Introduction
Liver cancer is one of the most common malignant
tumors worldwide, and its incidence is the second highest in China.
Liver cancer cannot be easily detected at an early stage due to the
lack of distinct symptoms and the scarcity of clinically specific
markers for serodiagnosis. Therefore, the majority of the patients
are diagnosed at advanced or late stages, resulting in distant
metastasis and a low 5-year survival rate (1). Thus, the metastasis and invasion of
liver cancer must be clinically investigated to prevent progression
of this disease and improve its prognosis.
The invasion and metastasis of malignant tumors are
regulated and controlled by various factors and mechanisms.
Epithelial-to-mesenchymal transition (EMT) is a key mechanism
participating in the invasion and metastasis of solid cancers, such
as colon, lung and pancreatic cancer (2–4).
However, the association between EMT and the onset and progression
of liver cancer has not been fully elucidated. In this context, Lee
et al (5) and Giannelli
et al (6) previously
reported that EMT is involved in the invasion and metastasis of
liver cancer cells.
A number of studies reported that transforming
growth factor β1 (TGFβ1) is a cytokine with multiple functions that
promotes EMT (7,8). The activation abnormalities in the
signal transducer and activator of transcription 3 (STAT3)
signaling pathway are associated with tumor onset and progression
(9). The activation of this
pathway is regulated and controlled by the upstream factor Janus
kinase (JAK). The activation of JAK/STAT3 signaling may directly
affects EMT and promotes the invasion and metastasis of tumor cells
in lung cancer and ovarian tumors (10). However, whether the EMT mediated
by the JAK/STAT3 signaling pathway promotes TGFβ1-induced invasion
and metastasis of liver cancer cells has not been clearly
determined.
The present study investigated the human liver
cancer line HepG2, in which invasion and metastasis were induced by
TGFβ1. The role of JAK/STAT3 signaling in mediating the involvement
of EMT in the invasion and metastasis of HepG2 cells induced by
TGFβ1 was also determined. Experiments were performed to confirm
whether Twist is a target of STAT3. Overall, the aim of this study
was to provide new experimental evidence and potential targets for
preventing the invasion and metastasis of liver cancer cells.
Materials and methods
Cell culture
The liver cancer cell line HepG2 was purchased from
Shanghai Institute of Biochemistry and Cell Biology, Chinese
Academy of Sciences (Shanghai, China). HepG2 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM)-high glucose containing
trypsin (cat no. SH30022.01B) supplemented with 10% fetal bovine
serum (FBS; cat no. SH30084.03) (both from HyClone, Logan, UT,
USA), 100 U/ml penicillin (cat no. ST488-1; Beyotime Institute of
Biotechnology, Shanghai, China) and 100 U/ml streptomycin (cat no.
ST488-2; Beyotime Institute of Biotechnology) at 37°C under 95% air
and 5% CO2.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted from the tissue samples using
TRIzol® reagent (Thermo Fisher Scientific, Waltham, MA,
USA), according to the manufacturer's instructions. Subsequently,
cDNA was synthesized using a TaqMan Reverse Transcription Reagents
kit (Thermo Fisher Scientific), according to the manufacturer's
protocol. The relative expression levels of mRNA were determined
using a Power SYBR-Green PCR Master Mix kit (Thermo Fisher
Scientific) and normalized to GAPDH. RT-PCR was performed using the
Applied Biosystems 7500 Fast Dx Real-Time PCR instrument (cat no.
