Introduction
Cisplatin [cis-diamminedichloroplatinum(II)] is a
potent chemotherapeutic agent, which is widely used for the
treatment of human cancers (1,2).
However, the clinical application of cisplatin is limited by
nephrotoxicity, leading to acute renal failure (3–6).
Cisplatin accumulates predominantly in the kidney, as this is the
major route for its excretion (7). Therefore, cisplatin nephrotoxicity
appears to be associated with its accumulation in the kidney.
Cisplatin nephrotoxicity is a complex multifactorial
process, including oxidative stress, mitogen-activated protein
kinases, p53 tumor suppressor and inflammation (8). In particular, reactive oxygen
species (ROS) and p53 have been shown to be important factors
contributing to cisplatin nephrotoxicity. Cisplatin induces
oxidative stress by generating ROS in renal cells, which leads to
the activation of apoptotic pathways (9–11).
p53 is activated during cisplatin nephrotoxicity and contributes to
renal cell injury and death (12). Of note, p53 may be activated by
ROS during cisplatin treatment of renal cells (13,14). Moreover, ROS scavengers and p53
inhibitors may prevent cisplatin-induced apoptosis by suppressing
the activation of p53 in renal cells (13–15). This suggests that regulation of
p53 activity may be an important target for alleviating cisplatin
nephrotoxicity.
Cisplatin nephrotoxicity is also associated with
endoplasmic reticulum (ER) stress (16). Pre-activation of ER stress
alleviates cisplatin-induced nephrotoxicity in various renal cell
lines (17). ER stress, which may
be observed under physiological or pathological conditions
(18), is caused by accumulation
of unfolded proteins in the ER. This accumulation attenuates mRNA
translation through activation of PKR-like ER kinase (PERK) and
subsequent phosphorylation of eukaryotic translation initiation
factor 2α (eIF2α) (19). ER
stress is involved in the stabilization of p53 through inhibiting
ubiquitin-mediated degradation of p53. Qu et al have
reported that ER stress prevents p53 stabilization and p53-mediated
apoptosis upon DNA damage (20).
Lee and Kim have also reported that ER stress-mediated eIF2α
phosphorylation attenuates cell death by inhibiting p53
stabilization in statin-induced apoptosis (21). Thus, it is most likely that eIF2α
phosphorylation plays an important role in the regulation of p53
expression or stability.
The aim of the present study was to investigate
whether cisplatin induces p53-mediated apoptosis and ER
stress-mediated eIF2α phosphorylation in human renal proximal
tubular HK-2 cells. In addition, since eIF2α phosphorylation has
been implicated in the stabilization of p53, which is due to p53
phosphorylation, the effect of ER stress-mediated eIF2α
phosphorylation on p53-mediated apoptosis induced by cisplatin was
investigated in HK-2 cells.
Materials and methods
Reagents and antibodies
Cisplatin, dimethyl sulfoxide (DMSO),
2′,7′-dichlorofluorescein diacetate (DCFH-DA),
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT),
N-acetyl-L-cysteine (NAC), propidium iodide (PI), protease
inhibitor cocktail, ribonuclease A (RNase A) and anti-β-actin
(sc-69879) antibody were all purchased from Sigma-Aldrich (St.
Louis, MO, USA). Pifithrin-α and Sal003 were purchased from
Calbiochem (La Jolla, CA, USA). Dulbecco's modified Eagle's medium
(DMEM/F12) (1:1), fetal bovine serum (FBS) and
antibiotic-antimycotic solution were all purchased from
Gibco/Thermo Fisher Scientific, Inc. (Grand Island, NY, USA).
Anti-rabbit cleaved-caspase-3 (Asp175) (no. 9661), anti-rabbit PARP
(no. 9542), anti-rabbit phospho-p53 (Ser15) (no. 9284), anti-rabbit
p53 (no. 9282), anti-rabbit phospho-eIF2α (Ser51) (no. 9721),
anti-rabbit eIF2α and anti-rabbit activating transcription factor 4
(ATF4) (no. 11815) antibodies were all purchased from Cell
Signaling Technology Inc. (Beverly, MA, USA). Anti-mouse heme
oxygenase-1 (HO-1) (ADI-OSA-110) antibody was purchased from Enzo
Life Sciences, Inc. (Farmingdale, NY, USA). Control siRNA and PERK
siRNA (h) were both purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA).
Cell culture
Immortalized human renal proximal tubular epithelial
(HK-2) cells were obtained from the American Type Culture
Collection (Manassas, VA, USA). The cells were maintained in
DMEM/F12 (1:1) supplemented with 10% FBS in 1:100 dilution of an
antibiotic-antimycotic solution at 37°C in a 5% CO2
incubator. Exponentially growing cells were seeded into a culture
dish at 1×105 cells/ml in complete medium for 24 h prior
to treatment with chemicals.
Cell viability assay
Cell viability was determined by the colorimetric
MTT metabolic activity assay. HK-2 cells were seeded at a density
of 5×104 cells/well in a 24-well plate. The cells were
treated with different concentrations of cisplatin for 24 h. After
treatment, 500 µl of MTT solution (0.5 mg/ml in serum-free
medium) was added to each well. After incubation for 4 h at 37°C in
the dark, the MTT-containing medium was removed by aspiration. The
generated blue formazan product was dissolved by the addition of
200 µl 100% DMSO per well. The amount of formazan was
determined at 570 nm using the SpectraMax 250 microplate reader
(Molecular Devices, Sunnyvale, CA, USA). The percentage of viable
cells was calculated as follows: Mean optical density (OD) of
treated cells/mean OD of control cells ×100.
Flow cytometric analysis
The percentage of apoptotic cells was quantitated by
PI staining and flow cytometric analysis, where the sub-G1 peak
represented apoptotic cells. The HK-2 cells exposed to cisplatin
were harvested, washed with ice-cold phosphate-buffered saline
(PBS; pH 7.4), and fixed in 1 ml of 70% ice-cold ethanol in PBS at
4°C for 1 h. After removing the ethanol by repeated washing in PBS,
cells were resuspended in 1 ml of PI/RNase A solution (PBS with 10
µg̸ml PI and 100 µg/ml RNase A) and incubated in the
dark at 37°C for 1 h. The sub-G1 DNA contents were quantified using
Fluorescence-Activated Cell Sorting Calibur and CellQuest Pro
software (both from BD Biosciences, San Jose, CA, USA).
