Introduction
Colon cancer is one of the most common malignant
tumors worldwide. In China, colon cancer is the fifth most common
type of cancer, and metastasis and recurrence of colon cancer are
the leading causes of mortality in most patients (1). At present, surgical resection is the
first-line treatment for patients with colon cancer (1,2);
however, surgical treatment is difficult to perform in patients
with recurrent cancer and metastasis (2). Numerous studies have reported that
molecular targeted therapy may further improve the survival of
patients with metastatic colon cancer (2–4).
For example, treatment with cetuximab, an anti-vascular endothelial
growth factor receptor antibody, alongside standard chemotherapy is
approved for first-line therapy in patients with K-ras mutations,
and for second-line therapy in patients with metastatic colon
cancer harboring wild-type K-ras (3,5).
Although numerous molecular targets have been identified for
individual-specific, targeted therapies in patients with colon
cancer, the mechanisms underlying these therapies remain unclear,
few agents are available, and the therapeutic efficacy is still not
satisfactory (5). Therefore, more
studies are required to fully elucidate the pathogenesis of colon
cancer and to identify potential therapeutic targets for this
disease.
Ras-related C3 botulinum toxin substrate 1 (RAC1) is
an important member of the Rho family of GTPases [Rac, Rho and cell
division cycle (CDC)42], which are molecular switches that regulate
key cellular activities, including cell proliferation, apoptosis,
gene expression and directional movement by cytoskeleton remodeling
(6–9). Numerous studies have demonstrated
that abnormal RAC1 signaling is associated with various human
diseases, including cancer, inflammation, neurodegenerative
disorders, kidney disorders and cardiovascular diseases (10). In addition, dysregulated RAC1
expression and activity has been detected in various types of
cancer cell, including colon cancer (11), gastric cancer (12), lung cancer (13) and breast cancer cells (14), and it has been reported to
modulate cancer cell proliferation, invasion, metastasis and
epithelial mesenchymal transition (EMT) by regulating several
cancer-associated signaling pathways, including p21-activated
kinases (PAK1-3), actin-binding LIM kinases (LIMK1 and LIMK2), Wnt,
phosphoinositol 3-kinase and nuclear factor (NF)-κB (10,15–18). Therefore, it has been hypothesized
that RAC1 represents an attractive therapeutic target, due to its
role in promoting cancer initiation (10,15–18).
RAC1 overexpression can initiate intestinal stem
cell proliferation and regeneration (19), thus resulting in accelerated
tumorigenic processes, reduced survival times (20), and disruption of RAC1-mediated
immune responses governing neutrophil chemotaxis and
apoptosis-associated carcinogenesis in ulcerative colitis (21). Overexpression of the RAC1 splice
variant RAC1b is also correlated with poor prognosis in patients
with metastatic colorectal cancer (11). Furthermore, increased RAC1 or
RAC1b expression can promote cellular transformation, thereby
enhancing colorectal cancer cell survival (22); conversely, silencing of RAC1
expression can induce apoptosis and cell cycle arrest, and inhibit
proliferation of colon cancer cells. Although previous studies have
demonstrated that abnormal RAC1 signaling is associated with the
pathogenesis of colon cancer, it remains unclear as to whether RAC1
signaling mechanisms regulate colon cancer.
In our previous study, RAC1 mRNA expression
was downregulated in HT-29 colon cancer cells following treatment
with the anticancer agent diallyl disulfide (DADS) (23,24). An additional study indicated that
DADS may suppress SW480 cell migration and invasion by
down-regulating the RAC1-Rho-associated protein kinase 1
(ROCK1)/PAK1-LIMK1-actin-depolymerizing factor/cofilin signaling
pathway (24).
Accordingly, the present study used RNA interference
(RNAi) technology to silence RAC1 gene expression in colon
cancer cells. Subsequently, cell proliferation, apoptosis and cell
cycle distribution were evaluated, in order to determine the role
of RAC1 in colon cancer cells. Gene expression profiles were
analyzed and bioinformatics analysis was performed to determine the
possible molecular mechanisms through which short hairpin
(sh)RNA-induced silencing of RAC1 modulated cell
proliferation in colon cancer.
Materials and methods
Cell lines and culture
The human colon cancer cell lines used in the
present study (i.e., HT-29, SW620 and HCT116 cells) and 293T cells
were purchased from China Typical Culture Center (Wuhan, China).
The cells were cultured in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 100 ml/l fetal bovine serum (FBS) (both
from Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 100
U/ml penicillin and 100 U/ml streptomycin at 37°C in a humidified
atmosphere containing 5% CO2.
Design and lentiviral packaging of RAC1
shRNA
Three pairs of shRNA sequences targeting the human
RAC1 gene were designed using the latest version of the
online RNAi design web tool (http://jura.wi.mit.edu/bioc/siRNA), as listed in
Table I. The negative control
duplexes of shRNA (shRNA-NC) were random sequences
(TTCTCCGAACGTGTCACGT), which did not target any known mammalian
gene, using the Blast website (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The shRNA
sequences were then cloned into the lentiviral vector GV248
(hU6-MCS-Ubi-EGFP-IRES-Puro; Shanghai GeneChem Co., Ltd., Shanghai,
China). Lentivirus (LV) (LV-shRNA-RAC1 and LV-shRNA-NC)
amplification and packaging was conducted according to the
lentiviral packaging protocol (Shanghai GeneChem Co., Ltd.).
Briefly, the 293T packaging cell line was cotransfected with GV248
carrying shRNA (LV-shRNA-RAC1 and LV-shRNA-NC) and pHelper
plasmids. The next day, medium was replaced with fresh DMEM and
culture was continued for 24 h at 37°C. The viral supernatant was
then collected, filtered, concentrated and stored in small aliquots
at −80°C for titration and cell infection.
 | Table IshRNA sequences targeting RAC1. |
Table I
shRNA sequences targeting RAC1.
| ID | Target | Sense | Antisense |
|---|
| RAC1-shRNA-1 |
5′-TTCTTAACATCACTGTCTT-3′ |
5′-ccTTCTTAACATCACTGTCTT-3′ |
5′-AAGACAGTGATGTTAAGAAgg-3′ |
| RAC1-shRNA-2 |
5′-CAAACAGATGTGTTCTTAA-3′ |
5′-cgCAAACAGATGTGTTCTTAA-3′ |
5′-TTAAGAACACATCTGTTTGcg-3′ |
| RAC1-shRNA-3 |
5′-TGAAGAAGAGGAAGAGAAA-3′ |
5′-cgTGAAGAAGAGGAAGAGAAA-3′ |
5′-TTTCTCTTCCTCTTCTTCAcg-3′ |
Infection of colon cancer cells with
LV-shRNA
Colon cancer cells were seeded in 6-well plates
(6×105 cells/well) and were incubated for 24 h in a
humidified atmosphere. The cells were divided into three groups: KD
group, NC group and control group (untreated colon cancer cells).
