Introduction
Modern surgical techniques for cataracts are
continuously improving; however, posterior capsule opacification
(PCO) remains common long-term complication that results in
decreased visual acuity following successful surgery (1,2).
Without posterior capsulorhexis, almost every child or infant
suffers a visual decline caused by PCO following cataract surgery,
while the incidence rate of PCO in adults is ≥50% at 2–5 years
post-surgery (3,4). Artificial intraocular lenses (IOLs)
have been developed for the prevention of PCO and have
well-designed sharp borders at the optic edge or on the capsular
tension rings, which may delay cell migration into the posterior
capsule (5,6). However, patients who have been
implanted with these lenses still undergo neodymium yttrium
aluminum garnet laser capsulotomy, the only effective treatment for
PCO, albeit at a lower rate (7).
This procedure is associated with rhegmatogenous retinal
detachment, cystoid macular edema and damage to the intraocular
lens (8), and is not suitable for
infants following congenital cataract surgery (3).
PCO has a complex pathogenesis and is induced by
cataract surgery as an inflammatory and wound-healing response
(2), which includes lens
epithelial cell (LEC) attachment, proliferation, spreading and
migration toward the capsular bag surface of the IOL. Damaged
ocular tissue releases chemokines, which attract cells of the
immune system to remove damaged tissue and to facilitate subsequent
tissue repair (9). Therefore,
post-surgical pharmacological inhibition of LEC proliferation and
migration, and the promotion of LEC apoptosis are possible options
for preventing PCO.
In the process of PCO development, the
phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin
(mTOR) signaling pathway serves a crucial role. mTOR is a
serine/threonine kinase at the nexus between oncogenic PI3K/AKT
signaling and critical downstream pathways that drive cell growth,
survival and resistance to therapeutic agents (10–12). Previously, numerous studies have
revealed that the activation of the AKT/mTOR signaling pathway is a
key regulator in the proliferation and migration of LECs following
stimulation by certain growth factors, including hepatocyte growth
factor (HGF), platelet-derived growth factor, insulin-like growth
factor (IGF), fibroblast growth factor (FGF) and integrin-linked
kinase (13–16).
PP242, as a new-generation mTOR inhibitor, is able
to completely suppress mTOR complex 1 (mTORC1) as well as
mTORC2-mediated signaling pathways by suppressing the AKT feedback
loop (17). Most importantly,
PP242 has been reported to markedly improve antitumor activity
in vivo and in vitro, and the effectiveness of this
drug in cancer treatment has been assessed in clinical trials
(18). Being more effective than
rapamycin, PP242 may be an attractive candidate for the
post-operative prevention and treatment of PCO following cataract
surgery.
The present study was performed to investigate the
capacity of PP242 to inhibit the crucial cellular events in the
formation of PCO, involving LEC proliferation, attachment and
migration, and the induction of LEC autophagy, apoptosis and/or
death compared with that of rapamycin. The present results may
contribute to the development of a novel strategy to prevent
PCO.
Materials and methods
Cell lines and reagents
The SRA01/04 cell line constituting human LECs
(HLECs) was purchased from The Cell Bank of the Chinese Academy of
Sciences (Shanghai, China). SRA01/04 cells were maintained in
Dulbecco’s modified Eagle’s medium (GE Healthcare Life Sciences,
Little Chalfont, UK) with 10% fetal bovine serum (FBS; GE
Healthcare Life Sciences), penicillin (100 U/ml) and streptomycin
(100 µg/ml). All cells were maintained at 37°C in a
humidified incubator with 95% air and 5% CO2.
PP242 was purchased from Selleck Chemicals (Houston,
TX, USA). The rabbit monoclonal anti-mTOR (cat. no. 2983; 1:1,000),
rabbit monoclonal anti-phosphorylated (p)-mTOR (Ser2448; cat. no.
5536; 1:1,000), rabbit monoclonal anti-p-p70S6K (Thr389; cat. no.
9234; 1:1,000), rabbit polyclonal anti-p-p70S6K (Ser371; cat. no.
9208; 1:1,000), rabbit polyclonal anti-p70S6K (cat. no. 9202;
1:1,000), rabbit monoclonal anti-p-4E-BP1 (Thr37/46; cat. no. 2855;
1:1,000), rabbit monoclonal anti-p-AKT (Ser473; cat. no. 4058,
1:1,000), rabbit monoclonal anti-AKT (cat. no. 4685, 1:1,000),
rabbit monoclonal anti-microtubule-associated protein light chain 3
(LC3) A/B (cat. no. 12741; 1:1,000), rabbit polyclonal anti-cleaved
caspase-3 (cat. no. 9661; 1:1,000), rabbit poly-clonal anti-cleaved
poly(ADP ribose) polymerase (PARP; cat. no. 9541; 1:1,000), and
mouse monoclonal anti-B-cell lymphoma 2 (Bcl-2; cat. no. 15071;
1:1,000) were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA). Rabbit monoclonal anti-Bcl-2-associated X
protein (Bax; cat. no. ab182733 1:1,000) was purchased from Abcam
(Cambridge, UK). Mouse monoclonal anti-p53 (cat. no. sc-47698;
1:1,000) and mouse monoclonal anti-cyclin D1 (cat. no. sc-70899;
1:1,000) were purchased from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Mouse monoclonal anti-α-tubulin (cat. no. AC012;
1:5,000), horseradish peroxidase (HRP) goat anti-rabbit
immunoglobulin G (IgG; H+L; cat. no. AS014; dilution, 1:5,000), HRP
goat anti-mouse IgG (H+L; cat. no. AS003; 1:5,000) were purchased
from ABclonal Biotech, Co., Ltd. (Wuhan, China).
Cell proliferation assays
A Cell Counting Kit-8 assay (CCK-8; Nanjing KeyGen
Biotech Co., Nanjing, China) was used to assess the effects of
PP242 on SRA01/04 cell growth according to the manufacturer’s
protocols. Cells were incubated at 37°C with 0, 100, 250, 500, 750
or 1,000 nM PP242 for 24 h. The absorbance was measured at a
wavelength of 450 nm using a plate reader (model 680; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Colony formation assay
SRA01/04 cells were counted and seeded into 3.5-cm
dishes at a density of 1,000 cells per dish in regular culture
medium containing various doses of rapamycin (0.5 or 1.0 µM)
or PP242 (0.5, 1.0 or 2.0 µM). The cells were cultured for
10 days, during which the medium was refreshed every 3 days, until
visible clones appeared. The cells were washed with PBS, fixed with
4% paraformaldehyde for 15 min at room temperature and stained with
Giemsa’s solution for 20 min at room temperature. Images of
colonies in each dish were captured using a digital camera.
Wound healing migration assay
Cells were incubated in the presence or absence of
PP242 (1,000 nM) at 37°C for 24 or 36 h. Following serum starvation
of confluent SRA01/04 cells for 2 h in 6-well plates, the media was
replaced with DMEM without supplementation; line-shaped wounds were
generated across the cell monolayers using a sterile pipette tip.
At 0, 24 and 36 h, images of the wounded cell layers were captured
under bright field using an Olympus IX-71 inverted fluorescence
microscope (Olympus Corp., Tokyo, Japan). ImageJ v1.51 software
(National Institutes of Health, Bethesda, MD, USA) was used to
quantify the closure of the wound gap area, which represented the
migratory potential of the cells.