Caspase-3 activity assay
The activity of caspase-3 was determined by the
caspase-3 colorimetric assay kit (Abcam, Cambridge, MA, USA)
according to the manufacturer's instructions. Briefly, cells were
harvested after treatment, washed with PBS, and then lysed in lysis
buffer. The cell lysates were centrifuged at 10,000 × g for 1 min,
and 50 µl of extracts containing 50 µg protein were
incubated with 50 µl of 2X reaction buffer and 5 µl
of 4 mM DEVD-pNA substrate at 37°C for 2 h in the dark. The
colorimetric release of p-nitroaniline from DEVD-pNA substrate was
measured at 405 nm using the SpectraMax 250 microplate reader. The
percentage of caspase-3 activity was calculated as follows: Mean OD
of treated cells/mean OD of control cells ×100.
siRNA transfection
Transfection of siRNAs was perfo rmed using
Lipofectamine 3000 reagent (Invitrogen/Thermo Fisher Scientific,
Carlsbad, CA, USA) according to the manufacturer's instructions.
HK-2 cells were seeded at a density of 2×105 cells in
60-mm dishes, cultured in complete medium for 24 h, and transfected
with control or PERK siRNAs. The siRNAs were diluted to 200 nM with
250 µl of Opti-MEM. Lipofectamine 3000 reagent was also
diluted 1:50 in Opti-MEM. Diluted siRNAs and Lipofectamine 3000
reagent were mixed in equal volumes and incubated for 15 min at
room temperature. The mixtures were added to culture dishes
containing 2 ml fresh serum-free medium, and incubated at 37°C in a
5% CO2 incubator for 24 h. Thereafter, complete medium
was added, the cells were further incubated for 24 h and subjected
to the experiments.
ROS production assay
The relative levels of ROS were determined by the
DCFH-DA fluorescence assay. HK-2 cells were seeded into 96-well
microplates (1×104 cells/well) for 24 h and incubated in
Hank's Balanced Salt Solution (HBSS; pH 7.4) containing 20
µM DCFH-DA at 37°C for 1 h. Following incubation, the cells
were washed with HBSS and treated with 40 µM cisplatin under
HBSS for the indicated time periods. The relative DCF fluorescence
intensity was determined with an excitation wavelength of 485 nm
and an emission wavelength of 538 nm using the SpectraMax M2
multi-mode microplate reader (Molecular Devices).
Western blot analysis
The cells were lysed in RIPA lysis buffer containing
1% halt protease and phosphatase inhibitor cocktails. Cell lysates
were centrifuged at 20,000 × g for 15 min at 4°C, and the protein
concentration was determined using a Bradford assay. Samples
containing 50 µg of total protein were resolved by SDS-PAGE
gel and transferred onto a nitrocellulose membrane for 3 h at 40 V.
The membranes were blocked with Tris-buffered saline with Tween-20
(20 mM Tris-HCl, pH 7.6; 150 mM NaCl; and 0.05% Tween-20)
containing 5% non-fat dry milk and probed with primary antibodies
(all 1:1,000 in 3% BSA in Tris-buffered saline with 0.05% Tween-20)
overnight at 4°C with gentle shaking. Protein spots were detected
using horseradish peroxidase-conjugated secondary antibodies (all
1:2,000 in 3% BSA in Tris-buffered saline with 0.05% Tween-20).
Immunoreactive bands were visualized using the SuperSignal West
Pico Chemiluminescent Substrate kit (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and then developed using the FluorChem E
system (ProteinSimple, San Jose, CA, USA). The density of the bands
was quantitated using Quantity One software, version 4.6.6 (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using Microsoft
Office Excel 2013 (Microsoft, Redmond, WA, USA) and data are
expressed as means ± standard deviation. The statistically
significant differences between two groups were calculated using
the Student's t-test. P-values <0.05 were considered to indicate
statistically significant differences.
Results
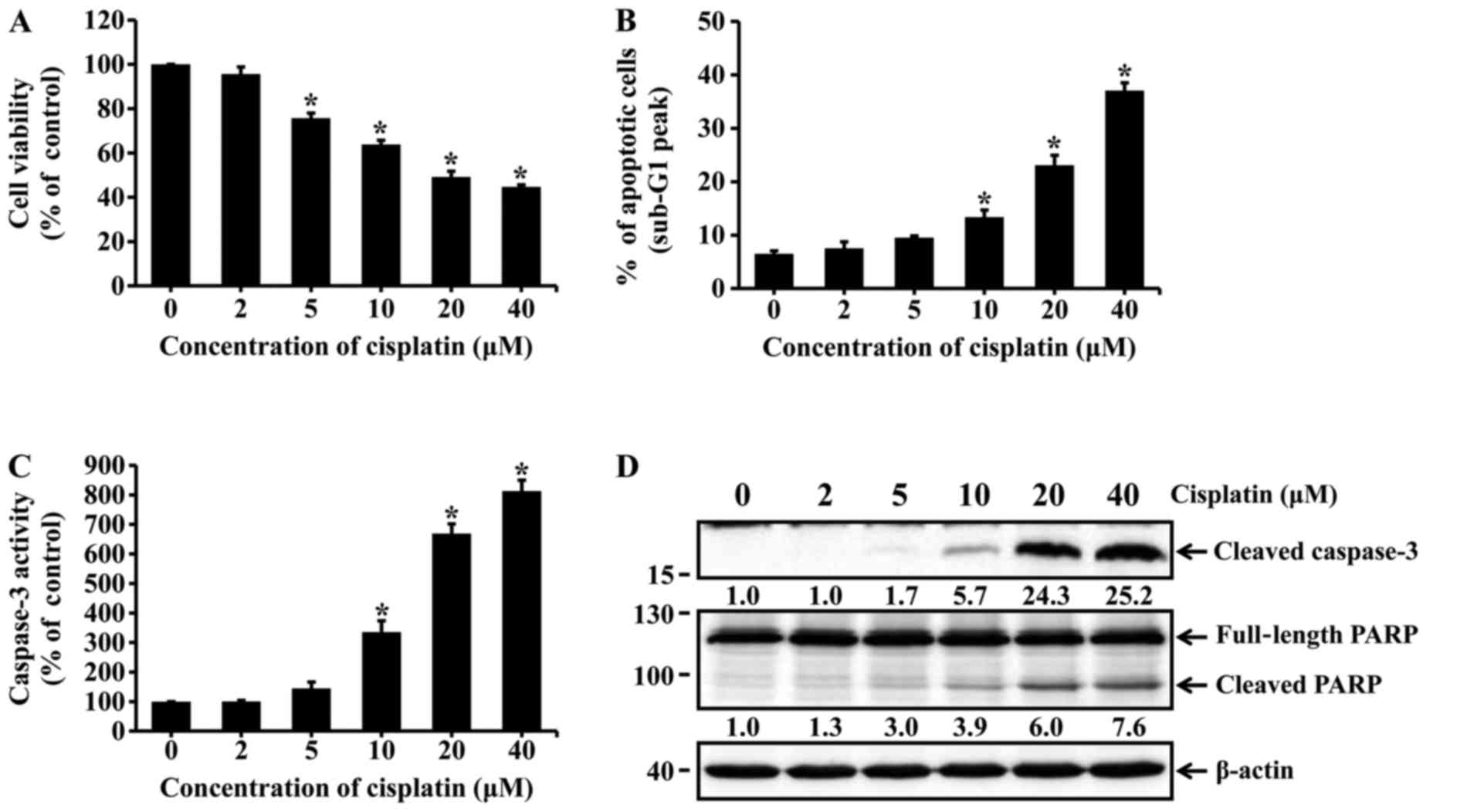
Cisplatin induces apoptotic cell death in
HK-2 cells
HK-2 cells were incubated with different
concentrations of cisplatin for 24 h. Cell viability was assessed
by the MTT assay. Apoptotic cells were determined using flow
cytometry with PI staining. The viability of HK-2 cells markedly
decreased with increasing concentrations of cisplatin (Fig. 1A). The percentage of apoptotic
cells was significantly increased among cisplatin-treated cells
(10, 20 and 40 µM cisplatin) compared with the untreated
cells (Fig. 1B). Caspase-3
activation and poly(ADP-ribose) polymerase (PARP) cleavage were
determined by using caspase-3 colorimetric assay kit and western
blot analysis. As shown in Fig. 1C
and D, treatment of HK-2 cells with cisplatin significantly
increased the activity of caspase-3 and the levels of cleaved
active caspase-3. Similarly, cisplatin treatment increased
proteolytic cleavage of PARP to form an 89-kDa fragment (Fig. 1D). These results indicate that
cisplatin induced apoptotic cell death in HK-2 cells.
