Introduction
Acute lung injury (ALI) and its severe form acute
respiratory distress syndrome (ARDS) are devastating clinical
syndromes that frequently lead to respiratory failure characterized
by neutrophil accumulation, diffuse endothelium and epithelial
damage, air-blood barrier disruption, and the subsequent
infiltration of peripheral inflammatory cells into lung tissues
(1). Currently, there is no
effective pharmacological therapy for ALI/ARDS, and the morbidity
and mortality rates of ARDS remain as high as 30–40% (2). The pathophysiological mechanism of
ALI/ARDS is believed to be associated with an uncontrolled and
excessive inflammatory response in lungs (3). An unchecked inflammatory reaction
and the presence of excessive inflammatory mediators can lead to
inappropriate immunity-related tissue damage, including ALI.
Alveolar macrophages (AMs) are the most abundant
cells in the alveoli, distal airspaces and conducting airways
(4). They form the first line of
defense against airborne particles and microbes. AMs account for
90% of the cells in bronchoalveolar lavage fluid; as the main
immune cells, they recognize pathogen-associated molecular patterns
and trigger innate immunity and host defenses (4,5).
Previous studies have demonstrated that macrophages contribute to
the modulation of inflammatory responses during ALI/ARDS as well as
to the resolution of inflammation and tissue repair in lungs
(5,6). With increased foreign antigen
stimulation, the macrophages become activated, which further leads
to inflammatory cytokine secretion, neutrophil recruitment and
T-effector cell interaction (7).
Lipopolysaccharides (LPS), an important component of the outer wall
of Gram-negative bacteria, are highly proinflammatory molecules
that can activate AMs, stimulating their production of inflammatory
mediators (8,9). LPS is recognized by LPS-binding
protein (LBP) and CD14, which then bind to Toll-like receptor 4
(TLR4). Following this, several adaptor proteins are recruited to
transduce multiple key downstream intracellular signaling pathways,
such as the mitogen-activated protein kinase (MAPK), IFN regulatory
factor 3 (IRF-3), and nuclear factor (NF)-κB cascades. Activation
of these signaling pathways eventually induces the production of
various cytokines, including tumor necrosis factor (TNF)-α,
interleukin (IL)-1β, interferon (IFN)-α and IFN-β (10,11). Despite their beneficial effects
during infection, excessive production of inflammatory mediators
may cause edema, cell necrosis, tissue damage and other
pathological changes (12).
Paralemmin-3 (PALM3) was first described in
Xenopus laevis as Xlgv7/Xlcaax-1 by Cornish et al
(13) and belongs to the
paralemmins (PALMs) protein family. The PALM protein family
includes PALM1, PALM2, PALM3 and palmdelphin (PALMD) (14), and these proteins are highly
expressed in the brain, kidney, adrenal gland and mammary gland, as
well as in breast cancers (15–17). Previous studies have implicated
PALM1 as having a role in cell shape control, plasma membrane
dynamics, cell motility, cancer cell invasiveness and metastatic
potential, cell migration and maturation modulation, and tumor
lymphangiogenesis (15,16,18). However, PALM3 is speculated to act
as an adaptor to link intrinsic membrane proteins to each other, to
the cytoskeleton, or to motor proteins (14,19). Previous work of the authors
revealed that LPS can upregulate PALM3 expression in alveolar
epithelial cells (19). In
addition, the study revealed that PALM3 expression is induced by
LPS and may be involved in the LPS-TLR4 signaling in alveolar
epithelial cells (A549 cells) (19). The downregulation of PALM3 is able
to ameliorate LPS-induced ALI in mice and reduce the LPS-induced
inflammatory response in alveolar epithelial cells (19,20). However, whether or not PALM3
participates in LPS-TLR4 signaling in AMs is still unclear.
Additionally, the manner in which PALM3 participates in TLR4 signal
transduction also warrants further investigation. Therefore, the
present study was conducted to investigate the role of PALM3 in
LPS-TLR4 signal transduction using a rat NR8383 macrophage model,
which can be stimulated with LPS to mimic an inflammatory state.
For this purpose, the authors aimed to modulate the expression of
PALM3 in AMs by using recombinant adenoviral vectors. To assess the
effects of PALM3 expression, the corresponding cytokine levels were
observed, as well as the activity of NF-κB and IRF-3 in AMs. They
detected the interaction of PALM3 with adaptor molecules in
LPS-TLR4 signaling by co-immunoprecipitation and confocal
microscopy.
Materials and methods
Cell culture
Cells (293) were obtained from Microbix Biosystems,
Inc. (Toronto, Canada) and were grown in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) at 37°C in a humidified chamber with 5%
CO2. The rat AM cell line NR8383 (American Type Culture
Collection, Manassas, VA, USA) was cultured in Ham's F12 medium
(Gibco; Thermo Fisher Scientific, Inc.), supplemented with 10%
fetal bovine serum at 37°C in a humidified atmosphere with 5%
CO2. Cells in the exponential growth phase were used in
the experiments described in the following section.
Generation of adenoviral vectors
An adenovirus was generated that contained a single
open reading frame encoding rat PALM3 (rPALM3).
First, cDNA coding for rPALM3 was synthesized by GeneChem
Co. Ltd. (Shanghai, China) and ligated into the adenoviral shuttle
plasmid pDC316 (Microbix Biosystems, Inc.) to generate the plasmid
pDC316-rPALM3. Recombinant viruses were then generated using 293
packaging cells co-transfected with pDC316-rPALM3 and a plasmid
containing cDNA for adenoviral proteins (pBHGlox; Microbix
Biosystems, Inc.) via the AdMax™ system (Microbix Biosystems,
Inc.). Plaques were isolated after ~14 days and expanded in 293
cells. Viral stocks were purified using CsCl gradients. The
acquired overexpression adenoviral vector was named Ad.rPALM3.
Meanwhile, a recombinant adenoviral vector expressing short hairpin
(sh)RNA for rat PALM3 was constructed using a similar method
and named Ad.shRNA. The sequence 5′-AGATCTTGATGGAGGGTTT-3′ was
chosen as the targeted sequence. The primers for the targeted
sequence were:
5′-CCGGGGAGATCTTGATGGAGGGTTTCTCGAGAAACCCTCCATCAAGATCTCCTTTTTG-3′
(sense) and
5′-AATTCAAAAAGGAGATCTTGATGGAGGGTTTCTCGAGAAACCCTCCATCAAGATCTCC-3′
(anti-sense) (GeneChem Co., Ltd.). An adenoviral vector, free of
any transgenes, was used as a control vector and named Ad.V
(GeneChem Co., Ltd.). The titers of the recombinant adenoviruses
were determined by means of the 50% tissue culture infectious dose.
The titers of Ad.rPALM3, Ad.shRNA and Ad.V were 5×109
plaque-forming units (PFU)/ml, 3×1010 PFU/ml, and
6×109 PFU/ml, respectively.
Experimental design
Rat AMs were routinely grown in Ham's F12 medium
supplemented with 10% fetal bovine serum and seeded in 6-well
plates at a density of 1×105 cells/well. After the cell
growth reached 50–60% confluence, rat AMs were treated with 0.5
µg/ml LPS (Escherichia coli 0111: B4; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) as described previously to simulate
ALI in an in vitro model (4,5).
The total RNA and protein were isolated before the treatment with
LPS and at 3, 6, 12, 24 and 48 h following the addition of LPS for
the analysis of PALM3 expression.
For some experiments, rat AMs were seeded in
appropriate plates and left untreated (normal) or were transfected
with the adenoviruses Ad.rPALM3, Ad.shRNA, or Ad.V when the cell
growth reached 50–60% confluence. At 48 h post-transfection, the
total RNA and protein were isolated from some of the cells for
detecting the PALM3 expression in cells transfected with the
recombinant adenoviruses. Also, at 48 h post-transfection, 0.5
µg/ml LPS was added into the cell culture medium of some
cells (normal + LPS, Ad.rPALM3 + LPS, Ad.shRNA + LPS and Ad.V +
LPS). The cell culture supernatant was collected both before the
LPS treatment and at 24 h following LPS stimulation for the
detection of cytokine levels, and the nuclear protein was isolated
for determining the activity of NF-κB and IRF-3.
For other experiments, rat AMs were seeded in
appropriate plates and, after the cells were treated with LPS (0.5
µg/ml) or phosphate-buffered saline (PBS) for 24 h, the
total protein was extracted for subsequent co-immunoprecipitation,
and cell monolayers were fixed with 4% paraformaldehyde for
immunofluorescence analysis.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Following the treatment, cells were harvested, and
total cellular RNA was isolated using TRIzol (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Reverse transcription (RT) of the lung samples was
performed using the Takara PrimeScript RT Reagent kit (Takara
Biotechnology Co., Ltd., Dalian, China). Then, RT-qPCR was
performed using the iTaq Universal SYBR Green Supermix (Takara
Biotechnology Co., Ltd.) according to the manufacturer's
instructions. Rat β-actin was selected as an internal
standard. The primers used for rat PALM3 were
5′-GAGGCAGGGATCTTGATGTC-3′ (sense) and 5′-GCCCAACACCCTCAAGACTA-3′
(antisense). The primers used for rat β-actin were
5′-GGAGATTACTGCCCTGGCTCCTA-3′ (sense) and
5′-GACTCATCGTACTCCTGCTTGCTG-3′ (antisense). The PCR conditions were
as follows: 95°C for 30 sec, then 35 cycles of 95°C for 5 sec, 60°C
for 30 sec. All reactions were performed in triplicate, and reports
were generated by Rotor-Gene Real-time Analysis Software 6.0
(Qiagen GmbH, Hilden, Germany). The relative expression of each
target gene (PALM3 and β-actin) was calculated by the
2−ΔΔCq method (21).
