Introduction
Familial exudative vitreoretinopathy (FEVR; OMIM
133780, 305390, 605750, 601813 and 613310) is a rare hereditary
retinal disorder first reported by Criswick and Schepens in 1969
(1). It is characterized by the
premature arrest of vascularization in the peripheral retina,
resulting in peripheral retinal neovascularization, subretinal
exudation, temporal radial retinal folds, macular dragging, and
tractional retinal detachment (1–6).
FEVR exhibits variable patterns of inheritance, including autosomal
dominant (AD), autosomal recessive (AR), and X-linked recessive
(7,8). The clinical manifestations of FEVR
vary significantly depending on age; from very mild symptoms to
complete blindness, even within the same family (4,9).
The diagnosis of FEVR is made based on the evidence of incomplete
peripheral retinal vascularization in one or two eyes in patients
born at pre-term or full-term, and must be differentiated from
retinopathy of prematurity (ROP), Coats' disease, incontinentia
pigmenti, and persistent fetal vasculature (2,10,11). The clinical staging of FEVR is
determined based on the presence of peripheral retinal avascularity
(stage I), retinal neovascularization (stage II), extramacular
retinal detachment (stage III), subtotal macula-involving retinal
detachment (stage IV), and total retinal detachment (stage V)
(2,10). Treatment for FEVR is usually
guided by the clinical stages. For the more advanced stages III-V,
surgery is often required to remove scar tissue and release
traction.
Currently, five genes have been identified to be
mutated in FEVR, including low-density lipoprotein receptor-related
protein 5 (LRP5; AD or AR; OMIM 603506), frizzled-4
(FZD4; AD or AR; OMIM 604579), tetraspanin-12
(TSPAN12; AD or AR; OMIM 613138), Norrie disease protein
(NDP; X-linked; OMIM300658) and Zinc Finger Protein 408
(ZNF408; AD; OMIM 616454) (2). Among them, LRP5, FZD4,
TSPAN12 and NDP are all involved in the Wnt/β-catenin
signaling pathway, indicating the key regulatory role of this
signaling pathway during retinal vascularization. In addition, the
Wnt/β-catenin signaling pathway communicates with other growth
factor signaling pathways, such as fibroblast growth factor and
transforming growth factor-β, which are critical for the regulation
of cell growth and differentiation during development (12–19). A recent study by Salvo et
al (20) sequenced 92 FEVR
patients by next-generation sequencing (NGS), and identified
mutations in LRP5, FZD4, TSPN12, NDP
and ZNF408 that account for 19.6, 15.2, 8.7, 6.5 and 1.1% of
the patients, respectively. Notably, the authors identified two
patients with mutations in more than one disease-associated gene
(20). However, the functional
consequences of these mutations remain unclear.
As FEVR is a clinically heterogeneous disorder,
molecular diagnosis provides useful information for disease
diagnosis and genetic counseling. Furthermore, identification of
genetic mutations in FEVR is the first stage of elucidating the
pathogenesis of this disease. The aim of the present study was to
characterize the clinical presentation of a family presenting with
bilateral severe FEVR, and to identify the underlying genetic
variations in this family.
Materials and methods
Study subjects and clinical
examinations
A family presenting with bilateral FEVR was
recruited for the present study. A total of five family members
underwent complete ophthalmic examinations in the Zhongshan
Ophthalmic Center, Sun Yat-sen University (Guangzhou, China).
Visual acuity was examined using the early treatment diabetic
retinopathy study chart (Precision Vision, Lasalle, IL, USA).
Anterior segment photography was performed using the BX 900 Slit
Lamp (Haag-Streit AG, Koeniz, Switzerland). Fundus photography and
fundus fluorescein angiography (FFA) were performed using the
Heidelberg Retina Angiograph (Heidelberg Engineering GmbH,
Heidelberg, Germany) and the Ret Cam imaging system (Clarity
Medical Systems, Inc., Pleasanton, CA, USA). The amplitudes of the
rod and cone responses were assessed using the Espion
electrophysiology system (Diagnosys LLC, Lowell, MA, USA) according
to the electroretinogram (ERG) standards of the International
Society for Clinical Electrophysiology of Vision (ISCEV; 2008
update). Physical examinations and B-scan ultrasonography were
performed to confirm the diagnosis and exclude systemic
diseases.
Target capture and NGS
A total volume of 15 ml venous blood samples from
the patient, the unaffected family members, and 200 unrelated
control subjects from the same population were collected. Genomic
DNA from peripheral blood leucocytes was extracted using the QIAamp
DNABlood Midi kit (cat. no. 51104, Qiagen GmbH, Hilden, Germany)
according to the manufacturer's instructions. The genomic DNA was
fragmented by the Covaris LE220 Sonicator (Covaris, Woburn, MA,
USA) to generate a paired-end library (200–250 bp). The library was
enriched by array hybridization as previously described (21), followed by elution and
post-capture amplification. The products were then subjected to the
Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., Santa Clara,
CA, USA) and StepOnePlus Real-Time PCR System (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) for estimation of the
enrichment magnitude. Subsequent to quality control, captured
library sequencing was performed on the Illumina HiSeq2500
Analyzers (Illumina, Inc., San Diego, CA, USA) for 90 cycles per
read to generate paired-end reads. Image analysis, error estimation
and base calling were performed using the Illumina Pipeline
software (version 1.3.4; Illumina, Inc.) to generate raw data
(22).
A capture panel of inherited retinal-disease genes
was previously designed and assessed in the present study. The
capture panel comprised 708,919 bp that covered all exons together
with the flanking exon and intron boundaries (±15 bp) of 175 genes,
including 138 genes causing common inherited non-syndromic eye
diseases and 54 genes causing syndromic eye diseases (22,23).
Data analysis and mutation
validation
To detect the potential variants in the family,
bioinformatics processing and data analysis were performed after
receiving the primary sequencing data. Previously published
filtering criteria were used to generate 'clean reads' for further
analysis (21). The 'clean reads'
(with a length of 90 bp) derived from targeted sequencing and
filtering were subsequently aligned to the human genome reference
(hg19) using the Burrows Wheeler Aligner (BWA) Multi-Vision
software package (version 0.7.12, http://bio-bwa.sourceforge.net) (24). Following alignment, the output
files were used to perform sequencing coverage and analysis of the
target region, single-nucleotidevariant (SNV), and
insertion/deletion (indel) calling. SOAPsnp software (version 1.03,
http://soap.genomics.org.cn/soapsnp.html) (24) and Samtools (version 1.6,
http://samtools.sourceforge.net)
(25) were used to detect SNVs
and indels. All SNVs and indels were filtered and estimated via
multiple databases, including the National Center for Biotechnology
Information (NCBI) dbSNP (https://www.ncbi.nlm.nih.gov/snp/), HapMap1000 human
genome dataset (http://www.internationalgenome.org/home), and a
database of 200 Chinese healthy adults based on the blood samples
of the 200 untreated control patients included in the present
study.