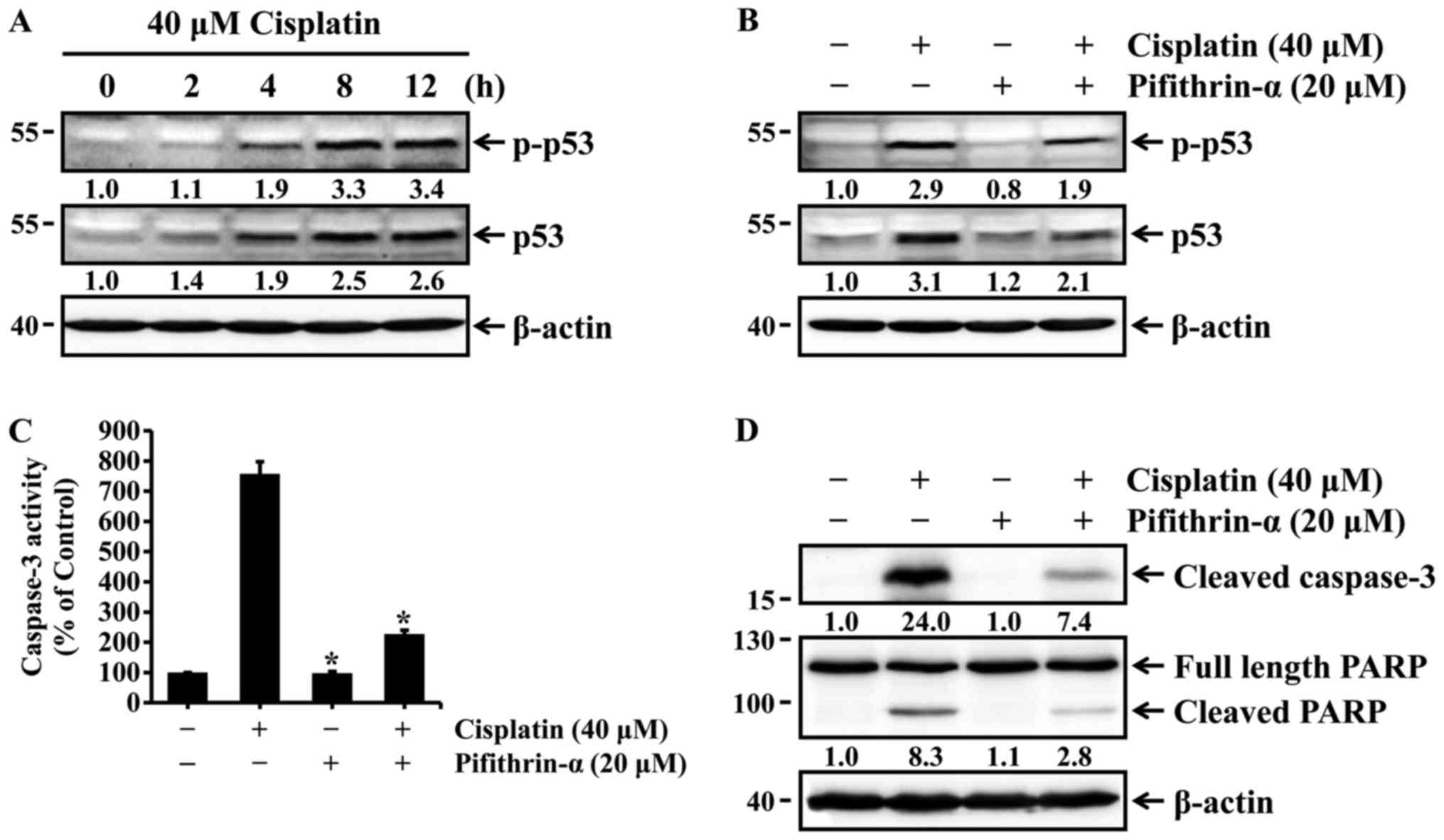
Cisplatin induces p53-mediated apoptosis
in HK-2 cells
The tumor suppressor p53 plays an important role in
regulating cisplatin-induced apoptosis of renal cells (13–15). Thus, we examined whether cisplatin
could induce the activation of p53 via phosphorylation at serine 15
in HK-2 cells. HK-2 cells were treated with 40 µM cisplatin
for different times, and the expression and phosphorylation status
of p53 were then determined by western blot analysis. The levels of
total and phosphorylated p53, which is directly associated with p53
activation, were increased in a time-dependent manner (Fig. 2A), whereas pifithrin-α, a specific
p53 inhibitor, reduced p53 expression and phosphorylation in cells
exposed to cisplatin (Fig. 2B).
We next examined whether p53 activation was associated with
cisplatin-induced apoptosis. HK-2 cells were treated with cisplatin
in the absence or presence of pifithrin-α, and caspase-3 activation
and PARP cleavage were then determined with the caspase-3
colorimetric assay kit and western blot analysis. As shown in
Fig. 2C and D, cisplatin-induced
activation of caspase-3 and cleavage of PARP were markedly
inhibited by pifithrin-α. These results indicate that p53
activation is required for cisplatin-induced apoptosis in HK-2
cells.