Transwell assay
SRA01/04 cells (3×104) were suspended in
100 µl serum-free medium and then applied to the upper
inserts of a Transwell plate (with 8.0-µm-pore polycarbonate
membranes; Corning Life Science, Corning, NY, USA) and 600
µl DMEM with 10% FBS was added to the lower chambers.
Following incubation with rapamycin (1.0 µM) or PP242 (0.5,
1.0 or 2.0 µM) at 37°C for 12 h, cells in the upper inserts
were removed with cotton swabs, while the cells on the lower side
of the membrane were then fixed with methanol for 10 min at room
temperature, stained with hematoxylin for 15 min at room
temperature and counted under a Nikon Eclipse Ni-U microscope
(Nikon Corp., Tokyo, Japan).
Immunocytochemistry assay
SRA01/04 cells were seeded into 24-well plates at
1×104 cells/well and cultured on glass coverslips
overnight. After serum starvation for 2 h, the cells were treated
with 0, 250 or 500 nM PP242 for 24 h. Following treatment, the
cells were rinsed once with PBS, fixed in fresh 4% paraformaldehyde
for 15 min at room temperature and permeabilized with 0.1% Triton
X-100. The cells were blocked with 5% bovine serum albumin
(Gen-View Scientific Inc., El Monte CA, USA) in PBS for 1 h at room
temperature and incubated overnight with anti-p-AKT (Ser473)
antibody (dilution, 1:200) at 4°C, followed by incubation with
Alexa Fluor® 488 goat anti-rabbit IgG (H+L) (1:500;
A-11034, dilution, Thermo Fisher Scientific, Inc., Waltham, MA,
USA) for 1 h at room temperature. The p-AKT (Ser473) protein was
detected using fluorescence microscopy at 450–490 nm.
Western blot analysis
To determine the expression profile of proteins,
whole-cell extracts were prepared from 1×106 cells in
radioimmunoprecipitation assay lysis buffer [50 mM Tris/HCl, pH
7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% Na-deoxycholate, 1 mM EDTA
and protease inhibitor cocktail (B14001, Bimake, Houston, TX,
USA)]. Protein concentrations were determined via a Bradford
protein assay. Aliquots of samples containing 30 µg
denatured protein were loaded on 10 or 15% polyacrylamide gels
respectively. Samples were separated by SDS-PAGE and transferred to
polyvinylidene fluoride (PVDF) membranes (Merck KGaA, Darmstadt,
Germany). The membranes were blocked with 5% nonfat dry milk in 20
mM Tris (pH 7.4), 137 mM NaCl and 0.05% Tween-20 for 1 h at room
temperature. The membranes were probed with the primary antibodies
overnight at 4°C in Tris-buffered saline with Tween-20. The
expression of mTOR, p-mTOR, p-AKT (Ser473), AKT, p-p70S6K (Thr389),
p-p70S6K (Ser371), p70S6K, p-4EBP1, cyclin D1, p53, Bax, Bcl-2,
cleaved caspase-3, cleaved-PARP, LC3 A/B, was detected using the
same specific primary antibodies as aforementioned and tubulin was
used as the loading control. All of the PVDF membranes were
analyzed by chemiluminescence (Tanon, Tanon Science &
Technology Co., Ltd., Shanghai, China).
Cell-cycle analysis by flow
cytometry
SRA01/04 cells were seeded into 6-well plates at
3×105 cells/well and incubated with different
concentrations of PP242 (0, 250, 500, 750 and 1,000 nM) for 48 h at
37°C. The cells were then collected, washed with PBS and suspended
in a staining buffer [10 µg/ml propidium iodide, 0.5% Tween
20 and 0.1% RNase A (ST578; Beyotime Institute of Biotechnology,
Shanghai, China) in PBS]. The cells were analyzed using a BD
FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) with
the analysis software program ModFit LT v3.0 (Verity Software
House, Inc., Topsham, ME, USA). The gating was set to exclude cell
debris, doublets and clumps.
Cell apoptosis analysis by flow
cytometry
SRA01/04 cells were seeded into 6-well plates at
3×105 cells/well and treated with PP242 (0, 0.5, 1, 1.5
and 2 µM) for 48 h in serum-free medium, washed with
ice-cold PBS and double-stained with annexin-V-allophycocyanin
(APC) and 7-aminoactinomycin D (7-AAD) to quantify the percentage
of apoptotic cells using a BD LSRFortessa flow cytometer (BD
Biosciences). Cells incubated in the binding buffer with only
annexin-V-APC or 7-AAD served as controls. The results were
analyzed with the software program BD FACSDiva 7.0 (BD
Biosciences).
Statistical analysis
Values are expressed as the mean ± standard
deviation of three independent experiments. Statistical
significance was analyzed using a Student’s t-test or a one-way
analysis of variance followed by a Tukey’s multiple comparisons
test using the SPSS 17.0 (SPSS Inc., Chicago, IL, USA) and Prism
5.0 software (GraphPad Software, Inc., LA Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Targeted inhibition of mTORC1/2 signaling
by the mTOR kinase inhibitor PP242 effectively suppresses LEC
proliferation and migration
The effects of targeted inhibition of mTORC1/2 on
LEC proliferation were examined using a CCK-8 assay. As presented
in Fig. 1A, PP242-treated
SRA01/04 cells proliferated more slowly compared with cells in the
control group. Treatment with 100–1,000 nM PP242 for 24 h reduced
the viability of the LEC lines in a dose-dependent manner
(P<0.01). In addition, the results of the colony formation assay
revealed that compared with rapamycin, PP242 inhibited the clone
forming ability of SRA01/04 cells more effectively (Fig. 1B). From the cell survival curve,
the half-maximal inhibitory concentration (IC50) of
PP242 on the SRA01/04 cell line was determined to be 2.159
µM. The difference in cell survival curves between rapamycin
and PP242 demonstrated that the inhibitory effect of PP242 on the
proliferation of SRA01/04 cells was stronger than that of rapamycin
(Fig. 1B).