Protein extraction and western
blotting
Following appropriate treatment, rat AMs were lysed
in lysis buffer [30 mM Tris-HCl (pH 8.0), 200 mM NaCl, 1% NP-40, 1
mM phenylmethylsulfonyl fluoride and protease inhibitor cocktail
(Sigma-Aldrich; Merck KGaA)]. The protein concentrations were
measured by the bicinchoninic acid (BCA) assay method
(Sigma-Aldrich; Merck KGaA). Equal amounts of protein (30
µg) were separated using SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane. The membranes were then blocked
and incubated overnight at 4°C with goat polyclonal anti-rPALM3
antibody (cat. no. sc-248213; polyclonal goat anti-rat; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) at a 1:500 dilution in TBST
(TBS with 0.1% Tween-20). After extensive washing with TBST, the
membranes were incubated with horseradish peroxidase-labeled rabbit
anti-goat secondary antibody at a 1:2,500 dilution in TBST (cat.
no. ZB-2306; Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd., Beijing, China) at room temperature for 120 min. The signals
were detected by enhanced chemiluminescence following the
manufacturer's instructions (Beyotime Institute of Biotechnology,
Haimen, China). The images were quantified by Quantity One
(version, 4.62; Bio-Rad Laboratories, Inc., Hercules, CA, USA)
software.
ELISA for the detection of cytokine
levels
The expression levels of TNF-α, IL-1β, IL-10, IFN-α,
IFN-β and macrophage migration inhibitory factor (MIF) were
measured by performing ELISAs. The NR8383 AMs were incubated in
24-well plates and transfected with Ad.rPALM3, Ad.shRNA or Ad.V. At
48 h following transfection, the AMs were treated with LPS (0.5
µg/ml). The cell culture supernatant was collected both
before the treatment with LPS and, at 24 h after the addition of
LPS, these samples were centrifuged at 1,000 × g for 15 min. The
levels of TNF-α (cat. no. RTA00), IL-1β (cat. no. RLB00) and IL-10
(cat. no. R1000) (R&D System, Inc., Minneapolis, MN, USA) and
of IFN-α (cat. no. CSB-E08637r), IFN-β (cat. no. CSB-E04845r) and
MIF (cat. no. CSB-E07293r) (Cusabio Biotech Co., Ltd., Wuhan,
China) in the supernatants were determined by using the
corresponding ELISA kit, according to the manufacturer's
instructions.
ELISA for the detection of NF-κB
activity
The NR8383 AMs were incubated in 6-well plates and
transfected with Ad.rPALM3, Ad.shRNA or Ad.V. At 48 h after
transfection, the AMs were treated with LPS (0.5 µg/ml) for
24 h. The nuclear extracts of AMs were then prepared by using the
Nuclear Extract kit (Active Motif, Carlsbad, CA, USA) according to
the manufacturer's protocol. The protein concentration of the
nuclear extracts was quantified by the BCA method (Sigma-Aldrich;
Merck KGaA). The NF-κB p65 DNA binding activity in the isolated
nuclear extracts was assessed by performing ELISAs using the
TransAM™ NF-κB Transcription Factor Assay kit according to the
manufacturer's protocol (Active Motif).
Western blot analysis of
phospho-IRF3
After the cells were treated as described above, the
nuclear extracts of AMs were prepared as described above, and the
procedure of western blot analysis was performed as described
above. The nuclear extracts were analyzed by immunoblotting with
rabbit polyclonal phosphor-specific anti-IRF-3 (1:500 in TBST; cat.
no. ab138449; Abcam, Cambridge, UK) antibody. Rabbit polyclonal
Histone H3 (cat. no. sc8654; 1:1,000 in TBST; Santa Cruz
Biotechnology, Inc.) was used as a lysate control.
Confocal immunofluorescence imaging
Rat AMs were plated on poly-L-lysine-coated glass
cover slides and cultured until they reached 50–60% confluence,
after which these cells were treated with LPS (0.5 µg/ml) or
PBS for 24 h. Following LPS stimulation, the cell monolayers were
fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1%
Triton X-100 for 30 min, and blocked with 5% bovine serum albumin
(BSA) for 1 h at room temperature. Slides were incubated overnight
at 4°C with a primary rat-specific anti-rPALM3 antibody (1:100 in
PBS with 1% BSA; cat. no. sc-248213; polyclonal goat anti-rat;
Santa Cruz Biotechnology, Inc.), anti-TLR4 antibody (1:50 in PBS
with 1% BSA; cat. no. sc-293072; monoclonal mouse anti-rat; Santa
Cruz Biotechnology, Inc.), anti-myeloid differentiation factor 88
(MyD88; HFL-296) antibody (1:50 in PBS with 1% BSA; cat. no.
sc-11356; monoclonal mouse anti-rat; Santa Cruz Biotechnology,
Inc.), anti-interleukin 1 receptor associated kinase (IRAK)-1
antibody (1:50 in PBS with 1% BSA; cat. no. NBP1-77068; polyclonal
rabbit anti-rat; Novus Biologicals, LLC, Littleton, CO, USA),
anti-tumor necrosis factor receptor associated factor (TRAF)-6
antibody (1:100 in PBS with 1% BSA; cat. no. NBP1-33357; polyclonal
rabbit anti-rat; Novus Biologicals, LLC), or anti-TICAM-2 (E-2)
antibody (1:50 in PBS with 1% BSA; cat. no. sc-376076; monoclonal
mouse anti-rat; Santa Cruz Biotechnology, Inc.). Following
incubation, the slides were washed three times with PBS. The slides
were then incubated at room temperature with a Cy3-conjugated
donkey anti-goat secondary antibody at a 1:1,000 dilution (cat. no.
A0502; Beyotime Institute of Biotechnology) for 90 min followed by
three washes with PBS. The slides were finally incubated with a
fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse
secondary antibody at a 1:1,000 dilution (cat. no. A0568; Beyotime
Institute of Biotechnology) or a FITC-conjugated goat anti-rabbit
secondary antibody at a 1:1,000 dilution (cat. no. A0562; Beyotime
Institute of Biotechnology) for 30 min. The slides were
subsequently counterstained for 5 min with
4′,6-diamidino-2-phenylindole (DAPI), and samples were observed
using a confocal Leica TCS SP5 (Leica Microsystems GmbH, Wetzlar,
Germany).
Co-immunoprecipitation
To examine protein-protein interactions,
co-immunoprecipitation assays were performed. The rat AMs were
treated with LPS (0.5 µg/ml) or PBS for 24 h. Then, the
cells were washed with PBS and scraped from the plate in the lysis
buffer described above. Aliquots of the resulting cell lysates
(containing 500 µg protein) were incubated with a primary
rat-specific anti-TLR4 (25)
antibody (cat. no. sc-293072; 1:100 in TBST; Santa Cruz
Biotechnology, Inc.), anti-MyD88 antibody (D80F5; 1:50 in TBST;
cat. no. 4283; monoclonal rabbit anti-rat; Cell Signaling
Technology, Inc., Danvers, MA, USA), anti-IRAK-1 antibody (cat. no.
NBP1-77068; 1:50 in TBST; Novus Biologicals, LLC), anti-TRAF-6
antibody (cat. no. NBP1-33357; 1:50 in TBST; Novus Biologicals,
LLC), anti-TICAM-2 (E-2) antibody (cat. no. sc-376076; 1:50 in
TBST; Santa Cruz Biotechnology, Inc.), or normal rat IgG (cat. no.
sc-2026; 1:50 in TBST; Santa Cruz Biotechnology, Inc.) at 4°C for 2
h and then with 20 µl protein A/G-agarose (Beyotime
Institute of Biotechnology) at 4°C with rocking overnight. The
pellets obtained after centrifugation at 14,000 × g for 5 sec were
washed five times with washing buffer [50 mM Tris (pH 7.5), 7 mM
MgCl2, 2 mM EDTA and 1 mM PMSF (phenylmethylsulfonyl
fluoride)]. The pellets were resolved by 1X SDS-PAGE loading buffer
and boiled for 10 min. Following centrifugation, the supernatants
were obtained as immunoprecipitates for western blotting analysis
by using anti-PALM3 antibody, anti-TLR4 antibody, anti-MyD88
antibody, anti-IRAK-1 antibody, anti-TRAF-6 antibody or
anti-TICAM-2 antibody.