The identified mutations were validated using
conventional polymerase chain reaction (PCR)-based sequencing
methods. Exon 40 of the ABCA4 gene (OMIM 601691) and the
exon 2 of the LRP5 gene were amplified by PCR using the
respective primers (Table I) as
previously described (26–28).
Briefly, the PCR reactions were conducted in a total volume of 50
µl using the following thermal cycling profile: One cycle at
94°C for 5 min, followed by 40 cycles at 94°C for 45 sec, 60–62°C
for 45 sec, 72°C for 45 sec, and a final extension step at 72°C for
10 min. The PCR products were sequenced from each direction with an
ABI3730 Automated Sequencer (Thermo Fisher Scientific, Inc.). The
sequencing results were analyzed using the SeqManII program of the
Lasergene package (DNAStar, Inc., Madison, WI, USA) and compared
with the reference sequences in the NCBI database (29,30).
 | Table IPrimer sets used for polymerase chain
reaction. |
Table I
Primer sets used for polymerase chain
reaction.
| Exon | Forward
(5′–3′) | Reverse
(5′–3′) | Product size
(bp) | Annealing
temperature (°C) |
|---|
| ATP binding
cassette subfamily A member 4 exon 40 |
CACAGGAGGGATGGAGGGC |
GCCTGCTGTGTCCTTTCTCTCA | 661 | 62 |
| LDL receptor
related protein 5 exon 2 |
CCGAATGTGGGAAGAAGGCT |
CTGAGTCCGTCCAGTACAGC | 521 | 60 |
Interpretation of the genetic
variants
To predict the effect of missense variants,
polymorphism phenotyping (PolyPhen) and sorting intolerant from
tolerant (SIFT) were used to predict the potential impact of a
single amino acid substitution on the protein structure and
function using straight forward physical and comparative
considerations (22,23,31). Variants were considered pathogenic
when at least one of the two programs predicted deleterious effect
of the amino acid substitution on the protein structure or
function. The Human Gene Mutation Database was used to screen
mutations reported in the published studies. In addition,
HomoloGene (https://www.ncbi.nlm.nih.gov/homologene) was used to
assess whether the mutated amino acid residues were conserved
across different species.
All experimental protocols were performed according
to the guidelines approved by the ethics committee of Zhongshan
Ophthalmic Center. Written informed consent forms were obtained
from all family members prior to the study. The tenets of the
Declaration of Helsinki were followed throughout the study.
Results
Clinical manifestations
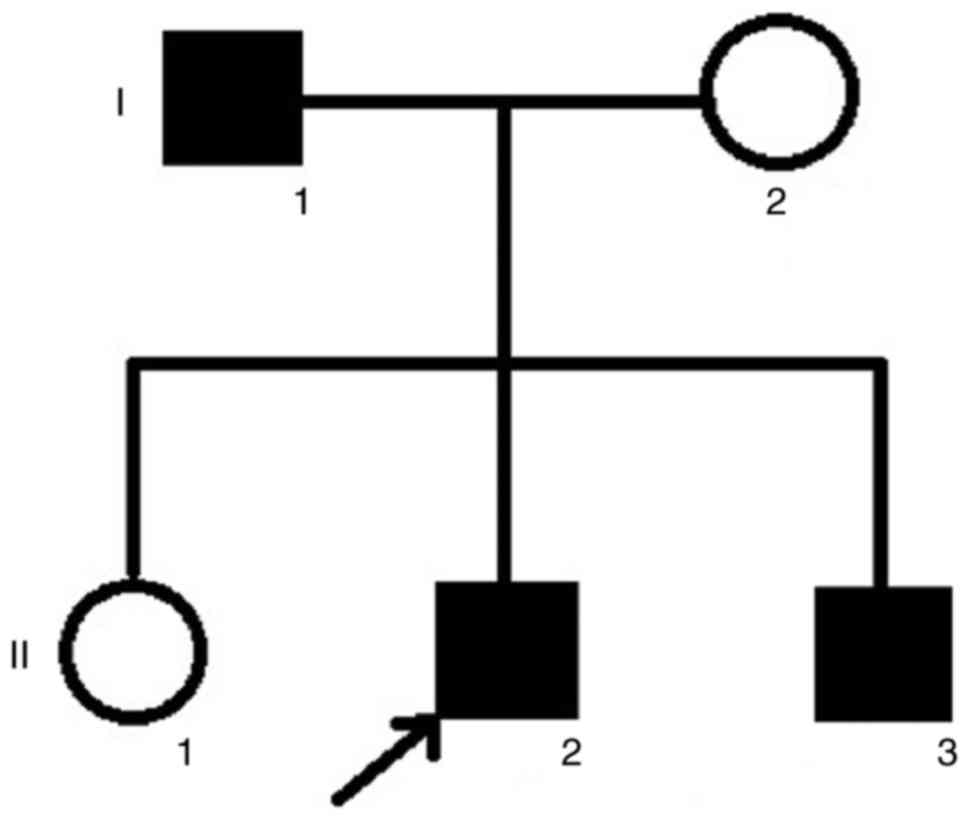
The family in the current study was from the
southern area of China. The pedigree indicated that the FEVR was
inherited in an AD manner (Fig.