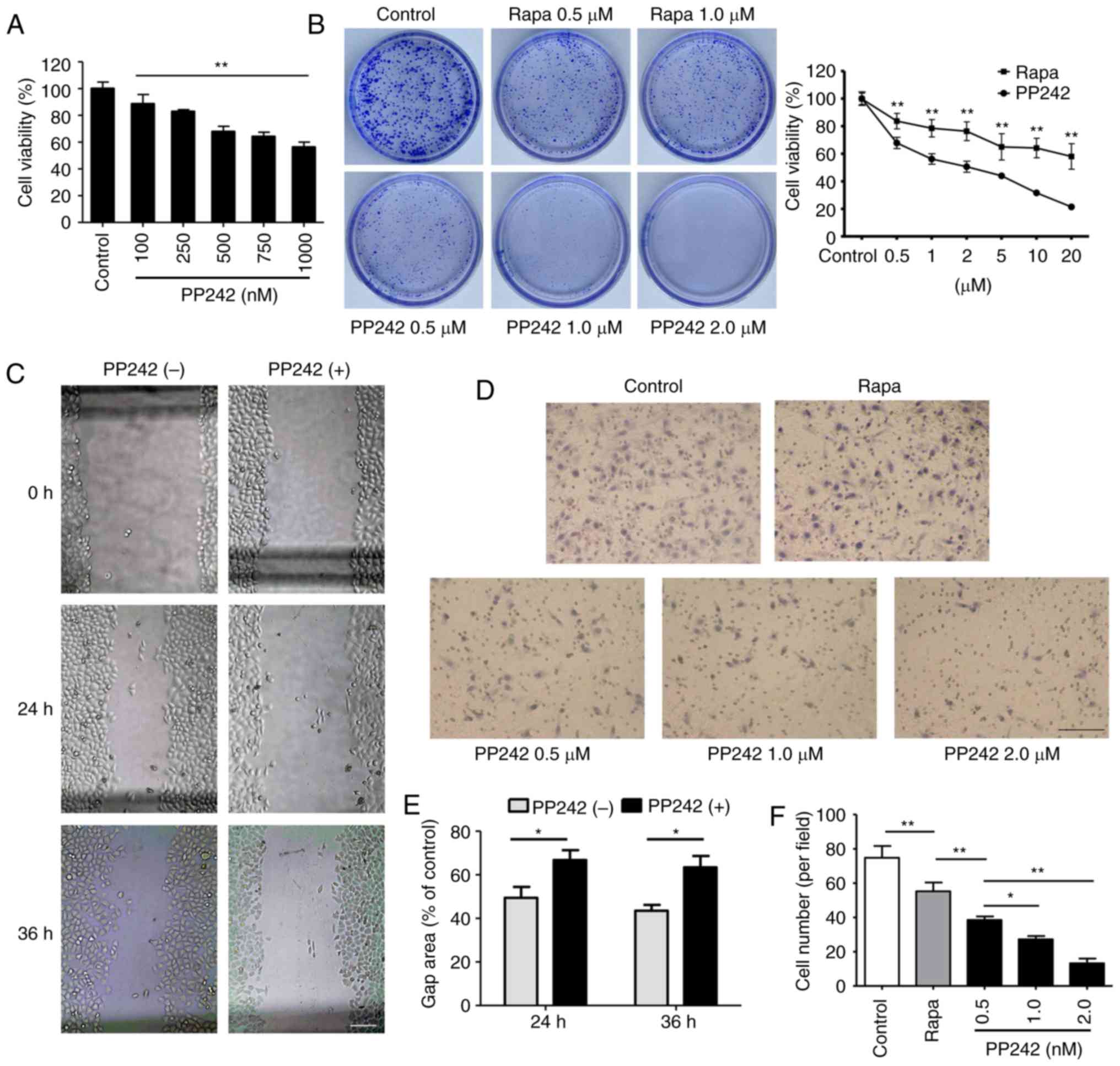 | Figure 1PP242 inhibits the proliferation and
migration of a human lens epithelial cell line. (A) PP242 inhibited
the survival of SRA01/04 cells in a dose-dependent manner. SRA01/04
cells were treated with various doses of PP242 (0, 100, 250, 500,
750 and 1,000 nM) for 24 h. A Cell Counting Kit-8 assay was used to
determine cell viability. (B) A plate colony formation assay (left
panel; dish diameter=3.5 cm) was used to examine the proliferation
of SRA01/04 cells following treatment with vehicle, Rapa (0.5 and
1.0 µM), or PP242 (0.5, 1.0 and 2.0 µM). Cell
survival curves (right panel) for Rapa and PP242 demonstrated that
the inhibitory effect of PP242 on the proliferation of SRA01/04
cells was stronger than that of Rapa. The half-maximal inhibitory
concentration of PP242 on the SRA01/04 cell line was determined to
be 2.159 µM. (C) A wound healing assay was used to examine
the migration of SRA01/04 cells following treatment with vehicle or
PP242 (1,000 nM, 24 or 36 h; scale bar=100 µm). (D) A
Transwell assay was performed on SRA01/04 cells treated with Rapa
(1.0 µM) or PP242 (0.5, 1.0 or 2.0 µM). (E)
Quantification of the wound gap area associated with the cell
migration ability. PP242 significantly restrained the migration of
SRA01/04 cells (scale bar=100 µm). (F) Quantification of the
migration ability of the cells (E). Values are expressed as the
mean ± standard deviation. *P<0.05, **P<0.01 vs.
control. Rapa, rapamycin. |
Subsequently, a wound healing assay and a Transwell
assay were performed to confirm the inhibitory effect of PP242 on
LEC migration. The cell migratory ability was evaluated at 24 and
36 h of exposure to 1,000 nM PP242. As presented in Fig. 1C and E, the larger gap area of the
wound demonstrated that SRA01/04 cells treated with PP242 migrated
more slowly compared with the cells in the control group
(P<0.05). Cell chemotactic motility was evaluated at 12 h of
exposure to rapamycin or PP242. The results of the present study
revealed significant decreases in the number of cells on the bottom
side of the Transwell inserts in the rapamycin and PP242 groups
compared with those in the control group. However, compared with
that in the rapamycin group, the number of cells which migrated to
the lower side of the membrane exhibited a more significant
decrease following treatment with PP242 (P<0.01; Fig. 1D and F).
PP242, but not rapamycin, inhibits mTORC1
as well as mTORC2 function in LECs
In the present study, the effects of PP242 on the
AKT/mTOR signaling pathway in LECs were detected via western blot
analysis. As expected, the formation of p-AKT (Ser473) was
significantly inhibited by PP242, indicating that PP242 may block
mTORC2 in the LECs. Western blot analysis revealed that the protein
expression levels of p-AKT (Ser473) decreased at 2 h following 500
nM PP242 treatment and the efficiency of inhibition increased with
increasing concentrations of PP242 (Fig. 2A–D). The phosphorylation of mTOR
was also detected, which demonstrated the same tendency as p-AKT
(Ser473) regarding PP242-mediated inhibition. The mTOR signaling
pathway has been reported to control cell growth primarily by
regulating cap-dependent translation and ribosome biogenesis via
the phosphorylation of p70S6K and 4EBP1, respectively, by mTORC1
(19). Therefore, p-p70S6K
(Thr389 and Ser371) and p-4EBP1 were assessed as indicators of
mTORC1 activation. The results revealed that mTORC1 was almost
completely inhibited by PP242 in the LECs at a concentration of 100
nM and a short incubation time of 2 h. An immunocytochemical
staining assay indicated that the fluorescence intensity of p-AKT
(Ser473) was also markedly decreased by PP242 treatment compared
with that in the control group (Fig.
2E). Conversely, the first-generation mTOR inhibitor rapamycin
did not reduce but increase p-AKT Ser473. In addition, rapamycin
did not significantly inhibit the activation of p-mTOR and also
failed to block the phosphorylation of 4EBP1 in the present study.
In addition, rapamycin inhibited the phosphorylation of p70S6K;
however, the results of the present study revealed that rapamycin
failed to inhibit mTOR pathway activity, but only partly inhibited
the activity of mTORC1 compared with that of PP242.
