiment to observe autophagy. In
the RAP+OGD group (Fig. 2A),
autophagy was enhanced, with numerous LC3-II-positive puncta, which
represented autophagosomes appearing in the cell soma, and the
expression levels of HIF-α and COX2 were decreased (Fig. 2). In contrast to the RAP+OGD
group, autophagy in the 3-MA+OGD group was significantly inhibited,
with an increase in the expression levels of HIF-α and COX2
(Fig. 2). The results of the
western blot analysis confirmed the results obtained from the
immunochemistry experiments (Fig.
3A–D).
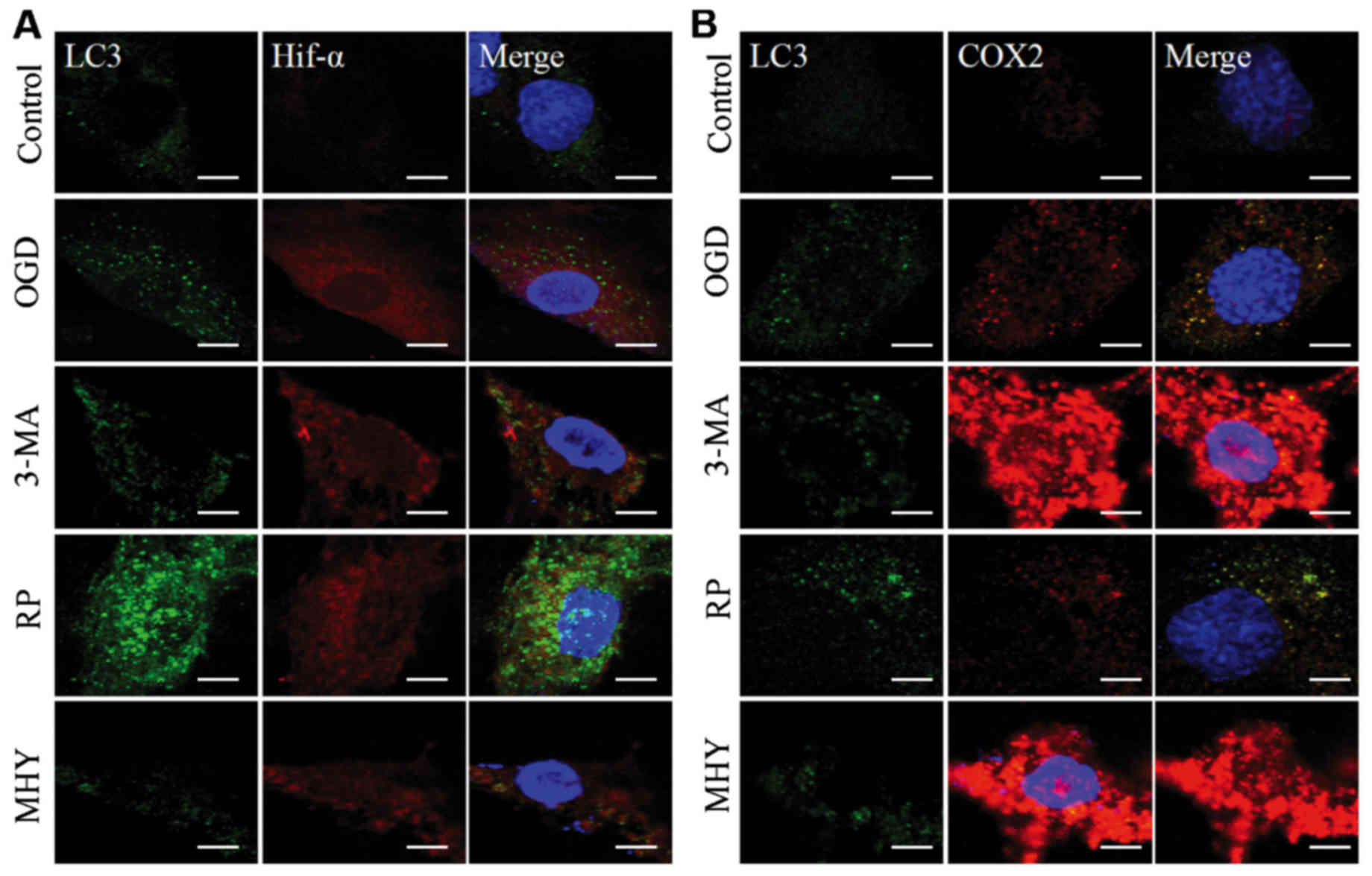 | Figure 2Chemical interventions of autophagy
and ischemic-hypoxic injury, assessed using immunochemical double
labeling. Compared with the control group, expression levels of
HIF-α and COX2 were altered by chemical intervention. (A) Chemical
intervention altered the expression of HIF-α and autophagosmes.
Following autophagy antagonist (3-MA) intervention, expression of
HIF-α was enhanced, compared with that in the OGD group, and was
decreased by the autophagy agonist, rapamycin. In the MHY treatment
group, expression of HIF-α was similar to that in the 3-MA group.
