Introduction
Mechanical and chemical stimuli, including changes
in pressure and changes in the levels of cytokines, hormones or
growth factors, trigger cardiac hypertrophy, which subsequently
leads to cardiomyopathy and heart failure (1,2).
Despite improvements in treatment strategies and the current
understanding of the pathology, heart failure continues to be the
leading cause of mortality worldwide. Pathological hypertrophy, as
well as other types of cardiac injury, may trigger cardiac
fibrosis, which in turn leads to adverse ventricular remodeling and
heart failure (3–5). Therefore, identifying therapeutics
that prevent the onset of pathological cardiac hypertrophy is of
great interest.
Aberrant expression of microRNAs (miRNAs/miRs),
small noncoding RNAs that regulate gene expression at the
post-transcriptional level (6–8),
serves a distinct role in disease pathology by disrupting signaling
cascades and gene-regulatory networks. For instance, miRNA has been
linked to adverse cardiac remodeling and fibrosis (4,9–11).
The current understanding of the exact role of miRNA in cardiac
hypertrophy has been increased by studies using mouse models
coupled with gain- and loss-of-function strategies (12,13).
In a previous study, through the use of miRNA
arrays, it was demonstrated that miR-327 levels were enhanced in
mouse left ventricle (LV) tissues following myocardial infarction
(MI), suggesting that miR-327 may be involved in cardiac
hypertrophy and fibrosis induced by pressure overload (3). In the present study, it was
demonstrated that miR-327 expression was increased in response to
transverse aortic constriction (TAC) in the LV of mice. Angiotensin
II-induced differentiation of cardiac fibroblasts into
myofibroblasts was increased by miR-327 overexpression. By
contrast, miR-327 knockdown inhibited angiotensin II-induced
differentiation. In the mouse model, downregulation of miR-327
using antagomiR prevented pressure overload-induced hypertrophy and
fibrosis. In addition, integrin β3 (ITGB3) was identified as a
target gene of miR-327. The present study suggests that cardiac
fibrosis is promoted by miR-327; therefore, therapeutics aimed
towards inhibiting miR-327 may be effective treatments for cardiac
fibrosis.
Materials and methods
Antibodies and reagents
Antibodies targeting GAPDH (cat. no. 5174),
phosphorylated extracellular signal-regulated protein kinases 1/2
(p-ERK1/2, cat. no. 4370), p-p38 (cat. no. 4511), p-c-Jun
N-terminal protein kinase (p-JNK, cat. no. 9251), matrix
metalloproteinase-9 (MMP-9, cat. no. 13667) and proliferating cell
nuclear antigen (PCNA, cat. no. 13110) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Collagen type I α 1
(Col1a1, cat. no. ab34710) and α-smooth muscle actin (α-SMA, cat.
no. ab32575) were purchased from Abcam (Cambridge, MA, USA).
Horseradish peroxidase (HRP)-conjugated secondary antibodies
targeting rabbit and mouse were from Cell Signaling Technology,
Inc. Alexa Fluor 488-conjugated streptavidin antibody (cat. no.
111-545-003) was from Jackson ImmunoResearch (West Grove, PA, USA)
and angiotensin II was obtained from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). miR-327 agomiR (cat. no. miR4000561),
antagomiR (cat. no. miR30000561) and scrambled controls (for
control of agomiR, cat. no. miR4000561; for control of antagomiR,
cat. no. miR4000561 were purchased from Guangzhou RiboBio Co., Ltd.
(Guangzhou, China).
Ethics statement
The Institutional Animal Care and Use Committee of
Nanjing Medical University (Nanjing, China) approved all animal
protocols. As described below, a cardiac hypertrophy and heart
failure model was developed using TAC surgery (14). All procedures involving animals
were in accordance with the Guide for the Care and Use of
Laboratory Animals published by the National Institutes of Health
(no. 85–23; revised 1996) and the study protocol was approved by
The Institutional Animal Care and Use Committee (IACUC) of Nanjing
Medical University (Nanjing, China; nos. IACUC-1701020 and
IACUC-14030149). The mice were used for TAC surgery, while the rats
were subjected to isolation of cardiac fibroblasts for in
vitro experiments.