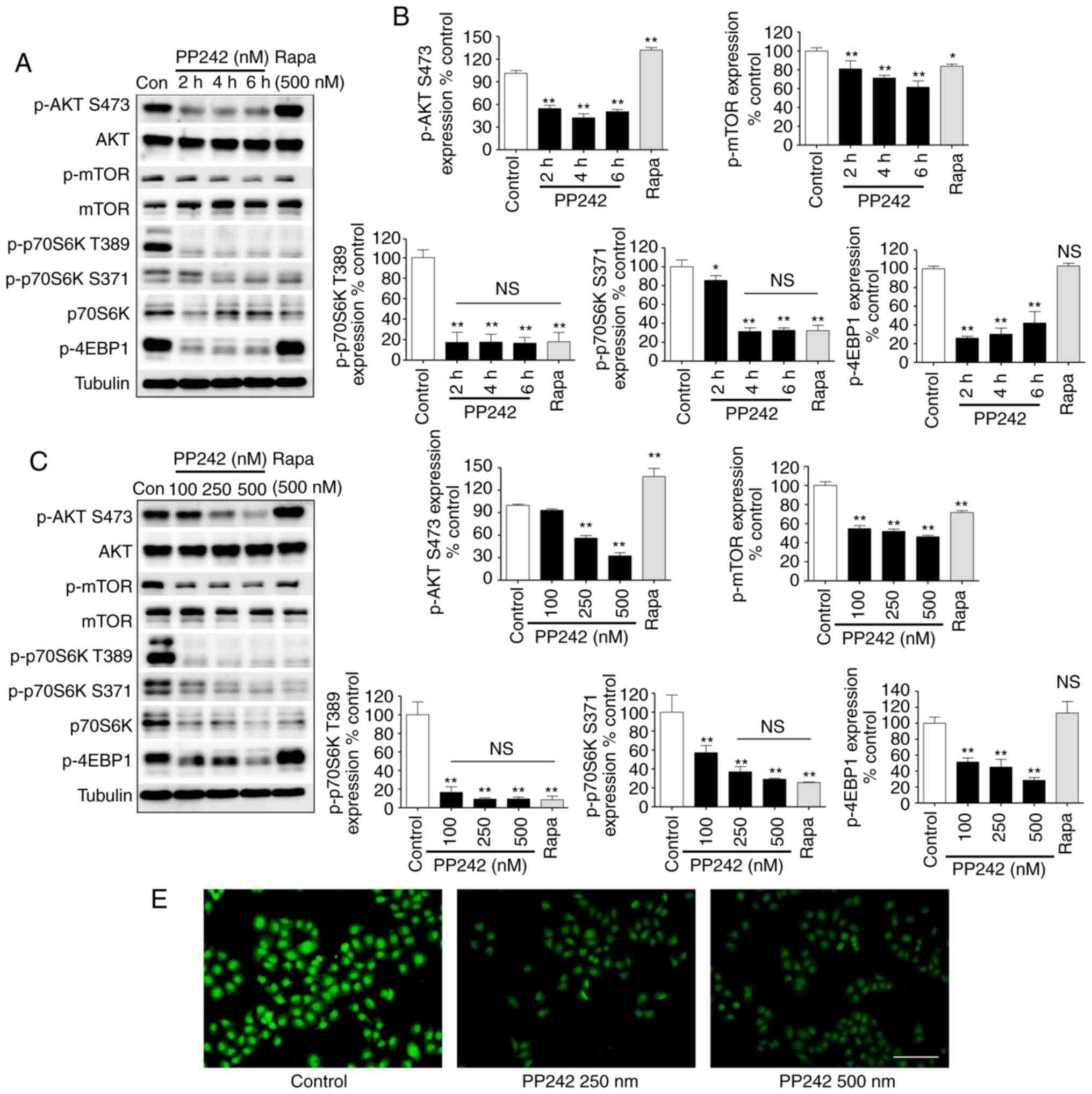 | Figure 2PP242 blocks the AKT/mTOR pathway by
inhibiting mTORC1 and mTORC2 signaling in lens epithelial cells.
(A) Targeted inhibition of mTORC1/2 signaling by PP242 in SRA01/04
cells exhibited a significant effect. SRA01/04 cells were incubated
with 500 nM Rapa for 6 h or the indicated times with 500 nM PP242.
Western blot analysis of p-AKT (Ser473), AKT, p-mTOR, mTOR,
p-p70S6K (Thr389 and Ser371), p70S6K and p-4EBP1 and (B)
quantitative analysis was performed. (C) Targeted inhibition of
mTORC1/2 signaling by PP242 in SRA01/04 cells exhibited a
concentration-dependent effect. SRA01/04 cells were incubated with
500 nM Rapa or the indicated doses of PP242 for 6 h. Cell lysates
were then subjected to western blot analysis to determine the
levels of p-AKT (Ser473), AKT, p-mTOR, mTOR, p-p70S6K (Thr389 and
Ser371), p70S6K and p-4EBP1 and (D) quantitative analyses of the
protein levels was performed. (E) Immunofluorescence analysis of
p-AKT (Ser473) (green) in SRA01/04 cells. Representative images
indicate that higher PP242 concentrations resulted in lower
fluorescence intensities. (scale bar=100 µm)
*P<0.05, **P<0.01 vs. control. Con,
control; ns, not significant; 4EBP1, 4E-binding protein 1;
mTORC1/2, mammalian target of rapamycin complex 1/2; p,
phosphorylated; P70S6K, ribosomal protein s6 kinase; Rapa,
rapamycin. |
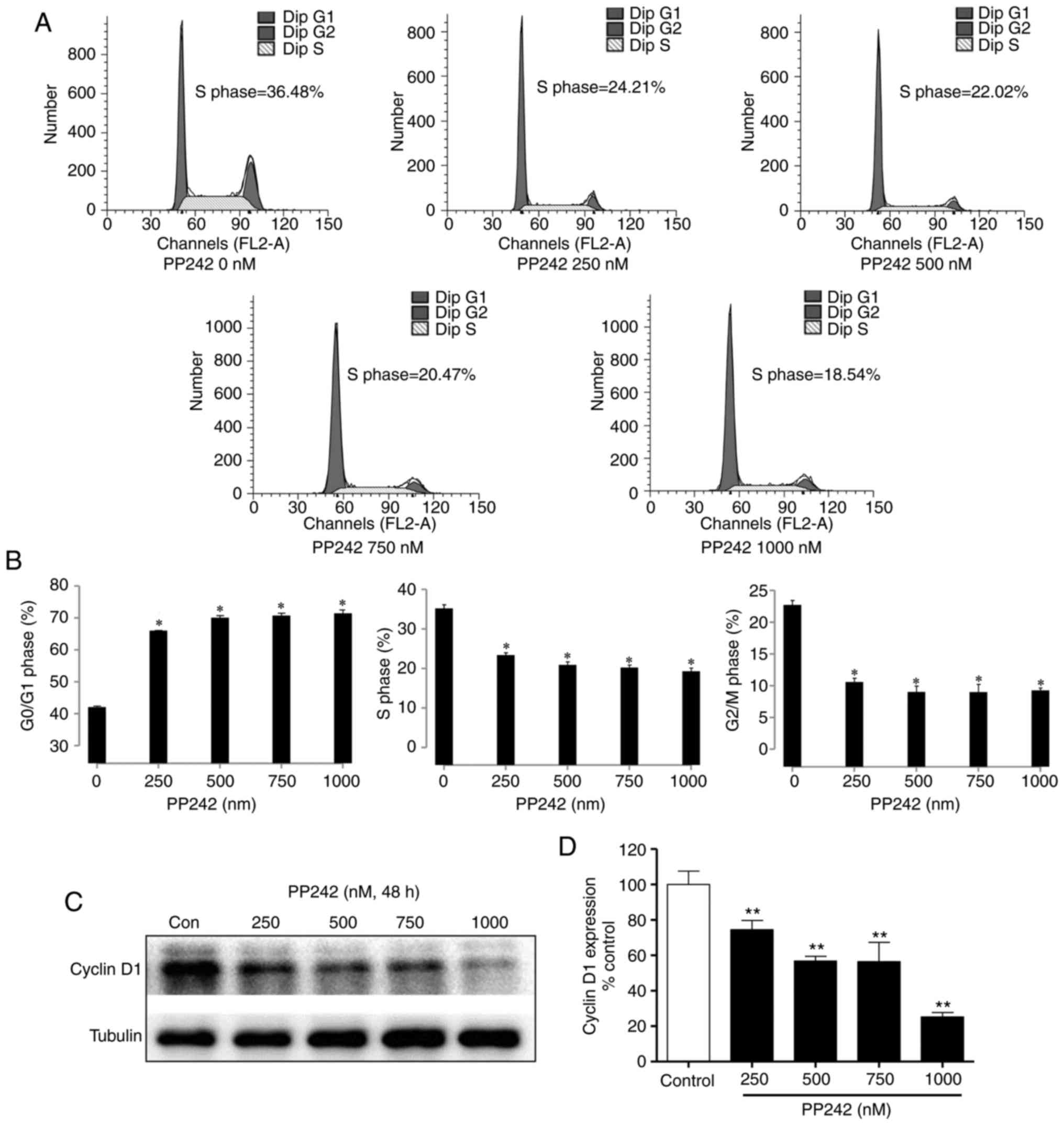
PP242 induces cell cycle arrest of LECs
in G0/G1-phase by downregulating cyclin D1
To investigate the effects of PP242 on the cell
cycle progression of LECs, SRA01/04 cells were analyzed by flow
cytometry. As presented in Fig. 3A
and B, treatment with PP242 (0, 250, 500, 750 and 1,000 nM) for
48 h induced marked cell cycle arrest in G0/G1-phase.
