Introduction
Hepatic fibrosis is a reversible stage of numerous
chronic liver diseases, which are associated with significant
morbidity and mortality (1).
Hepatic stellate cells (HSCs) are in close contact with hepatocytes
and sinusoidal endothelial cells within the space of Disse, and are
considered the primary cells that contribute to excessive
extracellular matrix (ECM) deposition during the pathogenesis of
hepatic fibrosis (2). Quiescent
HSCs are characterized by a lack of proliferation, and vitamin A
storage. In response to a fibrogenic stimulus, HSCs undergo an
activation process, which includes loss of vitamin A stores,
increased proliferation rate, increased ECM protein synthesis and
transformation into α-smooth muscle actin (α-SMA)-positive
myofibroblast-like cells (3,4).
Therefore, HSCs are considered a target for the treatment of
hepatic fibrosis (4). Oxidative
stress (OS) results from the increased production of reactive
oxygen species (ROS), and serves a crucial role in inducing HSC
activation and fibrogenic potential (5). ROS are able to stimulate expression
of the critical fibrosis-associated gene, transforming growth
factor (TGF)-β1, via activating the mitogen-activated protein
kinases (MAPKs) signaling pathway (6).
Gender has been identified as an independent risk
factor for the progression from fibrosis to cirrhosis (7), which has a male:female ratio ranging
between 2.3:1 and 2.6:1. It has previously been reported that
estradiol (E2) can attenuate dimethylnitrosamine (DMN)-
or carbon tetrachloride (CCl4)-induced liver fibrosis
(7,8), and significantly inhibit HSC
proliferation and transformation (9). Notably, E2 suppresses
hydrogen peroxide (H2O2)-induced activation
of cultured rat HSCs via decreasing lipid peroxide levels (10). However, despite these benefits,
the undesirable side effects of estrogen replacement therapy,
including increased risk of breast and endometrial cancers, limit
its clinical application; therefore, alternative drugs are required
(11,12).
Traditional Chinese medicine has been used for
thousands of years for the treatment of liver-related diseases.
Bupleurum-containing herbal prescriptions, including sho-saiko-to
(Xiao Chai Hu Tang) and Chaihu-Shugan-San, have been traditionally
used in Asian countries to treat various liver diseases (13–15). Saikosaponin-d (SSd) is one of the
major active pharmacological components extracted from Bupleurum
falcatum L., which has been reported to alleviate
CCl4-induced hepatocyte injury by inhibiting lipid
peroxidation (16). Furthermore,
it exhibits suppressive effects on hepatic fibrosis in rats, which
was induced by CCl4 injections in combination with
alcohol, high fat and low protein feeding, due to its protection
against inflammatory hepatocyte injury (17). It has also been reported that SSd
may inhibit proliferation and activation of HSC-T6 cells (18). Notably, our previous study
demonstrated that SSd can induce estrogen response
elements-luciferase activity in MCF-7 cells, thus suggesting that
SSd exerts estrogen-like activity (19). However, whether SSd could suppress
the activation of HSCs via the estrogen receptor (ER) signaling
pathway, and which ER subtype is regulated by SSd in HSC-T6 cells,
remains to be elucidated. Therefore, the present study aimed to
investigate the effects of SSd on OS-induced activation of HSCs, as
well as the underlying mechanisms associated with ERs.
Materials and methods
Materials
SSd (batch number: 110778-201409; purity, >95%)
was purchased from National Institutes for Food and Drug Control
(Beijing, China). SSd is quite stable at room temperature and
retains its activity following exposure to organic solvents,
including dimethyl sulfoxide (DMSO). ICI-182780, DMSO, bovine serum
albumin, E2 and phenol red-free Dulbecco's modified
Eagle's medium (DMEM) were purchased from Invitrogen; Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). Enhanced chemiluminescence
(ECL) kit was purchased from EMD Millipore (Billerica, MA, USA).
TRIzol® reagent was purchased from Invitrogen; Thermo
Fisher Scientific, Inc. ReverTra Ace-α® reverse
transcription (RT) kit and SYBR®-Green real-time
polymerase chain reaction (PCR) master mix were purchased from
Toyobo Life Science (Osaka, Japan). Fetal bovine serum (FBS) and
charcoal-stripped FBS (sFBS) were obtained from Gibco; Thermo
Fisher Scientific, Inc. The protein molecular weight marker was
purchased from Pierce; Thermo Fisher Scientific, Inc. Total and
phosphorylated MAPK primary antibodies [ERK (cat. no. 4695P), JNK
(cat. no. 9258P), P38 (cat. no. 8690P), p-ERK (cat. no. 4370P),
p-JNK (cat. no. 4668P), p-P38 (cat. no. 4511P)], β-actin antibody
(cat. no. 4970S) and horseradish peroxidase (HRP)-conjugated goat
anti-rabbit-and goat anti-mouse antibodies were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). α-SMA primary
antibody (cat. no. sc-32251) was purchased from Santa Cruz
Biotechnology, Inc. (Dallas, Texas, USA). Rat malondialdehyde (MDA)
ELISA test kit (cat. no. F16194), rat CuZn-superoxide dismutase
(SOD) ELISA test kit (cat. no. F16742), rat hydroxyproline (Hyp)
ELISA test kit (cat. no. F15649), rat collagen-1 (COL1) ELISA test
kit (cat. no. F5730), rat TIMP-1 ELISA test kit (cat. no. F16930)
and rat MMP-1 ELISA test kit (cat. no. F16160) were purchased from
Westang Biological Science and Technology Co., Ltd. (Shanghai,
China), and rat TGF-β1 ELISA test kit (cat. no. BMS623-3) was
purchased from eBioscience;Thermo Fisher Scientific, Inc.
Methylpiperidinopyrazole (MPP) dihydrochloride and
(R,R)-tetrahydrochrysene (THC) were purchased from Tocris
Bioscience (Bristol, UK). H2O2 solution was
purchased from Tianjin Dongfang Chemical Co. (Tianjin, China).