Statistical analysis
All data are presented as means ± standard error of
the mean. Comparison of means was performed by a one-way analysis
of variance, and the Student-Newman-Keuls test was used to analyze
comparisons between multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
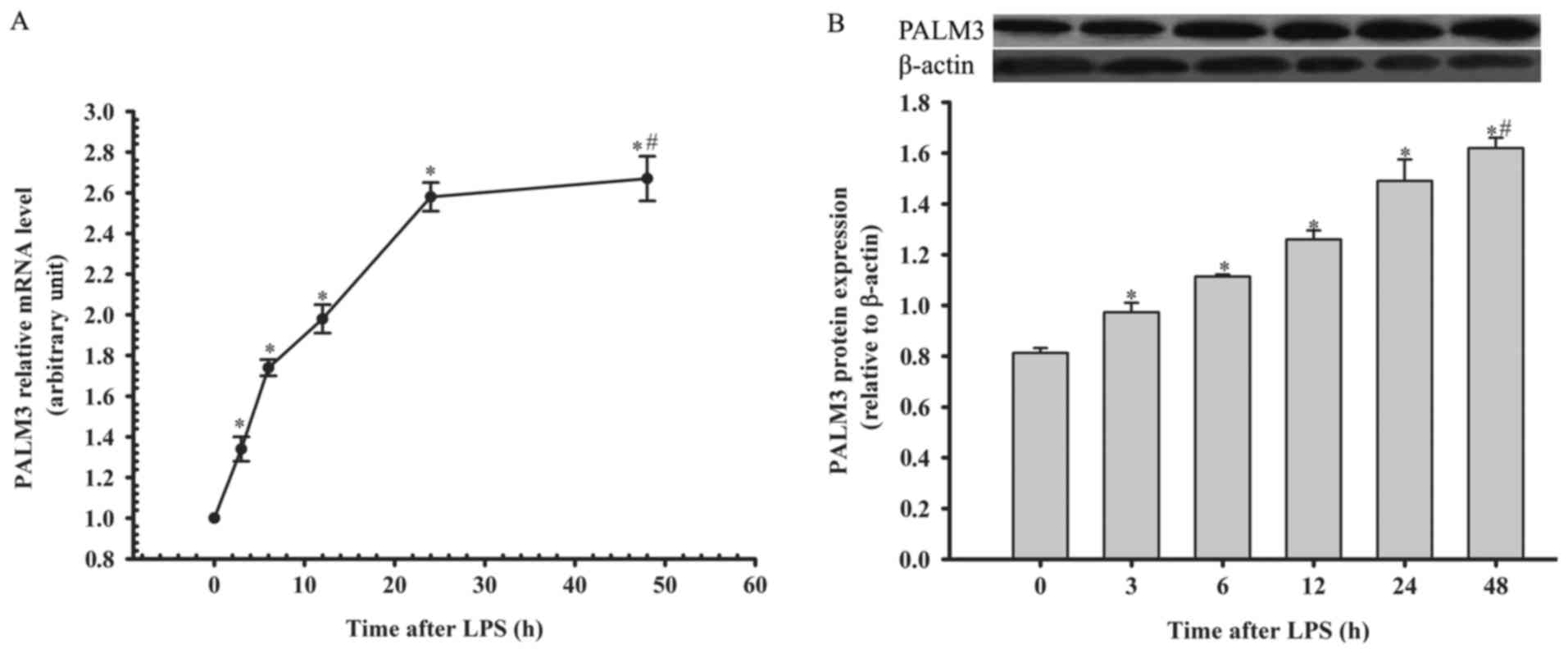
Expression of PALM3 after LPS stimulation
in rat AMs
The authors detected PALM3 gene and protein
expression in rat AMs by using RT-qPCR and western blot analysis at
0, 3, 6, 12, 24 and 48 h following LPS stimulation. The PALM3 gene
and protein expression in rat AMs were both upregulated following
the administration of LPS in a time-dependent manner (P<0.05 vs.
their respective 0 h time points; Fig. 1). However, there was no
significant difference in the PALM3 expression levels between the
24 and 48 h time points (P>0.05 for the 24 h time point vs. 48 h
time point; Fig. 1).
Expression of PALM3 after adenovirus
transfection in rat AMs
At 48 h following adenovirus transfection, the total
RNA and protein of rat AMs were isolated for an analysis of the
PALM3 expression. Compared with the normal (untransfected) and Ad.V
(Ad.V-transfected as a negative control) groups, the PALM3 mRNA and
protein levels in rat AMs in the Ad.rPALM3 (Ad.rPALM3-tranfected)
group were significantly enhanced (P<0.05 vs. normal and Ad.V
groups; Fig. 2A and B). However,
in the Ad.shRNA (Ad.shRNA-transfected) group, the PALM3 mRNA and
protein expression were significantly inhibited (P<0.05 vs.
normal and Ad.V groups; Fig. 2A and
B).
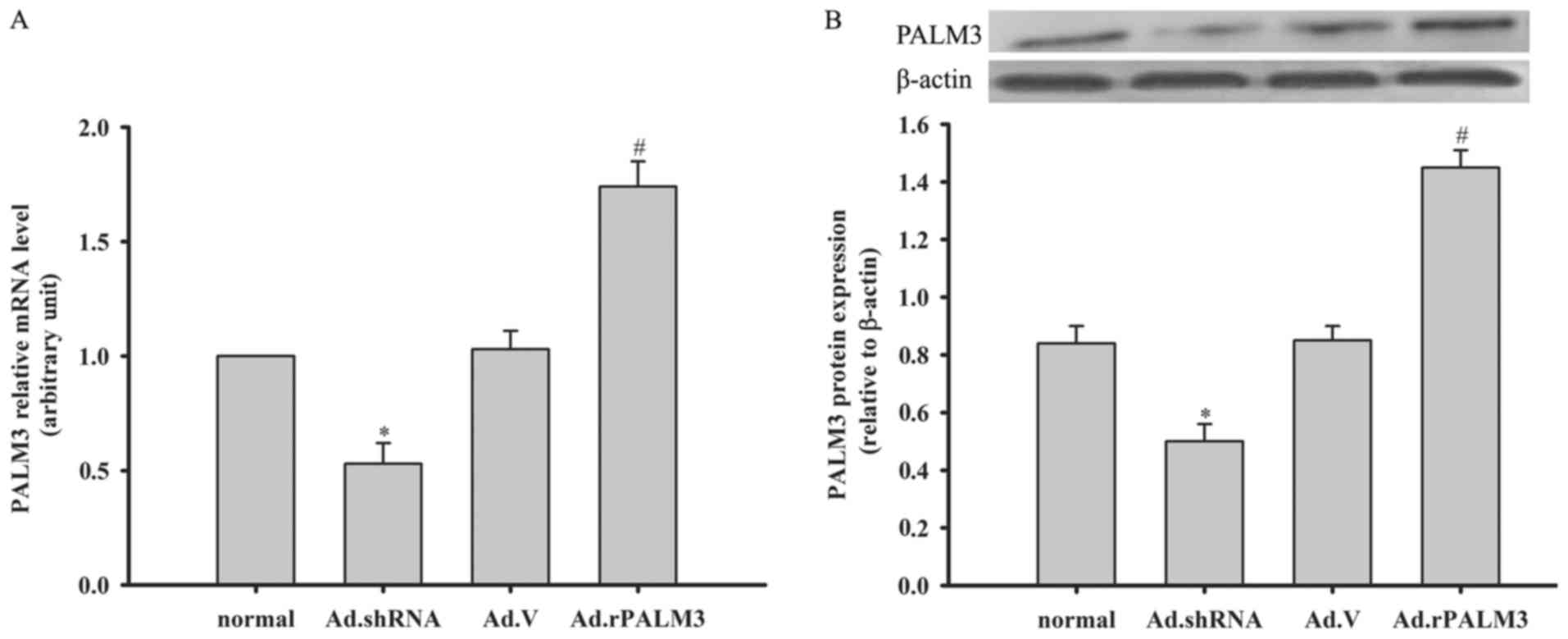 | Figure 2Expression of PALM3 after adenovirus
transfection in rat AMs. The (A) mRNA levels and (B) protein levels
of PALM3 in untransfected (normal) or adenovirus-transfected rat
AMs at 48 h following transfection with Ad.shRNA, Ad.V, or
Ad.rPALM3 were determined by reverse transcription-quantitative
polymerase chain reaction and western blotting, respectively. The
upper panels (B) show representative blots, and the lower panels
(B) represent densitometry analyses of the bands. Data are
expressed as the mean ± standard error of the mean of three
independent experiments. *P<0.05 vs. Ad.V and normal
groups; #P<0.05 vs. Ad.V and normal groups. AMs,
alveolar macrophages; shRNA, short hairpin RNA; Ad, adenovirus;
normal, untransfected cells; Ad.shRNA, Ad.shRNA-transfected cells;
Ad.V, Ad.V-transfected cells; Ad.rPALM3, Ad.rPALM3-transfected
cells. |
Effect of PALM3 on the production of
cytokines in LPS-stimulated rat AMs
The influence of PALM3 on the levels of TNF-α,
IL-1β, IL-10, IFN-α, IFN-β and MIF was measured by ELISAs in
LPS-stimulated NR8383 cells. The ELISA results show that the levels
of TNF-α, IL-1β, IL-10, IFN-α, IFN-β and MIF were very low in the
culture supernatant of LPS-unstimulated cells (Fig. 3). The administration of Ad.V,
Ad.shRNA or Ad.rPALM3 did not influence the level of cytokine
(TNF-α, IL-1β, IL-10, IFN-α, IFN-β and MIF) release before LPS
stimulation (P>0.05 vs. normal cells; Fig. 3). However, in response to LPS,
there was a marked increase in the levels of TNF-α, IL-1β, IL-10,
IFN-α, IFN-β, and MIF in all LPS-stimulated cells (P<0.05 vs.
unstimulated cells). Ad.rPALM3 pretreatment increased the amount of
proinflammatory cytokines (TNF-α, IL-1β, IFN-α, IFN-β and MIF) but
decreased the amount of an anti-inflammatory cytokine (IL-10)
released from rat AMs after LPS stimulation (P<0.05 vs. Ad.V +
LPS, normal + LPS, and Ad.shRNA + LPS groups; Fig. 3). Downregulation of PALM3 through
Ad.shRNA pretreatment inhibited LPS-induced expression of these
proinflammatory cytokines but promoted LPS-induced expression of an
anti-inflammatory cytokine (IL-10) (P<0.05 vs. Ad.V + LPS,
normal + LPS, and Ad.rPALM3 + LPS groups; Fig. 3). However, these changes in the
levels of cytokines were not observed in the Ad.V-transfected group
after LPS challenge (P>0.05 vs. normal + LPS group; Fig. 3).