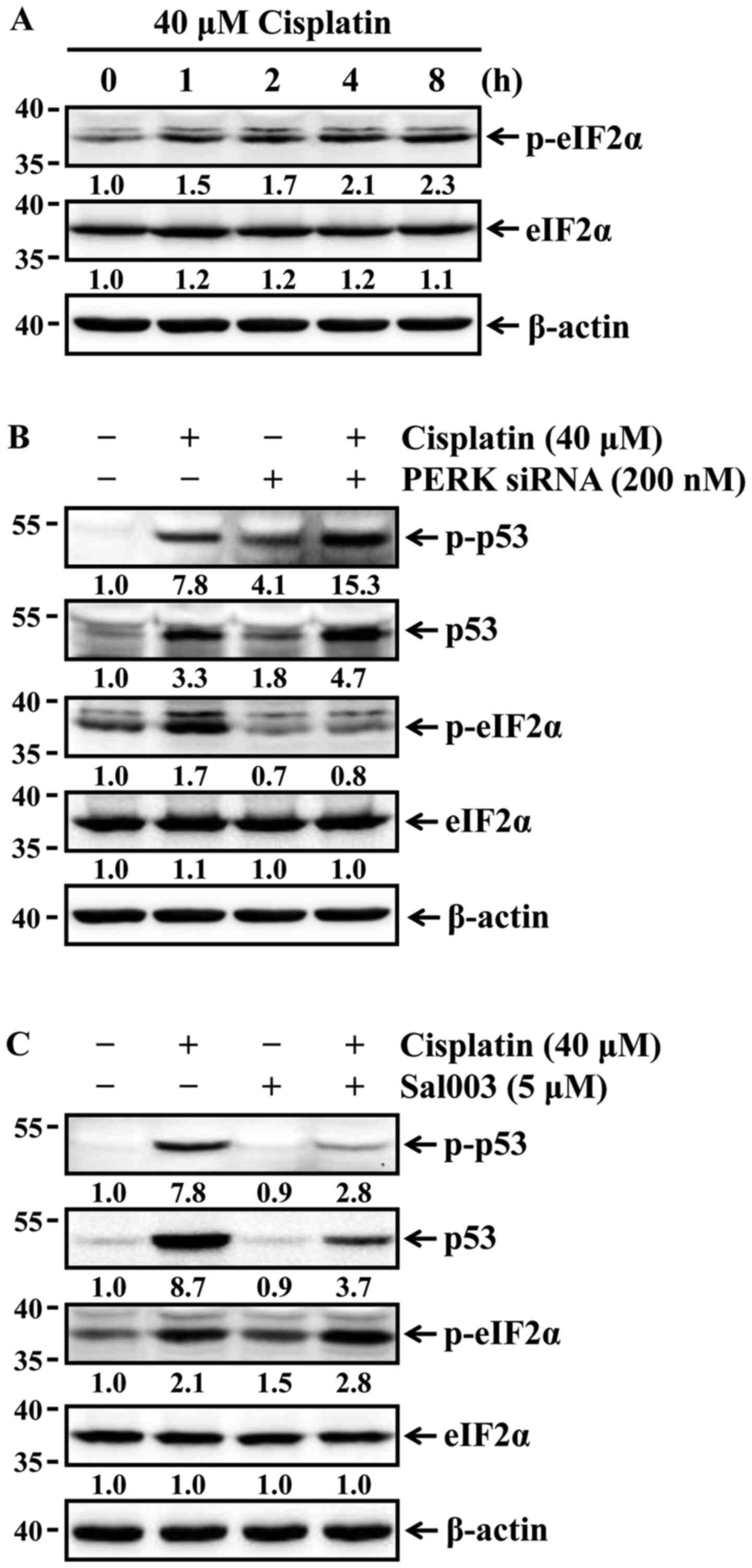
Phosphorylation of eIF2α suppresses
cisplatin-induced p53 activation and apoptosis in HK-2 cells
Lee and Kim reported that the stabilization of p53
is affected by ER stress-mediated eIF2α phosphorylation (21). p53 stability is associated with
the phosphorylation of p53, which leads to stabilization of p53 by
reducing its interaction with murine double minute 2, a negative
regulator of p53 (22). It was
first examined whether cisplatin was able to induce phosphorylation
of eIF2α in HK-2 cells. The cells were treated with 40 µM
cisplatin for the indicated times, and the phosphorylation status
of eIF2α was then determined by western blot analysis. The levels
of eIF2α phosphorylation were increased time-dependently (Fig. 3A). Changes in the expression and
phosphorylation status of p53 according to phosphorylation or
dephosphorylation of eIF2α were next examined in HK-2 cells exposed
to cisplatin. The cells were pretreated with Sal003 to inhibit
dephosphorylation of eIF2α, or transfected with siRNA against PERK
to block PERK-eIF2α phosphorylation prior to cisplatin treatment.
As shown in Fig. 3B, Sal003
enhanced cisplatin-induced phosphorylation of eIF2α, and reduced
expression and phosphorylation of p53 in cisplatin-treated cells.