EDTA-free digestive juices were purchased from Invitrogen; Thermo
Fisher Scientific, Inc.
Cell culture
Rat HSC-T6 cells (Cell Biological Research
Institution of Chinese Academy of Sciences, Shanghai, China) were
routinely cultured in DMEM supplemented with 5% FBS in an
atmosphere containing 5% CO2 at 37°C. Cells were grown
to 85% confluence and were then transferred to phenol red-free DMEM
supplemented with 5% sFBS for 2 days, in order to minimize
estrogenic activity of the serum. Cells were treated with SSd or
E2. SSd, E2, ICI-182780, MPP and THC were all
dissolved in DMSO. All were diluted in the medium immediately prior
to use (final concentration of DMSO, <0.1%). DMSO (<0.1%)
alone did not have any effect on the parameters measured.
Interaction with ICI-182780, MPP and
THC
Some phytoestrogens, including genistein and
daidzein, act as agonists and antagonists of ERs (20). In the present study, the effects
of SSd alone, as well as its interaction with ICI-182780, MPP and
THC, were examined. ICI-182780 is a pure ER antagonist; MPP is an
antagonist specific to ERα; THC is an antagonist specific to ERp.
HSC-T6 cells were divided into 4 groups as follows: vehicle group
(treated with DMSO at 37°C for 24 h); ICI group (treated with 1
μM ICI-182780 at 37°C for 24 h); MPP group (treated with 1
μM MPP at 37°C for 24 h); THC group (treated with 1
μM THC at 37°C for 24 h). Each group was divided into 4
subgroups as follows: control group (treated with DMSO at 37°C for
24 h, then DMSO at 37 °C for 4 h); OS group (treated with DMSO at
37°C for 24 h, then H2O2 at 37°C for 4 h);
SSd group (treated with 5 μM SSd at 37°C for 24 h, then
H2O2 at 37°C for 4 h); E2 group
(treated with 1 μM E2 at 37°C for 24 h, then
H2O2 at 37°C for 4 h). ER antagonist and drug
treatment were administrated at the same time.
MTT growth assay
HSC-T6 cells were washed twice with PBS, counted and
seeded into 96-well plates at a density of 0.8×104
cells/well. After 24 h, the cells completely attached to the wells.
Cell proliferation was assessed after 24 h [cells in the OS groups
were analyzed following 4 h induction with 0.2 mM
H2O2, as previously described (21,22)]. Briefly, cells were incubated with
100 μl 0.5 mg/ml MTT solution for 4 h at 37°C. The medium
was then discarded and 200 μl DMSO was added for 24 h at
room temperature. Absorbance was measured at 570 nm using an ELx800
universal microplate reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA). Cell numbers were obtained as absorbance values. The
results were expressed as proliferation of the treated cells
relative to the control group.
Detection of MDA, CuZn-SOD, tissue
inhibitor of metalloproteinases-1 (TIMP-1), matrix
metalloproteinase-1 (MMP-1), TGF-β1, Hyp and COL1 levels in cell
culture supernatants
HSC-T6 cells were seeded into 24-well plates
(8×104 cells/well) and cultured in phenol red-free DMEM
containing 5% sFBS. After 24 h, SSd and E2 were added in
the presence or absence of 1 μM ICI-182780, MPP or THC.
After 24 h, cell culture supernatants were collected [cells in the
OS groups were analyzed following 4 h induction with 0.2 mM
H2O2, as previously described (21,22)] and stored at −20°C. Subsequently,
supernatants were analyzed using ELISA kits according to the
manufacturer's protocols.
Detection of ROS in HSC-T6 cells
Intracellular ROS levels were measured by the
conversion of non-fluorescent 2,7-dichlorofluorescein diacetate
(DCFH-DA) into DCF. HSC-T6 cells were pretreated with SSd and
E2 in the presence or absence of ICI-182780, MPP or THC
for 24 h. Cells in OS groups were stimulated with
H2O2 for 4 h. Subsequently, cells were
incubated in 10 μM DCFH-DA for 20 min at 37°C. The cells
were washed three times, harvested and resuspended in PBS.
Fluorescence was detected using a BD FACSVerse™ flow cytometer (BD
FACSuite™, version LSR 2).
Western blot analysis
HSC-T6 cells were seeded into 6-well plates and
cultured in phenol red-free DMEM supplemented with 5% sFBS. Whole
cell extracts were prepared using the M-PER lysis buffer (cat. no.
78501; Thermo Fisher Scientific, Inc.). Lysates were then
centrifuged at 13,300 × g at 4°C for 15 min to remove insoluble
substances. Protein concentration was quantified using BCA assay
according to the manufacturer's protocols. Total protein extracts
(50 μg) from each sample were separated by 12% SDS-PAGE
followed by electrotransfer onto polyvinylidene difluoride
membranes (Immobilon-P; EMD Millipore). Membranes were blocked with
5% skimmed milk, and were incubated with rabbit polyclonal anti-rat
extracellular signal-regulated kinase (ERK)/c-Jun N-terminal kinase
(JNK)/p38 and phosphorylated (p)-ERK/p-JNK/p-p38 antibodies
(1:1,000), mouse monoclonal anti-rat α-SMA (1:500) or mouse
monoclonal anti-β-actin (1:2,000) diluted in 2% BSA at 4°C
overnight. Unbound primary antibodies were washed away using
Tris-buffered saline containing 0.1% Tween-20. Immune complexes
were probed using HRP-conjugated anti-mouse or anti-rabbit
secondary antibodies diluted in 5% skimmed milk, which were
incubated at room temperature for 2 h and were detected using an
ECL western blot procedure (EMD Millipore, Billerica, MA, USA).