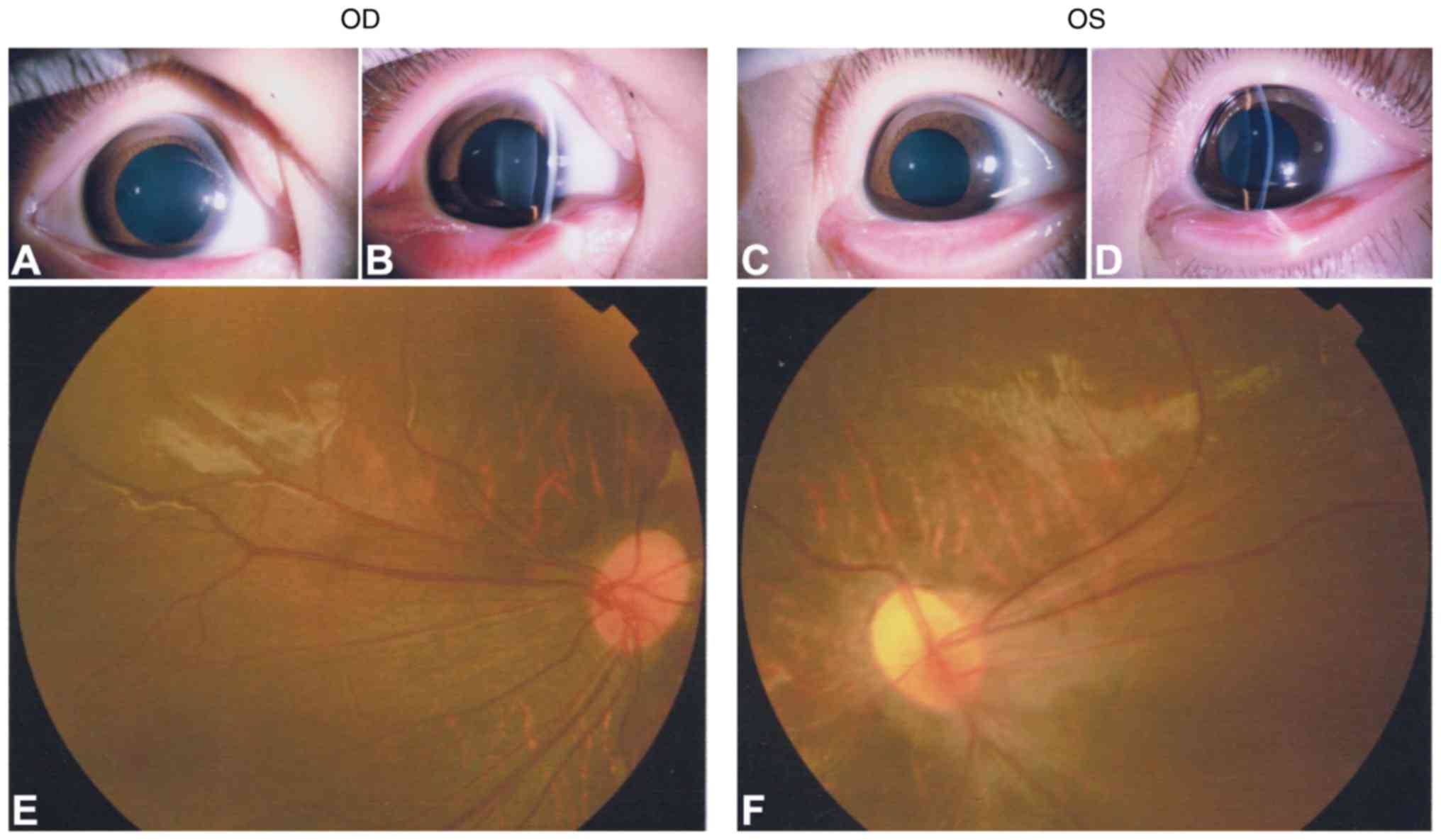
1). The affected father (I:1) exhibited a normal anterior
segment with transplant cornea and lens in each eye (Fig. 2A–D). Fundus photography and FFA
exhibited peripheral retinal degeneration (Fig. 2E and F; black arrows) and
brush-like peripheral vessels (Fig.
2G and H; white arrows), which are considered typical signs of
FEVR. The mother (I:2) and the 9-year-old daughter (II:1) presented
with normal anterior segments and normal retinal appearance without
leakage and degeneration (Figs. 3
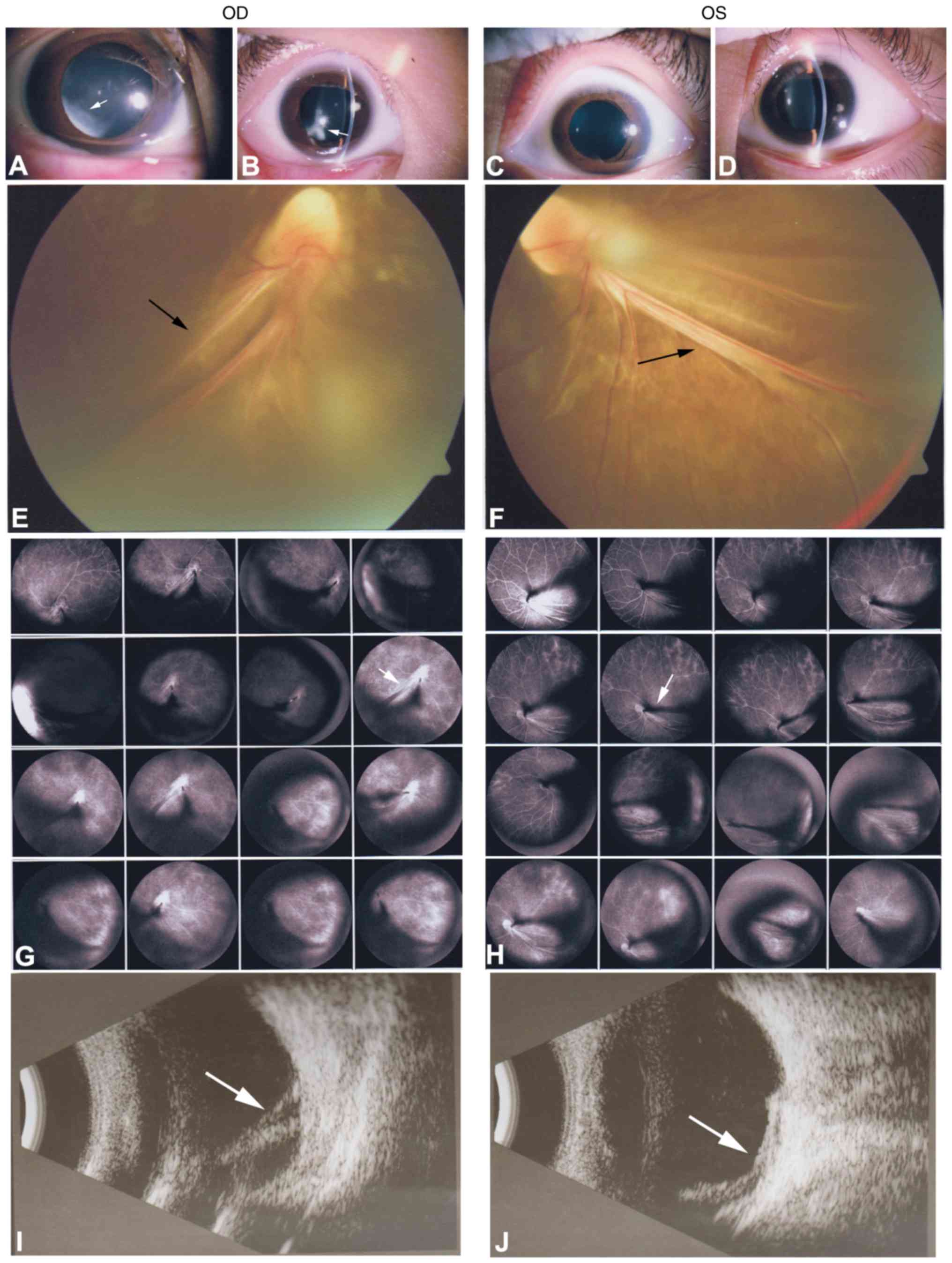
and 4). The 7-year-old son (II:2;
the proband) presented with local retinal detachment and low vision
at birth. He exhibited bilateral nystagmus. Anterior-segment
examination demonstrated local lens opacities in the right eye
(Fig. 5A and B; white arrows),
whereas the left eye was relatively normal (Fig. 5C and D). Fundus examination
exhibited knife-like retinal fold (falciform retinal fold), macular
dragging and retinal detachment (Fig.