Cells in the KD and NC groups were infected with LV-shRNA-RAC1 or
LV-shRNA-NC, respectively, at a multiplicity of infection of 10,
according to the manufacturer's protocol (Shanghai GeneChem Co.,
Ltd.). After 24 h at 37°C, the medium was replaced with fresh DMEM
and cells were cultured for a further 48 h. The lentivirus
contained the green fluorescent protein (GFP) , and the number of
GFP positive cells were counted by inverted fluorescence microscopy
(Olympus IX53; Olympus Co., Ltd., Shanghai, China). Finally, the
cells were harvested and prepared for subsequent analysis.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RT-qPCR was then performed. The RT-qPCR primers for
RAC1 and GAPDH (internal control) were synthesized by
Sangon Biotech Co., Ltd. (Shanghai, China). RT was performed using
a FastQuant RT kit (Tiangen Biotech Co., Ltd., Beijing, China)
according to the manufacturer's protocol. The PCR primers for
RAC1 and GAPDH were as follows: RAC1, forward
5′-ATGTCCGTGCAAAGTGGTATC-3′, reverse 5′-CTCGGATCGCTTCGTCAAACA-3′;
and GAPDH, forward 5′-GCCAAAAGGGTCATCATCTC-3′ and reverse
5′-GTAGAGGCAGGGATGATGTTC-3′. qPCR was performed using Taq PCR
MasterMix (Tiangen Biotech Co., Ltd.) and a ViiA™ system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The thermal cycling
conditions were as follows: Initial denaturation at 95°C for 60
sec, 40 cycles of amplification at 95°C for 20 sec, annealing and
extension at 60°C for 30 sec. GAPDH was used as an internal control
for PCR amplification. The data were analyzed using the
2−ΔΔCt method (25).
Proliferation assays
Cells were trypsinized 72 h postinfection and were
seeded in 96-well plates in triplicate (2×103
cells/well). Following adherence, the cells were exposed to DMEM
containing 0.5% FBS for 0, 12, 24, 48 or 72 h at 37°C, and the
effects of RAC1 knockdown on colon cancer cell proliferation
were evaluated by MTT colorimetric assays. Briefly, the medium was
removed and replaced with medium containing 5 mg/ml MTT. The cells
were then incubated for 4 h at 37°C, after which 100 μl
dimethyl sulfoxide solution was added. A microplate reader was used
to measure absorbance at 570 nm for each well. The growth
inhibition rate was calculated as follows: Growth inhibition rate =
1−A570 nm of treated cells/A570 nm.
Colony formation assay
Colon cancer cells from each group were plated in
6-well plates (1,000 cells/well) and were incubated for 15 days at
37°C in a humidified atmosphere; the medium was replaced every 3
days. On day 15, the cells were washed with PBS, fixed with 4%
paraformaldehyde for 30 min at 25°C and stained with Giemsa
(Tiangen Biotech Co., Ltd.) for 15 min. The number of colonies
containing >50 cells was counted under an inverted microscope
(Olympus Co., Ltd.).
Affymetrix microarray analysis
Gene chip assays and data analysis were performed by
Shanghai GeneChem Co., Ltd. using the GeneChip PrimeView Human Gene
Expression Array (cat. no. 901838; Affymetrix; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Briefly, three biological replicates collected from each group were
subjected to microarray analysis. Total RNA was extracted from
RAC1-knockdown cells and NC cells, and RNA quality and
quantity were measured using a NanoDrop spectrophotometer (ND-1000;
NanoDrop Technologies; Thermo Fisher Scientific, Inc., Wilmington,
DE, USA). RNA integrity was determined by gel electrophoresis, and
purified total RNA was subsequently subjected to DNase I treatment.
For microarray analysis, total RNA (300 ng) was first reverse
transcribed into a double-stranded cDNA template. Biotin-labeled
complementary RNA (cRNA) was then synthesized, amplified and
purified by in vitro transcription of the double-stranded
cDNA template using T7 RNA polymerase. The purified cRNA was
fragmented and prepared for hybridization onto the GeneChip
cartridge arrays. Hybridization, washing and staining were
performed using a GeneChip Hybridization Wash and Stain kit
(Affymetrix; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Scanning of hybridized arrays was
performed using a GeneChip Scanner 3000 (Affymetrix; Thermo Fisher
Scientific, Inc.). The data were analyzed with Microarray Suite
version 5.0 (MAS 5.0) using Partek Genomics Suite software
(Affymetrix; Thermo Fisher Scientific, Inc.). Expression values
underwent Robust Multiarray Averaging normalization and fold-change
values were then calculated using the least-squares mean between
samples. The significance of differences in gene expression in the
different groups (P-value) was estimated using Student's t-test.
Genes with changes in expression ≥2-fold (P<0.05) were regarded
as differentially expressed.
Cell cycle and apoptosis analysis
Colon cancer cells were harvested and fixed in 70%
ethanol at 4°C for 24 h after cells were grown to 80% conflu ence.
Fixed cells were washed with PBS and suspended in 1 ml propidium
iodide (PI) staining reagent (20 mg/l RNase A and 50 mg/l PI).
Samples were then incubated in the dark for 30 min at 25°C prior to
cell cycle analysis. Cell cycle distribution was determined and
analyzed using flow cytometry (FACSCalibur; Becton-Dickinson, San
Jose, CA, USA).
The apoptotic rate was determined using an Annexin
V-fluorescein isothiocyanate (FITC) detection kit (cat. no.
88-8007; eBioscience; Thermo Fisher Scientific, Inc.). Specific
binding of Annexin V-FITC was performed by incubating the cells for
15 min at room temperature in binding buffer (10 mM HEPES, 140 mM
NaCl, 2.5 mM CaCl2, pH 7.4) containing a saturating
concentration of Annexin V-FITC. After incubation, 1×106
colon cancer cells of each group were harvested and fixed in 70%
ethanol at 4°C for 24 h after cells were grown to 80% confluence.
Cells were analyzed using FlowJo software (Tree Star, Inc.,
Ashland, OR, USA).