4425757; Thermo Fisher Scientific) and the following gene-specific
primers (Sangon Biotech Co., Ltd., Shanghai, China): GAPDH: Sense,
5′-TGCCATCAACGACCCCTTCA-3′ and antisense,
5′-TGACCTTGCCCACAGCCTTG-3′; E-cadherin: Sense,
5′-AGCTATCCTTGCACCTCAGC-3′ and antisense, 5′-CCCAGGAGTTTGAG-3′;
N-cadherin: Sense, 5′-TCCTGCTCACCACCACTACTT-3′ and antisense,
5′-CTGACAATGACCCCACAGC-3′; Smad: Sense, 5′-ATAAGCAACCGCCTGAACAT-3′
and anti-sense, 5′-TTACCTGCCTCCTGAAGACC-3′; Twist: Sense,
5′-GCTGATTGGCACGACCTCT-3′ and antisense,
5′-CACCATCCTCACACCTCTGC-3′; and vimentin: Sense,
5′-CCAAACTTTTCCTCCCTGAACC-3′ and antisense,
5′-GTGATGCTGAGAAGTTTCGTTGA-3′. A control siRNA specific for the red
fluorescent protein, 5′-CCACTACCTGAGCACCCAG-3′, was used as the
negative control (sc-37007; Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA). All primers were designed using the National Center
for Biotechnology Information Primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). PCR
was performed under the following conditions: Denaturation at 50°C
for 2 min, followed by 38 cycles at 95°C for 15 sec and 60°C for 1
min. Gene expression was normalized to internal controls and fold
changes were calculated using the relative quantification method
(2−ΔΔCq) (11).
Western blot analysis
Cells were washed 3 times with ice-cold PBS and then
incubated on ice with 250 μl RIPA buffer (cat no. P0013;
Beyotime Institute of Biotechnology) with 2.5 μl
phenylmethylsulfonyl fluoride (cat no. ST506-2; Beyotime Institute
of Biotechnology) for 15–30 min. The cells were collected and
centrifuged at 13,000 × g for 10 min at 4°C. The protein
concentrations of the cell lysates were measured in duplicate using
a bicinchoninic acid protein assay kit (cat no. 23227; Thermo
Fisher Scientific). The protein lysates and 6X loading buffer were
mixed at a ratio of 4:1 and then boiled for 5 min at 100°C. Equal
amounts of total protein were separated by 10% sodium dodecyl
sulphate-polyacrylamide gel electrophoresis and transferred onto
polyvinylidene difluoride membranes (cat no. FFP39; Beyotime
Institute of Biotechnology). The membranes were immunoblotted with
monoclonal mouse β-actin (1:1,000; cat. no. sc-8432), goat
anti-human p-STAT3 (1:1,000; cat. no. sc-21876), monoclonal mouse
anti-human JAK (1:400; cat. no. sc-376996), monoclonal mouse
anti-human STAT3 (1:400; cat no. sc-293151), p-JAK (1:400; cat. no.
sc-16773), E-cadherin (1:500; cat. no. sc-21791), N-cadherin
(1:400; cat. no. sc-393933) and vimentin (1:400; cat. no.
sc-373717) antibodies (all from Santa Cruz Biotechnology, Inc.) at
4°C overnight. All antibodies were diluted with 0.5% bovine serum
albumin. Following incubation, the corresponding secondary antibody
conjugated with peroxidase and enhanced chemiluminescence reagents
(Beyo ECL Plus; cat. no. P0018; Beyotime Institute of
Biotechnology) were applied, and the blot was visualized (cat. no.
121-2550; Beijing Liuyi Biotechnology Co., Ltd., Beijing, China).
The amount of total protein was semiquantified as ratio to β-actin
on each gel.
Scattering assay
Scattering assay was performed as previously
described (7). HepG2 cells
(3×105/ml) were seeded into each well of a 24-well plate
(cat. no. 662102; Greiner Bio-One GmbH, Frickenhausen, Germany),
and incubated overnight at 37°C in an atmosphere of 5%
CO2. The cells were pretreated with 10 μM TGFβ1
for 48 h at 37°C for 48 h in 95% air and 5% CO2.
Representative images were captured at a magnification of ×20 using
the Eclipse TE2000-U inverted microscope (Nikon Corporation, Tokyo,
Japan).
Invasion and migration assay
The invasion assay was performed using Transwell
24-well plates with 8-μm pore polycarbonate membranes (BD
Biosciences, Franklin Lakes, NJ, USA). Briefly, the upper side of
the membranes was coated with Matrigel (20 μg/well) and the
membranes were then air-dried for 1 h at 37°C. The lower side of
the membranes was coated with 5 μg fibronectin, and the
treated or untreated HepG2 cells (2×105) in 200
μl of DMEM medium with 2.5% FBS were placed in the upper
chamber. The lower chamber was filled with DMEM medium with 10% FBS
as the chemoattractant. The invasion chamber was incubated for 8 h
at 37°C and 5% CO2. The cells on the upper surface of
the membrane were removed by gentle scrubbing with a cotton swab.