Furthermore, the siRNA-mediated knockdown of PERK completely
blocked cisplatin-induced phosphorylation of eIF2α, and promoted
expression of p53 and its phosphorylation in cisplatin-treated
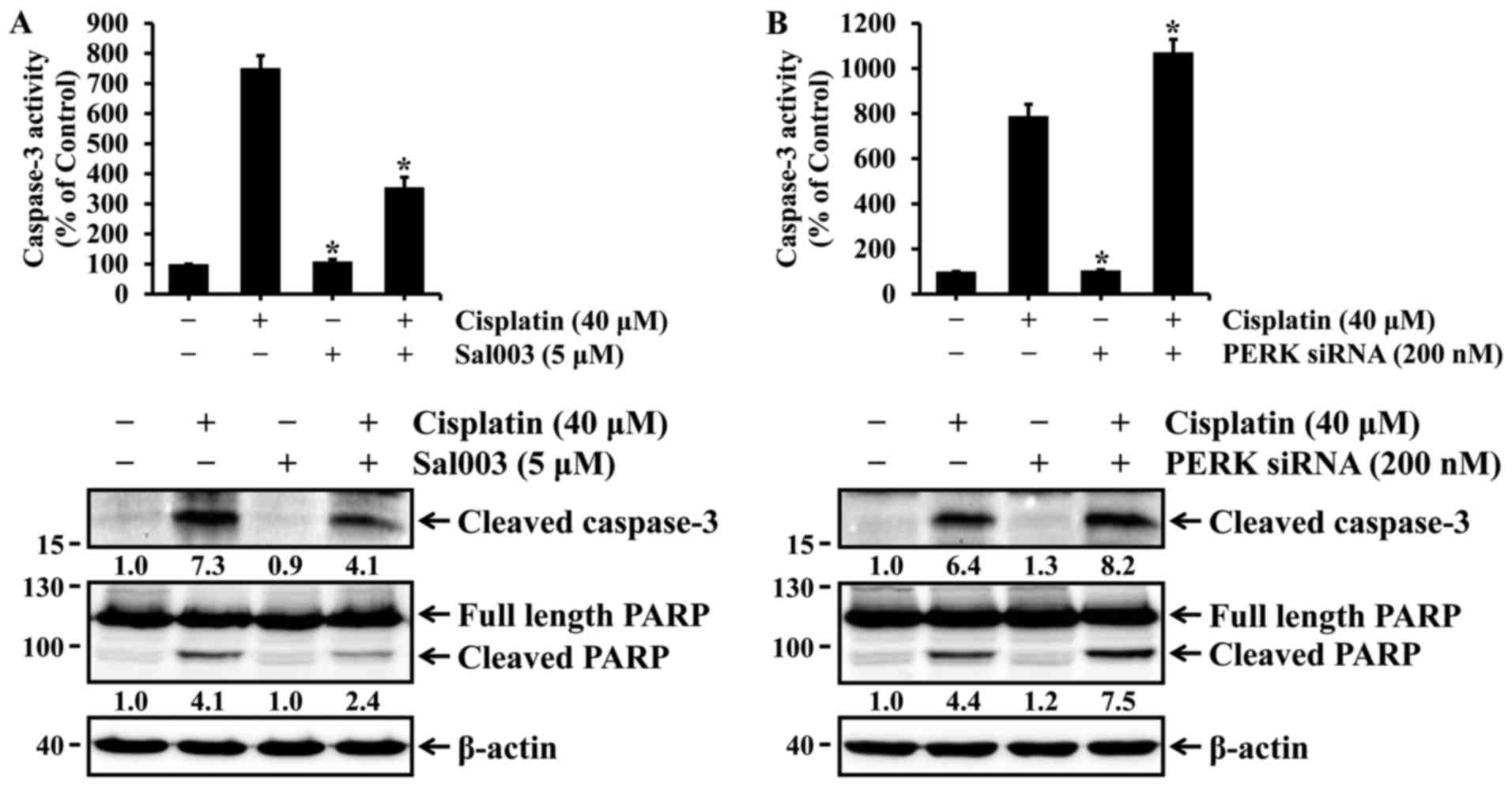
cells. Finally, it was investigated whether eIF2α-regulated p53
activation could be associated with cisplatin-induced apoptosis in
HK-2 cells. As shown in Fig. 4,
cisplatin-induced activation of caspase-3 and cleavage of PARP were
inhibited by Sal003, whereas they were promoted by PERK knockdown.
These results indicate that phosphorylation of eIF2α may confer
resistance to cisplatin-induced apoptosis, at least in part, via
p53 activation in HK-2 cells.
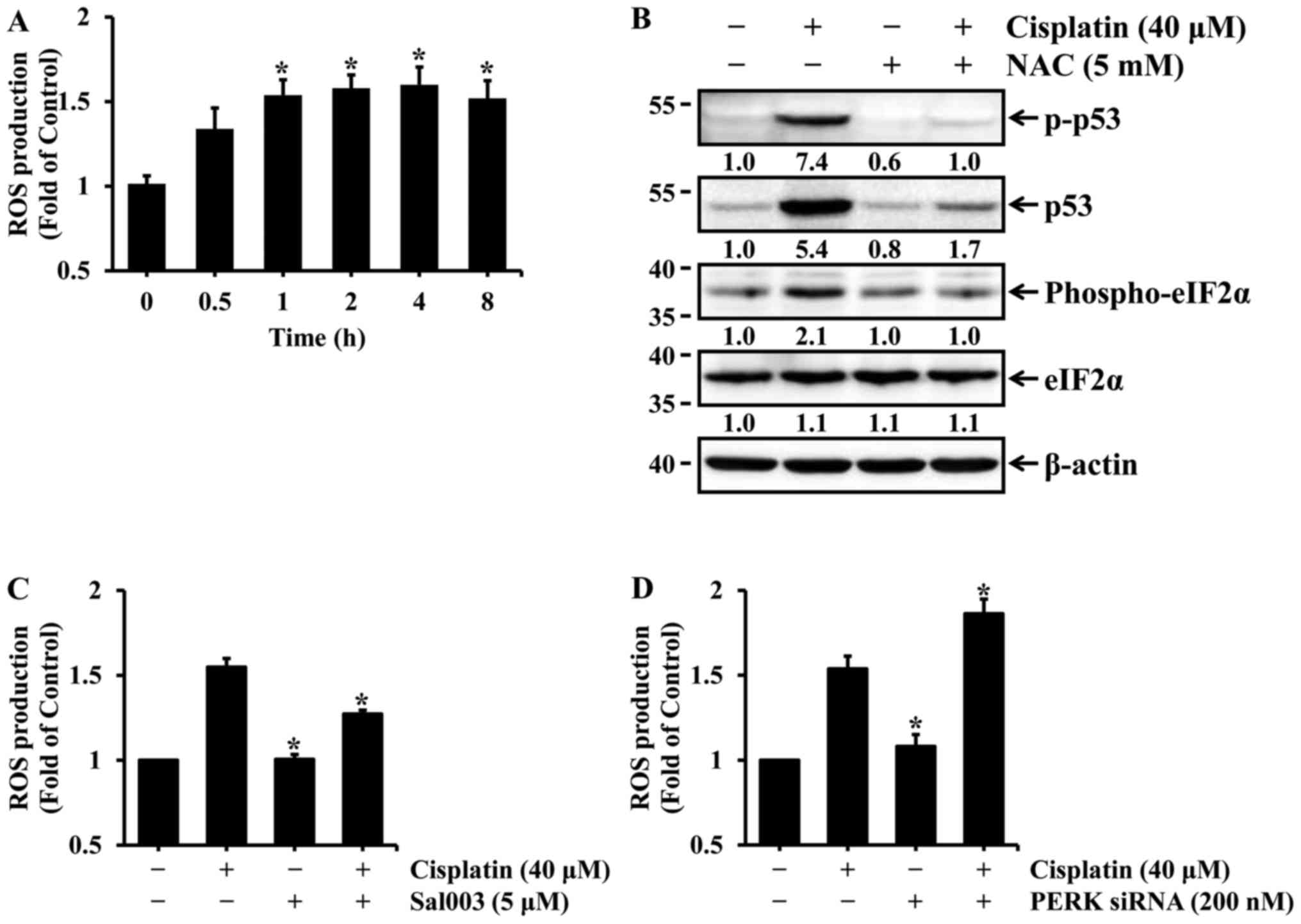
Oxidative stress is involved in
cisplatin-induced eIF2α phosphorylation and p53 activation in HK-2
cells
Similar to p53, ROS is also known to play an
important role in cisplatin-induced nephrotoxicity (13–15). Oxidative stress induced by ROS
production is involved in p53 activation in cisplatin-induced renal
cell injury. Therefore, it was examined whether cisplatin induced
the production of ROS in HK-2 cells and, if so, whether it
contributed to p53 activation. First, HK-2 cells were treated with
40 µM cisplatin for the indicated times, and intercellular
ROS production was then determined by measuring the fluorescence
intensity of DCF. As shown in Fig.