Fluorescence-assisted cell sorting (FACS) analysis revealed that a
48-h exposure to PP242 increased the G0/G1 phase population in a
dose-dependent manner (P<0.05). To investigate the underlying
mechanisms of the inhibition of the cell cycle of LEC by PP242, the
protein expression levels of cyclin D1, which serves an important
role in the progression of the cell cycle, were analyzed via
western blot analysis. As presented in Fig. 3C and D, PP242 significantly
decreased the cyclin D1 expression levels in the LECs. These
results suggested that PP242 suppressed cell proliferation and
inhibited cell cycle progression by downregulating cyclin D1
expression and causing G0/G1 phase arrest.
Effect of PP242 on p53, Bax and Bcl-2
protein expression in LECs
While p53 has long been recognized as a tumor
suppressor, it is indisputably important in normal cell functions
as a sequence-specific transcription factor that may inhibit cell
cycle progression, promote senescence or induce apoptotic cell
death following activation (20).
In the lens, p53 overexpression may counteract the proliferation of
nontumor cells. Previous studies have reported that in
lens-specific E1A expression transgenic mice, CREB-binding
protein/p300 activity induced LEC apoptosis was required for
activation of p53; p53 also regulated ectopic apoptosis in the lens
of Rb-deficient mice (21,22).
To further validate the signaling pathways involved in the
inhibition of LEC growth by PP242, the expression levels of p53 in
the LECs were analyzed. As presented in Fig. 4A and B, a significant and
time-dependent upregulation of p53 in the LECs was detected
following treatment with PP242. These results further confirmed
that PP242 not only inhibits cell cycle progression by
downregulating cyclin D1, but also upregulates p53 in LECs. An
increasing number of studies have demonstrated that Bax possesses
pro-apoptotic activities, which are activated by p53, and
associates with Bcl extra large protein in order to open
mitochondrial membrane pores, leading to the release of cytochrome
c into the cytoplasm (23). Cytochrome c then activates
the caspase cascade via apoptotic protease activating factor 1 and
caspase-3 (24). Conversely,
Bcl-2, which evolved as an important regulator of mitochondrial
integrity, is classified as an anti-apoptotic protein (25). As expected, the results of the
present study revealed that a gradual downregulation of the
anti-apoptotic Bcl-2 occurred with PP242 treatment, leading to an
increase in the pro-apoptotic activity of Bax. This result
suggested that PP242 may mediate apoptotic signaling via the
Bax/Bcl-2 pathway and that its effect is also associated with
increased levels of p53.
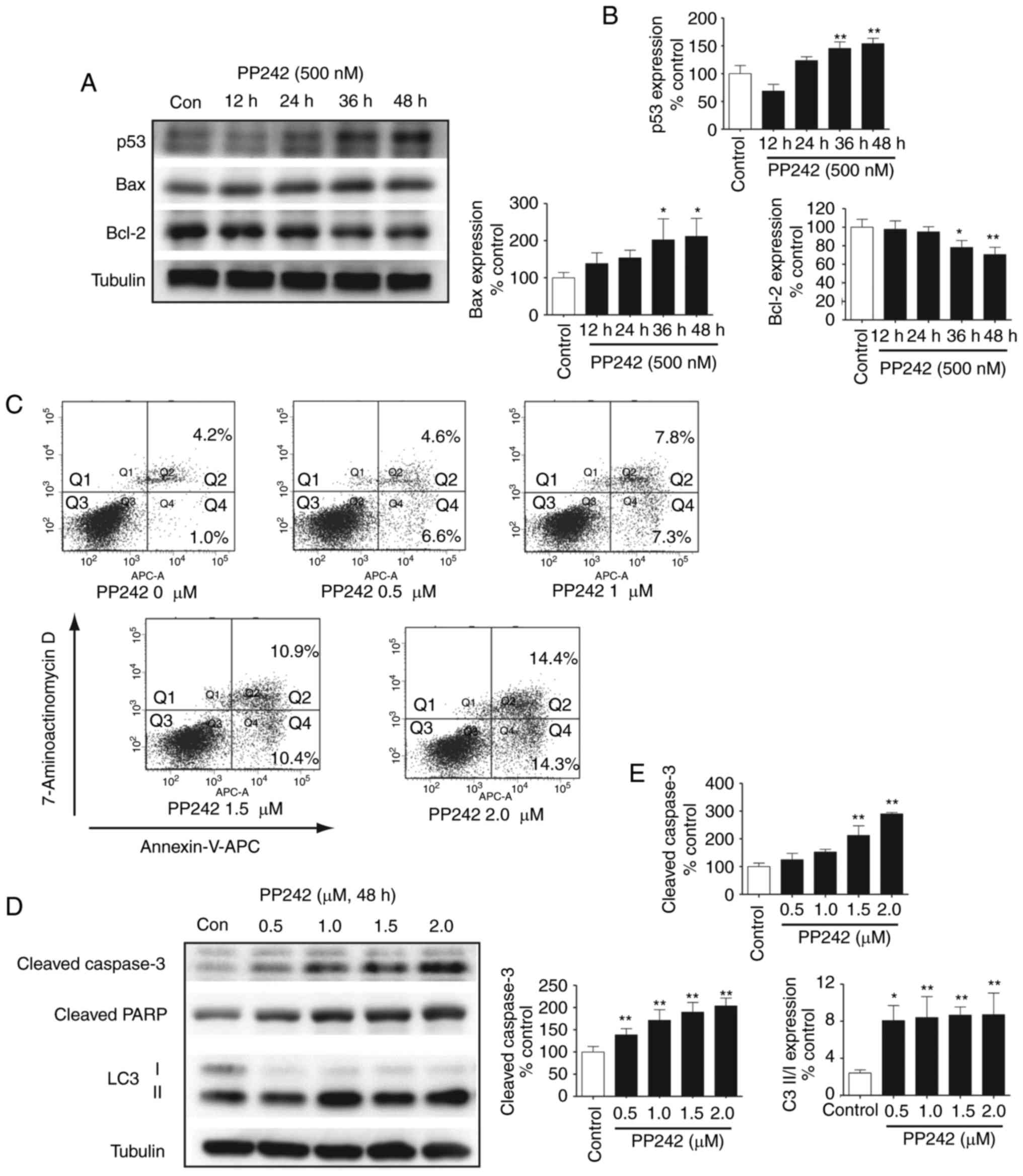 | Figure 4Increased caspase-3-dependent
apoptosis upon mTOR inhibition by PP242 treatment in LECs. (A)
Effect of PP242 on p53, Bax and Bcl-2 protein expression levels in
LECs. SRA01/04 cells were incubated with 500 nM PP242 for 12, 24,
36 and 48 h. Cell lysates were then subjected to western blotting
to determine the levels of p53, Bax and Bcl-2. (B) The
corresponding quantitative analysis indicated that the levels of
p53 and Bax were significantly increased by PP242 in a
time-dependent manner, while Bcl-2 gradually decreased. (C)
SRA01/04 cells were treated with PP242 (0, 0.5, 1, 1.5 and 2
µM) for 48 h in serum-free medium. Annexin-V-APC and
7-Aminoactinomycin D staining were used to quantify the percentage
of apoptotic cells. (D) Western blot analysis and (E) the
corresponding quantitative analyses revealed that the
apoptosis-associated proteins cleaved caspase-3 and cleaved PARP
exhibited significant increases induced by PP242 in a
dose-dependent manner. The increased LC3 II/I ratio revealed that
PP242 also induced autophagy via the inhibition of mTOR.