(B) Chemical intervention showed changes in the expression of COX2
and autophagosomes. Changes in the expression of COX2 were similar
to changes in HIF-α. Scale bar, 30 µm. OGD, oxygen-glucose
deprivation; HIF-α, hypoxia-inducible factor-α; COX2,
cyclooxygenase 2; 3-MA, 3-methyladenine; RP, rapamycin; MHY,
MHY1485. |
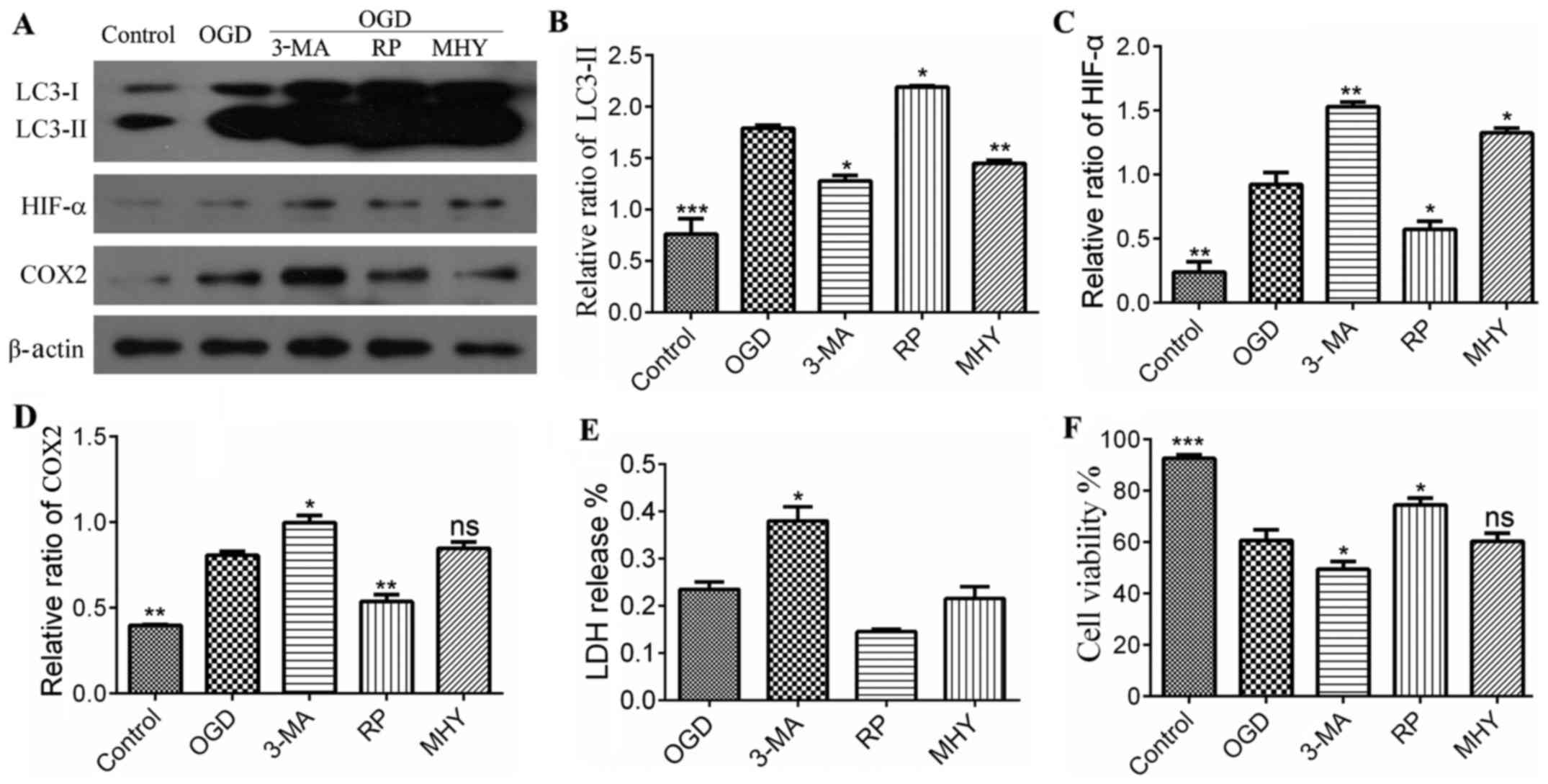 | Figure 3Statistical analysis of protein
expression in different treatment groups following immunoblotting.
(A) Expression of LC3, HIF-α and COX2. (B) Quantitative analyses of
LC3-II, showed significant difference between the control and
chemical intervention groups. *P<0.05,
**P<0.01 and ***P<0.001 vs. control
group (n=4). (C) Quantitative analysis of expression of HIF-α,
which showed significant differences between the control and
chemical intervention groups. *P<0.05 and
**P<0.01 vs. control group (n=4). (D) Quantitative
analysis of the expression of COX2, showed a significant difference
between the control and chemical intervention groups.
*P<0.05 and **P<0.01 vs. control group
(n=4). (E) Levels of LDH in were significantly increased in the
3-MA group, compared with the control. *P<0.05 vs.
control group. Data are presented as the mean ± standard error of
the mean of four independent experiments. (F) Cell viability in the
chemical intervention groups. Viability was increased in the OGD+RP
group, compared with the OGD group, and decreased in the 3-MA and
MHY groups, compared with the OGD group. Data are presented as the
mean ± standard deviation of four independent experiments.
*P<0.05 and ***P<0.001 vs. control
group. OGD, oxygen-glucose deprivation; COX2, cyclooxygenase 2;
HIF-α, hypoxia-inducible factor-α; 3-MA, 3-methyladenine; RP,
rapamycin; MHY, MHY1485; LDH, lactate dehydrogenase; ns, not
significant. |
Lysosomes are important in autophagy, as the protein
complexes and organelles in autophagosomes can be degraded by
lysosomes (22). Accordingly, the
lysosomal inhibitor MHY was used as an intervention for autophagy
in the OGD group in the present study. Following MHY intervention,
the autophagy in the PC12 cells was inhibited (Figs. 2 and 3A–D), suggesting that lysosomal
suppression alleviated IH injury. The results of the LDH assay also
showed that LDH release was increased in the 3-MA+OGD group and
decreased in the RAP+OGD group, compared with the OGD group with
statistical significance (Fig.
3E). Cell viability was decreased in the 3-MA+OGD group and
increased in the RAP+OGD group, compared with the OGD group with
statistical significance (Fig.
3F).
Wnt signaling pathway and autophagy
following IH
The Wnt pathway comprises canonical and noncanonical
pathways. The present study further examined whether the canonical
Wnt pathway was involved in IH-induced neuronal injury. The serial
key proteins of the Wnt pathway, Wnt1, Wnt3a, Dvl2, β-catenin,
C-myc and cyclin D1, were selected as targets for investigation
using western blot analysis. Compared with the control, the
expression levels of Wnt proteins in the PC12 cells were
significantly activated during IH, for example, expression of Wnt1
was increased in the OGD groups at 12 h (Fig. 4A and B), whereas the expression of
Wnt3a was elevated in the OGD groups at 6 h (Fig. 4A and C). However, the expression
levels of downstream proteins of the Wnt pathway, including
β-catenin and Dvl2, were initially increased compared with those in
the control, and then showed a decreasing trend with prolonging of
OGD time (Fig. 4D and E). Similar
results were observed in the expression levels of C-myc and cyclin
D1 (target protein) (Fig. 4F and
G). This suggested that the expression of downstream proteins
in the Wnt pathway was negatively correlated with the activation of
autophagy (Fig. 4A, D and E). As
shown in Fig. 5, the increases in
the expression levels of Wnt1 and Wnt3a were parallel with the
activation of autophagy (Fig.