5A, the levels of ROS production were significantly increased
after 1 h of cisplatin treatment, and were maintained during
prolonged cisplatin treatment. Previous studies have reported that
N-acetylcysteine (NAC), a ROS scavenger, suppresses
cisplatin-induced p53 activation in renal cells (13,15). Therefore, the effect of ROS
scavenging on cisplatin-induced p53 activation was examined in HK-2
cells. The cells were treated with cisplatin in the absence or
presence of NAC, a general antioxidant, and the levels of total and
phosphorylated p53 were then determined by western blot analysis.
The levels of p53 expression and phosphorylation were almost
completely blocked by NAC in cisplatin-treated cells (Fig. 5B). Since eIF2α is phosphorylated
mainly by PERK in response to oxidative stress, it was examined
whether cisplatin-induced oxidative stress affects the
phosphorylation of eIF2α in HK-2 cells. As shown in Fig. 5B, the blockade of ROS production
by NAC completely abolished the phosphorylation of eIF2α in cells
treated with cisplatin. Finally, changes in intracellular levels of
ROS according to phosphorylation or dephosphorylation of eIF2α were
examined in HK-2 cells exposed to cisplatin. As shown in Fig. 5C, cisplatin-induced ROS production
was suppressed by Sal003, whereas it was promoted by PERK
knockdown. These results indicate that oxidative stress may mediate
eIF2α phosphorylation as well as p53 activation induced by
cisplatin in HK-2 cells. In particular, phosphorylation of eIF2α
has been shown to exert a scavenging effect on cisplatin-induced
ROS production in HK-2 cells.
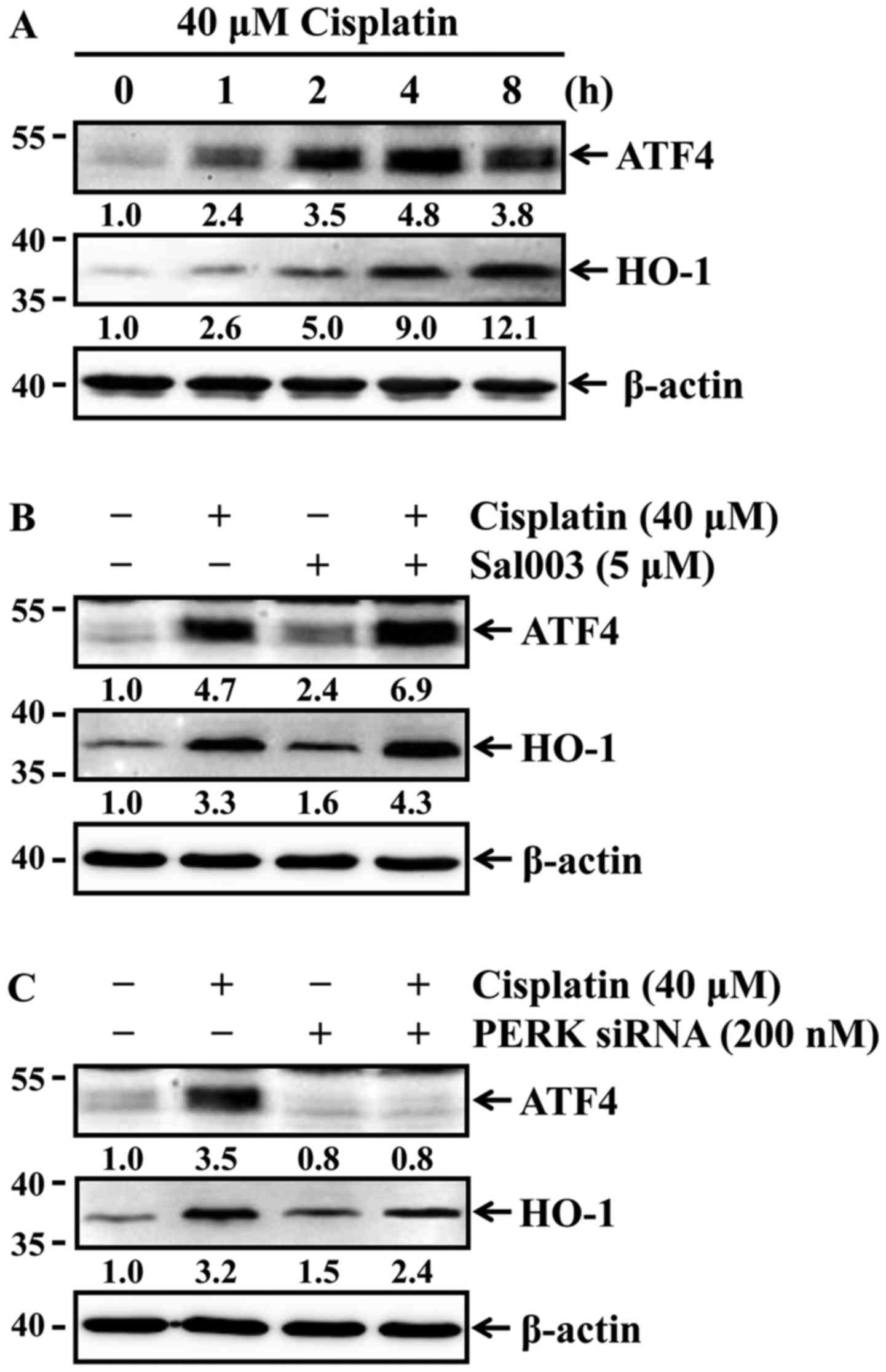
Phosphorylation of eIF2α induces ATF4 and
HO-1 expression in cisplatin-treated HK-2 cells
Phosphorylation of eIF2α by PERK selectively
enhances the translation of ATF4 that induces the expression of
genes involved in antioxidant response and apoptosis regulation
(23). HO-1, which is a redox-
sensitive microsomal enzyme, is known to be cytoprotective through
its potent antioxidant effect in cisplatin-induced acute renal
injury (8). Of note, ATF4 is able
to transcriptionally induce the expression of the antioxidant gene
HO-1 in response to oxidative stress (24,25). Accordingly, it was investigated
whether cisplatin was able to induce the expression of ATF4 and
HO-1 in HK-2 cells. The cells were treated with 40 µM
cisplatin for the indicated times, and the expression of ATF4 and
HO-1 was then determined by western blot analysis. Cisplatin
time-dependently induced the expression of both ATF4 and HO-1
(Fig. 6A). It was next examined
whether differences in the phosphorylation status of eIF2α affected
the expression levels of ATF4 and HO-1 in HK-2 cells exposed to
cisplatin. As shown in Fig. 6B and
C, cisplatin-induced ATF4 and HO-1 expression were elevated by
Sal003 (Fig. 6B), while their
expression was inhibited by PERK knockdown (Fig. 6C). These results indicate that
phosphorylation of eIF2α attenuates cisplatin-induced oxidative
stress through ATF4-mediated HO-1 expression in HK-2 cells.