Western blot analysis
Western blot analyses were performed as previously
described (8). Briefly, colon
cancer cells were harvested, rinsed twice with cold PBS and
incubated in lysis buffer (cat. no. P0013; Beyotime; Jiangsu,
China). Following centrifugation at 12,000 × g for 30 min at 4°C,
the amount of protein in the supernatant was determined using
bicinchoninic acid protein assay reagent. Equal amounts of protein
(30 μg) were completely vortexed with 2X SDS-gel buffer and
boiled for 5 min at 100°C to dissolve bound proteins. Whole-cell
lysates were then separated by 10% SDS-PAGE and were transferred to
polyvinylidene difluoride membranes, which were blocked with 50 g/l
nonfat milk at 25°C for 1.5 h. The association between RAC1 and
differential expression genes involved in cancer-related pathways
was investigated with Ingenuity Pathway Analysis (IPA), a web
delivered tool(www.ingenuity.com). Proteins were detected following
incubation of membranes with specific primary antibodies (Table II) at 4°C overnight, and
horseradish peroxidase (HRP)-conjugated secondary antibodies
(ab6721; dilution 1:2,000; Abcam, Shanghai, China) for 1 h at 25°C.
The membranes were then incubated in SuperSignal enhanced
chemiluminescence-HRP detection reagent (cat. no. P1108; Beyotime)
for 1 min, and semi-quantitative data were obtained using a
computing densitometer with a scientific imaging system (Tanon
5500; Shanghai Tian Neng Technology Co., Ltd., Shanghai, China).
GAPDH was used as a loading control for western blot analysis.
| Table IIList of primary antibodies used for
western blotting. |
Table II
List of primary antibodies used for
western blotting.
| Protein | Description | Vendor | Product code | Dilution | Molecular
weight |
|---|
| Cyclin D1 | Rabbit | Abcam | ab16663 | 1:100 | 33 kDa |
| BAD | Rabbit | Abcam | ab32445 | 1:2,000 | 23 kDa |
| RAC1 | Rabbit | Abcam | ab97568 | 1:200 | 21 kDa |
| GAPDH | Rabbit | Abcam | ab9485 | 1:2,000 | 37 kDa |
Statistical analysis
Data were analyzed using SPSS 18.0 software (SPSS,
Inc., Chicago, IL, USA). Measurement data (mRNA/protein levels,
cell cycle, cell apoptosis, colony formation and proliferation) are
presented as the means ± standard deviation from at least three
independent experiments. Data were analyzed by Student's t-test or
one-way analysis of variance followed by Fisher's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
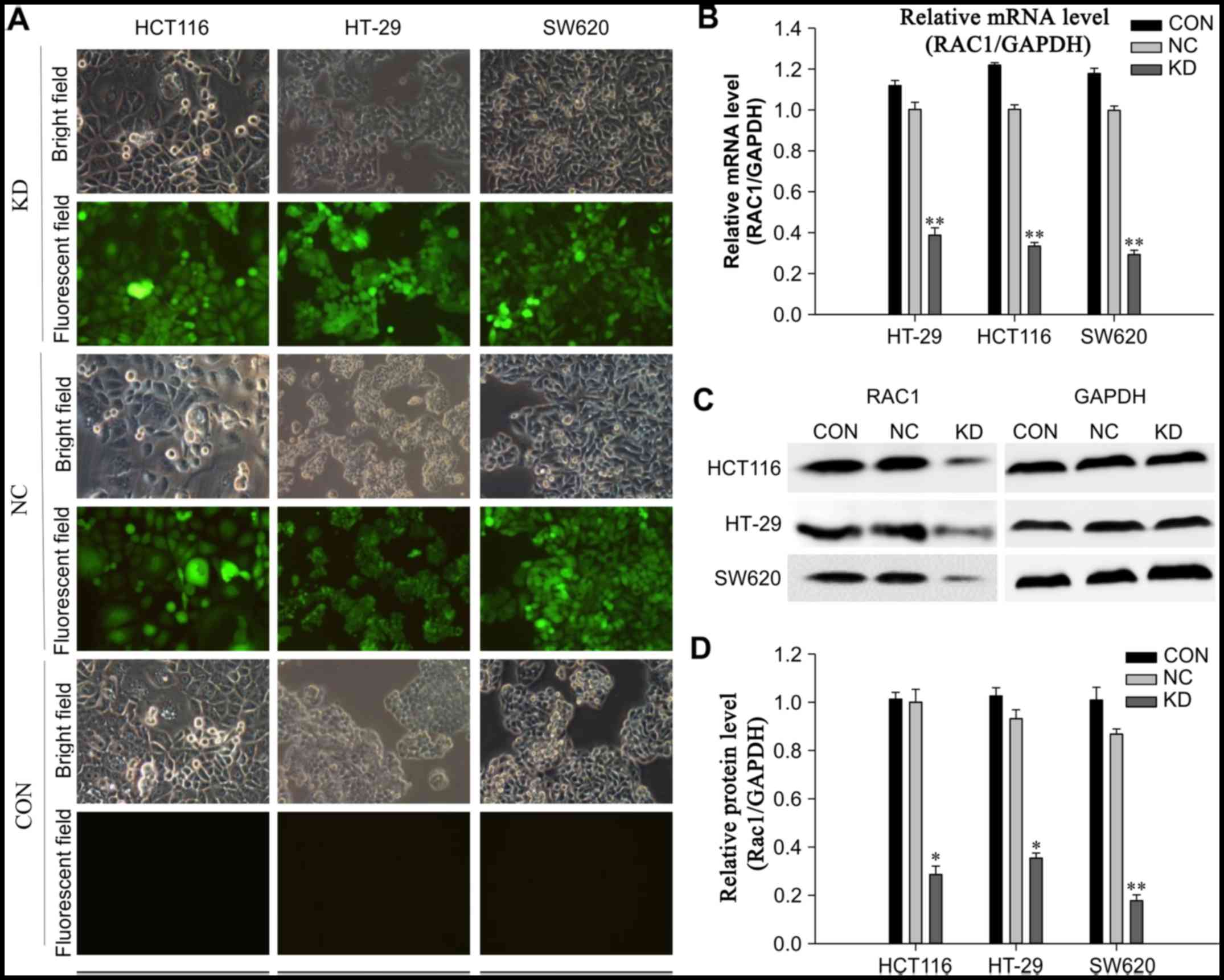
Effects of shRNA on RAC1 expression in
colon cancer cells
The expression levels of RAC1 were detected in three
colon cancer cell lines (HT-29, HCT116 and SW620). There were no
significant differences in the expression of RAC1, and all three
cell lines exhibited high levels of RAC1 mRNA and protein (Fig. 1). Conversely, the expression
levels of RAC1 were stably reduced using LV-mediated RNAi, and
infection efficiency of LV-shRNA-RAC1 and LV-NC was determined
using fluorescence microscopy. The results revealed that the
majority of cells (>95%) were successfully infected with shRNA
(Fig. 1A). The interference
efficiency was observed by RT-qPCR and western blotting 72 h
postinfection. The expression levels of RAC1 mRNA and protein were
significantly decreased in cells in the KD group compared with in
the NC and control groups (P<0.01). In the KD group of HCT116,
HT-29 and SW620 cells, RAC1 protein levels were reduced by 71.4,
64.6 and 82.2%, respectively, when compared with the NC group
(Fig. 1B–D). These results
indicated that LV-mediated RAC1 shRNA was able to
efficiently downregulate RAC1 expression in colon cancer cells.