5A–C), and the statistical data confirmed the observations in
the downstream proteins (P<0.05) (Fig. 5D and E). The results indicated
that the upstream proteins (Wnt1 and Wnt3a) were upregulated by the
activation of autophagy in the IH process, whereas down-stream
proteins (Dvl2, β-catenin, C-myc and cyclin D1) were downregulated
by the activation of autophagy.
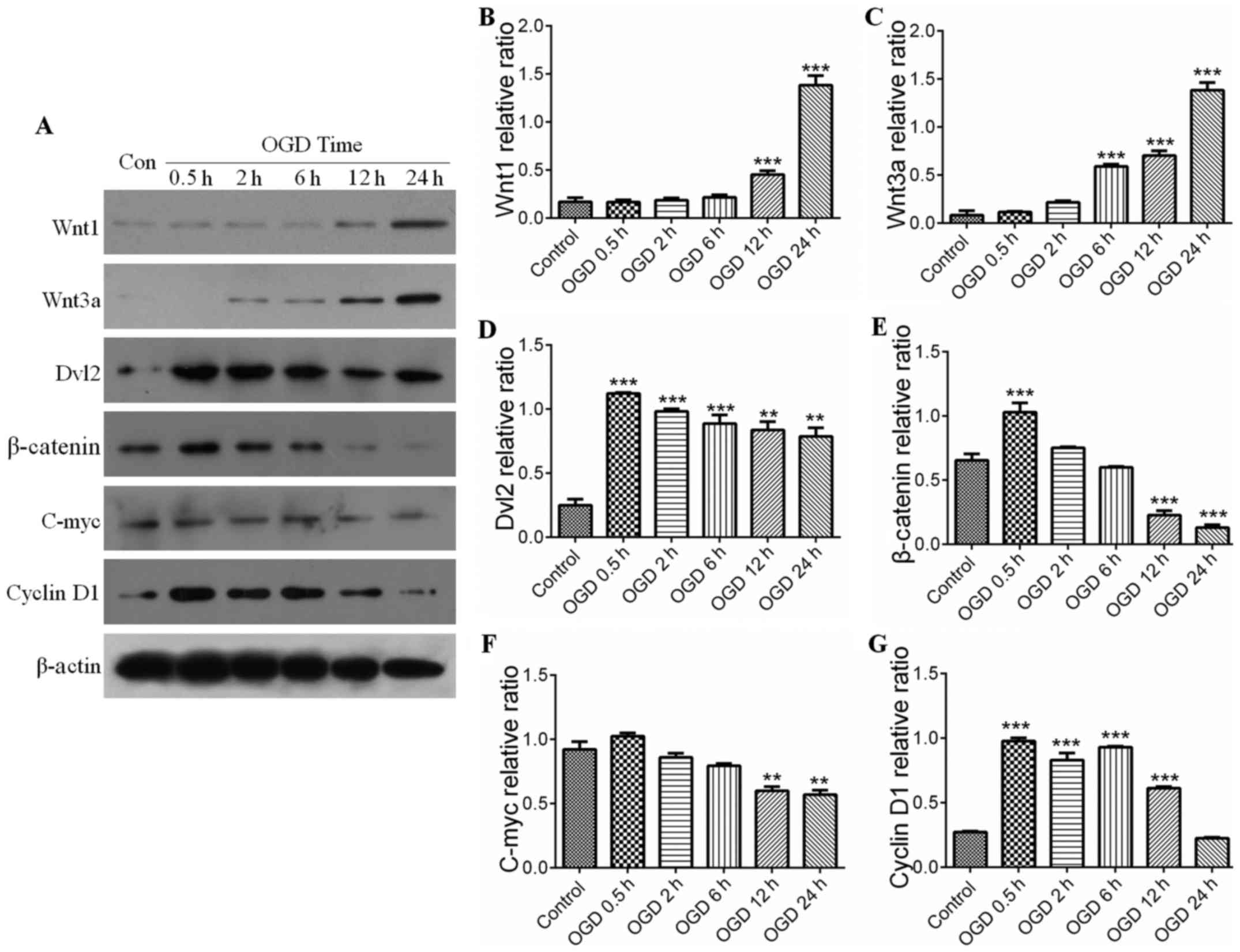 | Figure 4Wnt signaling pathway and
ischemic-hypoxic injury over time. (A) Expression levels of Wnt1,
Wnt3a, Dvl2, β-catenin, C-myc and cyclin D1 were shown by
immunoblotting. Expression levels of (B) Wnt1 and (C) Wnt3a in the
OGD groups were increased with time, with significant differences
in the OGD groups from 6 h. ***P<0.001 vs. control
group (n=4). Quantitative analyses showed the expression levels of
(D) Dvl2, (E) β-catenin, (F) C-myc and (G) cyclin D1 increased
initially compared with those in the control group, and then
decreased with increased OGD time. Quantitative analyses of Dvl2
and β-catenin showed there were significant differences in OGD at
0.5 h. **P<0.01 and ***P<0.001 vs.
control group (n=4). Quantitative analyses of cyclin D1 and C-myc
showed a significant difference between the OGD groups and control
group. **P<0.01 and ***P<0.001 vs.