5E and F; black arrows). FFA demonstrated peripheral retinal
avascularity with abnormal vessels and leakage (Fig. 5G and H; white arrows). A B-scan
indicated retinal detachment (Fig. 5I
and J; white arrows). ERG exhibited mild abnormal rod responses
and severe abnormal cone responses (Fig. 6). Another affected 6-year-old son
(II:3) presented with bilateral nystagmus, but normal anterior
segment (Fig. 7A–D). Fundus
examination revealed straight retinal blood vessels and the absence
of a macular arch ring (Fig. 7E and
F). The clinical manifestations of the family members are
summarized in Table II.
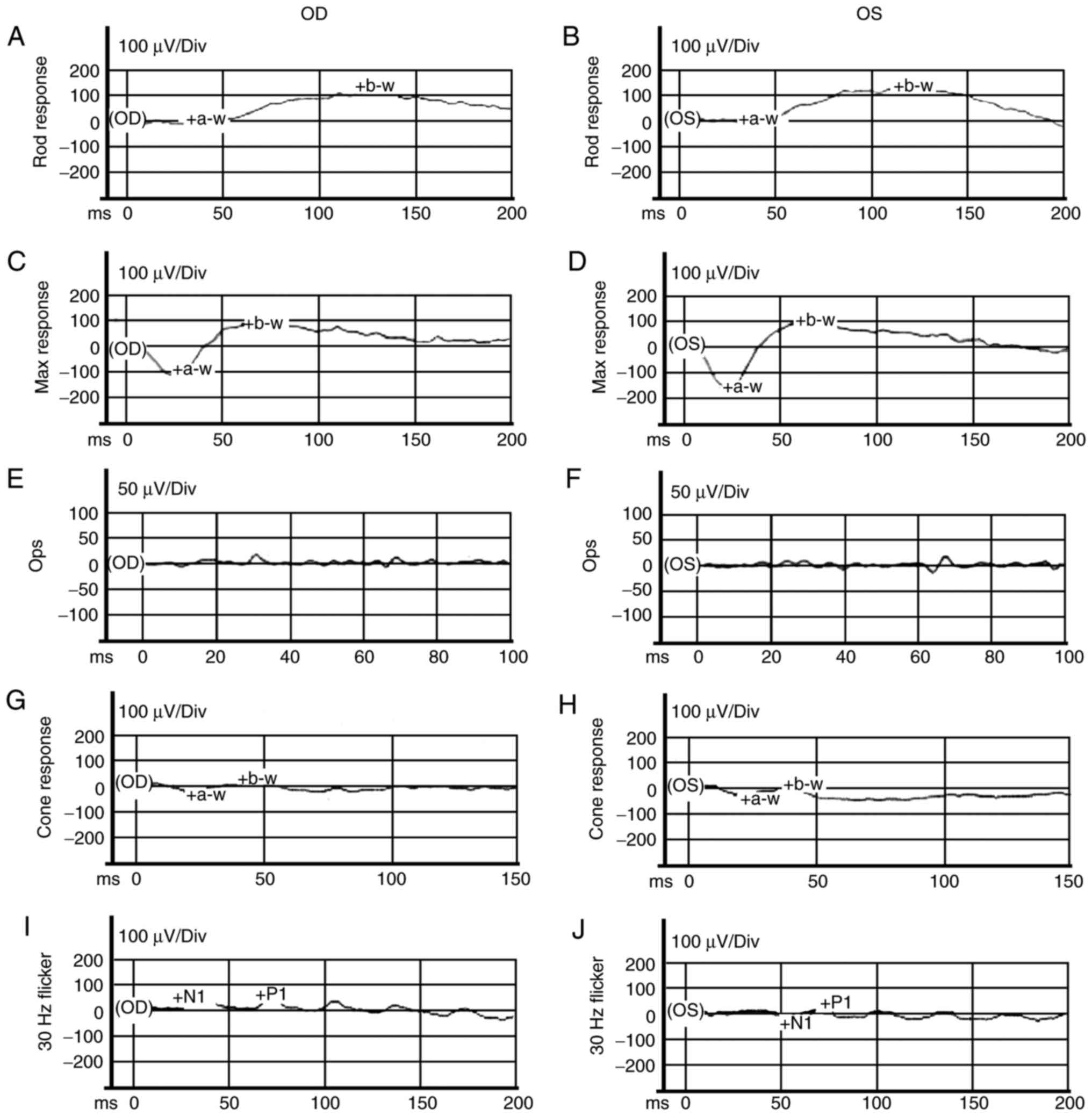 | Figure 6ERG examinations of the son (proband;
II:2). (A–F) ISCEV standard scotopic ERG. (G–J) ISCEV standard
photopic ERG. ERG indicated mild abnormal rod responses and severe
abnormal cone responses. ERG, electroretinogram; ISCEV,
International Society for Clinical Electrophysiology of Vision;
a-w, a-wave; b-w, b-wave; Div, division; N1, first negative wave;
P1, first positive wave; Ops, oscillatory potentials. OD, right
eye; OS, left eye. |
| Table IISummary of clinical manifestations
and mutations in the family. |
Table II
Summary of clinical manifestations
and mutations in the family.
| Patient | Sex | Age (years) | Clinical
manifestations
| Mutation |
|---|
| VA (BCVA) | Optometry | IOP | Lens/cornea | Fundus | FFA | B-scan | ERG |
|---|
| I:1 | M | 37 | OD:0.0 (0.0);
OS:0.0 (0.0) | N/A | Normal | Normal | Peripheral retinal
degeneration | Brush-like
peripheral vessels | N/A | N/A |
ABCA4(c.5693G>A);
LRP5(c.260T>G) |
| I:2 | F | 33 | OD:0.2 (0.0);
OS:0.2 (0.0) |
OD:−0.25DS&−0.75DC;
OS:+0.25DS&−1.50DC | Normal | Normal | Normal | Normal | N/A | N/A | – |