The membranes were fixed in a stationary liquid of 95% ethanol and
5% acetic acid for 30 min and stained with crystal violet. The
number of cells on the lower surface of the membrane in 5 random
visual fields (magnification, ×200) was then counted using an
Eclipse TE2000-U inverted microscope. Each assay was performed in
triplicate.
Wound healing assay
The wound healing assay was performed as previously
described: HepG2 cells (3×105/ml) were seeded into a
6-well plate (cat. no. 657160; Greiner Bio-One GmbH) in
serum-containing medium, and incubated at 37°C in an atmosphere of
5% CO2 in order to form a confluent monolayer. The
monolayer was scratched using a sterile plastic pipette tip (cat.
no. CLS4860; Sigma-Aldrich, Merck KGaA, St. Louis, MO, USA), and
washed with PBS to remove cell debris. Subsequently, fresh medium
was added, and 10 μM TGFβ1 or 0.1 ml DMSO was added to each
well. The scratched mono-layer was incubated at 37°C in an
atmosphere of 5% CO2 for 48 h. Wound closure was
measured in 6 random high-power fields at a magnification of ×200,
using Image-Pro®Express software, version 6 (Media
Cybernetics, Inc., Rockville, MD, USA) and an Eclipse TE2000-U
inverted microscope (11).
Statistical analysis
Data were analyzed using SPSS software, version 1.0
(SPSS, Inc., Chicago, IL, USA) and GraphPad Prism software, version
5.0 (GraphPad Software, Inc., La Jolla, CA, USA). Analysis of
variance was conducted followed by the Student's t-test. The data
are presented as mean ± standard deviation. P<0.05 was
considered to indicate statistically significant differences.
Results
TGFβ1 induces migration and invasion of
HepG2 cells
To determine the migration and invasion induced by
TGFβ1, liver cancer HepG2 cells were treated with different
concentrations of TGFβ1 for 48 h, and the migration and invasion of
cancer cells were assessed by wound closure assays and Matrigel
Transwell chamber invasion assays. The effects of TGFβ1 were
observed at concentrations as low as 5 μM. As the TGFβ1
concentration increased, the migration and invasion of HepG2 cells
also increased in a concentration-dependent manner, with the most
prominent effects observed at a concentration of 10 μM
(Fig. 1A and B). These results
indicated that TGFβ1 induced HepG2 cell migration and invasion in a
concentration-dependent manner. Hence, the concentration of 10
μM was selected for all further mechanistic studies.
 | Figure 1Migration and invasion induced by
TGFβ1 in HepG2 cells. (A) In wound-healing assay, HepG2 cells were
treated with TGFβ1 at different concentrations (5, 10 and 20
μM) for 48 h, and TGFβ1-induced cell motility was determined
by measuring wound closure at a magnification of ×200 with
Image-Pro® Express software. Scale bar, 10 μm.
(B) For the migration and invasion assays, HepG2 cells were treated
with TGFβ1 at different concentrations (5, 10 and 20 μM) for
48 h, and the cells were visualized by staining with toluidine blue
and counted in 6 random high-power fields at a magnification of
×200 using Image-Pro® Express software. Scale bar, 5
μm. The experiments were performed in triplicate. Data are
presented as mean ± standard deviation. **P<0.01 vs. control
group. TGFβ1, transforming growth factor β1; con, control. |
TGFβ1 induces EMT in HepG2 cells
TGFβ1 is a factor that promotes EMT in cancer cells,
as previously reported (7,8).