control group (n=4). All the target proteins were analyzed
quantitatively. Data are expressed as the mean ± standard error of
the mean of four independent experiments. OGD, oxygen-glucose
deprivation; Dvl2, dishevelled segment polarity protein 2; Con,
control. |
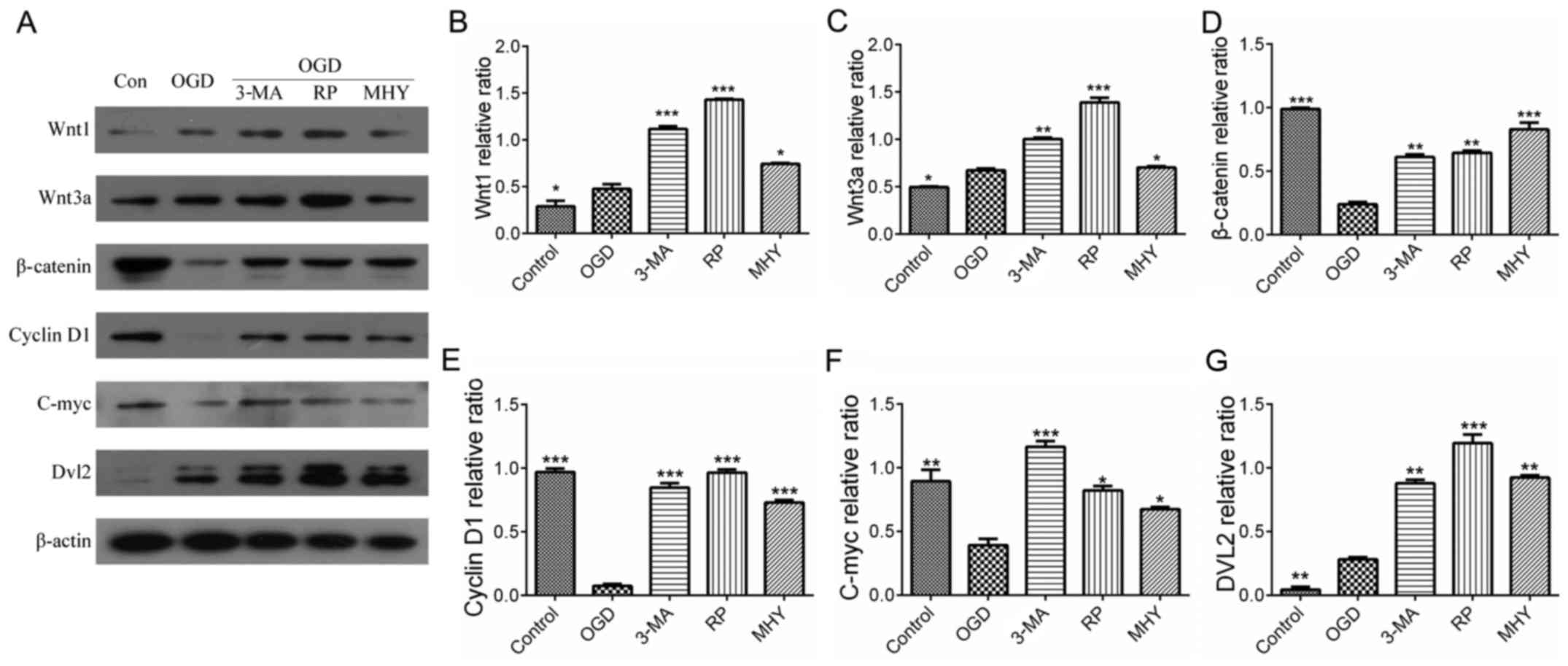 | Figure 5Wnt signaling pathway and autophagy
following chemical intervention. Results of immunoblotting and
statistical analysis. (A) Expression levels of Wnt1, Wnt3a, Dvl2,
β-catenin, C-myc and cyclin D1 with different chemical
interventions. Quantitative analyses of (B) Wnt1 and (C) Wnt3a
showed significant differences between the chemical intervention
groups and control group. *P<0.05,
**P<0.01 and ***P<0.001 vs. control
group (n=4). Quantitative analysis of (D) β-catenin and (E) cyclin
D1 showed significant differences between the chemical intervention
groups and control group. **P<0.01 and
***P<0.001 vs. control group (n=4). Quantitative
analysis of (F) C-myc and (G) Dvl2 showed there were significant
differences between the chemical intervention groups and control
group. *P<0.05, **P<0.01 and
***P<0.001 vs. control group (n=4). Data are
expressed as the mean ± standard error of the mean of four
independent experiments. OGD, oxygen-glucose deprivation; Dvl2,
dishevelled segment polarity protein 2; Con, control; 3-MA,
3-methyladenine; RP, rapamycin; MHY, MHY1485. |
Discussion
Stroke is a common disease for which there is a lack
of effective therapy (23–26).
Detailed investigations have focused on this complex disease and
advances have been made in the field. However, the mechanism
remains to be fully elucidated, particularly the functions of
autophagy during stroke (2,27).
Therefore, it is important to understand the mechanism and to
develop novel treatments for stroke. In the present study,
autophagy with respect to the Wnt signaling pathway in IH injury
was investigated, in order to produce data to provide novel insight
for stroke treatment.
Activation of autophagy with upregulation of
autophagic flux in IH injury. The process of autophagy involves a
series of autophagic membranous structures developing and evolving.
LC3 is an essential protein in the autopahgic process, which is
involved in the generation of autophagic vacuoles. Therefore, in
investigations of autophagy, LC3 is considered a marker of
autophagosomes (25). Cells
contain two types of LC3, which are 18 kDa LC3-I and16 kDa LC3-II.
The expression of LC3-II and ratio of LC3-II/LC3-I are positively
associated with the number of autophagsomes and autophagic activity
(21). Therefore, detecting the
expression of LC3-I and LC3-II using LC3 immunofluorescence
combined with western blot analysis enables assessment of the
activation of autophagy. Beclin l is the homologue of yeasty Atg6
in mammals, in the Golgi apparatus, which is a specific autophagic
protein. There is substantial evidence that Beclin 1 is involved
not only in the formation of autophagosomes, but also in regulating
the activity of autophagy (28,29). When autophagy is activated, the
expression of Beclin 1 is upregulated (30). p62 is the bridge between LC3 and
ubiquitination substrate; autophagic vacuoles engulfing
ubiquitination substrate require the assistance of p62 binding to
the target proteins, forming polymer ubiquitinated proteins, which
are engulfed by autophagosomes and then combine with lysosomes for
degradation. Therefore, p62 is considered a marker of autophagic
degradation. Under normal conditions, the levels of p62 increase
when autophagy is inhibited, whereas levels decrease when autophagy
is upregulated (31–37). In present study, the results of
the immunofluorescence evaluation of LC3 revealed that the
dispersed LC3-positive green dots gradually became aggregations of
numerous green dots in the OGD groups. It is generally recognized
that LC3-positive dots are diffuse in the cytoplasm when autophagy
is inactive and are in an aggregated state with the activation of
autophagy. The results of the present study indicated that
autophagy was upregulated in OGD-induced permanent IH injury. In
addition, the results of the western blot analysis confirmed that
the expression of LC3-II was enhanced as OGD time extended, and the
expression of Beclin 1 was similar to that of LC3. Therefore, the
results indicated that autophagy was activated in OGD-induced
permanent IH injury.