| II:1 | F | 9 | OD:0.2 (0.0);
OS:0.1 (0.0) |
OD:+0.00DS&+0.50DC;
OS:+0.00DS&+1.00DC | Normal | Normal | Normal | Normal | N/A | N/A |
ABCA4(c.5693G>A) |
| II:2 | M | 7 | OD:2.0 (2.0);
OS:2.0 (2.0) | OD:
−6.00DS&−2.00DC; OS: −2.00DS&−3.00DC | Normal | Lens opacities
(OD), bilateral nystagmus | Knife-like retinal
fold, macular dragging, and retinal detachment | Peripheral retinal
avascularity with leakage | Retinal
detachment | Abnormal rod and
cone responses |
ABCA4(c.5693G>A);
LRP5(c.260T>G) |
| II:3 | M | 6 | OD:1.0 (0.6);
OS:1.0 (0.6) | OD:
−2.75DS&−1.75DC; OS: −1.00DS&−2.00DC | Normal | Bilateral
nystagmus | Straight retinal
blood vessels and absence of macular arch ring | N/A | N/A | N/A |
ABCA4(c.5693G>A);
LRP5(c.260T>G) |
Mutation screening and bioinformatics
analysis of the mutations
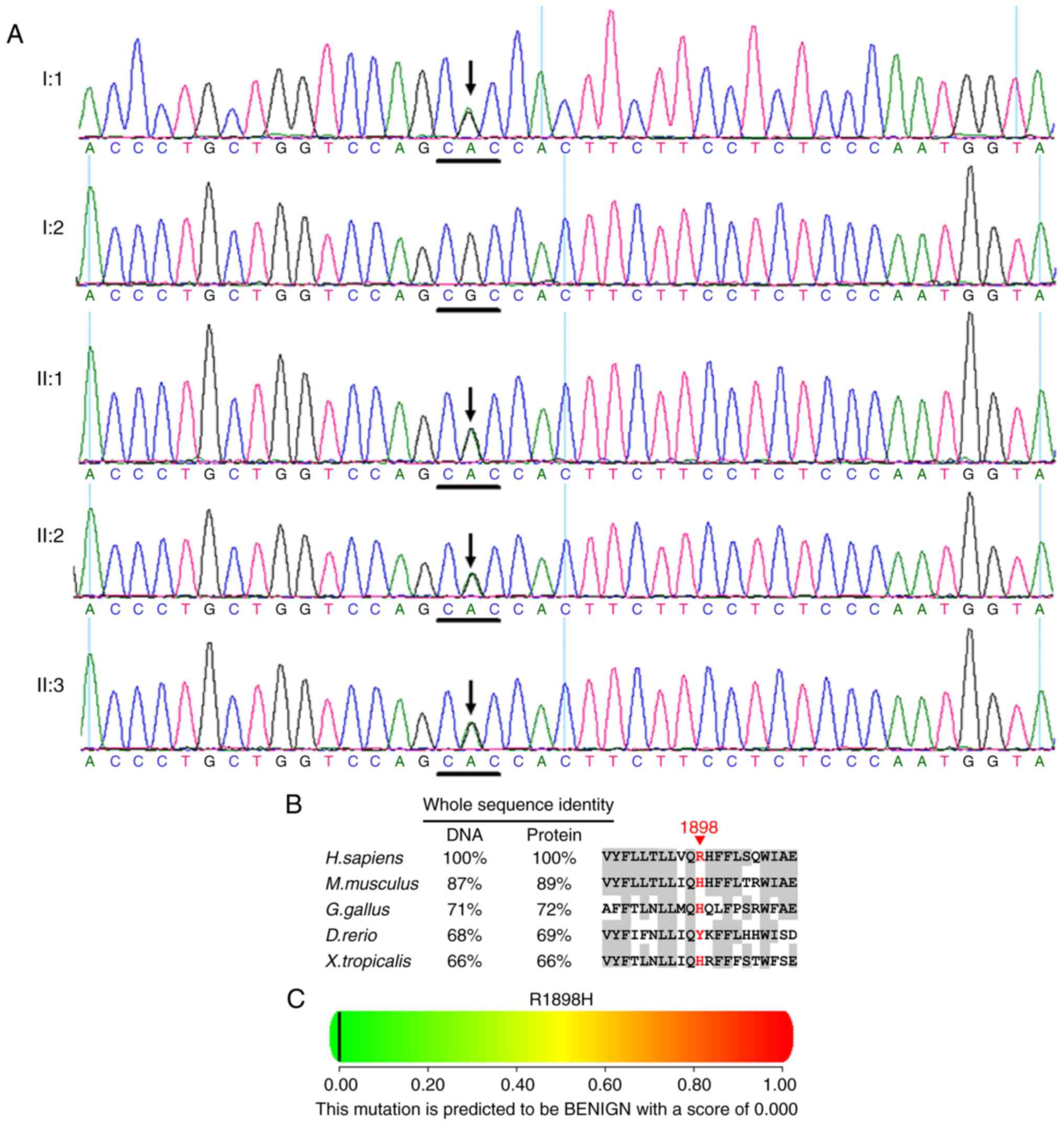
One recurrent heterozygous ABCA4 mutation
c.5693G>A (p.R1898H) in exon 40 was identified in the cases of
I:1, II:1, II:2 and II:3 (Fig. 8A
and Table II). Multiple sequence
alignment indicated that the residue at position 1898 of ABCA4 is
not highly conserved (Fig. 8B).
Polyphen (Fig. 8C) and SIFT
predicted that the amino acid substitution R1898H in the protein
ABCA4 is benign.
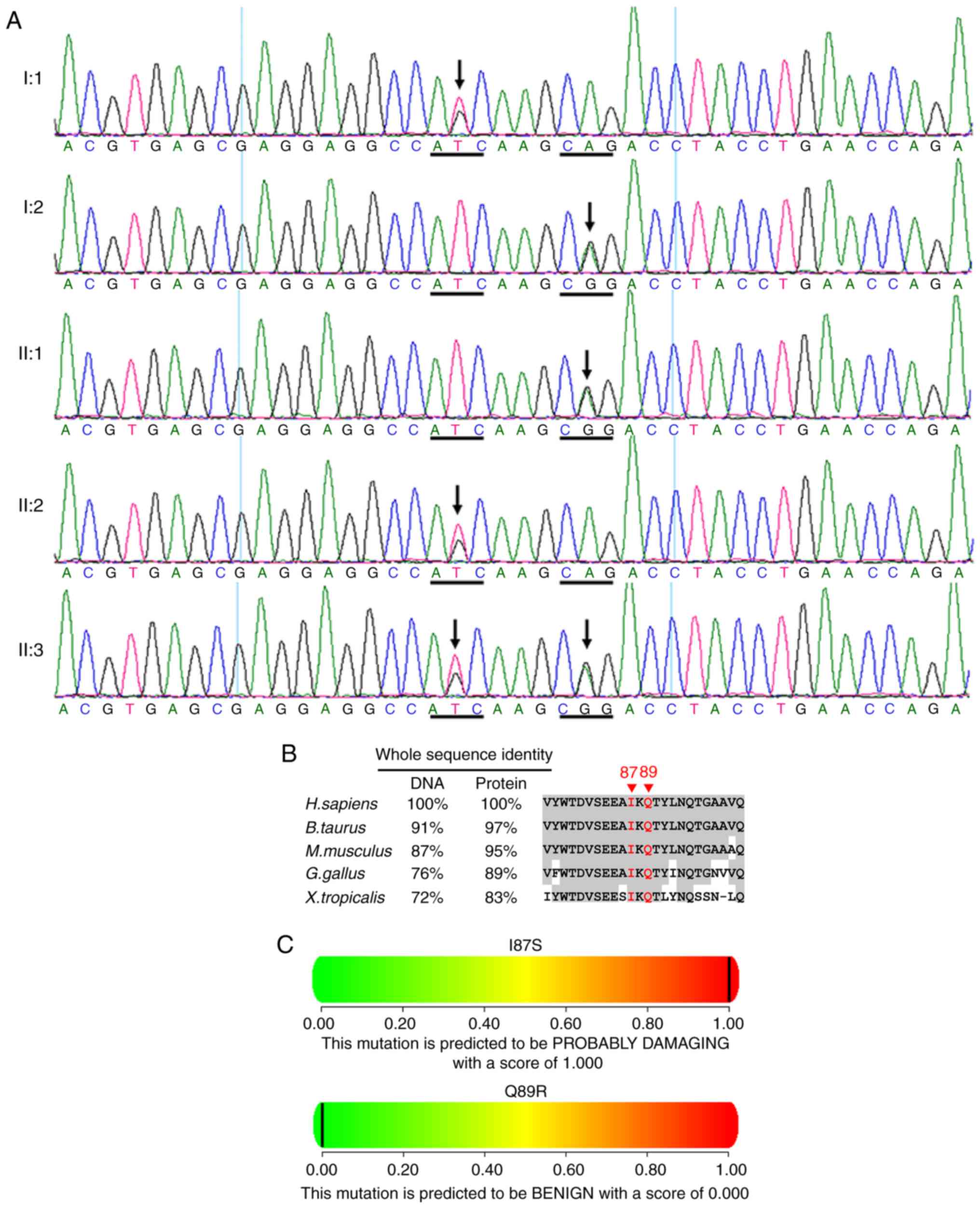
One novel heterozygous LRP5 mutation
c.260T>G (p.I87S) in exon 2 was identified in this family (I:1,
II:2 and II:3; Fig. 9 and
Table II). A single nucleotide
polymorphism c.266A>G (p.Q89R, rs41494349) in exon 2 was
identified in the cases of I:2, II:1 and II:3 (Fig. 9A) (13). The residues at position 87 and 89
of LRP5 are highly conserved across species (Fig. 9B). PolyPhen (Fig. 9C) and SIFT predicted that the
amino acid substitution I87S in the protein LRP5 is damaging.