Discussion
Nephrotoxicity is one of the main side effects that
limit the therapeutic efficacy of the antineoplastic drug cisplatin
(1–6). ROS and p53 have been found to be
leading causes of cisplatin nephrotoxicity (8–15).
Previous reported studies have demonstrated that cisplatin induces
apoptotic cell death through ROS-mediated p53 activation in renal
cells (13–15). In the present study, the roles of
ROS and p53 in cisplatin-induced apoptosis of human renal proximal
epithelial cells were investigated and the results demonstrated
that cisplatin induced oxidative stress by increasing the
production of intracellular ROS, which contributed to p53-mediated
apoptosis of HK-2 cells. Furthermore, cisplatin-induced ROS
production was involved in ER stress-mediated eIF2α
phosphorylation, which in turn suppressed p53 activation in HK-2
cells.
The phosphorylation of eIF2α primarily plays a
cytoprotective role against stress by attenuating global
translation (20). Previous
studies have reported that phosphorylation of eIF2α protected cells
against glucose deprivation stress and ER stress (26–28). Our results demonstrated that
phosphorylation of eIF2α protected HK-2 cells against
cisplatin-induced apoptosis through reduction of p53 activation. By
contrast, dephosphorylation of eIF2α aggravated cisplatin-induced
apoptosis in HK-2 cells by enhancing p53 activation. Similar to our
results, Lee and Kim reported that salubrinal, an inhibitor of
eIF2α dephosphorylation, attenuated statin-induced apoptosis by
inhibiting the stabilization of p53 (21). Thus, it is most likely that
phosphorylation of eIF2α contributes to the protection of HK-2
cells against cisplatin cytotoxicity. However, excessive
phosphorylation of eIF2α may negatively affect cytoprotection and
cell survival. It was demonstrated that salubrinal enhances
cisplatin-induced nephrotoxicity in mouse kidneys, indicating that
excessive eIF2α phosphorylation may be poorly tolerated by renal
cells and exacerbate cisplatin-induced apoptosis (29).
Oxidative stress refers to elevated intracellular
levels of ROS that may activate signaling pathways involving p53
phosphorylation and activation (30). Similar to previous studies
(13–15), our results demonstrated that
cisplatin induced apoptotic cell death in HK-2 cells through
ROS-mediated p53 activation. Oxidative stress may also lead to
phosphorylation of eIF2α via PERK activation, which enhances the
translation of ATF4 (31). The
phosphorylation of eIF2α has been shown to be crucial for the
selective translational induction of ATF4 during ER stress
(32). In accordance with those
findings, our results demonstrated that cisplatin-induced oxidative
stress was involved in eIF2α phosphorylation, thus leading to
increased ATF4 expression in HK-2 cells. It is noteworthy that ATF4
regulates the expression of several antioxidant genes in response
to oxidative stress, which plays a central role in antioxidant
responses (24). In particular,
ATF4 may induce the expression of the antioxidant gene HO-1 in
response to oxidative stress (24,25). Dey et al reported that ATF4
induces expression of HO-1 in response to matrix
detachment-activated oxidative stress in human fibrosarcoma cells
(24). He et al also
reported that ATF4 regulates basal and CdCl2-induced
expression of the HO-1 gene in a cell-specific manner (25). In the present study, cisplatin
induced HO-1 expression, which was partially regulated by the
eIF2α-ATF4 pathway in HK-2 cells. HO-1 has been shown to exert
renoprotective effects through a potent antioxidant pathway in
cisplatin-induced nephrotoxicity (8). Agarwal et al reported that
the early induction of HO-1 plays an important role in
cytoprotection against cisplatin-induced acute kidney injury
(33). Thus, it is most likely
that the phosphorylation of eIF2α suppresses cisplatin-induced p53
activation by attenuating oxidative stress via the ATF4-mediated
HO-1 expression in HK-2 cells, thereby resulting in protection
against apoptosis.
In conclusion, cisplatin-induced oxidative stress
leads to accumulation and activation of p53, leading to induction
of apoptosis in HK-2 cells, while interfering with p53 activation
through induction of eIF2α phosphorylation. Interference in p53
activation by eIF2α phosphorylation may be associated with
expression of ATF4 and HO-1 in HK-2 cells exposed to cisplatin. In
other words, the phosphorylation of eIF2α may inhibit
cisplatin-induced p53 activation and apoptosis by attenuating
oxidative stress via the ATF4-mediated HO-1 expression in HK-2
cells. Therefore, the phosphorylation of eIF2α may play an
important role in protective strategies to alleviate cisplatin
nephrotoxicity.
Acknowledgments
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant nos.
2017R1A5A2015805 and 2013R1A1A2064013).
References
|
1
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barabas K, Milner R, Lurie D and Adin C:
Cisplatin: A review of toxicities and therapeutic applications. Vet
Comp Oncol. 6:1–18. 2008. View Article : Google Scholar
|
|
3
|
Hanigan MH and Devarajan P: Cisplatin
nephrotoxicity: Molecular mechanisms. Cancer Ther. 1:47–61.
2003.
|
|
4
|
McWhinney SR, Goldberg RM and McLeod HL:
Platinum neurotoxicity pharmacogenetics. Mol Cancer Ther. 8:10–16.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rybak LP: Mechanisms of cisplatin
ototoxicity and progress in otoprotection. Curr Opin Otolaryngol
Head Neck Surg. 15:364–369. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cubeddu LX: Mechanisms by which cancer
chemotherapeutic drugs induce emesis. Semin Oncol. 19(Suppl 15):
2–13. 1992.PubMed/NCBI
|
|
7
|
Yao X, Panichpisal K, Kurtzman N and
Nugent K: Cisplatin nephrotoxicity: A review. Am J Med Sci.