Following the activation of autophagy,
autophagosomes gradually increase, however, evaluating whether the
autophagosomes can be degraded successfully or not requires
evaluation of a parameter, which can reflect the whole process of
autophagy. Autophagic flux is considered the most reliable and
widely recognized parameter reflecting the process of autophagy
(9). Evaluating autophagic flux
involves two aspects: Following the activation of autophagy,
whether mature autophagosomes increase gradually or whether
autophagosomes can combine with lysosomes and be degraded,
resulting in a decrease in p62. In the present study, the results
revealed that the ratio of LC3-II/I increased continuously,
indicating that mature autophagosomes were increased with the
activation of autophagy, and that autophagic flux was likely
upregulated. The expression of p62 was negatively associated with
increased OGD duration, which indicated that p62 was increasingly
degraded and that autophagic flux was upregulated. Using lysosomal
inhibitors to assess the LC3B-II/I ratio is considered the gold
standard parameter to evaluate autophagic flux (38). The results of the present study
showed that the ratio of LC3-II/I was markedly increased under
OGD-induced permanent IH injury following use of the lysosomal
inhibitor MHY1485. This indicated that autophagic flux was
upregulated. Taken together, the results indicated that autophagy
was activated and autophagic flux was upregulated in OGD-induced
permanent IH injury.
Autophagy has a neuroprotective effect in IH injury.
Whether IH induced-autophagy has a neuroprotective effect or
induces injury is debated (39).
Certain studies have revealed that autophagy is harmful during the
neural IH process (8,26,40), whereas other studies have
demonstrated that the protective role of autophagy protects cells
from death during injury (4,11,41–44). In previous studies on the role of
autophagy in IH encephalopathy, the majority focused on the role of
autophagy in IH reperfusion, whereas relatively few focused on the
role of autophagy in neuronal permanent IH injury, and the majority
of lacked evaluation of autophagic flux. Therefore, in order to
assess the role of autophagy in permanent IH injury and assess
autophagic flux, evaluation of the autophagic effect on permanent
IH injury is required. The present study demonstrated that
autophagy was activated with the upregulation of autophagic flux,
therefore, in order to further evaluate the change in IH injury by
autophagy, IH injury was investigated in further experiments. The
autophagy agonist rapamycin and antagonists 3-MA and MHY1485, were
used to enhance or inhibit autophagy to assess changes in IH injury
parameters, including the expression of pro-inflammatory factors
COX2 and HIF-α, the release of LDH and cell viability, to evaluate
the role of autophagy in permanent IH injuries. The results showed
that autophagy was enhanced with rapamycin administration and the
expression of LC3 was increased. The expression levels of HIF-α and
COX2 were decreased. Following the administration of 3-MA, the
expression of LC3 decreased, and the expression levels of HIF-α and
COX2 increased. This revealed that the upregulation of autophagy
alleviated inflammation and cell hypoxia, whereas the inhibition of
autophagy aggravated inflammation and cell hypoxia, indicating that
autophagy has a possible protective role in OGD-induced permanent
IH injury. On this basis, the present study aimed to understand
how, if autophagic activity is altered, LDH release and cell
viability change. It was found that the release of LDH was reduced
in the OGD+Rap group, compared with that in the OGD group, and LDH
release was increased in the OGD+3-MA group and OGD+MHY1485 group,
compared with that in the OGD group. As LDH release is a parameter
of cell injury, the results of the present study further confirmed
that autophagy had a protective role in OGD-induce permanent IH
injury. The results of the MTT assay showed that cell viability was
enhanced in the OGD+Rap group, compared with that in the OGD group,
and was decreased in the OGD+3-MA group and OGD+MHY1485 group,
compared with that in the OGD group. Taken together, it was
hypothesized that the upregulation of autophagy had a protective
role in OGD-induced permanent IH injury.
Studies have indicated that three factors determine
autophagic function, namely the level of activation, the time of
induction and whether autophagic flux is impaired or not. A
previous study found that, following the administration of the
autophagy antagonist 3-MA at different phases, the function of
autophagy was distinct. When 3-MA was administered at 24 h
following IH reperfusion, autophagy was inhibited and neuron
apoptosis was observed; however, when 3-MA was administered at
48–72 h following IH reperfusion, the inhibition of autophagy
protected the cells from death (8). The reason for this may be that
autophagy was excessively activated, resulting in a change in the
role of autophagy from protective to damaging with the increase in
IH reperfusion time. The present study focused on the time frame of
0.5–24 h OGD, and the results of 3-MA administration were similar
to the study described above. Therefore, in the present study, the
activation of autophagy was moderate, as autophagy was activated
within 24 h, therefore having a protective effect. Another study
reported that 3-MA administration prior to middle cerebral artery
occlusion led to aggregation following IH injury, whereas following
preprocessing with rapamycin, the protection was similar to that
following high pressure oxygen pretreatment, indicating that
autophagy was activated at the early phase and had a protective
effect (31,45). In the present study, the autophagy
agonist and antagonist were administered prior to OGD treatment of
the cells; therefore, autophagy was activated at the early phase of
IH injury, which was similar with the above study. Other studies
have reported that whether autophagic flux is damaged or not can
determine the function of autophagy (5). The present study demonstrated that
autophagic flux was not damaged and was upregulated. Therefore,
according to the results mentioned above, the present study showed
that autophagy had a protective effect, which may be associated
with the early activation of autophagy, its moderate activation,
and that autophagic flux was not damaged.
Wnt/β-catenin signaling pathway moderates autophagy
in PC12 cells during IH injury. In mammalian cells, the
Wnt/β-catenin signaling pathway has a regulatory role in nervous
system development, neuron proliferation and apoptosis. Wnt1
signaling proteins include two types: Wnt1 family and Wnt5a family
proteins (46). Wnt1 family
proteins include Wnt1, Wnt3a and Wnt7a, and the Wnt5a family
proteins include Wnt4 and Wnt5a. Wnt1 family proteins can activate
the canonical Wnt/β-catenin pathway, and it has been shown that it
is key in the nervous system and neural cell regulation by
improving mitochondrial function and reducing the damage from by
6-hydroxydopamine in SH-SY5Y cells, having a protective effect
(47). It has also been revealed
that Wnt1 can mediate neural protection under oxidative stress and
ischemic conditions (48).