PolyPhen (Fig. 9C) predicted that
the amino acid substitution Q89R in the protein LRP5 is benign.
These two mutations [c.5693G>A (p.R1898H) in
ABCA4 and c.260T>G (p.I87S) in LRP5] were not
observed in the 200 unrelated subjects from the same
population.
Discussion
FEVR exhibits highly variable expressivity in the
affected individuals even in the same family, which may be due to
the presence of genetic modifiers and environmental factors
(1). For example, alterations of
the oxygen levels in the intrauterine environment or exposure to
certain drugs, may predispose the affected individuals to develop
clinical symptoms (2).
Additionally, FEVR presents asymmetrically, where a single eye
develops retinal detachment and the due to local microenvironmental
differences during retinal other eye exhibits no observable
clinical signs, potentially vasculogenesis (2). In the present study, the proband
(II:2) exhibited various bilateral retinal folds, macula dragging
and retinal detachment, whereas the other individuals carrying the
same mutations exhibited relatively mild symptoms (I:1 and II:3).
The affected individuals (I:1, II:2 and II:3) presented with
brush-like peripheral vessels or avascularity, which are considered
as the hallmarks of FEVR (32,33). The proband (II:2) exhibited
decreased amplitudes and prolonged implicit times of the ERG
components, which were previously reported in the FEVR patients
(34). Notably, the FEVR patients
in the present study did not have a history of prematurity, which
is an important clinical feature that distinguishes FEVR from ROP,
as the vitreoretinal manifestations of these two diseases are very
similar (35).
In the present study, two mutations were identified
in the affected individuals of the family. To the best of our
knowledge, the ABCA4 mutation c.5693G>A (p.R1898H) has
not been reported in FEVR. The ABCA4 gene encodes a large
glycoprotein ABCA4 with 2,273 amino acids. The ABCA4 protein is
composed of two structurally-associated tandem-arranged halves. The
N and C halves are predicted to have a single membrane-spanning
segment followed by a large exocytoplasmic domain, a transmembrane
domain (TMD) and a nucleotide-binding domain (36,37). A highly conserved VFVNFA motif
near the C-terminus is essential for the cholesterol efflux and
apolipoprotein A-I binding activities of ABCA4 (Fig. 10A). Mutations in the ABCA4
gene are responsible for a large variety of retinal degenerative
diseases, including Stargardt disease, cone-rod degeneration,
retinitis pigmentosa, and age-related macular degeneration
(38,39). The mutation c.5693G>A
(p.R1898H) identified in the present study alters the amino acid
residue close to the TMD (Fig.
10A) (39). Two unrelated
pedigrees with Stargardt disease have been reported to carry this
mutation (6,40–42). However, bioinformatic analyses by
PolyPhen and SIFT predicted that this mutation is not harmful
(Fig. 8C) and the residue Arg
1898 on ABCA4 is not conserved across species (Fig. 8B). Therefore, detailed studies are
required to determine the functional consequences of this mutation.
Additionally, this mutation is associated with age-related macular
degeneration (AMD) (6,40,41). The present study was unable to
exclude the possibility that the individuals carrying this mutation
in this family are predisposed to the development of AMD;
therefore, follow-up studies are required.
 | Figure 10Schematic diagrams of the ABCA4 and
LRP5 protein indicating the location of the mutations. (A)
Predicted domains of ABCA4. ABCA4 protein consists of 2,273 amino
acids. Its structure contains two ECDs (ECD1 and ECD2), two CDs
(CD1 and CD2), 12 transmembrane segments, and a VFVNFA motif. Two
NBDs (NBD1 and NBD2) are in the CD1 and CD2, respectively. The
p.R1898H mutation is close to the last transmembrane segment (red
arrow). (B) Predicted domains of LRP5. The LRP5 protein consists of
1,615 amino acids. Its extracellular segment contains four YWTD
β-propeller domains and EGF-like domains. These domains are
followed by three LDLR-like ligand-binding domains. The mutation
p.I87S is in the first YWTD β-propeller domain. ABCA4, ATP binding
cassette subfamily A member 4; LRP5, LDL receptor related protein
5; ECD, exocytoplasmic domains; CD, cytoplasmic domain; NBD,
nucleotide binding domains; EGF, epidermal growth factor; LDLR, low
density lipoproteins receptor; M, membrane. |
The LRP5 mutation c.260T>G (p.I87S) in
exon 2 identified in this family is a novel mutation. Compared with
FZD4, TSPN12, NDP and ZNF408,
LRP5 is most frequently mutated in FEVR, likely due to its
large coding region (22,43–45). LRP5 and its homologue
LRP6 encode single-span transmembrane receptors, LRP5/LRP6
that interact with the seven-pass transmembrane receptor, FZD4 to
bind Wnt, activating the canonical Wnt-β-catenin signaling pathway
(46,47). Norrin, a protein encoded by
NDP, acts as a ligand and interacts with the FZD4-LRP5
complex, stabilizing the cytoplasmic β-catenin (48). Additionally, Norrin cooperates
with TSPAN12 to promote multimerization of FZD4 (49). Although the binding sites of LRP5
to FZD4 and Norrin have not been fully elucidated, it has been
proposed via comparative modeling that the missense mutations in
LRP5 may disrupt the protein-binding sites (15,50). The I87S mutation in LRP5 is in the
first YWTD β-propeller domain (Figs.
9B and 10B). This domain is
responsible for the binding of LRP5/LRP6 to the extracellular
ligands. Therefore, this mutation may compromise the Wnt-β-catenin
signaling, which is essential for retinal vascular development.