Band density was semi-quantified following densitometric analysis
of autoradiographs using a Bio-Rad GS-690 Scanner (Bio-Rad
Laboratories, Inc.). Optical density values from the experimental
groups were expressed as a mean percentage of control values, and
differences were calculated by normalizing the density of each band
to that of β-actin.
RT-quantitative (q)PCR
HSC-T6 cells were seeded onto 6-well plates and
cultured in phenol red-free DMEM containing 5% sFBS. After 24 h,
SSd and E2 were added, in the presence or absence of 1
μM ICI-182780, MPP or THC. Cells in the OS groups were
analyzed after 4 h induction with 0.2 mM
H2O2, as previously described (21,22). Total RNA was isolated using
TRIzol® reagent, according to the manufacturer's
protocol. Total RNA (3 μg) was used to generate cDNA in each
sample using Superscript II reverse transcriptase with oligo(dT)
primers (Toyobo Life Science, Osaka, Japan). qPCR was performed to
quantify gene expression levels. Each qPCR reaction contained 2
μl diluted cDNA sample, 10 μl SYBR-Green PCR master
mix, 0.5 μl 1 μM forward and reverse primers, and 7.5
μl ddH2O. For detection of TGF-p1 and GAPDH
(housekeeping gene) expression, the following primers were used:
TGF-β1, forward 5′-TTG ACT TCC GCA AGG ACC TCG G-3′, reverse 5′-GCG
CCC GGG TTA TGC TGC TGG T-3′; and GAPDH, forward 5′-CCT CTA TGC CAA
CAC AGT GC-3′ and reverse 5′-GTA CTC CTG CTT GCT GAT CC-3′ to yield
146 and 194 bp products within 35 PCR cycles, respectively. The
amplification process was conducted as follows: Pre-denaturation at
95°C for 3 min, denaturation at 95°C for 45 sec, annealing at 50°C
for 45 sec and elongation at 72°C for 45 sec, a final extension
step at 72°C for 10 min. qPCR was carried out using an ABI Prism
7900 HT Sequence Detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The ratio of TGF-β1 to GAPDH was used to
evaluate the significance of differences among the various groups.
PCR results were quantified by using the 2−ΔΔCq method
(23).
Statistical analysis
Results are presented as the means + standard
deviation. Data were analyzed using an analysis of variance
(one-way ANOVA; for multiple groups) with SPSS version 23.0. If
ANOVA revealed an overall effect, intergroup differences were
analyzed using Newman-Keuls test. Two-sided P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of SSd on the proliferation and
activation of HSC-T6 cells in the presence or absence of ER
antagonists
It has previously been reported that the protective
effects of E2 against hepatic fibrosis may be associated
with its inhibition of HSC activation (7–9).
As a phytoestrogen, SSd exerts similar effects (18). The results of an MTT assay
indicated that SSd and E2 significantly suppressed (n=4,
P<0.05) the proliferation of HSC-T6 cells induced by
H2O2 treatment (n=4, P<0.01) (Fig. 1). Western blot analysis indicated
that the expression levels of α-SMA, a fibrotic marker that is
highly expressed in activated HSCs, were reduced following
incubation with SSd and E2 (Fig. 2). Conversely, these effects were
suppressed by ICI-182780 and THC (n=4, P<0.05), but not by
MPP.
 | Figure 1Effects of SSd and E2 in the presence
or absence of ER antagonists on oxidative stress-induced HSC
proliferation. HSC-T6 cells were treated with DMSO, 1 μM E2
or 5 μM SSd, in the presence or absence of 1 μM
ICI-182780, MPP or THC for 24 h. Subsequently, oxidative stress was
induced by 0.2 mM H2O2 for 4 h. Cell
proliferation was detected using an MTT assay. Results are
presented as the means ± SD, n=4. **P<0.01 vs. the control group
with the same ER antagonist treatment (ICI-182780, MPP, THC or
DMSO); #P<0.05 vs. the H2O2 + DMSO group;
ΔP<0.05 vs. the H2O2 + E2 +
DMSO group; ◊P<0.05 vs. the