EMT is an important mechanism of cancer cell invasion and
metastasis. The downregulation of E-cadherin expression and the
upregulation of vimentin and N-cadherin expression are considered
to be markers of EMT. In the present study, TGFβ1 also induced cell
scattering (Fig. 2A), indicating
that TGFβ1 induces EMT, thereby increasing the migration and
invasion of HepG2 cells. To further investigate whether EMT is
involved in TGFβ1-induced scattering, migration and invasion of
HepG2 cells, the expression of E-cadherin, vimentin and N-cadherin
were first detected by qPCR and western blot analysis. As shown in
Fig. 2B and C, the expression of
vimentin and N-cadherin was upregulated, whereas that of E-cadherin
was downregulated following treatment with 10 μM TGFβ1.
These results demonstrated that TGFβ1-induced EMT promoted the
migration and invasion of HepG2 cells.
 | Figure 2EMT induced by TGFβ1 in HepG2 cells.
(A) TGFβ1 induced HepG2 cell scattering. Cells were incubated with
10 μM TGFβ1 for 48 h. Representative images were captured at
a magnification of ×200 using an Eclipse TE2000-U inverted
microscope. Scale bar, 10 μm. (B–D) qPCR and western blot
analysis of EMT markers in HepG2 cells after treatment with 10
μM TGFβ1 for 48 h. Data are presented as mean ± standard
deviation of 3 independent experiments. *P<0.05 vs.
control. (E and F) Western blot analysis revealed that TGFβ1
promoted the expression of JAK, p-JAK, STAT3, and p-STAT3. Data are
presented as mean ± standard deviation of three independent
experiments. *P<0.05 vs. control. TGFβ1, transforming
growth factor β1; con, control; EMT, epithelial-to-mesenchymal
transition; qPCR, quantitative polymerase chain reaction; JAK,
Janus kinase; STAT3, signal transducer and activator of
transcription 3. |
Moreover, JAK/STAT3 protein expression was detected
by western blot analysis and it was observed that TGFβ1 stimulated
the expression of p-JAK and p-STAT3. This finding indicates that
TGFβ1-induced EMT may be activated by JAK/STAT3 signaling.
JAK/STAT3 signaling is involved in
TGFβ1-induced EMT to increase migration and invasion of HepG2
cells
STAT3 is the key transcription factor regulating
cell proliferation and survival. STAT3 may be activated by
oncostatin M, interferons, interleukin-6 (IL-6) and epidermal
growth factor (EGF). It was recently reported that TGFβ1 induced
JAK/STAT3 signaling to increase migration and invasion in lung
carcinoma cells. Based on these reports, we hypothesized that
JAK/STAT3 signaling may be involved in TGFβ1-induced EMT to
increase the migration and invasion in HepG2 cells. To confirm this
hypothesis, HepG2 cells were incubated with the STAT3 inhibitor
AG490 prior to treatment with TGFβ1. As shown in Fig. 3A and C, AG490 significantly
suppressed the TGFβ1-induced upregulation of N-cadherin and
vimentin expression and the downregulation of E-cadherin
expression, compared with the TGFβ1 group. Furthermore, AG490
treatment significantly reduced the number of TGFβ1-induced
invasive and migratory cells (Fig. 3E
and F), which was consistent with the results obtained from
metastasized cell-wound closure (Fig.
3D).
 | Figure 3AG490 inhibits TGFβ1-induced EMT to
increase migration and invasion via JAK/STAT3 signaling in HepG2
cells (A–C). Effects of TGFβ1 and AG490 on EMT-related protein
expression in HepG2 cells. After the cells were treated with TGFβ1,
AG490 and TGFβ1 + AG490 for 48 h, reverse
transcription-quantitative polymerase chain reaction and western
blot analysis of EMT markers in HepG2 cells were performed. Data
are presented as mean ± standard deviation of three independent
experiments. *P<0.05 vs. TGFβ1 group. (D)
Wound-healing assay suggested that TGFβ1 markedly promoted cell
motility. The promoting effects of TGFβ1 were abolished by AG490
treatment in HepG2 cells. The experiments were performed in
triplicate. Data are presented as mean ± standard deviation.