Animals and TAC surgery
A total of 72 male C57BL/6 mice (age, 8 weeks;
weight, 22–24 g) obtained from the model animal research center of
Nanjing University (Nanjing, China) were stratified into groups of
6 animals each: Sham NC antagomiR, TAC NC antagomiR, Sham miR-327
antagomiR and TAC miR-327 antagomiR. Mice were maintained under
appropriate barrier conditions under a 12-h light/dark cycle,
constant temperature range from 20–22°C, humidity range from
50–60%, and received food and water ad libitum. TAC is the
primary method for inducing cardiac hypertrophy in mice and rats
(15). In brief, mice were deeply
anesthetized with 2% isoflurane, maintained under 1.5–2.0%
isoflurane and intubated using a volume-cycled ventilator, and a
midline incision was made above the sternum. The muscles were
carefully separated until the trachea was visualized. A chest
retractor was used to retract the sternum, and a partial left-side
thoracotomy was performed to the second rib using blunt-ended,
spring-action scissors. The lobes of the thymus were separated and
the aortic arch was cleaned of fat tissue using blunt-tipped,
45°-angled forceps. A sterile saline-soaked 7–0 silk suture was
placed between the innominate and left common carotid arteries
using 90°-curved forceps. A 27-gauge needle fragment was aligned
parallel to the transverse aorta, and the suture was loosely
tightened around the transverse aorta twice. The two knots were
tied against the needle in rapid succession. Following removal of
the needle, a 0.4-mm diameter constriction remained. The sham
surgery mice were subjected to the same procedure except for the
tightening around the transverse aorta.
Animal treatment
To determine whether miR-327 inhibition prevents
cardiac fibrosis in vivo, antagomiR, a
2′-O-methyl-5′cholesterol-modified miR-327 inhibitor, or a
scrambled control (aforementioned; Guangzhou RiboBio Co., Ltd.) was
administered to the mice at 80 mg/kg body weight via tail-vein
injection, daily for 3 days prior to TAC surgery (n=6 per
group).
Cardiac imaging
Transthoracic, two-dimensional M-mode
echocardiography was performed using a Vevo 770 high-resolution
in vivo imaging system with a 30-MHz transducer
(VisualSonics, Inc., Toronto, ON, Canada). Echocardiographic
studies were performed at 28 h following TAC surgery. The LV mass,
LV wall thickness, ejection fraction (EF) and percent of fractional
shortening were calculated as previously described (16).
Isolation of neonatal rat cardiomyocytes
and fibroblasts
Sprague Dawley rats (age, 1–3 days; weight, 5–6 g)
obtained from Beijing Vital River Laboratory Animal Technology Co.,
Ltd. (Beijing, China) were used to isolate cardiac fibroblasts. The
ventricular tissue was finely minced and digested in buffer (37°C,
with agitation, 30 min) containing trypsin (6 mg/ml, Sigma-Aldrich;
Merck KGaA) and collagenase type II (4 mg/ml, Worthington
Biochemical Corporation, Lakewood, NJ, USA) at a ratio of 3:2. The
resultant cell suspensions were pelleted by centrifugation (room
temperature, 120 g, 3 min), resuspended in Dulbecco's modified
Eagle's medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) with 100 U/ml penicillin, 100 µg/ml streptomycin and
10% fetal bovine serum (FBS; Sciencell Research Laboratories, Inc.,
San Diego, CA, USA), 5% horse serum (Hyclone; GE Healthcare, Little
Chalfont, UK) and seeded into culture plates. Fibroblasts were
allowed to attach for 2 h at 37°C and 5% CO2. The
unattached cardiomyocytes were then plated in culture plates coated
with 10 mg/ml gelatin (Sigma-Aldrich; Merck KGaA).
Cell transfection
Cardiac fibroblast experiments were performed in
low-serum (1% FBS) medium. At passage 3, cardiac fibroblasts
(5×104/ml) were transfected with miRNA agomiR (100 nM),
antagomiR (200 nM) or the scrambled controls (100 or 200 nM) for 48
h at 37°C. Subsequently, cells were treated with 1 µM
angiotensin II (Sigma-Aldrich; Merck KGaA) for 24 h at 37°C.