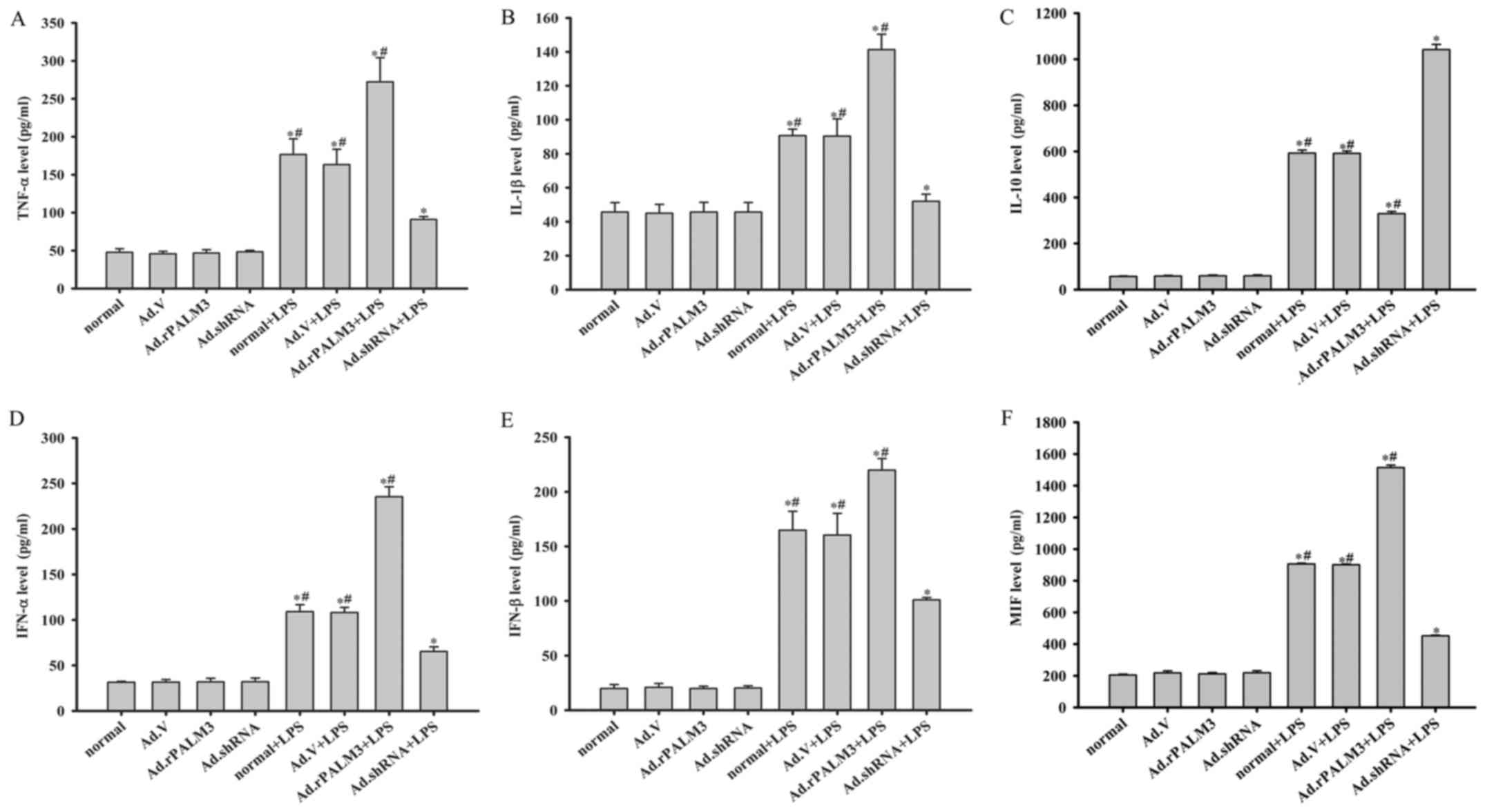 | Figure 3Effect of PALM3 on cytokine
production in LPS-stimulated rat AMs. Cells from each group of rat
AMs were treated with LPS (0.5 µg/ml) or left untreated. The
secretion levels of (A) TNF-α, (B) IL-1β, (C) IL-10, (D) IFN-α, (E)
IFN-β and (F) MIF were measured by using appropriate ELISA kits.
Data are expressed as the mean ± standard error of the mean of
three independent experiments. *P<0.05 vs. normal
group, Ad.V group, Ad.rPALM3 and Ad.shRNA groups;
#P<0.05 vs. Ad.shRNA + LPS group. LPS,
lipopolysaccharides; AMs, alveolar macrophages; LPS,
lipopolysaccharides; shRNA, short hairpin RNA; Ad, adenovirus;
normal, untransfected cells; Ad.shRNA, Ad.shRNA-transfected cells;
Ad.V, Ad.V-transfected cells; Ad.rPALM3, Ad.rPALM3-transfected
cells; normal + LPS, LPS-stimulated untransfected cells; Ad.V +
LPS, LPS-stimulated Ad.V-transfected cells; Ad.rPALM3 + LPS,
LPS-stimulated Ad.rPALM3-transfected cells; Ad.shRNA + LPS,
LPS-stimulated Ad.shRNA-transfected cells; TNF-α, tumor necrosis
factor-α; IL, interleukin; IFN, interferon; MIF, macrophage
migration inhibitory factor. |
Effect of PALM3 on the activity of NF-κB
in LPS-stimulated rat AMs
NF-κB is a critical transcription factor required
for the maximal expression of many cytokines (22). As presented in Fig. 4, adenovirus vectors, Ad.V,
Ad.rPALM3 and Ad.shRNA, had no impact on NF-κB activity before LPS
stimulation (P>0.05 vs. normal group) (Fig. 4A). All LPS-stimulated rat AMs
exhibited a marked increase in NF-κB-DNA-binding in comparison with
cells not treated with LPS (Fig.
4). Additionally, Ad.rPALM3 pretreatment further increased the
LPS-induced NF-κB activation (P<0.05 vs. Ad.V + LPS, normal +
LPS and Ad.shRNA + LPS groups; Fig.
4). However, Ad.shRNA pretreatment significantly attenuated the
LPS-induced increase in NF-κB activation compared with the Ad.V +
LPS and normal + LPS groups (P<0.05 vs. Ad.V + LPS, normal + LPS
and Ad.rPALM3 + LPS groups).
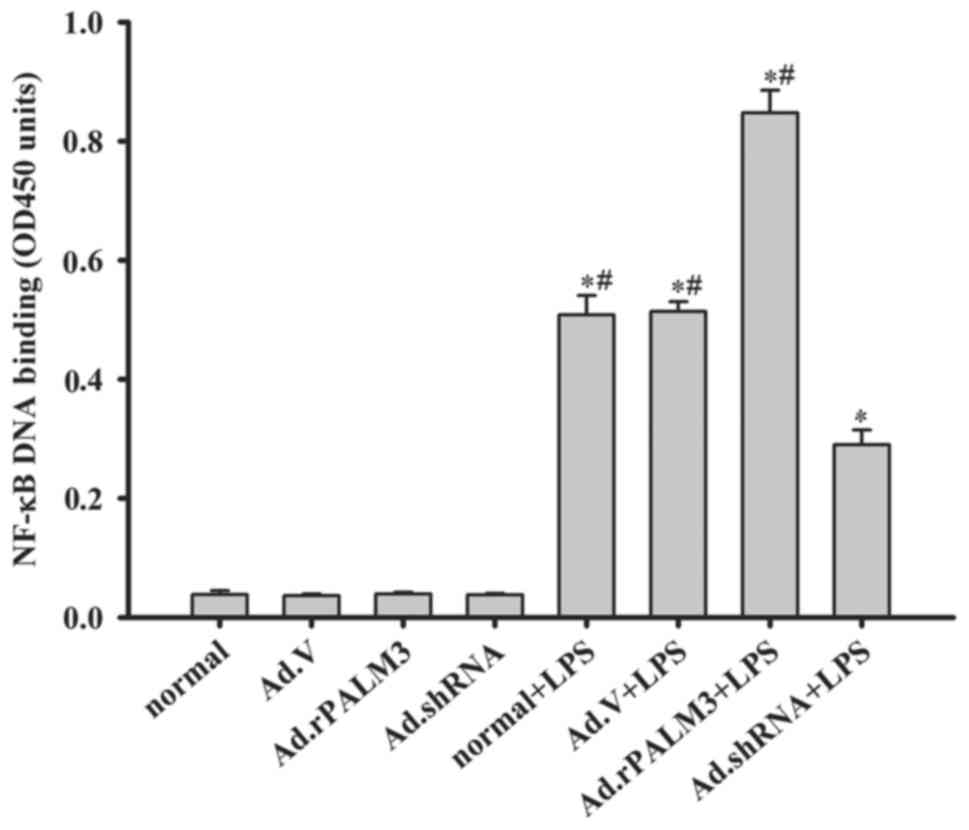 | Figure 4Effect of PALM3 on the NF-κB activity
in LPS-stimulated rat AMs. NF-κB activity was assessed by
performing ELISAs on isolated nuclear extracts of untransfected
(normal) or adenovirus-transfected rat AMs that had been stimulated
with LPS or left untreated. Data are expressed as the mean ±
standard error of the mean of three independent experiments.
*P<0.05 vs. normal group, Ad.V group, Ad.rPALM3, and
Ad.shRNA groups; #P<0.05 vs. Ad.shRNA + LPS group.
AMs, alveolar macrophages; NF-κB, nuclear factor-κB; OD, optical
density; LPS, lipopolysaccharides; shRNA, short hairpin RNA; Ad,
adenovirus; normal, untransfected cells; Ad.shRNA,
Ad.shRNA-transfected cells; Ad.V, Ad.V-transfected cells;
Ad.rPALM3, Ad.rPALM3-transfected cells; normal + LPS,
LPS-stimulated untransfected cells; Ad.V + LPS, LPS-stimulated
Ad.V-transfected cells; Ad.rPALM3 + LPS, LPS-stimulated
Ad.rPALM3-transfected cells; Ad.shRNA + LPS, LPS-stimulated
Ad.shRNA-transfected cells. |
Effect of PALM3 on the IRF3 activity in
LPS-stimulated rat AMs
IRF3 is also an important transcription factor in
cells and serves a role in the post-transcriptional regulation of
the proinflammatory cytokines TNF-α and IL-1β (23). In addition, the authors examined
the phospho-IRF-3 protein level in nuclear extracts by using
western blot analysis to detect the influence of PALM3 on
LPS-induced IRF-3 activation. The results demonstrated that the
phospho-IRF-3 protein levels in all LPS-stimulated rat AMs were
significantly higher than those in normal rat AMs (P<0.05 vs.
normal group; Fig. 5).