*P<0.05 vs. TGFβ1 group. (E) AG490 inhibited TGFβ1 to
promote migration and invasion in HepG2 cells. In migration and
invasion assays, cells were treated with TGFβ1, AG490 and TGFβ1 +
AG490 for 48 h. The promoting effects of TGFβ1 were abolished by
AG490 treatment in the HepG2 cells. Cells were visualized by
staining with toluidine blue and counted in six random high-power
fields at a magnification of ×200 using Image-Pro®
Express software. Scale bar, 10 μm. The experiments were
performed in triplicate. Data are presented as mean ± standard
deviation. *P<0.05 vs. TGFβ1 group. TGFβ1,
transforming growth factor β1; con, control; EMT,
epithelial-to-mesenchymal transition; JAK, Janus kinase; STAT3,
signal transducer and activator of transcription 3. |
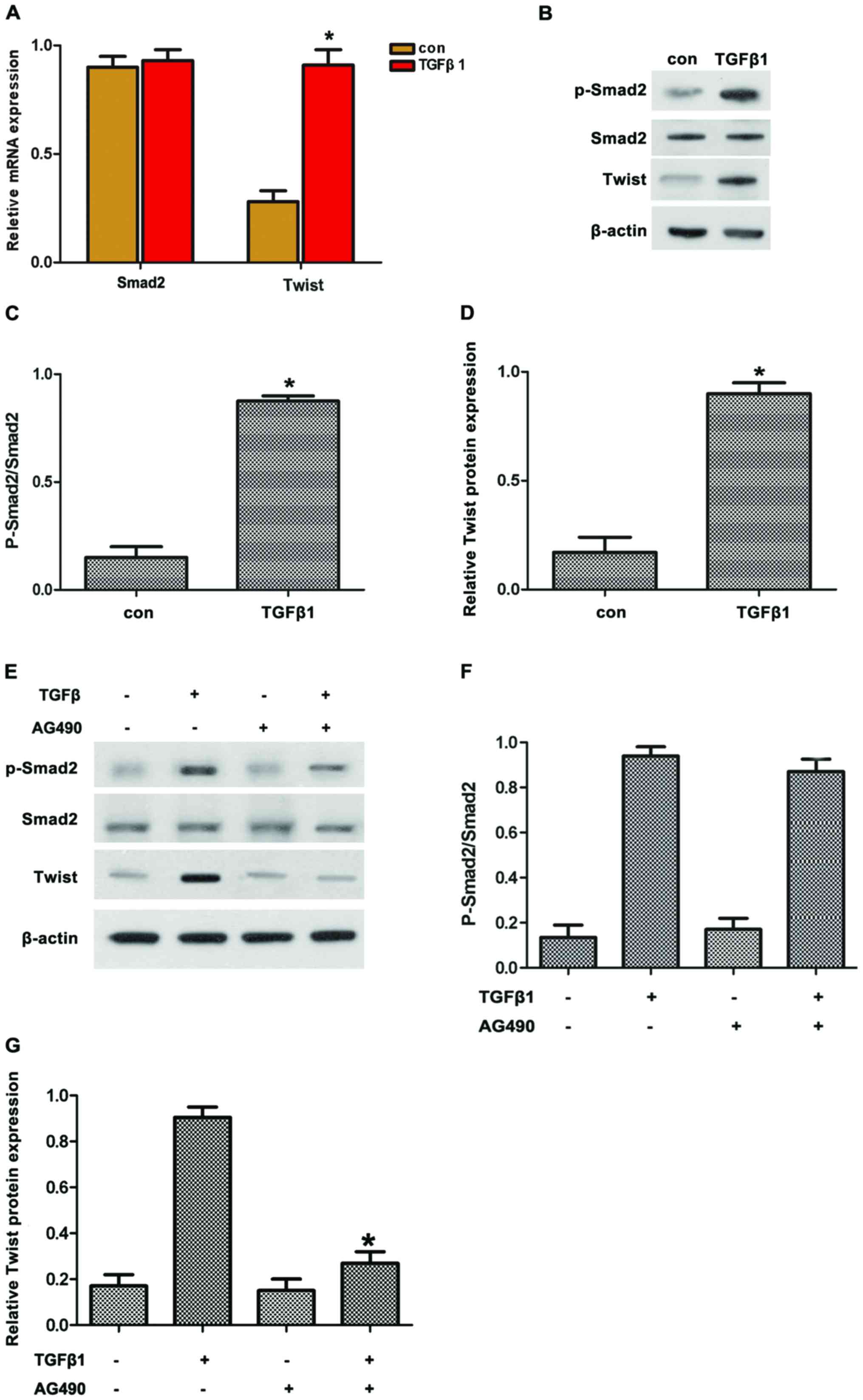
Twist is involved in TGFβ1-induced EMT
depending on STAT3
Smad2 and Twist are important factors regulating EMT
through TGFβ1. Thus, we hypothesized that TGFβ1 induced EMT by
upregulating Twist expression via JAK/STAT3 signaling. To confirm
this hypothesis, the expression of pSmad2 and Twist in HepG2 cells
treated with TGFβ1 was first detected. The results revealed that
TGFβ1 induced the protein expression of pSmad2 and Twist (Fig. 4A–D). By contrast, AG490 treatment
reversed the TGFβ1-induced protein expression of pSmad2 and Twist
(Fig. 4E–G). Along these lines,
TGFβ1 induced the protein expression of pSmad2 and Twist in
accordance with the activated JAK/STAT3 signaling.
To further determine whether Twist participated in
TGFβ1-induced EMT, HepG2 cells were transfected with siRNA of
Twist. As shown in Fig. 5A–C,
Twist knockdown significantly suppressed the TGFβ1-induced
expression of N-cadherin and vimentin, but reversed the
TGFβ1-inhibited expression of E-cadherin. The TGFβ1-induced
migration and invasion in HepG2 cells were also reversed through
Twist knockdown. Overall, these data strongly suggest that Twist
participates in TGFβ1-induced EMT to increase the migration and