H2O2 + SSd + DMSO group. DMSO, dimethyl
sulfoxide; E2, estradiol; ER, estrogen receptor;
H2O2, hydrogen peroxide; HSC, hepatic
stellate cell; MPP; methylpiperidinopyrazole; SSd, saikosaponin-d;
THC, (R,R)-tetrahydrochrysene. |
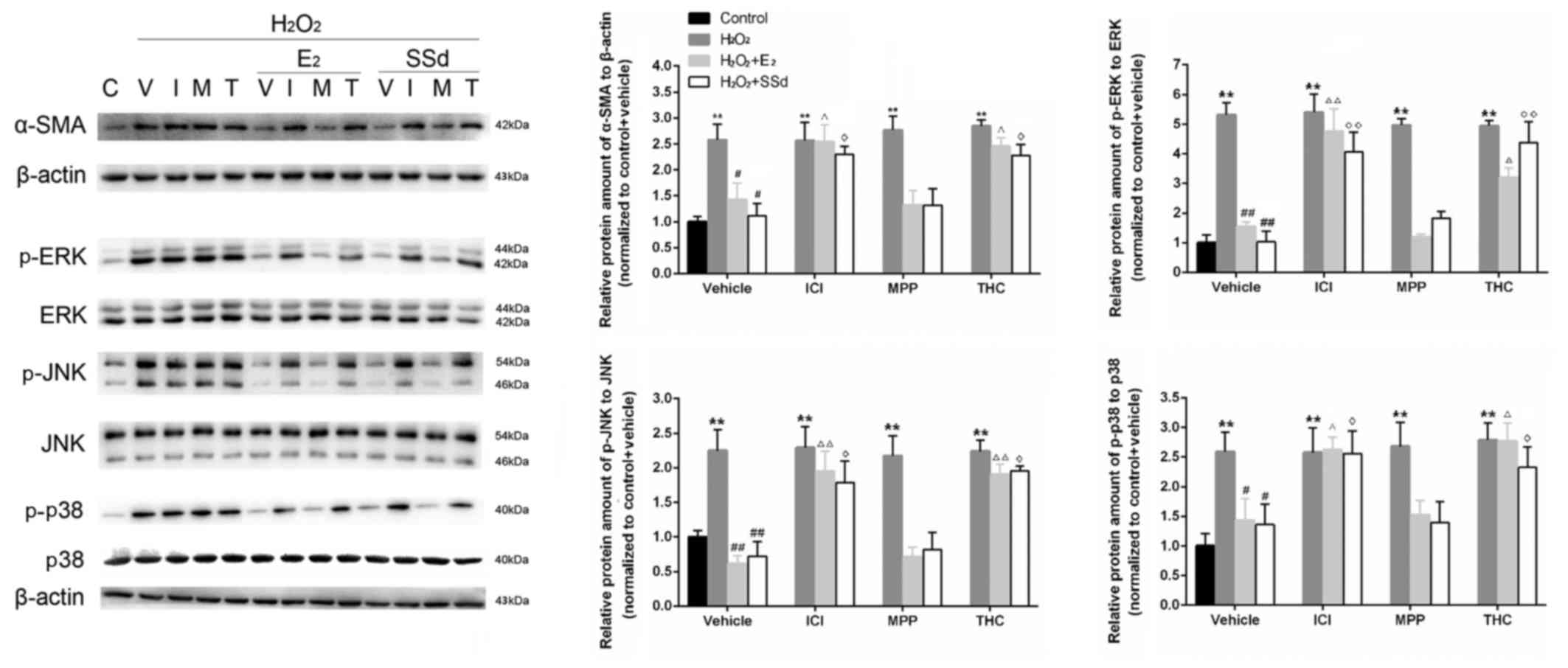 | Figure 2Effects of SSd and E2 in the presence
or absence of ER antagonists on oxidative stress-induced α-SMA,
ERK1/2, JNK and p38 expression. HSC-T6 cells were treated with
DMSO, 1 μM E2 or 5 μM SSd, in the presence or absence
of 1 μM ICI-182780, MPP or THC for 24 h. Subsequently,
oxidative stress was induced by 0.2 mM H2O2
for 4 h. Expression levels of α-SMA, and total and p-ERK1/2, JNK
and p38 were determined using western blot analysis. Levels of the
constitutively expressed protein, β-actin, were comparable in all
samples. Representative blotting images and relative
semi-quantification of protein levels are presented. C, control; V,
vehicle; I, ICI-182780; M, MPP; T, THC. Results are presented as
the means ± standard deviation, n=4. **P<0.01 vs. the control
group with the same ER antagonist treatment (ICI-182780, MPP THC or
DMSO); #P<0.05 and ##P<0.01 vs. the
H2O2 + DMSO group; ΔP<0.05 and
ΔΔP<0.01 vs. the H2O2 + E2 +
DMSO group; ◊P<0.05 and ◊◊P<0.01 vs.
the H2O2 + SSd + DMSO group. α-SMA, α-smooth
muscle actin; DMSO, dimethyl sulfoxide; E2, estradiol; ER, estrogen
receptor; ERK, extracellular signal-regulated kinase; H2O2,
hydrogen peroxide; HSC, hepatic stellate cell; JNK, c-Jun
N-terminal kinase; MPP; methylpiperidinopyrazole; p-,
phosphorylated; SSd, saikosaponin-d; THC,
(R,R)-tetrahydrochrysene. |
Effects of SSd on the synthesis and
degradation of ECM in the presence or absence of ER
antagonists
TGF-β1 is a key mediator that activates HSCs to
produce ECM (1,24). In addition, MMP and TIMP are two
important factors that regulate degradation of ECM (25). In the present study, SSd and
E2 were able to markedly decrease TGF-β1 (Fig. 3), Hyp, COL1 (Fig. 3A) and TIMP-1 expression (Fig. 4), and increase MMP-1 expression
(Fig. 4), thus inhibiting
H2O2-induced ECM formation. Conversely, the
effects of SSd and E2 were suppressed by ICI-182780 and
THC (n=4, P<0.05), but not MPP.
 | Figure 3Effects of SSd and E2 in the presence
or absence of ER antagonists on oxidative stress-induced
extracellular matrix synthesis. HSC-T6 cells were treated with
DMSO, 1 μM E2 or 5 μM SSd, in the presence or absence
of 1 μM ICI-182780, MPP or THC for 24 h. Subsequently,
oxidative stress was induced by 0.2 mM H2O2
for 4 h. (A) TGF-β1, Hyp, and COL1 contents in cell culture
supernatants were detected by ELISA. (B) TGF-β1 mRNA expression was
examined by quantitative polymerase chain reaction, with GAPDH mRNA
used as an internal control. Results are presented as the means ±
standard deviation, n=4. *P<0.05 and **P<0.01 vs. the control
group with the same ER antagonist treatment (ICI-182780, MPP, THC
or DMSO); #P<0.05 and ##P<0.01 vs. the
H2O2 + DMSO group; ΔP<0.05 and
ΔΔP<0.01 vs. the H2O2 + E2 +
DMSO group; ◊P<0.05 and ◊◊P<0.01 vs.
the H2O2 + SSd + DMSO group. COL1,
collagen-1; DMSO, dimethyl sulfoxide; E2, estradiol; ER,
estrogen receptor; H2O2, hydrogen peroxide;
HSC, hepatic stellate cell; Hyp, hydroxyproline; MPP; methylpi-
peridinopyrazole; SSd, saikosaponin-d; TGF-β1, transforming growth
factor-β1; THC, (R,R)-tetrahydrochrysene. |
 | Figure 4Effects of SSd and E2 in the presence
or absence of ER antagonists on oxidative stress-induced TIMP-1 and
MMP-1 expression. HSC-T6 cells were treated with DMSO, 1 μM
E2 or 5 μM SSd, in the presence or absence of 1 μM
ICI-182780, MPP or THC for 24 h. Subsequently, oxidative stress was
induced by 0.2 mM H2O2 for 4 h. TIMP-1 and
MMP-1 contents in cell culture supernatants were detected by ELISA.