*P<0.05, **P<0.01 vs. control. Con,
control; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X; LC3,
microtubule-associated protein light chain 3; LECs, lens epithelial
cells; mTOR, mammalian target of rapamycin; PARP, poly(ADP ribose)
polymerase; APC, allophycocyanin; Q, quadrant. |
The specific mTORC1/2 inhibitor PP242
causes apoptosis and induces autophagy in LECs
Following treatment with different concentrations of
PP242 (0, 0.5, 1, 1.5 and 2 µM) for 48 h, the induction of
apoptosis was verified using FACS analysis. The percentages of
early- and late-stage apoptotic cells are presented in the lower
right and upper right quadrants (Q4 and 2, respectively) of the
histograms (Fig. 4C). As
presented in Fig. 4C, only 5.2%
of the total cells in the control group were apoptotic, whereas in
the PP242-treated groups, the percentage of apoptotic cells
gradually increased with increasing concentrations of PP242. The
total percentage of apoptotic cells (Q2 + Q4) increased from 5.2%
in the PP242-untreated LEC cells to 11.2, 15.1, 21.3 and 28.7% in
the cells treated with 0, 0.5, 1, 1.5, and 2 µM PP242,
respectively, for 48 h. Western blot analysis was then used to
analyze signaling induction of LEC apoptosis by PP242. Treatment
with PP242 resulted in a marked increase in cleaved caspase-3 and
cleaved PARP. Existing evidence has reported that a number of mTOR
inhibitors activate autophagy in a variety of cancer cell types
(26). To determine whether the
mTORC1/2 inhibitor PP242 induced autophagy in LECs, the present
study analyzed LC3-I, which is an abundant cytoplasmic protein that
is cleaved and degraded, thereby forming LC3-II during the
initiation of autophagy. The results of Fig. 4D and E indicated an increase in
the ratio of LC3-II/LC3-I, further confirming an increase in
autophagy upon mTOR inhibition. Therefore, the results of the
present study suggested that PP242 induced LEC autophagy and
potentiated cell death.
Discussion
Understanding how PCO progresses from initiation to
end-stage fibrosis by investigating the principal factors and
mechanisms that promote the expansion of PCO is critical for
establishing novel therapies (27). The defining characteristic of PCO
progression is fibrosis, which includes hyperproliferation,
migration, matrix deposition, matrix contraction and
transdifferentiation of fibroblasts into myofibroblasts. The
activation of the AKT/mTOR signaling pathway serves a major role in
the initiation of PCO (28,29). Previous studies have demonstrated
that numerous growth factors, including transforming growth
factor-β, HGF, IGF and FGF (13–16,30), activate the AKT/mTOR signaling
pathway to induce epithelial-to-mesenchymal transformation, which
is a key process that may be triggered by an inflammatory response
(31), e.g. the response in the
eye following cataract surgery (2).
In cells, mTOR regulates numerous basic
characteristics of cell growth and division and is the catalytic
subunit of two functionally distinct complexes, namely mTORC1 and
mTORC2. mTORC1 is a primary regulator of protein translation
phosphorylates S6 kinase and the inhibitory 4EBPs. mTORC2
phosphorylates and regulates AKT, a downstream effector of the PI3K
signaling pathway, which mediates growth and survival signals.
Collectively, these complexes coordinate a variety of processes
that include protein translation, autophagy, proliferation,
survival and metabolism in response to nutrient, energy and growth
factor signals (32). Rapamycin
was originally identified in cancer research as an antifungal
compound that targeted mTORC1 and greatly facilitated the study of
mTOR signaling (33,34). In contrast to the in vitro
results of the present study, the clinical success of rapamycin has
been limited to a few rare cancers, including mantle cell lymphoma,
renal cell carcinoma and endometrial cancer (35). Regarding the prevention of PCO,
rapamycin was observed to inhibit the proliferation, migration and
fibronectin secretion of LECs in vitro and in vivo
(36–38); however, no long-term damage to the
corneal endothelium due to rapamycin has been reported. In
addition, rapamycin was less effective than PP242 in the inhibition
of proliferation and migration, and failed to inhibit the
phosphorylation of 4EBP1 in SRA01/04 cells in the present study.
This indicated that the effects of rapamycin in these LECs were
limited. In addition, this may also be the case in clinical trials
comparing cancer therapies. Compared with rapamycin, PP242
inhibited mTOR activation within SRA01/04 cells, while the
phosphorylation of mTOR failed to decrease significantly; however,
the expression of phosphorylated AKT S473 increased, demonstrating
that the AKT feedback loop was activated.
These limitations, including the incomplete
inhibition of mTORC1, the ineffectiveness toward mTORC2 and the AKT
feedback loop as reported in the present study, led to the
development of mTORC1/2 dual inhibitors, also known as
second-generation mTOR inhibitors (39). PP242 is an example of an
active-site inhibitor, which as identified by Feldman et al
(40), and which may be used to
investigate the selectivity of numerous inhibitors of PI3K scaffold
activity (32). In contrast to
rapamycin, which targets only certain functions of mTORC1, PP242
inhibits mTORC1 as well as mTORC2. Furthermore, PP242 also inhibits
PI3K in addition to inhibiting mTORC1 and mTORC2 (40). In the present study, PP242
effectively reduced LEC proliferation and migration in a
dose-dependent manner. The phosphorylation of AKT S473 was markedly
inhibited by PP242, which demonstrated that PP242 may inhibit
mTORC2 in the LECs. The significant downregulation of p-p70S6K
(Thr389 and Ser371) and p-4EBP1 indicated that mTORC1 was almost
completely blocked by PP242 in the LECs even at low concentrations
and for a short duration. The present study reported that the
action of PP242 in LECs was more effective than that of rapamycin,
similar to the results of previous studies on other cell types
(39,40).