invasion of HepG2 cells via JAK/STAT3 signaling.
Discussion
Invasion and metastasis are the leading causes of
death from liver cancer (12).
The onset and progression of liver cancer are regulated and
controlled by various factors, among which EMT is the key mechanism
promoting invasion and metastasis (13). A number of studies demonstrated
that molecular markers are altered during EMT in cancer cells; for
example, E-cadherin (an epithelial marker) and ZO-1 (a closely
connected protein) are downregulated, whereas the levels of
molecular markers derived from interstitial cells, including
vimentin and N-cadherin, are upregulated. Hence, adhesions among
tumor cells are reduced, thereby increasing the invasion and
metastasis of cancer cells (14–16). The occurrence of EMT is affected
by various factors. TGFβ1 is a key factor that induces and
participates in the entire process of EMT (17–19). In the present study, TGFβ1 was
found to upregulate vimentin and downregulate E-cadherin
expression. Moreover, TGFβ1 induced scattering, invasion and
metastasis of HepG2 cells. These results demonstrated that
TGFβ1-induced EMT, thereby promoting the invasion and metastasis of
HepG2 cells.
STAT3 is a signal transduction and transcription
activator. Abnormal regulation of the STAT3 signaling pathway is
associated with tumor occurrence and development (20). Following activation by cytokines
or growth factors, the activated JAK may collect STAT3 monomers to
produce homologous or heterogonous dimers; subsequently, nuclei and
specific DNA sequences regulate the transcription of target genes
(21). The abnormal expression
and activation of STAT3 in various tumor tissues and cell lines
(including liver cancer cells) are associated with the invasion and
metastasis of tumor cells (21).
EMT is the first focus in studies investigating the invasion and
metastasis of tumor cells. The activation of the STAT3 signaling
pathway is associated with EMT, invasion and metastasis of tumors.