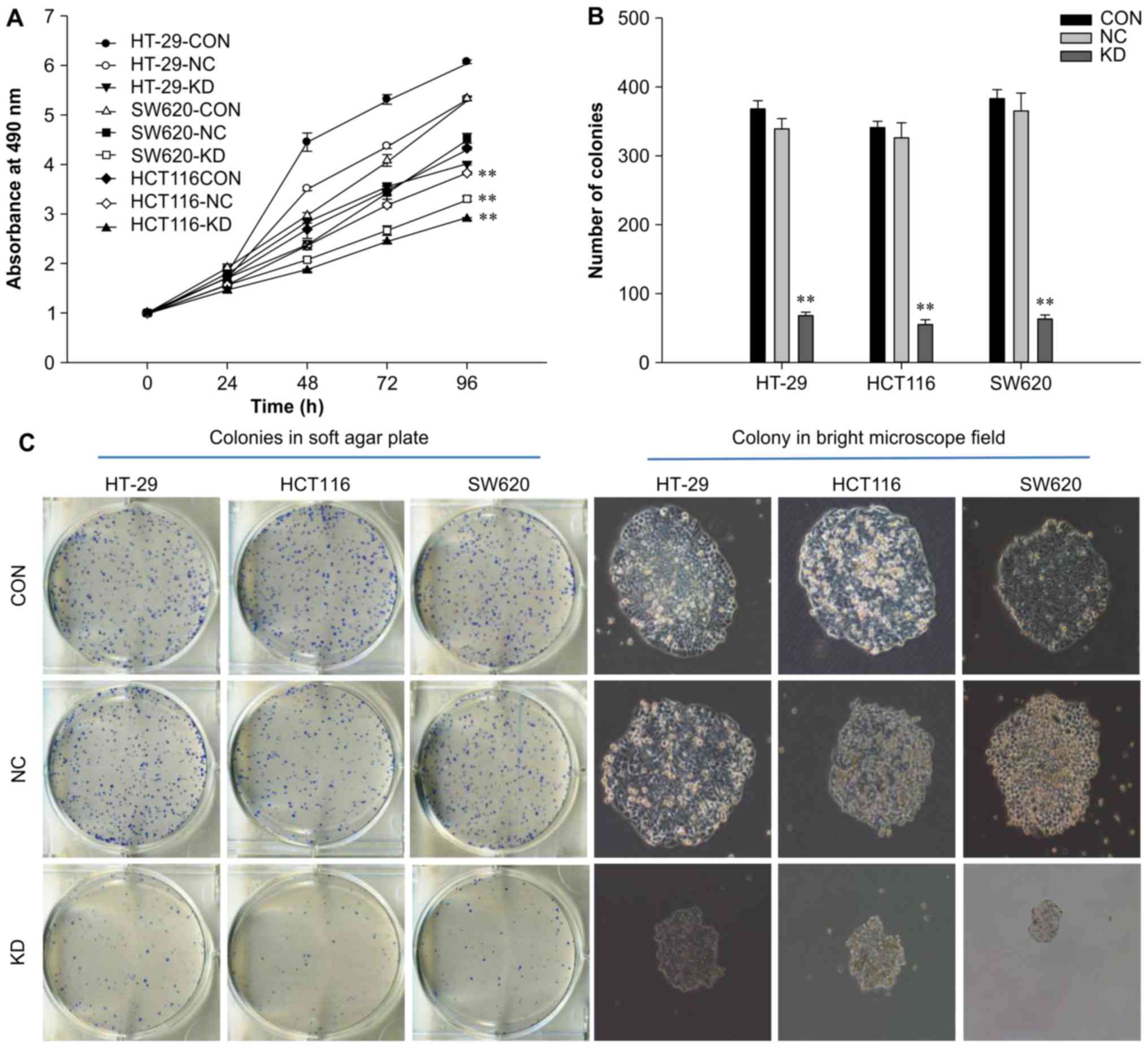
RAC1 knockdown suppresses the
proliferation and colony formation of colon cancer cells
MTT assays were used to determine whether silencing
RAC1 expression inhibited colon cancer cell viability.
Time-dependent inhibition of cell growth was determined in the KD
group, whereas no significant inhibitory effects were observed in
the NC and control groups (Fig.
2A). Cell proliferation was significantly inhibited after 4
days of infection (P<0.01), with proliferation decreased by
24.6, 11.4 and 27.2% in the KD group of HT-29, HCT116 and SW620
cells, respectively, when compared with the NC group. In addition,
following RAC1 knockdown, the colony-forming capacity of
colon cancer cells was significantly decreased by 60–80%
(P<0.01), and colony size was reduced compared with in the NC
and control groups (P<0.01; Fig.
2B and C). These data suggested that RAC1 may serve a critical
role in colon cancer cell proliferation, particularly in SW620 KD
group. Therefore, SW620 cells were selected for subsequent gene
chip analysis.