Wnt/β-catenin pathway activation and inhibition
determines the occurrence of growth-related physiological and
pathological processes and development, including maintaining
homeostasis under damage/stress conditions (49). In addition, there is crosstalk
between this pathway and other pathways, including the
phosphoinositide-3-kinase and receptor tyro-sine kinase pathway,
resulting in the Wnt/β-catenin pathway affecting the expression of
their downstream proteins (50).
Studies have revealed that the Wnt/β-catenin pathway is involved in
multiple neural activities and partial neurodegenerative disease,
including neural induction, synaptogenesis, intracephalic
neurogenesis, neuron repair and Alzheimer's disease regulation
(51–54). However, one of the primary
functions of autophagy is the maintenance of physiological and
pathological homeostasis in cells. It has been reported that
crosstalk exists between autophagy and the Wnt/β-catenin pathway
(55), however, in permanent IH
injury, the regulatory association between autophagy and the
Wnt/β-catenin pathway remains to be fully elucidated and warrants
investigation.
The present study investigated whether there is
crosstalk between Wnt/β-catenin pathway activation and autophagy in
OGD-induced permanent IH injury. Representative upstream and
downstream proteins of the Wnt/β-catenin pathway were examined, and
the results showed that the expression levels of upstream proteins
Wnt1 and Wnt3a were gradually increased with increased OGD time,
compared with those in the control group. The steady enhancement of
their expression indicated that the Wnt/β-catenin pathway began to
activate. These results were similar to those of a previous study
(56). However, the expression
levels of mid-downstream proteins, Dvl2, β-catenin, C-myc and
cyclin D1, were increased from OGD 0.5 h, compared with those in
the control group and exhibited a decreasing trend with increased
OGD time. These results differed from those of a previous study
(57). This was likely to be
associated with autophagy being involved in the regulation of this
process. A study by Gao et al showed that, in normal
conditions, autophagy inhibited the Wnt/β-catenin pathway,
predominantly due to LC3 and Dvl2 interacting to increase the
degradation of Dvl2 (55). In the
present study, it was demonstrated in permanent IH injury that
autophagy was activated from OGD 0.5 h, and exhibited OGD
time-dependency. This corresponded with the enhanced expression of
Dvl2 at OGD 0.5 h, which reduced gradually with increased OGD time.
In the Wnt/β-catenin pathway, the reduction of Dvl2 leads to the
combination of Dvl2 and Axin decreasing, which renders the
β-catenin degradation complex unable to depolymerize completely in
plasma, resulting in a decrease in the activation of downstream
proteins (55,58). The results of the present study
revealed the expression of downstream proteins of the Wnt pathway,
Dvl2 and β-catenin, were initially increased, compared with those
in the control, and then decreased with increased OGD time
(Fig. 4D and E). Similar results
were observed with C-myc and cyclin D1 (Fig. 4F and G). These results were as
expected, and indicated that autophagy likely degraded Dvl2 to
negatively regulate the Wnt/β-catenin pathway.
The present study further investigated whether
autophagy regulated the Wnt/β-catenin pathway. The autophagy
agonist and antagonist were used to regulate autophagic activity,
and the expression of the proteins of the Wnt/β-catenin pathway
mentioned above were examined to determine whether expression was
negatively associated with the degree of autophagic activation. The
results showed that the expression of upstream proteins Wnt1 and
Wnt3a were increased in accordance with the degree of autophagic
activation. The expression levels of Dvl2, β-catenin, C-myc and
cyclin D1 decreased as autophagy was upregulated, and their
expression levels increased when autophagy was downregulated, which
indicated that the expression of downstream proteins of the
Wnt/β-catenin pathway were negatively associated with the degree of
autophagic activation. It has been reported that β-catenin can
become the target protein of autolysosome degradation when
autophagy is activated (59).
Others have shown that β-catenin is selectively degraded, according
to the formation of a β-catenin-LC3 complex, which attenuates the
transcription of β-catenin to the downstream protein TCF and may
assist in coping with metabolic stress (60). Therefore, it was hypothesized that
autophagy negatively regulated the downstream proteins of the
Wnt/β-catenin pathway to assist in coping with IH stress, with
autophagy and the Wnt/β-catenin pathway having protective effects
in cells.
In conclusion, the present study revealed that
autophagy was upregulated with autophagic flux to protect cells
from damage and death following IH. The Wnt/β-catenin pathway was
involved in regulation during IH, and autophagy mediated the
negative regulation of the Wnt/β-catenin pathway. These findings
indicated that the Wnt/β-catenin pathway and autophagy offer
potential as therapeutic targets for the treatment of IH neural
injury.
Acknowledgments
This study was funded by the National Natural
Science Foundation of China (no. 1204311), the Henan Province
Higher Education Key Research Project funding Scheme (no.
18A310001).
References
|
1
|
Lozano R, Naghavi M, Foreman K, Lim S,
Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, et
al: Global and regional mortality from 235 causes of death for 20
age groups in 1990 and 2010: A systematic analysis for the Global
Burden of Disease Study 2010. Lancet. 380:2095–2128. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Donnan GA, Fisher M, Macleod M and Davis
SM: Stroke. Lancet. 371:1612–1623. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Han ZB, Ren H, Zhao H, Chi Y, Chen K, Zhou
B, Liu YJ, Zhang L, Xu B, Liu B, et al: Hypoxia-inducible factor
(HIF)-1 alpha directly enhances the transcriptional activity of
stem cell factor (SCF) in response to hypoxia and epidermal growth
factor (EGF). Carcinogenesis. 29:1853–1861. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wen Y, Zhai RG and Kim MD: The role of
autophagy in Nmnat-mediated protection against hypoxia-induced
dendrite degeneration. Mol Cell Neurosci. 52:140–151. 2013.
View Article : Google Scholar :
|
|
5
|
Yang Z, Zhao TZ, Zou YJ, Zhang JH and Feng
H: Hypoxia Induces autophagic cell death through hypoxia-inducible
factor 1α in microglia. PLoS One. 9:e965092014. View Article : Google Scholar
|
|
6
|
Ma L, Xu Y, Su J, Yu H, Kang J, Li H, Li
X, Xie Q, Yu C, Sun L, et al: Autophagic flux promotes cisplatin
resistance in human ovarian carcinoma cells through ATP-mediated
lysosomal function. Int J Oncol. 47:1890–1900. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clarke PGH and Puyal J: Autophagic cell
death exists. Autophagy. 8:867–869. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ginet V, Spiehlmann A, Rummel C, Rudinskiy
N, Grishchuk Y, Luthi-Carter R, Clarke PG, Truttmann AC and Puyal
J: Involvement of autophagy in hypoxic-excitotoxic neuronal death.