Colomiere et al (22)
demonstrated that the JAK̸STAT3 pathway is aberrantly activated in
ovarian cancer tissues. Furthermore, EMT in ovarian cancer cells
may be induced by EGF or IL-6 (23,24). These results indicated that the
action of EGF or IL-6 relies on the activation of JAK̸STAT3
signaling; EMT induced by EGF or IL-6 may be significantly
inhibited by treatment with the JAK̸STAT3 pathway inhibitor AG490,
and the invasion and metastasis of ovarian cancer cells may be
reduced. Xiong et al (25)
also reported that AG490 significantly suppressed STAT3 activation;
consequently, AG490 treatment upregulated E-cadherin expression but
reduced the invasion of tumor cells in colorectal cancer. In the
present study, the JAK/STAT3 pathway was activated when the
TGFβ1-induced EMT promoted the migration and invasion of HepG2
cells, whereas AG490 reversed these effects. Overall, the results
demonstrated that TGFβ1-induced EMT was inhibited, thereby
confirming the involvement of the JAK/STAT3 pathway in EMT
induction.
Lee et al (7) reported that JAK̸STAT3 pathway
activation promotes the expression of Twist and, thus, reduces the
EMT of breast cancer cells. Cheng et al (17) also reported that the activated
STAT3 may directly bind to the STAT3 binding site of the Twist
promoter in breast cancer cells. These studies support that STAT3
activation may regulate the expression of Twist, a key
transcription factor regulating EMT. In the present study, Twist
was downstream from the JAK/STAT3 pathway in HepG2 cells; thus,
AG490 treatment was applied for 2 h prior to incubation with TGFβ1.
Our results demonstrated that the expression of Twist was inhibited
by AG490. Overall, TGFβ1 induced the expression of Twist in
accordance with the JAK/STAT3 pathway activation. Furthermore, we
observed that Twist participated in the TGFβ1-induced EMT that
promoted invasion and metastasis in HepG2 cells. This finding is
consistent with the report of Liu et al (26). Thus, these results indicated that
TGFβ1 upregulated the expression of Twist via the JAK/STAT3
pathway, thereby promoting the invasion and metastasis of HepG2
cells.
The findings of the present study verified the
biological functions of TGFβ1 in liver cancer HepG2 cells and
provided evidence that the TGFβ1-induced EMT promoted the invasion
and metastasis of HepG2 cells in vitro. It was further
demonstrated that these actions may be mediated via the
JAK/STAT3/Twist signaling pathway. In conclusion, TGFβ1 appears to
be involved in the progression of liver cancer and represents a
potential molecular target for the treatment of this disease.
Acknowledgments
This study was supported by the Natural Science
Foundation of China (grant no. 81600342), the Medical Foundation of
Hui Zhou (grant no. 2015y134); the Medical Research Foundation of
Guangdong Province (grant no. A2015620); and the Graduate Student
Research Innovation Project of Hunan Province (grant no.
CX2013B396).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang HC, Hu CH, Tang MC, Wang WS, Chen PM
and Su Y: Thymosin beta4 triggers an epithelial-mesenchymal
transition in colorectal carcinoma by upregulating integrin-linked
kinase. Oncogene. 26:2781–2790. 2007. View Article : Google Scholar
|
|
3
|
Gjerdrum C, Tiron C, Høiby T, Stefansson
I, Haugen H, Sandal T, Collett K, Li S, McCormack E, Gjertsen BT,
et al: Axl is an essential epithelial-to-mesenchymal
transition-induced regulator of breast cancer metastasis and
patient survival. Proc Natl Acad Sci USA. 107:1124–1129. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Song Y, Washington MK and Crawford HC:
Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal
transition in pancreatic cancer. Cancer Res. 70:2115–2125. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee TK, Poon RT, Yuen AP, Ling MT, Kwok
WK, Wang XH, Wong YC, Guan XY, Man K, Chau KL, et al: Twist
overexpression correlates with hepatocellular carcinoma metastasis
through induction of epithelial-mesenchymal transition. Clin Cancer
Res. 12:5369–5376. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giannelli G, Fransvea E, Bergamini C,
Marinosci F and Antonaci S: Laminin-5 chains are expressed
differentially in metastatic and nonmetastatic hepatocellular
carcinoma. Clin Cancer Res. 9:3684–3691. 2003.PubMed/NCBI
|
|
7
|
Lee J, Choi JH and Joo CK: TGF-β1
regulates cell fate during epithelial-mesenchymal transition by
upregulating survivin. Cell Death Dis. 4:e7142013. View Article : Google Scholar
|
|
8
|
O'Connor JW and Gomez EW: Cell adhesion
and shape regulate TGF-beta1-induced epithelial-myofibroblast
transition via MRTF-A signaling. PLoS One. 8:e831882013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wendt MK, Balanis N, Carlin CR and
Schiemann WP: STAT3 and epithelial-mesenchymal transitions in
carcinomas. JAK-STAT. 3:e289752014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 421821:2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hills CE and Squires PE: The role of TGF-β
and epithelial-to mesenchymal transition in diabetic nephropathy.