334:115–124. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pabla N and Dong Z: Cisplatin
nephrotoxicity: Mechanisms and renoprotective strategies. Kidney
Int. 73:994–1007. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Casares C, Ramírez-Camacho R, Trinidad A,
Roldán A, Jorge E and García-Berrocal JR: Reactive oxygen species
in apoptosis induced by cisplatin: Review of physiopathological
mechanisms in animal models. Eur Arch Otorhinolaryngol.
269:2455–2459. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Conklin KA: Dietary antioxidants during
cancer chemotherapy: Impact on chemotherapeutic effectiveness and
development of side effects. Nutr Cancer. 37:1–18. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sahin K, Sahin N and Kucuk O: Lycopene and
chemotherapy toxicity. Nutr Cancer. 62:988–995. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bhatt K, Zhou L, Mi QS, Huang S, She JX
and Dong Z: MicroRNA-34a is induced via p53 during cisplatin
nephrotoxicity and contributes to cell survival. Mol Med.
16:409–416. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang M, Wei Q, Pabla N, Dong G, Wang CY,
Yang T, Smith SB and Dong Z: Effects of hydroxyl radical scavenging
on cisplatin-induced p53 activation, tubular cell apoptosis and
nephrotoxicity. Biochem Pharmacol. 73:1499–1510. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sánchez-Pérez Y, Morales-Bárcenas R,
García-Cuellar CM, López-Marure R, Calderon-Oliver M,
Pedraza-Chaverri J and Chirino YI: The alpha-mangostin prevention
on cisplatin-induced apoptotic death in LLC-PK1 cells is associated
to an inhibition of ROS production and p53 induction. Chem Biol
Interact. 188:144–150. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ju SM, Pae HO, Kim WS, Kang DG, Lee HS and
Jeon BH: Role of reactive oxygen species in p53 activation during
cisplatin-induced apoptosis of rat mesangial cells. Eur Rev Med
Pharmacol Sci. 18:1135–1141. 2014.PubMed/NCBI
|
|
16
|
Xu Y, Wang C and Li Z: A new strategy of
promoting cisplatin chemotherapeutic efficiency by targeting
endoplasmic reticulum stress. Mol Clin Oncol. 2:3–7. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peyrou M and Cribb AE: Effect of
endoplasmic reticulum stress preconditioning on cytotoxicity of
clinically relevant nephrotoxins in renal cell lines. Toxicol In
Vitro. 21:878–886. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cybulsky AV: Endoplasmic reticulum stress
in proteinuric kidney disease. Kidney Int. 77:187–193. 2010.
View Article : Google Scholar
|
|
19
|
Back SH, Scheuner D, Han J, Song B, Ribick
M, Wang J, Gildersleeve RD, Pennathur S and Kaufman RJ: Translation
attenuation through eIF2alpha phosphorylation prevents oxidative
stress and maintains the differentiated state in beta cells. Cell
Metab. 10:13–26. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qu L, Huang S, Baltzis D, Rivas-Estilla
AM, Pluquet O, Hatzoglou M, Koumenis C, Taya Y, Yoshimura A and
Koromilas AE: Endoplasmic reticulum stress induces p53 cytoplasmic
localization and prevents p53-dependent apoptosis by a pathway
involving glycogen synthase kinase-3beta. Genes Dev. 18:261–277.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee SK and Kim YS: Phosphorylation of
eIF2α attenuates statin-induced apoptosis by inhibiting the
stabilization and trans-location of p53 to the mitochondria. Int J
Oncol. 42:810–816. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
She QB, Chen N and Dong Z: ERKs and p38
kinase phosphorylate p53 protein at serine 15 in response to UV
radiation. J Biol Chem. 275:20444–20449. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hetz C, Chevet E and Harding HP: Targeting
the unfolded protein response in disease. Nat Rev Drug Discov.
12:703–719. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dey S, Sayers CM, Verginadis II, Lehman
SL, Cheng Y, Cerniglia GJ, Tuttle SW, Feldman MD, Zhang PJ, Fuchs
SY, et al: ATF4-dependent induction of heme oxygenase 1 prevents
anoikis and promotes metastasis. J Clin Invest. 125:2592–2608.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
He CH, Gong P, Hu B, Stewart D, Choi ME,
Choi AM and Alam J: Identification of activating transcription
factor 4 (ATF4) as an Nrf2-interacting protein. Implication for
heme oxygenase-1 gene regulation. J Biol Chem. 276:20858–20865.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scheuner D, Song B, McEwen E, Liu C,
Laybutt R, Gillespie P, Saunders T, Bonner-Weir S and Kaufman RJ:
Translational control is required for the unfolded protein response
and in vivo glucose homeostasis. Mol Cell. 7:1165–1176. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Muaddi H, Majumder M, Peidis P, Papadakis
AI, Holcik M, Scheuner D, Kaufman RJ, Hatzoglou M and Koromilas AE:
Phosphorylation of eIF2α at serine 51 is an important determinant
of cell survival and adaptation to glucose deficiency. Mol Biol
Cell. 21:3220–3231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boyce M, Bryant KF, Jousse C, Long K,
Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, et al: A
selective inhibitor of eIF2alpha dephosphorylation protects cells
from ER stress. Science. 307:935–939. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu CT, Sheu ML, Tsai KS, Chiang CK and Liu
SH: Salubrinal, an eIF2α dephosphorylation inhibitor, enhances
cisplatin-induced oxidative stress and nephrotoxicity in a mouse
model. Free Radic Biol Med. 51:671–680. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Martindale JL and Holbrook NJ: Cellular
response to oxidative stress: signaling for suicide and survival. J
Cell Physiol. 192:1–15. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Velasco G: Endoplasmic reticulum stressed
by pollution. Focus on 'Airborne particulate matter selectively
activates endoplasmic reticulum stress response in the lung and
liver tissues'. Am J Physiol Cell Physiol. 299:C727–C728. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Blais JD, Filipenko V, Bi M, Harding HP,
Ron D, Koumenis C, Wouters BG and Bell JC: Activating transcription
factor 4 is translationally regulated by hypoxic stress. Mol Cell
Biol. 24:7469–7482. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Agarwal A, Balla J, Alam J, Croatt AJ and
Nath KA: Induction of heme oxygenase in toxic renal injury: a
protective role in cisplatin nephrotoxicity in the rat. Kidney Int.
48:1298–1307. 1995. View Article : Google Scholar : PubMed/NCBI
|