Autophagy. 10:846–860. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kroemer G and Levine B: Autophagic cell
death: The story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Puyal J, Ginet V and Clarke PG: Multiple
interacting cell death mechanisms in the mediation of
excitotoxicity and ischemic brain damage: A challenge for
neuroprotection. Prog Neurobiol. 105:24–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei K, Wang P and Miao CY: A double-edged
sword with therapeutic potential: An updated role of autophagy in
ischemic cerebral injury. CNS Neurosci Ther. 18:879–886. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gomez-Cambronero J and Kantonen S: A river
runs through it: How autophagy, senescence, and phagocytosis could
be linked to phospholipase D by Wnt signaling. J Leukoc Biol.
96:779–784. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holla S, Kurowska-Stolarska M, Bayry J and
Balaji KN: Selective inhibition of IFNG-induced autophagy by
Mir155- and Mir31-responsive WNT5A and SHH signaling. Autophagy.
10:311–330. 2014. View Article : Google Scholar
|
|
14
|
Kim W, Kim M and Jho EH: Wnt/β-catenin
signalling: From plasma membrane to nucleus. Biochem J. 450:9–21.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Petherick KJ, Williams AC, Lane JD,
Ordóñez-Morán P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik
K, Paraskeva C, et al: Autolysosomal β-catenin degradation
regulates Wnt-autophagy-p62 crosstalk. EMBO J. 32:1903–1916. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Clevers H and Nusse R: Wnt/β-catenin
signaling and disease. Cell. 149:1192–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Inestrosa NC, Montecinos-Oliva C and
Fuenzalida M: Wnt signaling: Role in Alzheimer disease and
schizophrenia. J Neuroimmune Pharmacol. 7:788–807. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shruster A, Ben-Zur T, Melamed E and Offen
D: Wnt signaling enhances neurogenesis and improves neurological
function after focal ischemic injury. PLoS One. 7:e408432012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen JL, Lin HH, Kim KJ, Lin A, Ou JH and
Ann DK: PKC delta signaling: A dual role in regulating hypoxic
stress-induced autophagy and apoptosis. Autophagy. 5:244–246. 2009.
View Article : Google Scholar :
|
|
20
|
Petiot A, Ogier-Denis E, Blommaart EFC,
Meijer AJ and Codogno P: Distinct classes of phosphatidylinositol
3′-kinases are involved in signaling pathways that control
macroautophagy in HT-29 cells. J Biol Chem. 275:992–998. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bodhankar S, Chen Y, Vandenbark AA, Murphy
SJ and Offner H: PD-L1 enhances CNS inflammation and infarct volume
following experimental stroke in mice in opposition to PD-1. J
Neuroinflammation. 10:1112013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wong KS, Wang Y, Leng X, Mao C, Tang J,
Bath PM, Markus HS, Gorelick PB, Liu L, Lin W, et al: Early dual
versus mono antiplatelet therapy for acute non-cardioembolic
ischemic stroke or transient ischemic attack: An updated systematic
review and meta-analysis. Circulation. 128:1656–1666. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Z, Zhong L, Zhong S, Xian R and Yuan
B: Hypoxia induces microglia autophagy and neural inflammation
injury in focal cerebral ischemia model. Exp Mol Pathol.
98:219–224. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang X, Yuan Y, Jiang L, Zhang J, Gao J,
Shen Z, Zheng Y, Deng T, Yan H, Li W, et al: Endoplasmic reticulum
stress induced by tunicamycin and thapsigargin protects against
transient ischemic brain injury: Involvement of PARK2-dependent
mitophagy. Autophagy. 10:1801–1813. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pellegrini L, Bennis Y, Guillet B, Velly
L, Bruder N and Pisano P: Cell therapy for stroke: From myth to
reality. Rev Neurol (Paris). 169:291–306. 2013.In French.
View Article : Google Scholar
|
|
28
|
Edinger AL and Thompson CB: Defective
autophagy leads to cancer. Cancer Cell. 4:422–424. 2003. View Article : Google Scholar
|
|
29
|
Scarlatti F, Maffei R, Beau I, Codogno P
and Ghidoni R: Role of non-canonical Beclin 1-independent autophagy
in cell death induced by resveratrol in human breast cancer cells.
Cell Death Differ. 15:1318–1329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liang XH, Jackson S, Seaman M, Brown K,
Kempkes B, Hibshoosh H and Levine B: Induction of autophagy and
inhibition of tumorigenesis by beclin 1. Nature. 402:672–676. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bartlett BJ, Isakson P, Lewerenz J,
Sanchez H, Kotzebue RW, Cumming RC, Harris GL, Nezis IP, Schubert
DR, Simonsen A, et al: P62, Ref(2)P and ubiquitinated proteins are
conserved markers of neuronal aging, aggregate formation and
progressive autophagic defects. Autophagy. 7:572–583. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cui J, Bai XY, Shi S, Cui S, Hong Q, Cai G
and Chen X: Age-related changes in the function of autophagy in rat
kidneys. Age (Dordr). 34:329–339. 2012. View Article : Google Scholar
|
|
33
|
Komatsu M and Ichimura Y: Physiological
significance of selective degradation of p62 by autophagy. FEBS
Lett. 584:1374–1378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Masiero E, Agatea L, Mammucari C, Blaauw
B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S and
Sandri M: Autophagy is required to maintain muscle mass. Cell
Metab. 10:507–515. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nezis IP, Simonsen A, Sagona AP, Finley K,
Gaumer S, Contamine D, Rusten TE, Stenmark H and Brech A: Ref(2)P,
the Drosophila melanogaster homologue of mammalian p62, is required
for the formation of protein aggregates in adult brain. J Cell
Biol. 180:1065–1071. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nixon RA, Wegiel J, Kumar A, Yu WH,
Peterhoff C, Cataldo A and Cuervo AM: Extensive involvement of
autophagy in Alzheimer disease: An immuno-electron microscopy
study. J Neuropathol Exp Neurol. 64:113–122. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang L, Gao C, Yao S and Xie B: Blocking
autophagic flux enhances matrine-induced apoptosis in human
hepatoma cells. Int J Mol Sci. 14:23212–23230. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar
|
|
39
|
Gabryel B, Kost A and Kasprowska D:
Neuronal autophagy in cerebral ischemia - a potential target for
neuroprotective strategies? Pharmacol Rep. 64:1–15. 2012.