Cytokine Growth Factor Rev. 22:131–139. 2011.PubMed/NCBI
|
|
12
|
Książkiewicz M, Markiewicz A and Zaczek
AJ: Epithelial-mesenchymal transition: A hallmark in metastasis
formation linking circulating tumor cells and cancer stem cells.
Pathobiology. 79:195–208. 2012. View Article : Google Scholar
|
|
13
|
Levy DE and Darnell JE Jr: Stats:
transcriptional control and biological irnpact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu Y, Sarkissyan M and Vadgama JV:
Epithelial-Mesenchymal Transition and Breast Cancer. J Clin Med.
5:2–18. 2016. View Article : Google Scholar
|
|
15
|
Brivio S, Cadamuro M, Fabris L and
Strazzabosco M: Epithelial-to-mesenchymal transition and cancer
invasiveness: What can we learn from cholangiocarcinoma. J Clin
Med. 4:2028–2041. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nalluri SM, O'Connor JW and Gomez EW:
Cytoskeletal signaling in TGFβ-induced epithelial-mesenchymal
transition. Cytoskeleton. 72:557–569. 2015. View Article : Google Scholar
|
|
17
|
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola
D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH: Twist is
transcriptionally induced by activation of STAT3 and mediates STAT3
oncogenic function. J Biol Chem. 283:14665–14673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang S, Sun WY, Wu JJ, Gu YJ and Wei W:
Decreased expression of the type III TGF-β receptor enhances
metastasis and invasion in hepatocellullar carcinoma progression.
Oncol Rep. 35:2373–2381. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yeh YH, Wang SW, Yeh YC, Hsiao HF and Li
TK: Rhapontigenin inhibits TGF-β-mediated epithelial mesenchymal
transition via the I3K/AKT/mTOR pathway and is not associated with
HIF-1α degradation. Oncol Rep. 35:2887–2895. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Piccirillo R and Giavazzi R: Inactivating
STAT3: Bad for tumor, good for muscle. Cell Cycle. 14:939–940.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wake MS and Watson CJ: STAT3 the oncogene
- still eluding therapy. FEBS J. 282:2600–2611. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Colomiere M, Ward AC, Riley C, Trenerry
MK, Cameron-Smith D, Findlay J, Ackland L and Ahmed N: Cross talk
of signals between EGFR and IL-6R through JAK2/STAT3 mediate
epithelial-mesenchymal transition in ovarian carcinomas. Br J
Cancer. 100:134–144. 2009. View Article : Google Scholar
|
|
23
|
Li CJ, Li YC, Zhang DR and Pan JH: Signal
transducers and activators of transcription 3 function in lung
cancer. J Cancer Res Ther. 9(Suppl 2): S67–S73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu A, Zhao F, Wang J, Zhao Y, Luo Z, Gao
Y and Shi J: Regulation of TRPM7 function by IL-6 through the JAK2
STAT3 signaling pathway. PLoS One. 11:e0152120–e0152132. 2016.
View Article : Google Scholar
|
|
25
|
Xiong H, Zhang ZG, Tian XQ, Sun DF, Liang
QC, Zhang YJ, Lu R, Chen YX and Fang JY: Inhibition of JAK1,
2/STAT3 signaling induces apoptosis, cell cycle arrest, and reduces
tumor cell invasion in colorectal cancer cells. Neoplasia.
10:287–297. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014. View Article : Google Scholar : PubMed/NCBI
|