RAC1 knockdown induces differential gene
expression
To explore the molecular effectors associated with
RAC1 activity in colon cancer, microarray analysis was performed to
determine the mRNA profiles of SW620 cells following RAC1
knockdown (Fig. 3). From this
analysis, 1,215 transcripts were identified as having ≥1.5-fold
differential expression; 604 genes were upregulated, whereas 611
genes were downregulated after RAC1 silencing (Fig. 3B). The results of unsupervised
hierarchical clustering of gene expression levels demonstrated that
SW620 cells with RAC1 knockdown were easily distinguishable
from SW620 cells without RAC1 knockdown, and six samples
were essentially partitioned into two groups: The first containing
knockdown cells and the other containing cells without RAC1
knockdown (Fig. 3A). All
differentially expressed genes were divided into groups according
to their biological functions using Gene Ontology (GO) terms and
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (https://david.ncifcrf.gov/). The main functional
groups and molecular pathways included cell cycle and apoptosis,
cell adhesion, metabolic process, mitosis and cancer-related
pathways. Of these, the relationship between cell cycle and
RAC1 silencing was the most significant according to GO
analysis (P=P=5.19E 16,-log10(5.19E 16)=15.28); >140 genes,
including cyclin D1 (CCND1), cyclin-dependent kinase 1
(CDK1), cyclin B1 (CCNB1), B-cell lymphoma
(Bcl)-2-associated agonist of cell death (BAD), caspase-8
(CASP8) and MYC proto-oncogene, bHLH transcription factor
(MYC), were involved in this process, and the most
significant differences in expression were found in cell
adhesion-related genes, some of which are listed in Table III. Through KEGG analysis, it
was demonstrated that silencing RAC1 expression inhibited
the proliferation of colon cancer cells, potentially via p53
signaling, mitogen-activated protein kinase (MAPK) signaling or
cancer-related pathways. The top 10 pathways are listed in Fig. 3C according to their P-values. From
these data, the present study further investigated the association
between RAC1 and 29 genes involved in cancer-related
pathways through bioinformatics analysis and prediction. The
results indicated that RAC1 regulated the expression of
these genes by direct or indirect interactions (Fig. 4).
| Table IIIDifferentially expressed genes
associated with the cell cycle, apoptosis and cell adhesion
following Ras-related C3 botulinum toxin substrate 1 knockdown. |
Table III
Differentially expressed genes
associated with the cell cycle, apoptosis and cell adhesion
following Ras-related C3 botulinum toxin substrate 1 knockdown.
| Gene name | GenBank no. | Fold-change | Function |
|---|
| CCND1 | NM_053056 | −2.4881644 | Cell cycle |
| CCNB2 | NM_004701 | 3.625082 | Cell cycle |
| CHEK2 | NM_145862 | 1.6333724 | Cell cycle |
| CDC25C | NM_022809 | 4.5017295 | Cell cycle |
| BUB1 | NM_004336 | 2.8708594 | Cell cycle |
| SMC1A | NM_006306 | −1.6359816 | Cell cycle |
| PTTG1 | NM_004219 | 2.4099076 | Cell cycle |
| ESPL1 | NM_012291 | 2.1459587 | Cell cycle |
| MCM2 | NM_004526 | −1.723211 | Cell cycle |
| MCM6 | NM_005915 | −1.8482218 | Cell cycle |
| ORC3 | NM_181837 | 1.6491874 | Cell cycle |
| STAG2 | NM_006603 | 1.7200525 | Cell cycle |
| BUB1B | NM_001211 | 1.8905722 | Cell cycle |
| TTK | NM_003318 | 2.1164258 | Cell cycle |
| BUB3 | NM_004725 | 1.5317589 | Cell cycle |
| MYC | NM_002467 | −1.549336 | Cell cycle, Cell
apoptosis |
| CASP8 | NM_033356 | 2.0469344 | Cell apoptosis |
| ARHGDIB | NM_001175 | −1.9277692 | Cell apoptosis |
| CASP7 | NM_033338 | 1.9895566 | Cell apoptosis |
| CASP2 | NM_032982 | −1.7764742 | Cell apoptosis |
| CASP4 | NM_001225 | 1.9777244 | Cell apoptosis |
| BAD | NM_004322 | 1.6885499 | Cell apoptosis,
Focal adhesion |
| JUN | NM_002228 | −1.892828 | Focal adhesion |
| LAMA3 | NM_198129 | −2.1677043 | Focal adhesion |
| LAMB1 | NM_002291 | 1.8277249 | Focal adhesion |
| LAMB3 | NM_000228 | −2.2665424 | Focal adhesion |
| PAK2 | NM_002577 | −1.6881523 | Focal adhesion |
| ITGB7 | NM_000889 | −2.1417882 | Focal adhesion |
| ITGB8 | NM_002214 | −6.589815 | Focal adhesion |
| COL6A1 | NM_001848 | −1.8742688 | Focal adhesion |
| MYL5 | NM_002477 | −2.0736258 | Focal adhesion |
RAC1 knockdown induces cell cycle arrest
and apoptosis
To further validate the effects of RAC1
knockdown on the viability of SW620 colon cancer cells, flow
cytometry was used to detect alterations in cell cycle progression
and apop-tosis following RAC1 knockdown (Figs. 5 and 6). The results demonstrated that the
apoptotic rate of the KD group (10.45%) was significantly higher
compared with in the NC and control groups (5.45 and 4.42%,
respectively; P<0.01) (Fig. 6A and
B). Compared with the NC group, the proportion of cells in S
phase in the KD group was significantly decreased from 45.65 to
32.68% (P<0.01), whereas the ratio of cells in the
G0/G1 phase was significantly increased from
43.91 to 54.14% (P<0.01) (Fig. 5A
and B). However, there were no significant differences between
the NC and control groups, thus indicating that RAC1
knockdown interfered with cell cycle progression, leading to cell
cycle arrest in G1 phase in the KD group.
To further investigate the molecular mechanisms
through which RAC1 knockdown inhibited the proliferation of
colon cancer cells, potentially via G0/G1
cell cycle arrest and increased apoptosis, cyclin D1 and BAD
protein expression levels were evaluated by western blotting after
RAC1 knockdown in SW620 cells. Notably, BAD expression was
upregulated, whereas cyclin D1 expression was downregulated in the
KD group compared with in the NC and control groups (Figs. 5C and D, and 6C and D). These data suggested that
RAC1 knockdown may suppress the proliferation of colon
cancer cells by inducing apoptosis and cell cycle arrest,
potentially through upregulation of BAD and downregulation of
cyclin D1.
Discussion
Colorectal cancer is the third most common malignant
tumor worldwide and the second leading cause of cancer-associated
mortality in developed countries (1,26).
Despite marked improvements in the diagnosis and treatment of colon
cancer, the incidence and mortality rates of this disease are still
increasing (1,26). Personalized medicine may lead to
improvements in the survival rates of patients with colon cancer;
however, additional studies are required for the identification of
additional potential therapeutic targets, and the development of
targeted agents with high efficacy and low toxicity (27,28). Numerous studies have reported that
abnormal RAC1 signaling is associated with various human diseases,
and that RAC1 expression and activity are dysregulated in numerous
types of cancer, including colon cancer (10,15). Overexpression of RAC1 triggers
tumor initiation and is associated with the metastasis and invasion
of cancer cells; therefore, RAC1 may be a potent therapeutic target
for patients with malignant tumors (10,15).