Additionally, Ad.rPALM3 pretreatment further increased the
phospho-IRF-3 protein level in rat AMs after LPS stimulation
(P<0.05 vs. Ad.V + LPS, normal + LPS, and Ad.shRNA + LPS groups;
Fig. 5). However, Ad.shRNA
pretreatment significantly attenuated the phospho-IRF-3 protein
level in rat AMs after LPS stimulation, compared with the Ad.V +
LPS and normal + LPS groups (P<0.05 vs. Ad.V + LPS, normal + LPS
and Ad.rPALM3 + LPS groups). Pretreatment with Ad.V had no impact
on the LPS-induced increase in phospho-IRF-3 protein level
(P>0.05 vs. normal group; Fig.
5).
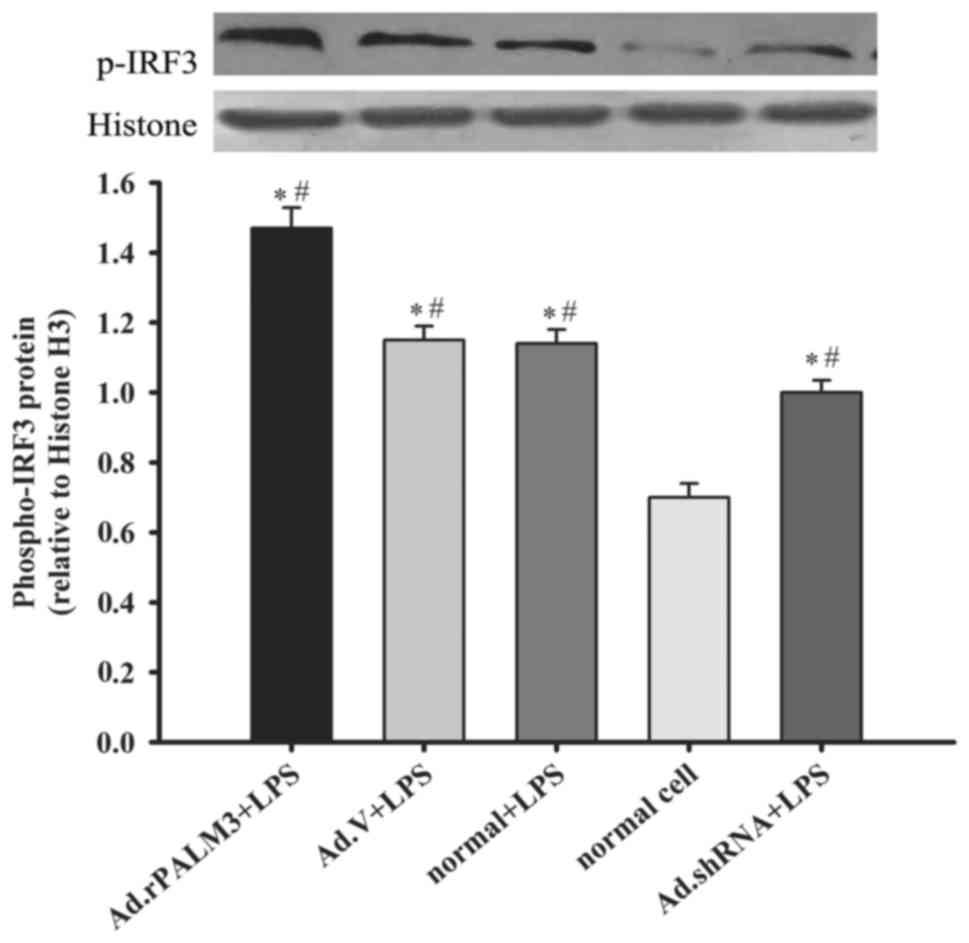 | Figure 5Effect of PALM3 on the IRF-3 activity
in LPS-stimulated rat AMs. The phosphor-IRF-3 protein levels in
nuclear extracts of LPS-stimulated untransfected (normal) or
adenovirus-transfected rat AMs were determined by western blotting.
The upper panel shows a representative blot, and the lower panel
represents densitometry analyses of the bands. Data are expressed
as the mean ± standard error of the mean of three independent
experiments. *P<0.05 vs. normal group;
#P<0.05 vs. Ad.shRNA + LPS group. AMs, alveolar
macrophages; LPS, lipopolysaccharides; shRNA, short hairpin RNA;
Ad, adenovirus; normal, untransfected cells; normal + LPS,
LPS-stimulated untransfected cells; Ad.V + LPS, LPS-stimulated
Ad.V-transfected cells; Ad.rPALM3 + LPS, LPS-stimulated
Ad.rPALM3-transfected cells; Ad.shRNA + LPS, LPS-stimulated
Ad.shRNA-transfected cells. |
Co-localization of TLR4, MyD88, IRAK-1,
TRAF-6, and TICAM-2 with PALM3
The cellular localizations of TLR4, MyD88, IRAK-1,
TRAF-6, TICAM-2 and PALM3 in NR8383 cells were observed by
performing confocal microscopy. Cells were incubated at 4°C with
anti-TLR4, anti-MyD88, anti-IRAK-1, anti-TRAF-6, anti-TICAM-2 and
anti-PALM3 antibodies. Corresponding fluorescence-conjugated
secondary antibodies were used to visualize the localization of
TLR4, MyD88, IRAK-1, TRAF-6, TICAM-2 and PALM3. The resulting
fluorescence signals revealed that, before LPS stimulation, both
TLR4 and PALM3 were primarily located on the cell membrane
(Fig. 6A1 and A2). When the
TLR4-FITC and PALM3-Cy3 localization patterns were superimposed,
co-localization of these signals was observed (Fig. 6A3). These results indicated that
TLR4 and PALM3 co-localized on the plasma membranes of rat AMs
before LPS stimulation. Following LPS stimulation, some
TLR4-specific and PALM3-specific signals were still observed in the
plasma membrane, but they were additionally observed co-localizing
in the cell cytoplasm (Fig.
6B1–3). This result suggested that, like the LPS-induced
internalization of TLR4, PALM3 also translocated into the cytoplasm
in response to LPS stimulation.
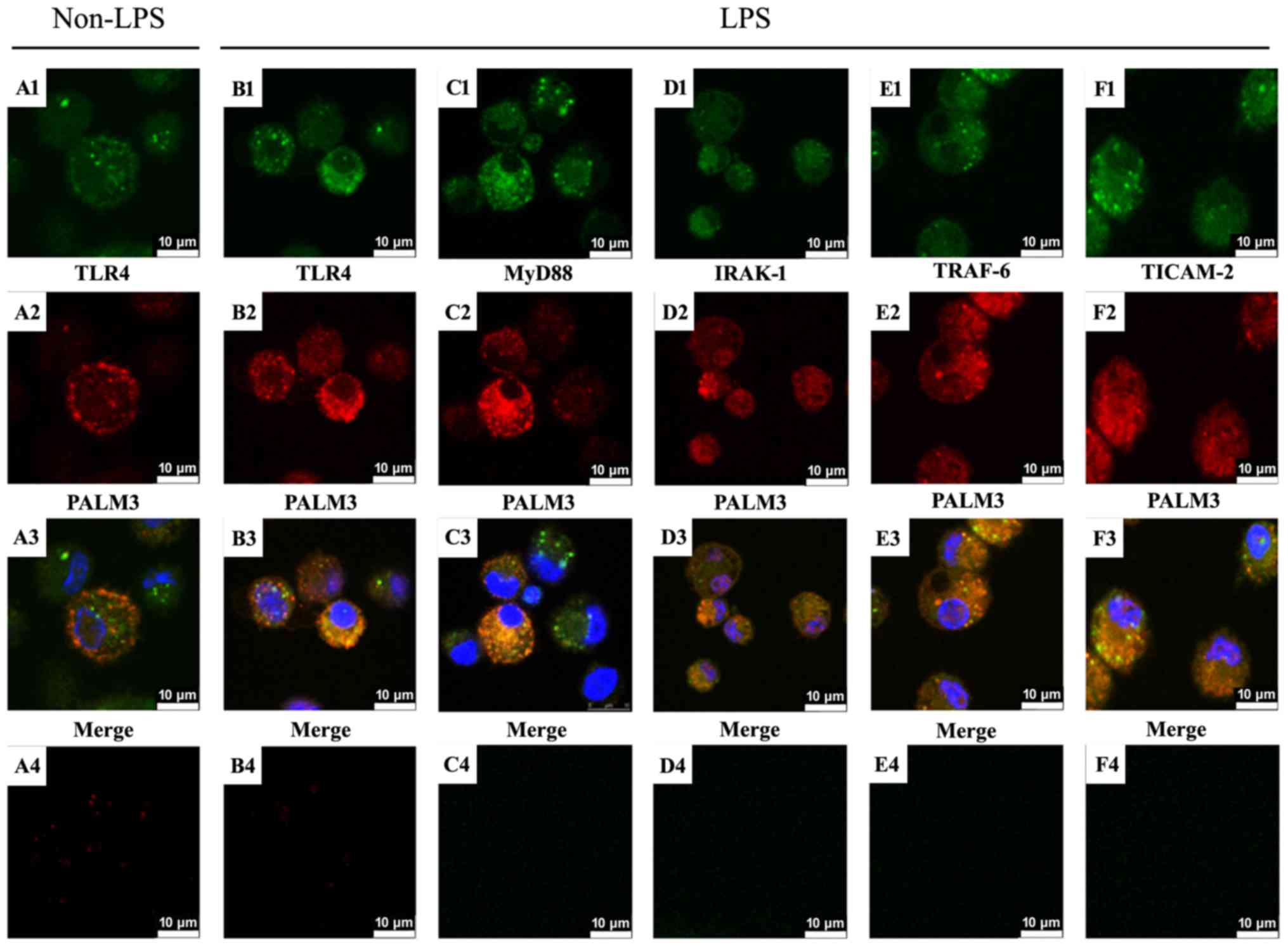 | Figure 6Localization of PALM3 and LPS-TLR4
signaling adaptor proteins in rat AMs. Rat AMs were incubated with
anti-PALM3 antibody along with anti-TLR4, anti-MyD88, anti-IRAK-1,
anti-TRAF-6, or anti-TICAM-2 antibodies, followed by incubation
with corresponding fluorescein isothiocyanate-conjugated or
Cy3-conjugated secondary antibodies. Nuclei were counterstained
with DAPI. The TLR4-, MyD88-, IRAK-1-, TRAF-6- and TICAM-2-specific
signals resulting from the immunofluorescence staining are shown in
green (A1, B1, C1, D1, E1, F1), and the PALM3-specific signals are
shown in red (A2, B2, C2, D2, E2, F2). The resulting merged images
highlight co-localizations (shown in yellow) of the signals for
TLR4, MyD88, IRAK-1, TRAF-6, or TICAM-2 and PALM3 (A3, B3, C3, D3,
E3, F3). Negative controls, which were not treated with primary
antibodies, are shown in A4, B4, C4, D4, E4, F4. LPS,
lipo-polysaccharides; TLR4, Toll-like receptor 4; AMs, alveolar
macrophages; MyD88, myeloid differentiation factor 88; IRAK,
interleukin 1 receptor associated kinase; TRAF, tumor necrosis
factor receptor associated factor; TICAM2, Toll-like receptor
adaptor molecule 2. |
The immunofluorescence results also demonstrated
that MyD88, IRAK-1, TRAF-6 and TICAM-2 localized in both the cell
cytoplasm and plasma membrane after LPS stimulation (Fig. 6C1–F1). Moreover, as presented in
Fig. 6C2–F2, PALM3-specific
signals similarly localized not only on the plasma membrane but
also in the cell cytoplasm in the presence of LPS. When the
MyD88-specific, IRAK-1-specific, TRAF-6-specific or
TICAM-2-specific and PALM3-specific signals were superimposed, it
was observed that PALM3 co-localized with MyD88, IRAK-1, TRAF-6,
and TICAM-2 (Fig. 6C3–F3).