Results are presented as the means ± standard deviation, n=4.
**P<0.01 vs. the control group with the same ER antagonist
treatment (ICI-182780, MPP THC or DMSO); #P<0.05 and
##P<0.01 vs. the H2O2 + DMSO
group; ΔP<0.05 vs. the H2O2 +
E2 + DMSO group; ◊P<0.05 vs.
H2O2 + SSd + DMSO group. DMSO, dimethyl
sulfoxide; E2, estradiol; ER, estrogen receptor; H2O2, hydrogen
peroxide; HSC, hepatic stellate cell; MMP-1, matrix
metalloproteinase-1; MPP; methylpiperidinopyrazole; SSd,
saikosaponin-d; THC, (R,R)- tetrahydrochrysene; TIMP-1, tissue
inhibitor of metalloproteinases-1. |
Effects of SSd on
H2O2-induced OS in the presence or absence of
ER antagonists
OS is recognized as having a crucial role in HSC
activation (5). Recent studies
indicated that the liver-protective and antifibrotic effects of SSd
may be attributed to its antioxidative capacity (26,27). Consequently, the antioxidative
effects of SSd on H2O2-induced OS in HSC-T6
cells were explored. Flow cytometry revealed that the control group
had low levels of ROS and that ER antagonists alone had no
significant effect. Conversely, groups treated with
H2O2 exhibited a significant increase in ROS
(n=3, P<0.01), which could be reversed by SSd and E2
(n=3, P<0.01) (Fig. 5A). MDA,
which is an end product of lipid peroxidation, and the endogenous
antioxidant CuZn-SOD, which indirectly reflects OS status, were
detected by ELISA. The results indicated that increased MDA content
induced by H2O2 (n=4, P<0.01) (Fig. 5B) was reduced by SSd and
E2 (n=4, P<0.05), whereas CuZn-SOD content (Fig. 5B) was increased by SSd and
E2 (n=4, P<0.01). However, the suppressive effects of
SSd on OS could be inhibited by ICI-182780 and THC (n=4,
P<0.05), but not MPP.
 | Figure 5Effects of SSd and E2 in
the presence or absence of ER antagonists on oxidative
stress-induced ROS generation, and MDA and CuZn-SOD activity.
HSC-T6 cells were treated with DMSO, 1 μM E2 or 5
μM SSd, in the presence or absence of 1 μM
ICI-182780, MPP or THC for 24 h. Subsequently, oxidative stress was
induced by 0.2 mM H2O2 for 4 h. (A) ROS
generation was detected by flow cytometry. (B) MDA and SOD contents
in cell culture supernatants were detected by ELISA. Representative
flow cytometry images and quantification are presented. Results are
presented as the means ± standard deviation, n=3 (A) and n=4 (B).
**P<0.01 vs. the control group with the same ER
antagonist treatment (ICI-182780, MPP, THC or DMSO);
#P<0.05 and ##P<0.01 vs. the
H2O2 + DMSO group; ΔP<0.05 and
ΔΔP<0.01 vs. the H2O2 +
E2 + DMSO group; ◊P<0.05 vs. the
H2O2 + SSd + DMSO group. CuZn-SOD,
CuZn-superoxide dismutase; DMSO, dimethyl sulfoxide; E2,
estradiol; ER, estrogen receptor; H2O2,
hydrogen peroxide; HSC, hepatic stellate cell; MDA,
malondialdehyde; MPP; methylpiperidinopyrazole; ROS, reactive
oxygen species; SSd, saikosaponin-d; THC,
(R,R)-tetrahydrochrysene. |
Effects of SSd on the MAPK signaling
pathway in the presence or absence of ER antagonists
ROS can upregulate the expression of critical
fibrosis-associated genes via activation of signal transduction
pathways, including MAPKs (28,29). In the present study, the
phosphorylation of three MAPK proteins (p-ERK, p-JNK and p-p38) was
examined by western blot analysis, in order to provide further
evidence regarding the antifibrotic effects of SSd. As shown in
Fig. 2, the relative expression
levels of p-ERK, p-JNK and p-p38 to their respective total proteins
were increased following H2O2 treatment (n=4,
P<0.01), whereas SSd and E2 were able to
significantly downregulate expression levels (n=4, P<0.05).
Conversely, these effects were suppressed by coadministration with
ICI-182780 and THC (n=4, P<0.05), but not MPP.
Discussion
Liver fibrosis and the final stage of liver
fibrosis, cirrhosis, represent the final common pathway of
virtually all chronic liver diseases (30–32). SSd has been reported to possess
antifibrotic activity; however, there is currently little
information regarding the effects of SSd on HSCs. Therefore, the
present study aimed to investigate the effects of SSd on the
proliferation and activation of HSCs, and the underlying mechanisms
associated with ERs. The results strongly suggested that SSd may
suppress OS-induced activation of HSCs in an ERβ-dependent manner.
This finding may be of potential clinical interest, since previous
studies have indicated that liver fibrosis is potentially
reversible with the reduction of HSC activation (1,25).
Sex hormones may affect the development of hepatic
fibrosis and cirrhosis (33).
E2 is an endogenous fibrosuppressant, which has been
reported to attenuate liver fibrosis in DMN- or
CCl4-induced rat models (7,8).
In addition, previous studies have demonstrated that the protective
effects of E2 against hepatic fibrosis may be associated
with its ability to inhibit HSC activation (7–9).