Immunohistochemistry
At 4 weeks following TAC surgery, mice were deeply
anesthetized with 2% isoflurane, maintained at 1.5–2% isoflurane
and intubated using a volume-cycled ventilator. The mice were
perfused with PBS, followed by 4% buffered paraformaldehyde. The
hearts were isolated, subjected to PBS perfusion and formalin-fixed
overnight. The head and apex of the heart were divided, tissues
were embedded in paraffin blocks and tissue sections (n=20–25 per
heart; thickness, 4 µm) were prepared. To assess fibrosis,
the sections were stained with Masson's Trichrome and picrosirius
red according to standard procedures (16,17). Collagen and non-collagen
components were red- and orange-stained, respectively. The
percentage of the tissue stained was determined (in 5 fields per
sample) and statistically significant differences between groups
were determined.
Neonatal rat cardiac fibroblasts (NRCF) were fixed
in 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 in
PBS for 10 min. NRCF were blocked with 5% normal serum that matched
the species used to generate the secondary antibody and incubated
with α-SMA primary antibody (1:500 dilution) overnight at 4°C,
followed by fluorochrome-conjugated secondary antibodies (1:500
dilution) for 1 h at room temperature. DAPI was used to
counterstain nuclei. Images were captured with a Carl Zeiss
Axioskop microscope (Carl Zeiss AG, Oberkochen, Germany).
Cell proliferation assay
Cell proliferation was measured in real-time using
the xCELLigence system (Roche Applied Science, Penzberg, Germany)
(18). This system measures
electrical impendence caused by adherent cells to evaluate
proliferation in real time (19).
Cells were seeded at 2,000 cells per well and allowed to attach for
12 h. The cell number, and therefore cell proliferation, was
correlated with the relative change in impedance when measurements
were made with and without cells through a unit-less parameter
referred to as the Cell Index (20). The Cell Index at each time-point
(0, 1, 6, 12, 18, 24, 30, 36, 42, 48, 54, 60 and 66 h) was
normalized to the value recorded at time-point 0/baseline, which
was immediately following transfection with miRNA agomiR (100 nM),
antagomiR (200 nM) or the scrambled controls (100 or 200 nM) for 24
h at 37°C, and treatment with 1 µM angiotensin II for 24 h
at 37°C.
Western blot analysis
The phosphorylated and total protein content from
cultured cells and heart tissue of mice was measured via western
blot analysis, as previously described (14). Lysis buffer [(for cells:
Nonidet-P40 Cell Lysis Buffer (Thermo Fisher Scientific, Inc); for
tissue: T-PER tissue protein extraction reagent (cat. no. 78510;
Thermo Fisher Scientific, Inc.), 1 mM phenylmethylsulfonylfluoride,
phosphatase inhibitor cocktail (cat. no. 04906845001; Roche
Diagnostics, Basel, Switzerland) and protease inhibitor cocktail
(Pierce™ Protease Inhibitor Tablets, cat. no. 88265; Thermo Fisher
Scientific, Inc.)] was used for lysis of cells and mammalian tissue
and protein extraction. A bicinchoninic acid protein assay kit
(Pierce; Thermo Fisher Scientific, Inc.) was used to determine the
total protein concentration. Protein (30 mg) was subjected to
10–15% SDS-PAGE followed by electrophoresis and transfer onto
nitrocellulose membranes (cat. no. 03010040001, Roche Diagnostics).
Membranes were blocked with Tris-buffered saline containing
Tween-20 and 5% bovine serum albumin for 2 h in room temperature.
and then incubated with GAPDH (1:1,000 dilution), p-ERK1/2 (1:1,000
dilution), p-p38 (1:1,000 dilution), p-JNK (1:1,000 dilution),
MMP-9 (1:1,000 dilution) and PCNA (1:1,000 dilution), Col1a1 (1:500
dilution) and α-SMA (1:1,000 dilution) antibodies overnight at 4°C.
Membranes were then washed and incubated with HRP-labeled secondary
antibodies (1:5,000 dilution) for 2 h at room temperature.