Positive staining signals were not observed in the negative
controls, which lacked the addition of primary antibody (Fig. 6A4–F4).
PALM3 interacted with the adaptors of
LPS-TLR4 signaling
As TLR4, MyD88, IRAK-1, TRAF-6 and Toll-IL-1
receptor containing adapter molecule (TIACM)-2 are believed to be
the key adaptor proteins in LPS-TLR4 signaling, the authors used
co-immunoprecipitation assays to examine if PALM3 can interact with
these adaptors of LPS-TLR4 signaling. Following LPS stimulation,
PALM3 protein was detected by western blot analysis after
co-precipitation with anti-TLR4, anti-MyD88, anti-IRAK-1,
anti-TRAF-6 and anti-TICAM-2 antibodies (Fig. 7). However, when the same assays
were conducted on non-LPS-stimulated cells, PALM3 protein was only
detected after co-precipitation with an anti-TLR4 antibody
(Fig. 7). Notably, PALM3 protein
was not detected after co-precipitation with normal nonspecific IgG
(Fig. 7). These results suggested
that PALM3 interacts with TLR4 under normal conditions, but it also
interacts with MyD88, IRAK-1, TRAF-6 and TICAM-2 in a
ligand-dependent manner.
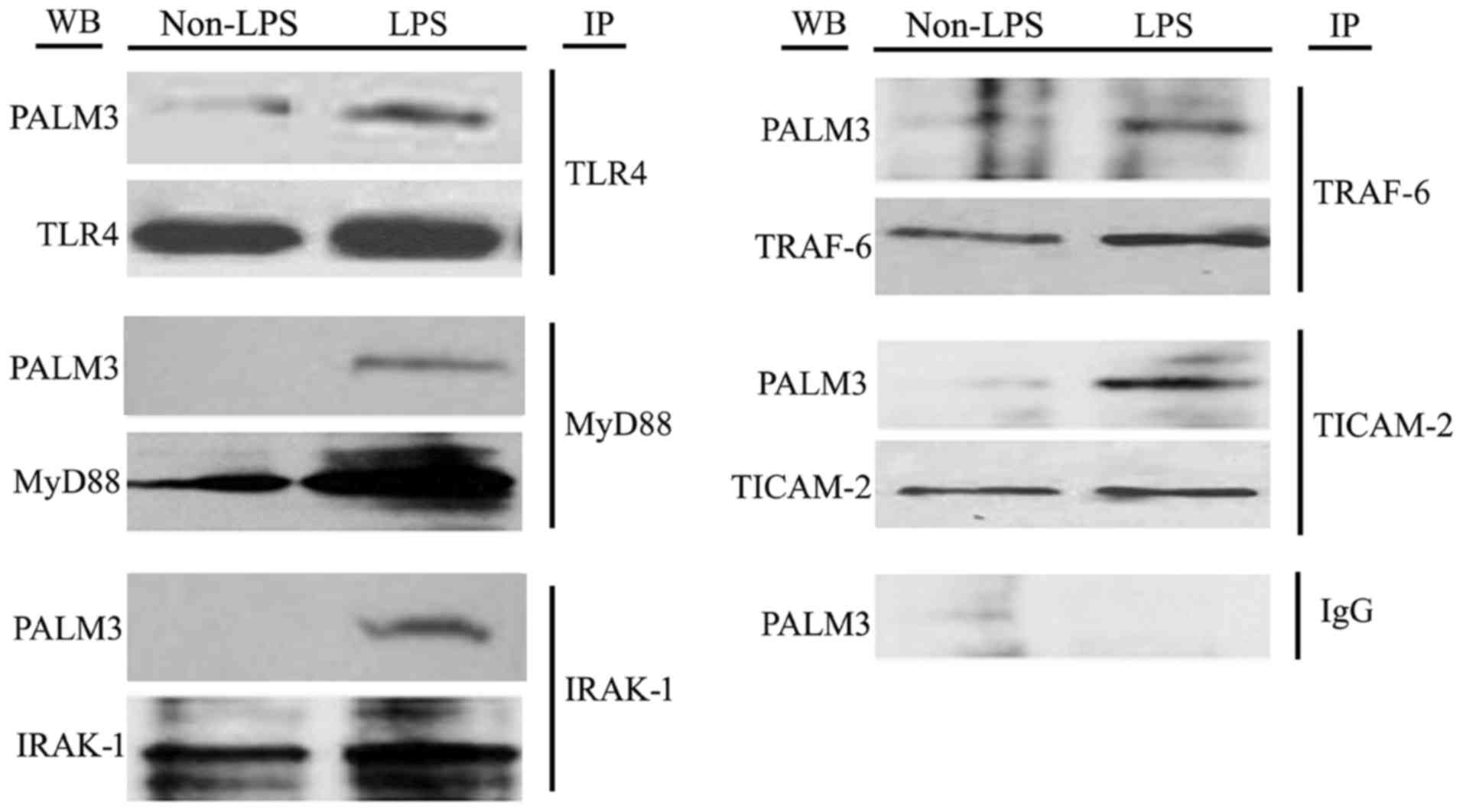 | Figure 7Co-immunoprecipitation assays of
TLR4, MyD88, IRAK-1, TRAF-6 and TICAM-2 with PALM3 in
LPS-stimulated rat AMs. IP, immunoprecipitation: the total cell
lysates were immunoprecipitated with anti-TLR4, anti-MyD88,
anti-IRAK-1, anti-TRAF-6, anti-TICAM-2 or normal IgG antibodies;
WB, western blot analysis: the immunocomplexes were examined by
western blot analysis using the indicated antibodies; TLR4,
Toll-like receptor 4; AMs, alveolar macrophages; MyD88, myeloid
differentiation factor 88; IRAK, interleukin 1 receptor associated
kinase; TRAF, tumor necrosis factor receptor associated factor;
TICAM2, Toll-like receptor adaptor molecule 2; LPS,
lipopolysaccharide; AMs, alveolar macrophages; IgG, immunoglobulin
G. |
Discussion
In the present study, it was demonstrated that the
PALM3 expression level was upregulated by the administration of
LPS, and modulating the expression level of PALM3 could affect both
the production of cytokines (TNF-α, IL-1β, IL-10, IFN-α, IFN-β and
MIF) and the activation of NF-κB and IRF-3 in rat AMs after LPS
stimulation. Additionally, PALM3 was found to interact with TLR4,
IRAK-1, TRAF-6 and TIACM-2 during LPS-TLR4 signaling in rat AMs. To
the best of the authors' knowledge, the current study is the first
to demonstrate that PALM3 contributes to the LPS-induced
inflammatory response and is involved in LPS-TLR4 signaling in
AMs.
ALI/ARDS is a lung inflammation disorder with a high
mortality rate (2), and its
typical pathological characteristic is an excessive lung
inflammation response (24).
LPS-TLR4 signaling is one of the most important inflammatory
response signal pathways (25).
As AMs are the major effector cells that participate in the
initiation and development of ALI (4,5,26),
modulating the excessive inflammatory response to
pathogen-associated molecular patterns in AMs during systemic
inflammatory response syndrome is a therapeutic target for treating
ALI/ARDS. PALM3 is a novel interactive partner of single
immunoglobulin IL-1 receptor-related molecule (SIGIRR) and may
function as an adaptor in LPS-TLR4 signaling (19). Downregulation of PALM3 has been
reported to benefit mice with LPS-induced ALI by means of
suppressing the inflammatory response, ameliorating histological
tissue injury, and decreasing the permeability of the alveolar
capillary barrier (20).