E2 inhibits intracellular pathways and activation
processes stimulated by H2O2 in cultured rat
HSCs via its suppressive effect on lipid peroxidation (10). Previous studies have suggested
that phytoestrogens, including isoflavones, resveratrol and
genistein, may suppress the progression of liver fibrosis due to
their weak estrogen-like activities (34–36). Our previous study demonstrated
that SSd exerts estrogen-like actions via activation of the ER
signaling pathway (19). The
present study further verified that the suppressive effects of SSd
on H2O2-induced activation of HSC-T6 cells
could be reversed by coincubation with ER antagonists, thus
confirming that the effects of SSd may be ER-dependent.
OS and ROS are predominant factors in HSC activation
(37). Previous studies have
reported that SSd possesses marked antioxidant activity (16,17,26,27). Consistently, the present study
indicated that SSd could inhibit H2O2-induced
OS in HSC-T6 cells, as evidenced by decreased ROS and MDA
generation, and increased CuZn-SOD activity.
ROS can upregulate the expression of critical
fibrosis-associated genes via activation of signal transduction
pathways and transcription factors, including MAPKs, activator
protein-1 and nuclear factor-κB (5). Suppression of ERK activation is
associated with complete inhibition of HSC proliferation in
vitro (38). In the liver,
ERK1 is associated with TGF-β1-induced fibrotic signaling in HSCs
(39), whereas ERK2 has a key
role in hepatocyte survival (40). In addition, JNK inhibition not
only prevents TGF-β-induced murine HSC activation and decreases
TGF-β signaling in human HSCs in vitro, but also
significantly reduces CCl4-induced liver fibrosis in
vivo (41). p38 MAPK is also
associated with ECM synthesis and degradation. It has previously
been reported that phosphorylation of p38 MAPK is augmented in
activated HSCs, which is involved in TGF-β1-downregulated MMP-13
expression, as well as in upregulated COL1 expression (24,29). In the present study, OS increased
the expression levels of p-ERK, p-JNK and p-p38, whereas SSd and
E2 could significantly downregulate these protein levels
and hence inhibit activation of the MAPK pathway. These results are
in agreement with the aforementioned involvement of the MAPK
pathway. In addition, TGF-β1 is a key mediator that activates HSCs
to produce ECM (1,24), whereas MMPs and TIMPs regulate ECM
degradation (25). In the present
study, SSd and E2 were able to decrease the expression
levels of TGF-β1, Hyp, COL1 and TIMP-1, and increase MMP-1 levels.
These effects may induce positive remodeling of liver fibrosis.
The physiological effects of estrogens are mediated
through two receptor subtypes, ERα and ERβ. It has been reported
that only ERβ, not ERα, is expressed in primary cultured rat HSCs
(42). However, our previous
study detected both ERα and ERβ proteins by immunofluorescence and
western blot analysis in the rat HSC line, HSC-T6 (43). This discrepancy regarding the
presence of ERα may be due to the use of different HSC lines.
Furthermore, the effects of genistein, a phytoestrogen, on OS and
mitochondria may be due, at least in part, to increased ERβ
presence, and may be due to upregulation of ERβ induced by
genistein (44). It has also been
demonstrated that 17β-E2 protects ARPE-19 cells from OS
via an ERβ-dependent mechanism (45). These results suggested that
activation of ERβ may be associated with the reduction of ROS
production. Conversely, ERα activation has been revealed to inhibit
high-glucose-induced proliferation of vascular smooth muscle cells
by downregulating ROS-mediated ERK activation, thus suggesting that
selective activation of ERα is required for reducing OS (46). In the present study, the effects
of SSd together with ICI-182780 (pure ER antagonist), MPP (ERα
antagonist) or THC (ERβ antagonist) were explored, in order to
determine which ER subtype contributes to the suppression of
OS-induced HSC-T6 activation. The results revealed that the
suppressive effects of SSd on H2O2-induced
activation of HSC-T6 cells could be inhibited by coincubation with
ICI-182780 or THC, but not MPP. These results strongly suggested
that SSd suppresses OS-induced activation of HSCs, and this effect
is largely dependent on modulation of ERβ.
In conclusion, the present study is the first, to
the best of our knowledge, to suggest that the suppressive effects
of SSd towards OS-induced HSC activation depend on ERβ activity,
and may be at least partially attributed to inhibition of the
ROS-MAPK signaling pathway. These results provided novel evidence
regarding the target of SSd and may establish an experimental basis
for the development of novel drugs for the treatment of hepatic
fibrosis.
Abbreviations:
|
HSCs
|
hepatic stellate cells
|
|
ECM
|
extracellular matrix
|
|
ROS
|
reactive oxygen species
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
SSd
|
saikosaponin-d
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
|
TIMP-1
|
tissue inhibitor of
metalloproteinases-1
|
|
MMP-1
|
matrix metalloproteinase-1
|
|
Hyp
|
hydroxyproline
|
|
COL1
|
collagen-1
|
|
TGF-β1
|
transforming growth factor-β1
|
|
α-SMA
|
α-smooth muscle actin
|
|
ER
|
estrogen receptor
|
Acknowledgments
The present study was supported by grants from the
National Natural and Science Foundation of China (grant no.
81573775), the Shanghai Natural Science Foundation (grant no.
09ZR1429700) and Shanghai Outstanding Academic Leader of Health
System (grant no. XBR2013120).
References
|
1
|
Bataller R and Brenner DA: Liver fibrosis.