Furthermore, in addition to studying the inhibition
of proliferation and migration by PP242, alterations in the cell
cycle of PP242-treated LECs were assessed by flow cytometry. The
present study revealed that PP242 induced the cell cycle in LECs by
downregulating cyclin D1. In normal cells, p53 is known as a tumor
suppressor gene that controls responses to numerous different
cellular stresses, including DNA damage, hypoxia and oncogene
activation (41). In the present
study, the levels of p53 markedly increased in a time-dependent
manner following PP242 treatment, suggesting that PP242 may inhibit
cell growth by regulating p53. In addition, the gradual
downregulation of anti-apoptotic Bcl-2 was also observed in
response to PP242 treatment, leading to an increase in the
pro-apoptotic activity of Bax. The marked increase in Bax
expression following PP242 treatment of the LEC, along with the
reduction of Bcl-2 expression, confirmed that following treatment
of the LECs with PP242, the cells ceased to proliferate and
underwent apoptosis. The cells were unprotected from the apoptotic
stimuli of Bax by the attenuation of expression of the Bcl-2
protein. Accumulating evidence revealed that abnormal growth or
apoptotic cell death in the lens epithelium significantly alters
cataract formation (42,43). Hence, the apoptosis of HLECs is
one of the most crucial pathologic factors of cataracts. On the
contrary, the apoptosis or cell death of residual HLECs following
cataract surgery is an effective strategy in preventing PCO. The
intraocular HLECs at the equator and under the anterior lens
capsule may be removed as much as possible in cataract surgery.
Despite advanced technologies and equipment in cataract surgical
procedures and IOL design, it is difficult to remove HLECs. This
situation promotes the use of post-operative pharmacotherapy for
inhibiting the proliferation of HLECs and promoting apoptosis or
cell death. Post-operative pharmacotherapy is expected to become an
important means of treatment of PCO. In the present study, FACS
analysis demonstrated an increased caspase-3-dependent apoptosis
upon the inhibition of mTOR by PP242 treatment of LECs. The results
of present study indicated that the dual mTORC1/2 inhibitor PP242
suppressed LEC proliferation and migration. Collectively, due to
its ability to induce LEC apoptosis, PP242 may be considered as a
candidate drug for the prevention of PCO.
As mTOR controls numerous cellular processes,
including autophagy (44), this
process was also taken into consideration in the present study.
Autophagy protects cells, but this process may also mediate cell
death or develop as a primary response to stress inducing cell
death, depending on the specific circumstances (45,46). mTOR promotes cell death and
results in deterioration in certain diseases. In mammalian cells,
autophagy genes or the autophagy signaling pathway are necessary
for a non-apoptotic cell death pathway; it is possible that the
autophagy pathway may act as a tumor suppressor by causing
autophagic cell death (47). It
was observed in the present study that PP242 induced and increased
the conversion of LC3-I to LC3-II, accompanied by increased levels
of cleaved-caspase-3 and cleaved-PARP in the LECs, indicating that
autophagy is involved in PP242-mediated LEC death. Further study is
required to determine the complex regulatory role of autophagy and
mTOR signaling in the death of LECs.
The present study revealed that PP242 markedly
inhibited mTORC1 and mTORC2-mediated signaling activation,
suppressed the proliferation and migration of LECs, and induced
cell apoptosis in vitro. Further in vivo experimental
study of PCO is required to support the potential clinical
application of PP242. PP242 has been tested in cancer therapy in
vivo. Atreya et al (48) reported that 5.8 µM PP242
may be detected in the plasma following an oral dose of 2.5 mg
PP242 in mice. This indicated that compared with the
IC50 (2.159 µM) of PP242 in the SRA01/04 cell
line tested in the present study in vitro, the limitation
that the IC50 of the inhibitor may exceed
pharmacologically relevant concentrations is unlikely to apply to
PP242. In addition, partial or temporary TORC1/2 kinase inhibition
is tolerated by normal lymphocytes (49). Local therapies, including eye
drops or intraocular injection, are prefered for eye disease
treatments. Therefore, localized therapy using this route of PP242
administration may maximally limit the risk of PP242 toxicity to
other organs. Based on the aforementioned evidence, PP242 may have
potential for in vivo application in the future. In
conclusion, the active-site inhibitor PP242 requires further
investigation as a potential strategy for drug treatment in the
prevention of PCO.
Acknowledgments
The present study was supported by the Liaoning
Province Science and Technology Plan Project (grant no. 2013225303)
and the Liaoning Provincial Natural Science Foundation (grant no.
2013021029).
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Apple DJ, Solomon KD, Tetz MR, Assia EI,
Holland EY, Legler UF, Tsai JC, Castaneda VE, Hoggatt JP and
Kostick AM: Posterior capsule opacification. Surv Ophthalmol.
37:73–116. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wormstone IM, Wang L and Liu CS: Posterior
capsule opacification. Exp Eye Res. 88:257–269. 2009. View Article : Google Scholar
|
|
3
|
Awasthi N, Guo S and Wagner BJ: Posterior
capsular opacification: A problem reduced but not yet eradicated.
Arch Ophthalmol. 127:555–562. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chan E, Mahroo OA and Spalton DJ:
Complications of cataract surgery. Clin Exp Optom. 93:379–389.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nishi O: Influence of intraocular lens
material and design on the development of posterior capsule
opacification. Ophthalmologe. 102:572–578. 2005.In German.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nishi O, Yamamoto N, Nishi K and Nishi Y:
Contact inhibition of migrating lens epithelial cells at the
capsular bend created by a sharp-edged intraocular lens after
cataract surgery. J Cataract Refract Surg. 33:1065–1070. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Apple DJ, Peng Q, Visessook N, Werner L,
Pandey SK, Escobar-Gomez M, Ram J and Auffarth GU: Eradication of
posterior capsule opacification: Documentation of a marked decrease
in Nd:YAG laser posterior capsulotomy rates noted in an analysis of
5416 pseudophakic human eyes obtained postmortem. Ophthalmology.
108:505–518. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Javitt JC, Tielsch JM, Canner JK, Kolb MM,
Sommer A and Steinberg EP: National outcomes of cataract
extraction. Increased risk of retinal complications associated with
Nd:YAG laser capsulotomy. The cataract patient outcomes research
team. Ophthalmology. 99:1487–1498. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nibourg LM, Gelens E, Kuijer R, Hooymans
JM, van Kooten TG and Koopmans SA: Prevention of posterior capsular
opacification. Exp Eye Res. 136:100–115. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zoncu R, Efeyan A and Sabatini DM: mTOR:
From growth signal integration to cancer, diabetes and ageing. Nat
Rev Mol Cell Biol. 12:21–35. 2011. View
Article : Google Scholar
|
|
11
|
Guertin DA and Sabatini DM: An expanding
role for mTOR in cancer. Trends Mol Med. 11:353–361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cargnello M, Tcherkezian J and Roux PP:
The expanding role of mTOR in cancer cell growth and proliferation.
Mutagenesis. 30:169–176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tian F, Dong L, Zhou Y, Shao Y, Li W,
Zhang H and Wang F: Rapamycin-induced apoptosis in HGF-stimulated
lens epithelial cells by AKT/mTOR, ERK and JAK2/STAT3 pathways. Int
J Mol Sci. 15:13833–13848. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong W, Cheng BH, Jia SB and Tang LS:
Involvement of the PI3K/Akt signaling pathway in platelet-derived
growth factor-induced migration of human lens epithelial cells.
Curr Eye Res. 35:389–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wederell ED and de Iongh RU: Extracellular
matrix and integrin signaling in lens development and cataract.
Semin Cell Dev Biol. 17:759–776. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Teo ZL, McQueen-Miscamble L, Turner K,
Martinez G, Madakashira B, Dedhar S, Robinson ML and de Iongh RU:
Integrin linked kinase (ILK) is required for lens epithelial cell
survival, proliferation and differentiation. Exp Eye Res.