Additionally, the preliminary data also showed that the
downregulation of PALM3 protected rats from LPS-induced ALI, and
its mechanisms were partially associated with the modulation of
cytokine secretion and inhibition of NF-κB and IRF3 activation. In
the present study, the authors indicated that the downregulation of
PALM3 significantly suppressed the inflammatory response to LPS in
AMs. Therefore, inhibiting the LPS-induced inflammatory response in
AMs may be part of the mechanism for the protective effect of
PALM3-knockdown on ALI.
At present, little is understood regarding the
biological function of the PALM3. Previous studies have
demonstrated that PALM3 may act as an adaptor to link intrinsic
membrane proteins to each other, to the cytoskeleton, or to motor
proteins (14,19). Previous work of the authors
revealed that LPS upregulated the PALM3 expression in alveolar
epithelial cells (19).
Furthermore, the present results show that PALM3 expression was
detected and also upregulated in a time-dependent manner in rat AMs
following LPS stimulation, which is similar to previous results in
alveolar epithelial cells (19).
The PALM3 expression pattern is similar to the expression patterns
of TLR4, TIR domain-containing adaptor inducing IFN-β (TRIF) and
MyD88 (27,28), and the upregulation of PALM3 may
be related to its functional involvement in LPS-TLR4 signal
transduction, similar to the adaptors in TLR signaling (19). To elucidate the role of PALM3 in
LPS-TLR4 signaling in AMs, the authors used a recombinant
adenovirus expressing shRNA for PALM3 (Ad.shRNA) and a
recombinant adenovirus carrying the rat PALM3 gene
(Ad.rPALM3) to modulate the expression of PALM3 in rat AMs. The
results of western blotting and RT-qPCR analyses demonstrate that
using these recombinant adenoviral vectors successfully modulated
the PALM3 gene transcript in AMs.
Previous studies have demonstrated that the
down-regulation of PALM3 inhibits the LPS-induced release of
inflammatory cytokines in alveolar epithelial cells and had a
protective effect against LPS-induced ALI in mice (19,20). In the current study, we identified
a similar result in rat AMs. That is, the downregulation of PALM3
by transfection with Ad.shRNA suppressed the release of
proinflammatory cytokines (TNF-α, IL-1β, IFN-α, IFN-β and MIF), but
the upregulation of PALM3 by transfection with Ad.rPALM3 promoted
the production of proinflammatory cytokines (TNF-α, IL-1β, IFN-α,
IFN-β and MIF) in LPS-stimulated AMs. The increase in the
production of proinflammatory cytokines in Ad.rPALM3-pretreated
cells may be due to the promotion of LPS-TLR4 signal transduction
by the enhanced expression of PALM3. Correspondingly, the
downregulation of PALM3 may decrease the promotion effect of PALM3
on proinflammatory cytokine secretion. In contrast, modulating the
PALM3 expression level had the opposite effect on the secretion of
the anti-inflammatory cytokine IL-10. Moreover, the authors
examined the effect of PALM3 on the activation of NF-κB and IRF-3.
The results demonstrated that the activity levels of transcription
factor IRF-3 and NF-κB were significantly strengthened in
Ad.rPALM3-pretreated cells following LPS stimulation, compared with
the Ad.V-pretreated cells. Correspondingly, the downregulation of
PALM3 by pretreatment with Ad.shRNA suppressed the
NF-κB-DNA-binding activity and decreased the phospho-IRF3 protein
levels in the nucleus of rat AMs. These changes in the IRF-3 and
NF-κB activity support the idea that enhanced PALM3 expression may
promote the transduction of LPS-TLR4 signaling while interfering
with PALM3 expression might impede this signal transduction.
Determining the detailed molecular mechanisms of this process will
require further investigations.
Protein-protein interaction is an important
molecular mechanism in signal transduction (29). LPS can induce the dimerization of
TLR4, which results in conformational changes of the TLR4 homodimer
that induce the recruitment of adaptor proteins containing
Toll/interleukin-1 receptor-like (TIR) domains. In the
MyD88-dependent pathway, the TLR4 TIR domains recruit TIR
domain-containing adaptor proteins MyD88-adaptor-like (MAL) and
MyD88, and in the MyD88-independent pathway, these domains recruit
TIR domain-containing adaptor inducing IFN-β (TRIF) and TICAM-2
(11). The MyD88-dependent
pathway involves the recruitment and activation of IRAKs and
TRAF-6, and the activation of this pathway induces the activation
and translocation in the nucleus of transcription factors, such as
NF-κB and activator protein-1 (AP-1) (11). The MyD88-independent pathway
involves TICAM-1 and TICAM-2 adaptor proteins, and the activation
of this signaling pathway leads to the activation and translocation
in the nucleus of the transcription factor IRF3 (30). Furthermore, the MyD88-dependent
pathway induces the production of proinflammatory cytokines, while
the MyD88-independent pathway induces the production of type I
interferons (11).
In the previous study, the authors found that PALM3
can interact with SIGIRR, a negative regulator of TLR signaling
(19). Based on what is currently
known about SIGIRR, the authors presumed that, as an interaction
partner of SIGIRR, PALM3 may be involved in TIR signaling.
Additionally, because PALM3 can function as an adaptor that links
intrinsic membrane proteins to each other (14), it was also suspected that PALM3
may function as an adaptor in TLR signaling. Herein, the results of
co-immunoprecipitation assays showed that PALM3 interacted with
TLR4, IRAK-1, TRAF-6 and TIACM-2 in rat AMs following LPS
stimulation. Moreover, the interaction between PALM3 and TLR4 was
independent of LPS stimulation. Meanwhile, the results of
immunofluorescence staining revealed that PALM3 and TLR4 proteins
co-localize in rat AMs whether or not the cells are stimulated by
LPS, demonstrating that PALM3 is located in the cell membrane under
normal conditions and can internalize into the cell cytoplasm with
the translocation of TLR4 after LPS stimulation. Together, these
results suggested that the interaction between PALM3 and TLR4
exists in the natural state, independent of LPS stimulation.
Past work has indicated that the adaptor proteins
MyD88, IRAK-1, TRAF-6 and TIACM-2 are located in the cell cytoplasm
in the natural state (11,31),
and these adaptors can be recruited to the cell membrane through
homophilic interactions between TIR domains in the cytoplasmic tail
of TLR4 and those present on the adaptors after LPS stimulation
(11). Our findings are similar
to the results of these previous studies (11,31). The adaptor proteins MyD88, IRAK-1,
TRAF-6 and TIACM-2 were localized in the cell cytoplasm and on the
plasma membrane in the presence of LPS. This supports the spatial
possibility that an interaction between PALM3 and the adaptor
proteins, i.e. MyD88, IRAK-1, TRAF-6 and TIACM-2, exists in the
presence of LPS. All of these data support the idea that PALM3 can
interact with TLR4, MyD88, IRAK-1, TRAF-6 and TIACM-2 in rat AMs.
Notably, MyD88, IRAK-1 and TRAF-6 are adaptor proteins of the
MyD88-dependent pathway, but TIACM-2 is an adaptor protein of the
MyD88-independent pathway (11,30). Therefore, the current results
suggested that PALM3 may function as an adaptor that participates
in both the MyD88-dependent and MyD88-independent pathways.
However, whether or not PALM3 serves the same role in LPS-TLR4
signaling and possess similar regulation effects in an ALI animal
model warrants further investigation. Additionally, the authors
used a rat NR8383 macrophage cell line to investigate the role of
PALM3 in LPS-TLR4 signaling in the present study. Further
experiments in primary AMs isolated from an ALI animal model will
be needed to confirm these findings on the regulation effect of
PALM3 on LPS-TLR4 signaling.
TLR4 belongs to a family of pattern recognition
receptors and has been recognized as the sensing receptor for LPS
(11). In the present study, the
results showed that PALM3 co-localized and interacted with TLR4
protein in rat AMs in an LPS-independent manner. In contrast, the
interaction between PALM3 and the adaptor proteins, MyD88, IRAK-1,
TRAF-6 and TIACM-2, depended on the presence of LPS. In addition,
previous studies have demonstrated that PALM3 interacts with SIGIRR
(a negative regulator of TLR signaling) (19) and functions as an adaptor that
links intrinsic membrane proteins to each other (14). Therefore, it was proposed that
PALM3 may act as an 'adaptor' or a 'bridge' between TLR4 and the
proteins MyD88, IRAK-1, TRAF-6 and TIACM-2 in the LPS-TLR4 signal
transduction pathway, which may enhance the interaction between
TLR4 and these adaptor proteins. Subsequently, this interaction
enhancement may further lead to inflammation. The results do not
reveal whether or not the interaction between PALM3 and the adaptor
proteins, MyD88, IRAK-1, TRAF-6 and TIACM-2, depends on TLR4. This
question remains to be confirmed by experiments using small
interfering RNA or an inhibitor to downregulate TLR4. Although it
is believed that this issue is an important subject worth studying
in LPS-TLR4 signaling, it was not the focus of the present study.
Moreover, the mechanisms relevant to the interactions between the
adaptors and LPS-induced inflammatory signaling are very complex,
beyond simply the effect of TLR4. Therefore, a separate, but more
extensive, study of this topic is planned in the future. In
conclusion, the authors have demonstrated that PALM3 can interact
with TLR4, MyD88, IRAK-1, TRAF-6 and TIACM-2 after LPS stimulation
and that upregulation of PALM3 aggravates the LPS-induced
inflammation, while downregulation of PALM3 suppresses the
LPS-induced inflammation in rat AMs. Therefore, the authors
hypothesized that PALM3 may function as an adaptor to participate
in the transduction of LPS-TLR4 signaling and contribute to
LPS-induced inflammatory responses in AMs. Modulating PALM3
expression may be a potential novel target for treating
macrophage-associated inflammatory diseases. Future research will
focus on the issues emerging from this study and on the detailed
mechanisms by which PALM3 participates in LPS-TLR4 signal
transduction.
Acknowledgments
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no. 81300050)
and the Innovative Cultivation Foundation of Navy General Hospital
of PLA (grant no. CXPY201417).