J Clin Invest. 115:209–218. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sánchez-Valle V, Chávez-Tapia NC, Uribe M
and Méndez-Sánchez N: Role of oxidative stress and molecular
changes in liver fibrosis: A review. Curr Med Chem. 19:4850–4860.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li JT, Liao ZX, Ping J, Xu D and Wang H:
Molecular mechanism of hepatic stellate cell activation and
antifibrotic therapeutic strategies. J Gastroenterol. 43:419–428.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schon HT, Bartneck M, Borkham-Kamphorst E,
Nattermann J, Lammers T, Tacke F and Weiskirchen R: Pharmacological
Intervention in Hepatic Stellate Cell Activation and Hepatic
Fibrosis. Front Pharmacol. 7:332016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ghatak S, Biswas A, Dhali GK, Chowdhury A,
Boyer JL and Santra A: Oxidative stress and hepatic stellate cell
activation are key events in arsenic induced liver fibrosis in
mice. Toxicol Appl Pharmacol. 251:59–69. 2011. View Article : Google Scholar
|
|
6
|
Mormone E, George J and Nieto N: Molecular
pathogenesis of hepatic fibrosis and current therapeutic6
approaches. Chem Biol Interact. 193:225–231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yasuda M, Shimizu I, Shiba M and Ito S:
Suppressive effects of estradiol on dimethylnitrosamine-induced
fibrosis of the liver in rats. Hepatology. 29:719–727. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu JW, Gong J, Chang XM, Luo JY, Dong L,
Hao ZM, Jia A and Xu GP: Estrogen reduces CCl4-induced
liver fibrosis in rats. World J Gastroenterol. 8:883–887. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shimizu I, Mizobuchi Y, Yasuda M, Shiba M,
Ma YR, Horie T, Liu F and Ito S: Inhibitory effect of oestradiol on
activation of rat hepatic stellate cells in vivo and in vitro. Gut.
44:127–136. 1999. View Article : Google Scholar
|
|
10
|
Itagaki T, Shimizu I, Cheng X, Yuan Y,
Oshio A, Tamaki K, Fukuno H, Honda H, Okamura Y and Ito S: Opposing
effects of oestradiol and progesterone on intracellular pathways
and activation processes in the oxidative stress induced activation
of cultured rat hepatic stellate cells. Gut. 54:1782–1789. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cuzick J: Hormone replacement therapy and
the risk of breast cancer. Eur J Cancer. 44:2344–2349. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barnes PJ: Corticosteroids: The drugs to
beat. Eur J Pharmacol. 533:2–14. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JK, Kim JH and Shin HK: Therapeutic
effects of the oriental herbal medicine Sho-saiko-to on liver
cirrhosis and carcinoma. Hepatol Res. 41:825–837. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng G, Kurtz RC, Vickers A, Lau N, Yeung
KS, Shia J and Cassileth B: A single arm phase II study of a
Far-Eastern traditional herbal formulation (sho-sai-ko-to or
xiao-chai-hu-tang) in chronic hepatitis C patients. J
Ethnopharmacol. 136:83–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Qin F, Liu JY and Yuan JH:
Chaihu-Shugan-San, an oriental herbal preparation, for the
treatment of chronic gastritis: A meta-analysis of randomized
controlled trials. J Ethnopharmacol. 146:433–439. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fan J, Li X, Li P, Li N, Wang T, Shen H,
Siow Y, Choy P and Gong Y: Saikosaponin-d attenuates the
development of liver fibrosis by preventing hepatocyte injury.
Biochem Cell Biol. 85:189–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dang SS, Wang BF, Cheng YA, Song P, Liu ZG
and Li ZF: Inhibitory effects of saikosaponin-d on
CCl4-induced hepatic fibrogenesis in rats. World J
Gastroenterol. 13:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen MF, Huang CC, Liu PS, Chen CH and
Shiu LY: Saikosaponin a and saikosaponin d inhibit proliferation
and migratory activity of rat HSC-T6 cells. J Med Food. 16:793–800.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang P, Ren J, Tang J, Zhang D, Li B and
Li Y: Estrogen-like activities of saikosaponin-d in vitro: A pilot
study. Eur J Pharmacol. 626:159–165. 2010. View Article : Google Scholar
|
|
20
|
Richter DU, Mylonas I, Toth B, Scholz C,
Briese V, Friese K and Jeschke U: Effects of phytoestrogens
genistein and daidzein on progesterone and estrogen (estradiol)
production of human term trophoblast cells in vitro. Gynecol
Endocrinol. 25:32–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gille JJ and Joenje H: Cell culture models
for oxidative stress: Superoxide and hydrogen peroxide versus
normobaric hyperoxia. Mutat Res. 275:405–414. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiyoshima T, Enoki N, Kobayashi I, Sakai
T, Nagata K, Wada H, Fujiwara H, Ookuma Y and Sakai H: Oxidative
stress caused by a low concentration of hydrogen peroxide induces
senescence-like changes in mouse gingival fibroblasts. Int J Mol
Med. 30:1007–1012. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Coates D, Zafar S and Milne T:
Quantitative real-time gene profiling of human alveolar
osteoblasts. Methods Mol Biol. 1537:447–459. 2017. View Article : Google Scholar
|
|
24
|
Lechuga CG, Hernández-Nazara ZH, Domínguez
Rosales JA, Morris ER, Rincón AR, Rivas-Estilla AM, Esteban-Gamboa
A and Rojkind M: TGF-beta1 modulates matrix metalloproteinase-13
expression in hepatic stellate cells by complex mechanisms
involving p38MAPK, PI3-kinase, AKT, and p70S6k. Am J Physiol
Gastrointest Liver Physiol. 287:G974–G987. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Iredale JP: Hepatic stellate cell behavior
during resolution of liver injury. Semin Liver Dis. 21:427–436.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang BZ, Guo XT, Chen JW, Zhao Y, Cong X,
Jiang ZL, Cao RF, Cui K, Gao SS and Tian WR: Saikosaponin-D
attenuates heat stress-induced oxidative damage in LLC-PK1 cells by
increasing the expression of anti-oxidant enzymes and HSP72. Am J
Chin Med. 42:1261–1277. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao L, Zhang H, Bao J, Liu J and Ji Z:
Saikosaponin-d protects renal tubular epithelial cell against high
glucose induced injury through modulation of SIRT3. Int J Clin Exp
Med. 8:6472–6481. 2015.PubMed/NCBI
|
|
28
|
Hong IH, Park SJ, Goo MJ, Lee HR, Park JK,
Ki MR, Kim SH, Lee EM, Kim AY and Jeong KS: JNK1 and JNK2 regulate
α-SMA in hepatic stellate cells during CCl4-induced
fibrosis in the rat liver. Pathol Int. 63:483–491. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Varela-Rey M, Montiel-Duarte C,
Osés-Prieto JA, López-Zabalza MJ, Jaffrèzou JP, Rojkind M and
Iraburu MJ: p38 MAPK mediates the regulation of alpha1(I)
procollagen mRNA levels by TNF-alpha and TGF-beta in a cell line of
rat hepatic stellate cells(1). FEBS Lett. 528:133–138. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Friedman SL: Liver fibrosis - from bench
to bedside. J Hepatol. 38(Suppl 1): S38–S53. 2003. View Article : Google Scholar
|
|
31
|
Bhaskar ME: Management of cirrhosis and
ascites. N Engl J Med. 351:300–301; author reply 300-301, 2004.