View Article : Google Scholar
|
|
40
|
Chang CF, Huang HJ, Lee HC, Hung KC, Wu RT
and Lin AM: Melatonin attenuates kainic acid-induced neurotoxicity
in mouse hippocampus via inhibition of autophagy and α-synuclein
aggregation. J Pineal Res. 52:312–321. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Carloni S, Buonocore G, Longini M,
Proietti F and Balduini W: Inhibition of rapamycin-induced
autophagy causes necrotic cell death associated with Bax/Bad
mitochondrial translocation. Neuroscience. 203:160–169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Papadakis M, Hadley G, Xilouri M, Hoyte
LC, Nagel S, McMenamin MM, Tsaknakis G, Watt SM, Drakesmith CW,
Chen R, et al: Tsc1 (hamartin) confers neuroprotection against
ischemia by inducing autophagy. Nat Med. 19:351–357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Scherz-Shouval R, Weidberg H, Gonen C,
Wilder S, Elazar Z and Oren M: p53-dependent regulation of
autophagy protein LC3 supports cancer cell survival under prolonged
starvation. Proc Natl Acad Sci USA. 107:18511–18516. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang P, Guan YF, Du H, Zhai QW, Su DF and
Miao CY: Induction of autophagy contributes to the neuroprotection
of nicotinamide phosphoribosyltransferase in cerebral ischemia.
Autophagy. 8:77–87. 2012. View Article : Google Scholar
|
|
45
|
Yan W, Zhang H, Bai X, Lu Y, Dong H and
Xiong L: Autophagy activation is involved in neuroprotection
induced by hyperbaric oxygen preconditioning against focal cerebral
ischemia in rats. Brain Res. 1402:109–121. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gordon MD and Nusse R: Wnt signaling:
Multiple pathways, multiple receptors, and multiple transcription
factors. J Biol Chem. 281:22429–22433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wei L, Ding L, Mo MS, Lei M, Zhang L, Chen
K and Xu P: Wnt3a protects SH-SY5Y cells against 6-hydroxydopamine
toxicity by restoration of mitochondria function. Transl
Neurodegener. 4:112015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang J, Götz S, Vogt Weisenhorn DM,
Simeone A, Wurst W and Prakash N: A WNT1-regulated developmental
gene cascade prevents dopaminergic neurodegeneration in adult
En1(+/−) mice. Neurobiol Dis. 82:32–45. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
L'episcopo F, Serapide MF, Tirolo C, Testa
N, Caniglia S, Morale MC, Pluchino S and Marchetti B: A Wnt1
regulated Frizzled-1/β-Catenin signaling pathway as a candidate
regulatory circuit controlling mesencephalic dopaminergic
neuron-astrocyte crosstalk: Therapeutical relevance for neuron
survival and neuroprotection. Mol Neurodegener. 6:49. 2011.
View Article : Google Scholar
|
|
50
|
Demagny H and De Robertis EM: Smad4/DPC4:
A barrier against tumor progression driven by RTK/Ras/Erk and
Wnt/GSK3 signaling. Mol Cell Oncol. 3:e9891332015. View Article : Google Scholar
|
|
51
|
Hooper C, Killick R and Lovestone S: The
GSK3 hypothesis of Alzheimer's disease. J Neurochem. 104:1433–1439.
2008. View Article : Google Scholar
|
|
52
|
Magdesian MH, Carvalho MM, Mendes FA,
Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J and Ferreira ST:
Amyloid-beta binds to the extracellular cysteine-rich domain of
Frizzled and inhibits Wnt/beta-catenin signaling. J Biol Chem.
283:9359–9368. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sebastião AM, Colino-Oliveira M,
Assaife-Lopes N, Dias RB and Ribeiro JA: Lipid rafts, synaptic
transmission and plasticity: Impact in age-related
neurodegenerative diseases. Neuropharmacology. 64:97–107. 2013.
View Article : Google Scholar
|
|
54
|
Viti J, Gulacsi A and Lillien L: Wnt
regulation of progenitor maturation in the cortex depends on Shh or
fibroblast growth factor 2. J Neurosci. 23:5919–5927.
2003.PubMed/NCBI
|
|
55
|
Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T,
Fu W, Zhang J, Wu W, Zhang X, et al: Autophagy negatively regulates
Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol.
12:781–790. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chong ZZ, Shang YC, Hou J and Maiese K:
Wnt1 neuroprotection translates into improved neurological function
during oxidant stress and cerebral ischemia through AKT1 and
mitochondrial apoptotic pathways. Oxid Med Cell Longev. 3:153–165.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Varela-Nallar L, Rojas-Abalos M, Abbott
AC, Moya EA, Iturriaga R and Inestrosa NC: Chronic hypoxia induces
the activation of the Wnt/β-catenin signaling pathway and
stimulates hippocampal neurogenesis in wild-type and APPswe-PS1ΔE9
transgenic mice in vivo. Front Cell Neurosci. 8:172014. View Article : Google Scholar
|
|
58
|
Zhou KK, Benyajati S, Le Y, Cheng R, Zhang
W and Ma JX: Interruption of Wnt signaling in Müller cells
ameliorates ischemia-induced retinal neovascularization. PLoS One.
9:e1084542014. View Article : Google Scholar
|
|
59
|
Zhang Y, Wang F, Han L, Wu Y, Li S, Yang
X, Wang Y, Ren F, Zhai Y, Wang D, et al: GABARAPL1 negatively
regulates Wnt/beta-catenin signaling by mediating Dvl2 degradation
through the autophagy pathway. Cell Physiol Biochem. 27:503–512.
2011. View Article : Google Scholar
|
|
60
|
Choi SW, Song JK, Yim YS, Yun HG and Chun
KH: Glucose deprivation triggers protein kinase C-dependent
β-catenin proteasomal degradation. J Biol Chem. 290:9863–9873.
2015. View Article : Google Scholar : PubMed/NCBI
|