Downregulation of RAC1 can inhibit the proliferation
of various cancer cells, including gastric cancer (12), colon cancer (20) and breast cancer cells (29), among others (10,13,30). In the present study, the
expression of RAC1 was silenced by LV-mediated RNAi.
Subsequently, the proliferation and colony-forming abilities of
HT-29, HCT116 and SW620 colon cancer cells were significantly
inhibited, which is consistent with previous data. These findings
indicated that RAC1 may serve an important role in maintaining the
growth of colon cancer cells.
To improve understanding of the molecular mechanisms
through which shRNA-induced silencing of RAC1 inhibits
proliferation of colon cancer cells, a gene expression micro-array
analysis was conducted. The results demonstrated that >1,200
genes involved in RAC1 silencing were differentially expressed,
including 604 upregulated and 611 downregulated genes. These genes
were divided into various groups according to their biological
function using GO terms and KEGG analysis. The main functional
groups and signaling pathways included cell cycle, apoptosis, cell
adhesion, metabolic process, mitosis, p53 signaling pathway and
cancer-related pathways.
Notably, RAC1 has been reported to serve an
important role in the regulation of various signaling pathways
involved in apoptosis and cell cycle progression, and RAC1 has been
revealed to facilitate tumor cell proliferation (31,32). Consistent with these findings, the
present results demonstrated that RAC1 knockdown in SW620
cells induced differential expression of >140 genes associated
with the cell cycle and apoptosis, including CCNB1,
CCND1, Fos proto-oncogene, AP-1 transcription factor subunit
(FOS) and MYC, which were downregulated, and
CASP8, BAD and β-catenin, which were upregulated, and
genes involved in p53 signaling, MAPK signaling and cancer-related
pathways.
Cancer may become autonomous if the genes that drive
the cell cycle and apoptosis become dysregulated (33,34). Cyclin/CDK complexes phosphorylate
crucial target proteins that drive the cell through the cell cycle
(33,35). Cyclin D1, which is encoded by the
CCND1 gene, forms a complex with CDK4 or CDK6, whose
activity is required for the G1/S transition of the cell
cycle (33,35). The CCNB1 gene encodes the
cyclin B1 protein, which has regulatory functions in mitosis. In
addition, this protein forms a complex with p34 (CDC2), leading to
generation of the maturation-promoting factor, which is expressed
predominantly during G2/M phase (33,35). Increased MYC protein expression
can activate cyclin/CDK complexes, drive cells into the cell cycle
and promote cell proliferation in numerous human cancers (36,37). The tumor suppressor p53 can arrest
growth by halting the cell cycle at G1/S checkpoint and
initiate apoptosis by regulating the expression of cyclin D1,
cyclin B1 and MYC (38). Cyclin
D1 and cyclin B1 overexpression, potentially through inactivity of
the p53 gene or activity of the MYC gene, has been
reported to be correlated with tumor progression (39), shorter survival in patients with
cancer and increased metastasis (36–39). Previous studies have reported that
decreased expression of cyclin D1 (40,41), MYC (42,43) and cyclin B1 (43) using anticancer agents or RNAi may
lead to G1 or G2/M cell cycle arrest.
Activation of the GTPase RAC1 can promote tumor cell proliferation
through the abnormal expression of c-Jun, MYC and RAS, and
silencing of RAC1 expression leads to cell cycle arrest,
potentially through pathways involving PAK1 (44), cyclin D1 (29) and NF-κB (45) in breast cancer, lung cancer and
colon cancer (15). The present
findings confirmed these previous results, demonstrating that
RAC1 knockdown induced SW620 cell cycle arrest at
G1 phase, and this process may be associated with the
differential expression of numerous genes, including CCND1,
CCNB1, CDC12 and MYC. Notably, the
downregulation of cyclin D1 was further validated by western
blotting, thus suggesting that silencing of RAC1 expression
may induce G1 arrest through this pathway.
A large family of genes that regulate apoptosis has
been identified, and caspase-8 and BAD are important members of
this family (34). Caspase-8 can
activate downstream caspases that cause cell death, and BAD
proteins are able to promote apoptosis by neutralizing the activity
of anti-apoptotic proteins, such as Bcl-2 and Bcl-extra large. MYC
protein can either activate CDK and cyclin genes, or suppress the
transcription of apoptosis-associated genes (36,46). Suppression of RAC1 expression has
been reported to decrease the phosphorylation of MYC and inhibit
cell proliferation (47).
Furthermore, increased p38 MAPK expression leads to upregulation of
FOS activity through a RAC1-dependent and CDC42-independent
pathway, and co-expression of the dominant-negative mutants of FOS,
p38 and RAC1 blocks MYC-mediated apoptosis (48). The present data demonstrated that
knockdown of the RAC1 gene may induce apoptosis of SW620
colon cancer cells; numerous genes that are potentially involved in
this process were identified by gene chip analysis, of which BAD
upregulation was further confirmed by western blotting. These
results may indicate that silencing RAC1 expression can
induce cell apoptosis through a pathway involving BAD.
Previous studies have reported that RAC1 is involved
in cancer invasion and metastasis through controlling cell motility
and cell adhesion (10,15). For example, RAC1 hyperactivation
or overexpression drives the mesenchymal mode of cell migration by
stimulating the formation of actin-rich membrane extensions
(49,50); promotes tumor-associated
angiogenesis by regulating the balance of inhibitors and
stimulators of endothelial cell proliferation, endothelial cell
migration and capillary formation molecules (15,51,52); controls the expression and release
of matrix metalloproteinases required for proteolytic degradation
of the extracellular matrix (ECM) (10,53,54); and induces EMT by mediating
cellular plasticity and ECM modulation (10,30,55). In the present study, after
silencing RAC1 expression, some differentially expressed
genes involved in cancer-related pathways were detected, including
laminin subunit α3, Jun proto-oncogene, AP-1 transcription factor
subunit, MYC, laminin subunit β1, PAK2 and
ROCK2, which were downregulated, and thrombospondin 1 and
BAD, which were upregulated; these genes are involved in
cell motility and adhesion. Therefore, the present findings further
supported that RAC1 promoted tumor invasion and metastasis through
regulating cell motility and adhesion.