Glossary
Abbreviations
Abbreviations:
|
ALI
|
acute lung injury
|
|
AM
|
alveolar macrophage
|
|
AP-1
|
activator protein-1
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
IFN
|
interferon
|
|
IL
|
interleukin
|
|
IRAK
|
interleukin 1 receptor associated
kinase
|
|
IRF-3
|
IFN regulatory factor 3
|
|
LBP
|
LPS-binding protein
|
|
LPS
|
lipopolysaccharide
|
|
MAL
|
MyD88-adaptor-like
|
|
TLR
|
Toll-like receptor
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MyD88
|
myeloid differentiation factor 88
|
|
NF-κB
|
nuclear transcription factor κB
|
|
PALM3
|
parelemmin-3
|
|
SIGIRR
|
single immunoglobulin IL-1
receptor-related molecule
|
|
TIACM
|
Toll-IL-1 receptor containing adapter
molecule
|
|
TNF
|
tumor necrosis factor
|
|
TRAF
|
tumor necrosis factor receptor
associated factor
|
|
TRIF
|
TIR domain-containing adaptor inducing
IFN-β
|
References
|
1
|
Wang Q, Wang J, Hu M, Yang Y, Guo L and Xu
J, Lei C, Jiao Y and Xu J: Uncoupling protein 2 increases
susceptibility to lipopolysaccharide-induced acute lung injury in
mice. Mediators Inflamm. 2016:91542302016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Q, Zheng X, Cheng Y, Zhang YL, Wen
HX, Tao Z, Li H, Hao Y, Gao Y, Yang LM, et al: Resolvin D1
stimulates alveolar fluid clearance through alveolar epithelial
sodium channel, Na,K-ATPase via ALX/cAMP/PI3K pathway in
lipopolysaccharide-induced acute lung injury. J Immunol.
192:3765–3777. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu T, Wang DX, Zhang W, Liao XQ, Guan X,
Bo H, Sun JY, Huang NW, He J, Zhang YK, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-κB. PLoS One. 8:e564072013. View Article : Google Scholar
|
|
4
|
Han J, Li C, Liu H, Fen D, Hu W, Liu Y,
Guan C and Luo ZQ: Inhibition of lipopolysaccharide-mediated rat
alveolar macrophage activation in vitro by antiflammin-1. Cell Biol
Int. 32:1108–1115. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu DD, Pan PH, Liu B, Su XL, Zhang LM, Tan
HY, Cao Z, Zhou ZR, Li HT, Li HS, et al: Inhibition of alveolar
macrophage pyroptosis reduces lipopolysaccharide-induced acute lung
injury in mice. Chin Med J (Engl). 128:2638–2645. 2015. View Article : Google Scholar
|
|
6
|
Johnston LK, Rims CR, Gill SE, McGuire JK
and Manicone AM: Pulmonary macrophage subpopulations in the
induction and resolution of acute lung injury. Am J Respir Cell Mol
Biol. 47:417–426. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aggarwal NR, King LS and D'Alessio FR:
Diverse macrophage populations mediate acute lung inflammation and
resolution. Am J Physiol Lung Cell Mol Physiol. 306:L709–L725.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dagvadorj J, Shimada K, Chen S, Jones HD,
Tumurkhuu G, Zhang W, Wawrowsky KA, Crother TR and Arditi M:
Lipopolysaccharide induces alveolar macrophage necrosis via CD14
and the P2X7 receptor leading to interleukin-1α release. Immunity.
42:640–653. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takashima K, Matsushima M, Hashimoto K,
Nose H, Sato M, Hashimoto N, Hasegawa Y and Kawabe T: Protective
effects of intratracheally administered quercetin on
lipopolysaccharide-induced acute lung injury. Respir Res.
15:1502014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xi F, Liu Y, Wang X, Kong W and Zhao F:
LYATK1 potently inhibits LPS-mediated pro-inflammatory response.
Biochem Biophys Res Commun. 470:1–8. 2016. View Article : Google Scholar
|
|
11
|
Molteni M, Gemma S and Rossetti C: The
role of Toll-like receptor 4 in infectious and noninfectious
inflammation. Mediators Inflamm. 2016:69789362016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bordon Y: Inflammation: TNF snuffs out
steroids. Nat Rev Immunol. 14:2142014.PubMed/NCBI
|
|
13
|
Cornish JA, Kloc M, Decker GL, Reddy BA
and Etkin LD: Xlcaax-1 is localized to the basolateral membrane of
kidney tubule and other polarized epithelia during Xenopus
development. Dev Biol. 150:108–120. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu B, Petrasch-Parwez E, Laue MM and
Kilimann MW: Molecular characterization and immunohistochemical
localization of palmdelphin, a cytosolic isoform of the paralemmin
protein family implicated in membrane dynamics. Eur J Cell Biol.
84:853–866. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kutzleb C, Sanders G, Yamamoto R, Wang X,
Lichte B, Petrasch-Parwez E and Kilimann MW: Paralemmin, a
prenyl-palmitoyl-anchored phosphoprotein abundant in neurons and
implicated in plasma membrane dynamics and cell process formation.
J Cell Biol. 143:795–813. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Albrecht I, Bieri R, Leu A, Granacher P,
Hagmann J, Kilimann MW and Christofori G: Paralemmin-1 is expressed
in lymphatic endothelial cells and modulates cell migration, cell
maturation and tumor lymphangiogenesis. Angiogenesis. 16:795–807.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Turk CM, Fagan-Solis KD, Williams KE,
Gozgit JM, Smith-Schneider S, Marconi SA, Otis CN, Crisi GM,
Anderton DL, Kilimann MW, et al: Paralemmin-1 is over-expressed in
estrogen-receptor positive breast cancers. Cancer Cell Int.
12:172012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kutzleb C, Petrasch-Parwez E and Kilimann
MW: Cellular and subcellular localization of paralemmin-1, a
protein involved in cell shape control, in the rat brain, adrenal
gland and kidney. Histochem Cell Biol. 127:13–30. 2007. View Article : Google Scholar
|
|
19
|
Chen X, Wu X, Zhao Y, Wang G, Feng J, Li Q
and Qian G: A novel binding protein of single immunoglobulin IL-1
receptor-related molecule: Paralemmin-3. Biochem Biophys Res
Commun. 404:1029–1033. 2011. View Article : Google Scholar
|
|
20
|
Li S, Guo L, Zhao Y, Qian P, Lv X, Qian L,
Wang Q, Qian G, Yao W and Wu X: Silencing of paralemmin-3 protects
mice from lipopolysaccharide-induced acute lung injury. Peptides.
76:65–72. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Rao R, Nagarkatti P and Nagarkatti M: Role
of miRNA in the regulation of inflammatory genes in staphylococcal
enterotoxin B-induced acute inflammatory lung injury and mortality.
Toxicol Sci. 144:284–297. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu LM, Tu WJ, Zhu T, Wang XT, Tan ZL,
Zhong H, Gao DY and Liang DY: IRF3 is an important molecule in the
UII/UT system and mediates immune inflammatory injury in acute
liver failure. Oncotarget. 7:49027–49041. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qi T, Xu F, Yan X, Li S and Li H:
Sulforaphane exerts anti-inflammatory effects against
lipopolysaccharide-induced acute lung injury in mice through the
Nrf2/ARE pathway. Int J Mol Med. 37:182–188. 2016. View Article : Google Scholar
|
|
25
|
Sugiyama K, Muroi M, Kinoshita M, Hamada
O, Minai Y, Sugita-Konishi Y, Kamata Y and Tanamoto K: NF-κB
activation via MyD88-dependent Toll-like receptor signaling is
inhibited by trichothecene mycotoxin deoxynivalenol. J Toxicol Sci.
41:273–279. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fernandez-Bustamante A, Agazio A, Wilson
P, Elkins N, Domaleski L, He Q, Baer KA, Moss AF, Wischmeyer PE and
Repine JE: Brief glutamine pretreatment increases alveolar
macrophage CD163/heme oxygenase-1/p38-MAPK dephosphorylation
pathway and decreases capillary damage but not neutrophil
recruitment in IL-1/LPS-insufflated rats. PLoS One.
10:e01307642015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shan Y, Lin N, Yang X, Tan J, Zhao R, Dong
S and Wang S: Sulphoraphane inhibited the expressions of
intercellular adhesion molecule-1 and vascular cell adhesion
molecule-1 through MyD88-dependent toll-like receptor-4 pathway in
cultured endothelial cells. Nutr Metab Cardiovasc Dis. 22:215–222.
2012. View Article : Google Scholar
|
|
28
|
Van Linthout S, Spillmann F, Graiani G,
Miteva K, Peng J, Van Craeyveld E, Meloni M, Tölle M, Escher F,
Subasigüller A, et al: Down-regulation of endothelial TLR4
signalling after apo A-I gene transfer contributes to improved
survival in an experimental model of lipopolysaccharide-induced
inflammation. J Mol Med (Berl). 89:151–160. 2011. View Article : Google Scholar
|
|
29
|
Safari-Alighiarloo N, Taghizadeh M,
Rezaei-Tavirani M, Goliaei B and Peyvandi AA: Protein-protein
interaction networks (PPI) and complex diseases. Gastroenterol
Hepatol Bed Bench. 7:17–31. 2014.PubMed/NCBI
|
|
30
|
Oshiumi H, Sasai M, Shida K, Fujita T,
Matsumoto M and Seya T: TIR-containing adapter molecule (TICAM)-2,
a bridging adapter recruiting to toll-like receptor 4 TICAM-1 that
induces interferon-beta. J Biol Chem. 278:49751–49762. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wald D, Qin J, Zhao Z, Qian Y, Naramura M,
Tian L, Towne J, Sims JE, Stark GR and Li X: SIGIRR, a negative
regulator of Toll-like receptor-interleukin 1 receptor signaling.
Nat Immunol. 4:920–927. 2003. View
Article : Google Scholar : PubMed/NCBI
|