PubMed/NCBI
|
|
32
|
O'Beirne JP, Foxton MR and Heneghan MA:
Management of cirrhosis and ascites. N Engl J Med. 351:300–301;
author reply 300-301, 2004. PubMed/NCBI
|
|
33
|
Xu JW, Gong J, Chang XM, Luo JY, Dong L,
Jia A and Xu GP: Effects of estradiol on liver estrogen
receptor-alpha and its mRNA expression in hepatic fibrosis in rats.
World J Gastroenterol. 10:250–254. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li JF, Chen BC, Lai DD, Jia ZR, Andersson
R, Zhang B, Yao JG and Yu Z: Soy isoflavone delays the progression
of thioacetamide-induced liver fibrosis in rats. Scand J
Gastroenterol. 46:341–349. 2011. View Article : Google Scholar
|
|
35
|
Lee ES, Shin MO, Yoon S and Moon JO:
Resveratrol inhibits dimethylnitrosamine-induced hepatic fibrosis
in rats. Arch Pharm Res. 33:925–932. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Demiroren K, Dogan Y, Kocamaz H, Ozercan
IH, Ilhan S, Ustundag B and Bahcecioglu IH: Protective effects of
L-carnitine, N-acetylcysteine and genistein in an experimental
model of liver fibrosis. Clin Res Hepatol Gastroenterol. 38:63–72.
2014. View Article : Google Scholar
|
|
37
|
Novo E, Busletta C, Bonzo LV, Povero D,
Paternostro C, Mareschi K, Ferrero I, David E, Bertolani C,
Caligiuri A, et al: Intracellular reactive oxygen species are
required for directional migration of resident and bone
marrow-derived hepatic pro-fibrogenic cells. J Hepatol. 54:964–974.
2011. View Article : Google Scholar
|
|
38
|
Marra F, Arrighi MC, Fazi M, Caligiuri A,
Pinzani M, Romanelli RG, Efsen E, Laffi G and Gentilini P:
Extracellular signal-regulated kinase activation differentially
regulates platelet-derived growth factor's actions in hepatic
stellate cells, and is induced by in vivo liver injury in the rat.
Hepatology. 30:951–958. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhong W, Shen WF, Ning BF, Hu PF, Lin Y,
Yue HY, Yin C, Hou JL, Chen YX, Zhang JP, et al: Inhibition of
extracellular signal-regulated kinase 1 by adenovirus mediated
small interfering RNA attenuates hepatic fibrosis in rats.
Hepatology. 50:1524–1536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Frémin C, Bessard A, Ezan F, Gailhouste L,
Régeard M, Le Seyec J, Gilot D, Pagès G, Pouysségur J, Langouët S,
et al: Multiple division cycles and long-term survival of
hepatocytes are distinctly regulated by extracellular
signal-regulated kinases ERK1 and ERK2. Hepatology. 49:930–939.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kluwe J, Pradere JP, Gwak GY, Mencin A, De
Minicis S, Österreicher CH, Colmenero J, Bataller R and Schwabe RF:
Modulation of hepatic fibrosis by c-Jun-N-terminal kinase
inhibition. Gastroenterology. 138:347–359. 2010. View Article : Google Scholar :
|
|
42
|
Zhou Y, Shimizu I, Lu G, Itonaga M,
Okamura Y, Shono M, Honda H, Inoue S, Muramatsu M and Ito S:
Hepatic stellate cells contain the functional estrogen receptor
beta but not the estrogen receptor alpha in male and female rats.
Biochem Biophys Res Commun. 286:1059–1065. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, Liu J, Lin L, Que R, Shen Y and Tao
Z: Regulation of saikosaponin-d on the level of of ERα and ERβ
protein in hepatic stellate cells. Trad Chin Drug Res Clin
Pharmacol. 27:58–61. 2016.In Chinese.
|
|
44
|
Nadal- Serrano M, Pons DG, Sastre- Serra
J, Blanquer-Rosselló MM, Roca P and Oliver J: Genistein modulates
oxidative stress in breast cancer cell lines according to ERα/ERβ
ratio: Effects on mitochondrial functionality, sirtuins, uncoupling
protein 2 and antioxidant enzymes. Int J Biochem Cell Biol.
45:2045–2051. 2013. View Article : Google Scholar
|
|
45
|
Giddabasappa A, Bauler M, Yepuru M, Chaum
E, Dalton JT and Eswaraka J: 17-β estradiol protects ARPE-19 cells
from oxidative stress through estrogen receptor-β. Invest
Ophthalmol Vis Sci. 51:5278–5287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ortmann J, Veit M, Zingg S, Di Santo S,
Traupe T, Yang Z, Völzmann J, Dubey RK, Christen S and Baumgartner
I: Estrogen receptor-a but not -β or GPER inhibits high
glucose-induced human VSMC proliferation: Potential role of ROS and
ERK. J Clin Endocrinol Metab. 96:220–228. 2011. View Article : Google Scholar
|