In conclusion, silencing RAC1 expression
inhibited colon cancer cell growth, promoted cell cycle arrest and
enhanced apoptosis; these functions may be associated with the
differential expression of numerous genes involved in various
biological functions, including downregulation of CCND1 and
upregulation of BAD. Further studies regarding these
differentially expressed genes may allow a better understanding of
the effects of RAC1 signaling dysregulation on the promotion of
colon carcinogenesis.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant no. 81260321). The
obtained micro-array data were deposited in the Gene Expression
Omnibus database (accession no. GSE78093).
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sideris M and Papagrigoriadis S: Molecular
biomarkers and classification models in the evaluation of the
prognosis of colorectal cancer. Anticancer Res. 34:2061–2068.
2014.PubMed/NCBI
|
|
3
|
Holubec L, Polivka J Jr, Safanda M, Karas
M and Liska V: The role of cetuximab in the induction of anticancer
immune response in colorectal cancer treatment. Anticancer Res.
36:4421–4426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen BJ, Wu YL, Tanaka Y and Zhang W:
Small molecules targeting c-Myc oncogene: Promising anti-cancer
therapeutics. Int J Biol Sci. 10:1084–1096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Raskov H, Pommergaard HC, Burcharth J and
Rosenberg J: Colorectal carcinogenesis–update and perspectives.
World J Gastroenterol. 20:18151–18164. 2014. View Article : Google Scholar
|
|
6
|
Tejada-Simon MV: Modulation of actin
dynamics by Rac1 to target cognitive function. J Neurochem.
133:767–779. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fritz G and Henninger C: Rho GTPases:
Novel players in the regulation of the DNA damage response.
Biomolecules. 5:2417–2434. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lawson CD and Burridge K: The on-off
relationship of Rho and Rac during integrin-mediated adhesion and
cell migration. Small GTPases. 5:e279582014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pick E: Role of the Rho GTPase Rac in the
activation of the phagocyte NADPH oxidase: Outsourcing a key task.
Small GTPases. 5:e279522014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marei H and Malliri A: Rac1 in human
diseases: The therapeutic potential of targeting Rac1 signaling
regulatory mechanisms. Small GTPases. Jul 21–2016.Epub ahead of
print. PubMed/NCBI
|
|
11
|
Alonso-Espinaco V, Cuatrecasas M, Alonso
V, Escudero P, Marmol M, Horndler C, Ortego J, Gallego R,
Codony-Servat J, Garcia-Albeniz X, et al: RAC1b overexpression
correlates with poor prognosis in KRAS/BRAF WT metastatic
colorectal cancer patients treated with first-line FOLFOX/XELOX
chemotherapy. Eur J Cancer. 50:1973–1981. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ji J, Feng X, Shi M, Cai Q, Yu Y, Zhu Z
and Zhang J: Rac1 is correlated with aggressiveness and a potential
therapeutic target for gastric cancer. Int J Oncol. 46:1343–1353.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen QY, Xu LQ, Jiao DM, Yao QH, Wang YY,
Hu HZ, Wu YQ, Song J, Yan J and Wu LJ: Silencing of Rac1 modifies
lung cancer cell migration, invasion and actin cytoskeleton
rearrangements and enhances chemosensitivity to antitumor drugs.
Int J Mol Med. 28:769–776. 2011.PubMed/NCBI
|
|
14
|
Hein AL, Post CM, Sheinin YM, Lakshmanan
I, Natarajan A, Enke CA, Batra SK, Ouellette MM and Yan Y: RAC1
GTPase promotes the survival of breast cancer cells in response to
hyper-fractionated radiation treatment. Oncogene. 35:6319–6329.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bid HK, Roberts RD, Manchanda PK and
Houghton PJ: RAC1: An emerging therapeutic option for targeting
cancer angiogenesis and metastasis. Mol Cancer Ther. 12:1925–1934.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lv Z, Hu M, Zhen J, Lin J, Wang Q and Wang
R: Rac1/PAK1 signaling promotes epithelial-mesenchymal transition
of podocytes in vitro via triggering β-catenin transcriptional
activity under high glucose conditions. Int J Biochem Cell Biol.
45:255–264. 2013. View Article : Google Scholar
|
|
17
|
Kallergi G, Agelaki S, Markomanolaki H,
Georgoulias V and Stournaras C: Activation of FAK/PI3K/Rac1
signaling controls actin reorganization and inhibits cell motility
in human cancer cells. Cell Physiol Biochem. 20:977–986. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Myant KB, Cammareri P, McGhee EJ, Ridgway
RA, Huels DJ, Cordero JB, Schwitalla S, Kalna G, Ogg EL, Athineos
D, et al: ROS production and NF-κB activation triggered by RAC1
facilitate WNT-driven intestinal stem cell proliferation and
colorectal cancer initiation. Cell Stem Cell. 12:761–773. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Myant KB, Scopelliti A, Haque S, Vidal M,
Sansom OJ and Cordero JB: Rac1 drives intestinal stem cell
proliferation and regeneration. Cell Cycle. 12:2973–2977. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Espina C, Céspedes MV, García-Cabezas MA,
Gómez del Pulgar MT, Boluda A, Oroz LG, Benitah SA, Cejas P, Nistal
M, Mangues R, et al: A critical role for Rac1 in tumor progression
of human colorectal adenocarcinoma cells. Am J Pathol. 172:156–166.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu C, Zhang S, Song L, Wang Y, Hwaiz R,
Luo L and Thorlacius H: Rac1 signaling regulates
neutrophil-dependent tissue damage in experimental colitis. Eur J
Pharmacol. 741:90–96. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li G, Ying L, Wang H, Wei SS, Chen J, Chen
YH, Xu WP, Jie QQ, Zhou Q, Li YG, et al: Rac1b enhances cell
survival through activation of the JNK2/c-JUN/Cyclin-D1 and
AKT2/MCL1 pathways. Oncotarget. 7:17970–17985. 2016.PubMed/NCBI
|
|
23
|
Huang YS, Xie N, Su Q, Su J, Huang C and
Liao QJ: Diallyl disulfide inhibits the proliferation of HT-29
human colon cancer cells by inducing differentially expressed
genes. Mol Med Rep. 4:553–559. 2011.PubMed/NCBI
|
|
24
|
Zhou Y, Su J, Shi L, Liao Q and Su Q: DADS
downregulates the Rac1/ROCK1/PAK1-LIMK1-ADF/cofilin signaling
pathway, inhibiting cell migration and invasion. Oncol Rep.
29:605–612. 2013. View Article : Google Scholar
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real time quantitative PCR and
the 2(Delta DeltaC(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pohl M and Schmiegel W: Therapeutic
strategies in diseases of the digestive tract - 2015 and beyond
targeted therapies in colon cancer today and tomorrow. Dig Dis.