In conclusion, a detailed characterization of one
Chinese family with bilateral FEVR was performed, and two trans
heterozygous mutations were identified in ABCA4 and
LRP5. These findings expand the mutation spectrums of
ABCA4 and LRP5, and will be valuable for genetic
counseling and development of therapeutic interventions for FEVR
patients. Although our understanding of the function of ABCA4 and
LRP5 proteins remains limited, the discovery of these mutant
variants provides an opportunity and rationale for in-depth
mechanistic studies, and may help to reveal the critical physiology
underlying associated retinal development disorders in general.
Acknowledgments
The authors would like to thank the patients, their
families and the control volunteers for participating in the
present study. The study was supported by the National Natural
Science Foundation of China (grant nos. 81500709, 81570862 and
81670872) and the State Scholarship Fund from the China Scholarship
Council.
References
|
1
|
Criswick V and Schepens C: Familial
exudative vitreoretinopathy. Am J Ophthalmol. 68:578–594. 1969.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gilmour D: Familial exudative
vitreoretinopathy and related retinopathies. Eye. 29:1–14. 2015.
View Article : Google Scholar :
|
|
3
|
Riveiro-Alvarez R, Trujillo-Tiebas MJ,
Gimenez-Pardo A, Garcia-Hoyos M, Cantalapiedra D, Lorda-Sanchez I,
Rodríguez de Alba M, Ramos C and Ayuso C: Genotype-phenotype
variations in five Spanish families with Norrie disease or X-linked
FEVR. Mol Vis. 11:705–712. 2005.PubMed/NCBI
|
|
4
|
Sızmaz S, Yonekawa Y and Trese MT:
Familial exudative vitreoretinopathy. Turk J Ophthalmol.
45:164–168. 2015.
|
|
5
|
Khwarg JW, Bourla D, Gonzales CA and
Schwartz SD: Familial exudative vitreoretinopathy and macular hole
exhibited in same individual. Semin Ophthalmol. 22:85–86. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lewis RA, Shroyer NF, Singh N, Allikmets
R, Hutchinson A, Li Y, Lupski JR, Leppert M and Dean M:
Genotype/phenotype analysis of a photoreceptor-specific ATP-binding
cassette transporter gene, ABCR, in Stargardt disease. Am J Hum
Gene. 64:422–434. 1999. View
Article : Google Scholar
|
|
7
|
Tang M, Ding X, Li J, Hu A, Yuan M, Yang
Y, Zhan Z, Li Z and Lu L: Novel mutations in FZD4 and
phenotype-genotype correlation in Chinese patients with familial
exudative vitreoretinopathy. Mol Vis. 22:917–932. 2016.PubMed/NCBI
|
|
8
|
Li JK, Fei P, Li Y, Huang QJ, Zhang Q,
Zhang X, Rao YQ, Li J and Zhao P: Identification of novel KIF11
mutations in patients with familial exudative vitreoretinopathy and
a phenotypic analysis. Sci Rep. 6:265642016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benson WE: Familial exudative
vitreoretinopathy. Trans Am Ophthalmol Soc. 93:473–521.
1995.PubMed/NCBI
|
|
10
|
Ranchod TM, Ho LY, Drenser KA, Capone A
and Trese MT: Clinical presentation of familial exudative
vitreoretinopathy. Ophthalmology. 118:2070–2075. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kondo H, Kusaka S, Yoshinaga A, Uchio E,
Tawara A and Tahira T: Genetic variants of FZD4 and LRP5 genes in
patients with advanced retinopathy of prematurity. Mol Vis.
19(476): 4852013.
|
|
12
|
Buchtova M, Oralova V, Aklian A, Masek J,
Vesela I, Ouyang Z, Obadalova T, Konecna Z, Spoustova T,
Pospisilova T, et al: Fibroblast growth factor and canonical
WNT/β-catenin signaling cooperate in suppression of chondrocyte
differentiation in experimental models of FGFR signaling in
cartilage. Biochim Biophys Acta. 1852:839–850. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hiyama A, Sakai D, Tanaka M, Arai F,
Nakajima D, Abe K and Mochida J: The relationship between the
Wnt/β-catenin and TGF-β/BMP signals in the intervertebral disc
cell. J Cell Physiol. 226:1139–1148. 2011. View Article : Google Scholar
|
|
14
|
Lu GQ, Wu ZB, Chu XY, Bi ZG and Fan WX: An
investigation of crosstalk between Wnt/β-catenin and transforming
growth factor-β signaling in androgenetic alopecia. Medicine
(Baltimore). 95:pp. e42972016, View Article : Google Scholar
|
|
15
|
Fujimura N: WNT/β-Catenin signaling in
vertebrate eye development. Front Cell Develop Biol. 4:1382016.
View Article : Google Scholar
|
|
16
|
Zhang Y, Morgan R, Chen C, Cai Y, Clark E,
Khan WN, Shin SU, Cho HM, Al Bayati A, Pimentel A and Rosenblatt
JD: Mammary-tumor-educated B cells acquire LAP/TGF-beta and PD-L1
expression and suppress anti-tumor immune responses. Int Immunol.
28:423–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tan X, Zhu Y, Chen C, Chen X, Qin Y, Qu B,
Luo L, Lin H, Wu M, Chen W and Liu Y: Sprouty2 suppresses
epithelial-mesenchymal transition of human lens epithelial cells
through blockade of Smad2 and ERK1/2 pathways. PLoS On.
11:e01592752016. View Article : Google Scholar
|
|
18
|
Qin Y, Zhu Y, Luo F, Chen C, Chen X and Wu
M: Killing two birds with one stone: Dual blockade of integrin and
FGF signaling through targeting syndecan-4 in postoperative
capsular opacification. Cell Death Dis. 8:pp. e29202017, View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan X, Chen C, Zhu Y, Deng J, Qiu X, Huang
S, Shang F, Cheng B and Liu Y: Proteotoxic stress desensitizes
TGF-beta signaling through receptor downregulation in retinal
pigment epithelial cells. Curr Mol Med. 17:189–199. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Salvo J, Lyubasyuk V, Xu M, Wang H, Wang
F, Nguyen D, Wang K, Luo H, Wen C, Shi C, et al: Next-generation
sequencing and novel variant determination in a cohort of 92
familial exudative vitreoretinopathy patients. Invest Ophthalmol
Vis Sci. 56:1937–1946. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PLoS One. 6:e295002011. View Article : Google Scholar
|
|
22
|
Lin Li T, Gao Y, Chen H, Zhu C, Liu Y,
Lian B, Li Y, Zhou Y, Jiang WH, et al: Two heterozygous mutations
identified in one Chinese patient with bilateral macular coloboma.
Mol Med Rep. 16:2505–2510. 2017.PubMed/NCBI
|
|
23
|
Avila-Fernandez A, Perez-Carro R, Corton
M, Lopez-Molina MI, Campello L, Garanto A, Fernandez-Sanchez L,
Duijkers L, Lopez-Martinez MA, Riveiro-Alvarez R, et al:
Whole-exome sequencing reveals ZNF408 as a new gene associated with
autosomal recessive retinitis pigmentosa with vitreal alterations.