121:130–142. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sparks CA and Guertin DA: Targeting mTOR:
Prospects for mTOR complex 2 inhibitors in cancer therapy.
Oncogene. 29:3733–3744. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schenone S, Brullo C, Musumeci F, Radi M
and Botta M: ATP-competitive inhibitors of mTOR: An update. Curr
Med Chem. 18:2995–3014. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Weber JD and Gutmann DH: Deconvoluting
mTOR biology. Cell Cycle. 11:236–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vousden KH and Prives C: Blinded by the
Light: The growing complexity of p53. Cell. 137:413–431. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen Q, Ash JD, Branton P, Fromm L and
Overbeek PA: Inhibition of crystallin expression and induction of
apoptosis by lens-specific E1A expression in transgenic mice.
Oncogene. 21:1028–1037. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Morgenbesser SD, Williams BO, Jacks T and
DePinho RA: p53-dependent apoptosis produced by Rb-deficiency in
the developing mouse lens. Nature. 371:72–74. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malecaze F, Decha A, Serre B, Penary M,
Duboue M, Berg D, Levade T, Lubsen NH, Kremer EJ and Couderc B:
Prevention of posterior capsule opacification by the induction of
therapeutic apoptosis of residual lens cells. Gene Ther.
13:440–448. 2006. View Article : Google Scholar
|
|
24
|
Pataer A, Fang B, Yu R, Kagawa S, Hunt KK,
McDonnell TJ, Roth JA and Swisher SG: Adenoviral Bak overexpression
mediates caspase-dependent tumor killing. Cancer Res. 60:788–792.
2000.PubMed/NCBI
|
|
25
|
Zheng JH, Viacava Follis A, Kriwacki RW
and Moldoveanu T: Discoveries and controversies in BCL-2
protein-mediated apoptosis. FEBS J. 283:2690–2700. 2016. View Article : Google Scholar
|
|
26
|
Munson MJ and Ganley IG: MTOR, PIK3C3, and
autophagy: Signaling the beginning from the end. Autophagy.
11:2375–2376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wormstone IM and Eldred JA: Experimental
models for posterior capsule opacification research. Exp Eye Res.
142:2–12. 2016. View Article : Google Scholar
|
|
28
|
Liegl R, Wertheimer C, Kernt M, Docheva D,
Kampik A and Eibl-Lindner KH: Attenuation of human lens epithelial
cell spreading, migration and contraction via downregulation of the
PI3K/Akt pathway. Graefes Arch Clin Exp Ophthalmol. 252:285–292.
2014. View Article : Google Scholar
|
|
29
|
Laplante M and Sabatini DM: mTOR signaling
at a glance. J Cell Sci. 122:3589–3594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo R, Meng Q, Guo H, Xiao L, Yang X, Cui
Y and Huang Y: TGF-β2 induces epithelial-mesenchymal transition in
cultured human lens epithelial cells through activation of the
PI3K/Akt/mTOR signaling pathway. Mol Med Rep. 13:1105–1110. 2016.
View Article : Google Scholar
|
|
31
|
López-Novoa JM and Nieto MA: Inflammation
and EMT: An alliance towards organ fibrosis and cancer progression.
EMBO Mol Med. 1:303–314. 2009. View Article : Google Scholar
|
|
32
|
Liu Q, Thoreen C, Wang J, Sabatini D and
Gray NS: mTOR mediated anti-cancer drug discovery. Drug Discov
Today Ther Strateg. 6:47–55. 2009. View Article : Google Scholar
|
|
33
|
Sehgal SN, Baker H and Vézina C: Rapamycin
(AY-22,989), a new antifungal antibiotic. II. Fermentation,
isolation and characterization. J Antibiot (Tokyo). 28:727–732.
1975. View Article : Google Scholar
|
|
34
|
Rao RD, Buckner JC and Sarkaria JN:
Mammalian target of rapamycin (mTOR) inhibitors as anti-cancer
agents. Curr Cancer Drug Targets. 4:621–635. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Houghton PJ: Everolimus. Clin Cancer Res.
16:1368–1372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu H, Feng G, Wu L, Fu S, Liu P, Yang W
and Zhang X: The effects of rapamycin on lens epithelial cell
proliferation, migration, and matrix formation: An in vitro study.
Mol Vis. 16:1646–1653. 2010.PubMed/NCBI
|
|
37
|
Liu H, Wu L, Fu S, Hou Y, Liu P, Cui H,
Liu J, Xing L and Zhang X: Polylactide-glycoli acid and rapamycin
coating intraocular lens prevent posterior capsular opacification
in rabbit eyes. Graefes Arch Clin Exp Ophthalmol. 247:801–807.
2009. View Article : Google Scholar
|
|
38
|
Wang Z and Wang Z: Effects of rapamycin on
expression of Bcl-2 and Bax in human lens epithelial cells and cell
cycle in rats. J Huazhong Univ Sci Technolog Med Sci. 31:555–559.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou HY and Huang SL: Current development
of the second generation of mTOR inhibitors as anticancer agents.
Chin J Cancer. 31:8–18. 2012.
|
|
40
|
Feldman ME, Apsel B, Uotila A, Loewith R,
Knight ZA, Ruggero D and Shokat KM: Active-site inhibitors of mTOR
target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol.
7:e382009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Muller PA and Vousden KH: Mutant p53 in
cancer: New functions and therapeutic opportunities. Cancer Cell.
25:304–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Osnes-Ringen Ø, Berg KH, Moe MC,
Zetterström C, Røger M and Nicolaissen B: Cell death pattern in
lens epithelium of cataract patients. Acta Ophthalmol. 94:514–520.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li Y, Liu S, Zhang F, Jiang P, Wu X and
Liang Y: Expression of the microRNAs hsa-miR-15a and hsa-miR-16-1
in lens epithelial cells of patients with age-related cataract. Int
J Clin Exp Med. 8:2405–2410. 2015.PubMed/NCBI
|
|
44
|
Lin Z, McDermott A, Shao L, Kannan A,
Morgan M, Stack BC Jr, Moreno M, Davis DA, Cornelius LA and Gao L:
Chronic mTOR activation promotes cell survival in Merkel cell
carcinoma. Cancer Lett. 344:272–281. 2014. View Article : Google Scholar :
|
|
45
|
Baehrecke EH: Autophagic programmed cell
death in Drosophila. Cell Death Differ. 10:940–945. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tresse E, Kosta A, Luciani MF and Golstein
P: From autophagic to necrotic cell death in Dictyostelium. Semin
Cancer Biol. 17:94–100. 2007. View Article : Google Scholar
|
|
47
|
Yu L, Alva A, Su H, Dutt P, Freundt E,
Welsh S, Baehrecke EH and Lenardo MJ: Regulation of an ATG7-beclin
1 program of autophagic cell death by caspase-8. Science.
304:1500–1502. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Atreya CE, Ducker GS, Feldman ME,
Bergsland EK, Warren RS and Shokat KM: Combination of
ATP-competitive mammalian target of rapamycin inhibitors with
standard chemotherapy for colorectal cancer. Invest New Drugs.
30:2219–2225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Janes MR, Limon JJ, So L, Chen J, Lim RJ,
Chavez MA, Vu C, Lilly MB, Mallya S, Ong ST, et al: Effective and
selective targeting of leukemia cells using a TORC1/2 kinase
inhibitor. Nat Med. 16:205–213. 2010. View Article : Google Scholar : PubMed/NCBI
|