34:574–579. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Zhang J, Li L, Xu X, Zhang Y, Teng
Z and Wu F: Identification of molecular targets for predicting
colon adenocarcinoma. Med Sci Monit. 22:460–468. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yoshida T, Zhang Y, Rivera Rosado LA, Chen
J, Khan T, Moon SY and Zhang B: Blockade of Rac1 activity induces
G1 cell cycle arrest or apoptosis in breast cancer cells through
downregulation of cyclin D1, survivin, and X-linked inhibitor of
apoptosis protein. Mol Cancer Ther. 9:1657–1668. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leng R, Liao G, Wang H, Kuang J and Tang
L: Rac1 expression in epithelial ovarian cancer: Effect on cell EMT
and clinical outcome. Med Oncol. 32:3292015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leve F and Morgado-Díaz JA: Rho GTPase
signaling in the development of colorectal cancer. J Cell Biochem.
113:2549–2559. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rathinam R, Berrier A and Alahari SK: Role
of Rho GTPases and their regulators in cancer progression. Front
Biosci (Landmark Ed). 16:2561–2571. 2011. View Article : Google Scholar
|
|
33
|
Champeris Tsaniras S, Kanellakis N,
Symeonidou IE, Nikolopoulou P, Lygerou Z and Taraviras S: Licensing
of DNA replication, cancer, pluripotency and differentiation: An
interlinked world. Semin Cell Dev Biol. 30:174–180. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wong RS: Apoptosis in cancer: From
pathogenesis to treatment. J Exp Clin Cancer Res. 30:872011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Orlando DA, Lin CY, Bernard A, Wang JY,
Socolar JE, Iversen ES, Hartemink AJ and Haase SB: Global control
of cell-cycle transcription by coupled CDK and network oscillators.
Nature. 453:944–947. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dang CV and Lewis BC: Role of oncogenic
transcription factor c-Myc in cell cycle regulation, apoptosis and
metabolism. J Biomed Sci. 4:269–278. 1997. View Article : Google Scholar
|
|
37
|
Sipos F, Firneisz G and Műzes G:
Therapeutic aspects of c-MYC signaling in inflammatory and
cancerous colonic diseases. World J Gastroenterol. 22:7938–7950.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Surget S, Khoury MP and Bourdon JC:
Uncovering the role of P53 splice variants in human malignancy: A
clinical perspective. Onco Targets Ther. 7:57–68. 2013.
|
|
39
|
Diehl JA: Cycling to cancer with cyclin
D1. Cancer Biol Ther. 1:226–231. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mohanty S, Mohanty A, Sandoval N, Tran T,
Bedell V, Wu J, Scuto A, Murata-Collins J, Weisenburger DD and Ngo
VN: Cyclin D1 depletion induces DNA damage in mantle cell lymphoma
lines. Leuk Lymphoma. 58:676–688. 2017. View Article : Google Scholar
|
|
41
|
Zhou J, Li LU, Fang LI, Xie H, Yao W, Zhou
X, Xiong Z, Wang LI, Li Z and Luo F: Quercetin reduces cyclin D1
activity and induces G1 phase arrest in HepG2 cells. Oncol Lett.
12:516–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Posternak V and Cole MD: Strategically
targeting MYC in cancer. F1000Res. 5:F10002016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rajput S, Khera N, Guo Z, Hoog J, Li S and
Ma CX: Inhibition of cyclin dependent kinase 9 by dinaciclib
suppresses cyclin B1 expression and tumor growth in triple negative
breast cancer. Oncotarget. 7:56864–56875. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Oleinik V, Helke KL, Kistner-Griffin E,
Krupenko I and Krupenko SA: Rho GTPases RhoA and Rac1 mediate
effects of dietary folate on metastatic potential of A549 cancer
cells through the control of cofilin phosphorylation. J Biol Chem.
289:26383–26394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gastonguay A, Berg T, Hauser AD, Schuld N,
Lorimer E and Williams CL: The role of Rac1 in the regulation of
NF-κB activity, cell proliferation, and cell migration in non-small
cell lung carcinoma. Cancer Biol Ther. 13:647–656. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
McMahon SB: MYC and the control of
apoptosis. Cold Spring Harb Perspect Med. 4:a0144072014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Nikolova E, Mitev V, Minner F, Deroanne CF
and Poumay Y: The inhibition of the expression of the small Rho
GTPase Rac1 induces differentiation with no effect on cell
proliferation in growing human adult keratinocytes. J Cell Biochem.
103:857–864. 2008. View Article : Google Scholar
|
|
48
|
Kalra N and Kumar V: c-Fos is a mediator
of the c-myc-induced apoptotic signaling in serum-deprived hepatoma
cells via the p38 mitogen-activated protein kinase pathway. J Biol
Chem. 279:25313–25319. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Parri M and Chiarugi P: Rac and Rho
GTPases in cancer cell motility control. Cell Commun Signal.
8:232010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sanz-Moreno V, Gadea G, Ahn J, Paterson H,
Marra P, Pinner S, Sahai E and Marshall CJ: Rac activation and
inactivation control plasticity of tumor cell movement. Cell.
135:510–523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Vader P, van der Meel R, Symons MH, Fens
MH, Pieters E, Wilschut KJ, Storm G, Jarzabek M, Gallagher WM,
Schiffelers RM, et al: Examining the role of Rac1 in tumor
angiogenesis and growth: A clinically relevant RNAi-mediated
approach. Angiogenesis. 14:457–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Brantley-Sieders DM, Zhuang G, Vaught D,
Freeman T, Hwang Y, Hicks D and Chen J: Host deficiency in Vav2/3
guanine nucleotide exchange factors impairs tumor growth, survival,
and angiogenesis in vivo. Mol Cancer Res. 7:615–623. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Westermarck J and Kähäri VM: Regulation of
matrix metalloproteinase expression in tumor invasion. FASEB J.
13:781–792. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jin G, Sah RL, Li YS, Lotz M, Shyy JY and
Chien S: Biomechanical regulation of matrix metalloproteinase-9 in
cultured chondrocytes. J Orthop Res. 18:899–908. 2000. View Article : Google Scholar
|
|
55
|
Yang WH, Lan HY, Huang CH, Tai SK, Tzeng
CH, Kao SY, Wu KJ, Hung MC and Yang MH: RAC1 activation mediates
Twist1-induced cancer cell migration. Nat Cell Biol. 14:366–374.
2012. View Article : Google Scholar : PubMed/NCBI
|