Hum Mol Genet. 24:4037–4048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li R, Li Y, Fang X, Yang H and Wang J,
Kristiansen K and Wang J: SNP detection for massively parallel
whole-genome resequencing. Genome Res. 19:1124–1132. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup: The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin Y, Ai S, Chen C, Liu X, Luo L, Ye S,
Liang X, Zhu Y, Yang H and Liu Y: Ala344 Pro mutation in the FGFR2
gene and related clinical findings in one Chinese family with
Crouzon syndrome. Mol Vis. 18:1278–1282. 2012.
|
|
27
|
Lin Y, Liang X, Ai S, Chen C, Liu X, Luo
L, Ye S, Li B, Liu Y and Yang H: FGFR2 molecular analysis and
related clinical findings in one Chinese family with Crouzon
syndrome. Mol Vis. 18:449–454. 2012.PubMed/NCBI
|
|
28
|
Lin Y, Liu X, Yu S, Luo L, Liang X, Wang
Z, Chen C, Zhu Y, Ye S, Yan H and Liu Y: AX6 analysis of two
sporadic patients from southern China with classic aniridia. Mol
Vis. 18:2190–2194. 2012.
|
|
29
|
Lin Y, Li T, Gao H, Lian Y, Chen C, Zhu Y,
Li Y, Liu B, Zhou W, Jiang H, et al: Bestrophin 1 gene analysis and
associated clinical findings in a Chinese patient with Best
vitelliform macular dystrophy. Mol Med Rep. 16:4751–4755.
2017.PubMed/NCBI
|
|
30
|
Lin Y, Gao H, Ai S, Eswarakumar JV, Chen
C, Zhu Y, Li T, Liu B, Liu X, Luo L, et al: C278F mutation in FGFR2
gene causes two different types of syndromic craniosynostosis in
two Chinese patients. Mol Med Rep. 16:5333–5337. 2017.PubMed/NCBI
|
|
31
|
Nikopoulos K, Gilissen C, Hoischen A, van
Nouhuys CE, Boonstra FN, Blokland EA, Arts P, Wieskamp N, Strom TM,
Ayuso C, et al: Next-generation sequencing of a 40 Mb linkage
interval reveals TSPAN12 mutations in patients with familial
exudative vitreoretinopathy. Am J Hum Genet. 86:240–247. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Miyakubo H, Hashimoto K and Miyakubo S:
Retinal vascular pattern in familial exudative vitreoretinopathy.
Ophthalmology. 91:1524–1530. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Nouhuys CE: Signs, complications, and
platelet aggregation in familial exudative vitreoretinopathy. Am J
Ophthalmol. 111:34–41. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yaguchi Y, Katagiri S, Fukushima Y, Yokoi
T, Nishina S, Kondo M and Azuma N: Electroretinographic effects of
retinal dragging and retinal folds in eyes with familial exudative
vitreoretinopathy. Sci Rep. 6:305232016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dickinson JL, Sale MM, Passmore A,
FitzGerald LM, Wheatley CM, Burdon KP, Craig JE, Tengtrisorn S,
Carden SM, Maclean H and Mackey DA: Mutations in the NDP gene:
Contribution to Norrie disease, familial exudative
vitreoretinopathy and retinopathy of prematurity. Clin Exp
Ophthalmol. 34:682–688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shroyer NF, Lewis RA, Allikmets R, Singh
N, Dean M, Leppert M and Lupski JR: The rod photoreceptor
ATP-binding cassette transporter gene, ABCR, and retinal disease:
From monogenic to multifactorial. Vision Res. 39:2537–2544. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Molday RS and Zhang K: Defective lipid
transport and biosynthesis in recessive and dominant Stargardt
macular degeneration. Prog Lipid Res. 49:476–492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koenekoop RK: The gene for Stargardt
disease, ABCA4, is a major retinal gene: A mini-review. Ophthalmic
Genet. 24:75–80. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun H, Smallwood PM and Nathans J:
Biochemical defects in ABCR protein variants associated with human
retinopathies. Nat Genet. 26:242–246. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shroyer NF, Lewis RA and Lupski JR:
Complex inheritance of ABCR mutations in Stargardt disease: Linkage
disequilibrium, complex alleles, and pseudodominance. Hum Genet.
106:244–248. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rivera A, White K, Stöhr H, Steiner K,
Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP,
Apfelstedt-Sylla E and Weber BH: A comprehensive survey of sequence
variation in the ABCA4 (ABCR) gene in Stargardt disease and
age-related macular degeneration. Am J Hum Genet. 67:800–813. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Molday RS: Photoreceptor membrane
proteins, phototransduction, and retinal degenerative diseases. The
Friedenwald Lecture. Invest Ophthalmol Vis Sci. 39:2491–2513.
1998.PubMed/NCBI
|
|
43
|
Pefkianaki M, Hasanreisoglu M, Suchy SF
and Shields CL: Familial exudative vitreoretinopathy with a novel
LRP5 mutation. J Pediat Ophthalmol Strabismus. 53:pp. e39–e42.
2016, PubMed/NCBI
|
|
44
|
Musada GR, Syed H, Jalali S, Chakrabarti S
and Kaur I: Mutation spectrum of the FZD-4, TSPAN12 AND ZNF408
genes in Indian FEVR patients. BMC Ophthalmol. 16:902016.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang H, Li S, Xiao X, Wang P, Guo X and
Zhang Q: Identification of FZD4 and LRP5 mutations in 11 of 49
families with familial exudative vitreoretinopathy. Mol Vis.
18:2438–2446. 2012.PubMed/NCBI
|
|
46
|
Tamai K, Semenov M, Kato Y, Spokony R, Liu
C, Katsuyama Y, Hess F, Saint-Jeannet JP and He X:
LDL-receptor-related proteins in Wnt signal transduction. Nature.
407:530–535. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Pinson KI, Brennan J, Monkley S, Avery BJ
and Skarnes WC: An LDL-receptor-related protein mediates Wnt
signalling in mice. Nature. 407:535–538. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xu Q, Wang Y, Dabdoub A, Smallwood PM,
Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K and
Nathans J: Vascular development in the retina and inner ear:
Control by Norrin and Frizzled-4, a high-affinity ligand-receptor
pair. Cell. 116:883–895. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Junge HJ, Yang S, Burton JB, Paes K, Shu
X, French DM, Costa M, Rice DS and Ye W: TSPAN12 regulates retinal
vascular development by promoting Norrinbut not Wnt-induced
FZD4/beta-catenin signaling. Cell. 139:299–311. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Toomes C, Bottomley HM, Jackson RM, Towns
KV, Scott S, Mackey DA, Craig JE, Jiang L, Yang Z, Trembath R, et
al: Mutations in LRP5 or FZD4 underlie the common familial
exudative vitreoretinopathy locus on chromosome 11q. Am J Hum
Genet. 74:721–730. 2004. View
Article : Google Scholar : PubMed/NCBI
|