Detection was performed using clarity western ECL substrate
(Bio-Rad Laboratories, Hercules, CA, USA). Images were acquired and
quantification analyses were performed using a ChemiDocMP system
(Bio-Rad Laboratories).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The expression of Col1a1, Col3a1, transforming
growth factor-β (TGF-β), MMP-9 and α-SMA was determined using
RT-qPCR. Total RNA was extracted using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. In total, 0.5 ng RNA was used as a template for the
synthesis of complementary (c)DNA using a first strand synthesis
kit (cat. no. K1612; Invitrogen; Thermo Fisher Scientific, Inc.).
qPCR analysis (ABI PRISM 7900 sequence detection system; Applied
Biosystems; Thermo Fisher Scientific, Inc.) was performed on cDNA
using PowerUp™ SYBR® Green Master Mix (cat. no. A25742;
Thermo Fisher Scientific, Inc.). The PCR conditions were 50°C for 2
min and 95°C for 10 min, followed by 50 cycles of 95°C for 15 sec
and 60°C for 75 sec. The 2−ΔΔCq method was used to
calculate the relative expression level of the transcripts
(21). The mRNA expression of the
target gene was normalized to endogenous GAPDH expression and
represented as a fold-change relative to the control. Primers used
for the amplification were as follows: Rat COL1 forward,
5′-CCCAAGGAAAAGAAGCACGTC-3′ and reverse,
5′-AGGTCAGCTGGATAGCGACATC-3′; rat α-SMA forward,
5′-GTCCCAGACATCAGGGAGTAA-3′ and reverse,
5′-TCGGATACTTCAGCGTCAGGA-3′; rat MMP9 forward,
5′-CCTCTGCATGAAGACGACAT-3′ and reverse, 5′-GAGGTGCAGTGGGACACATA-3′;
rat GAPDH forward, 5′-GGCACAGTCAAGGCTGAGAATG-3′ and reverse,
5′-GGCACAGTCAAGGCTGAGAAT G-3′; rat ITGB3 forward,
5′-TGCAACAATGGTAACGGAAC-3′ and reverse, 5′-CCTGCTGAGAGGGTCGATAG-3′;
mouse COL1 forward, 5′-GCTCCTCTTAGGGGCCACT-3′ and reverse,
5′-ATTGGGGACCCTTAGGCCAT-3′; mouse COL3 forward,
5′-CTGTAACATGGAAACTGGGGAAA-3′ and reverse,
5′-CCATAGCTGAACTGAAAACCACC-3′; mouse TGF-β forward,
5′-GGTCGCATCAAGGTGGTCTTT-3′ and reverse,
5′-GTGGTGGTATTCCCCTTCTGG-3′; mouse GAPDH forward,
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse,
5′-TGTAGACCATGTAGTTGAGGTCA-3′.
For quantitative miRNA analysis, the Bulge-Loop™
miRNA qPCR Primer Set (Guangzhou RiboBio, Guangzhou, Guangdong,
China) was used with the Takara SYBR Premix Ex Taq™ (Tli RNaseH
Plus; Takara Bio Inc., Otsu, Japan) on an ABI-7900 Real-Time PCR
Detection System (Applied Biosystems; Thermo Fisher Scientific,
Inc.). A cDNA library was generated via RT using 20 µl
reaction system: 4 µl 5X reaction buffer, 1 µl RNA
sample, 8 µl RNase-free water, 2 µl dNTPs, 2
µl enzyme inhibitor, and 0.5 µl MMLV reverse
transcriptase (Takara Bio, Inc.) and 2.5 µl specific RT
primers of miRNA (Guangzhou RiboBio Co., Ltd.). The RT conditions
were as follows: 42°C for 60 min followed by 95°C for 5 min, then
immediate cooling to 4°C. Primers used for the amplification of
miR-327 were as follows: mmu-miR-327 5′-ACUUGAGGGGCAUGAGGAU-3′;
rno-miR-327, 5′-CCUUGAGGGGCAUGAGGGU-3′; U6 (cat. no. ssD0904071008,
Guangzhou RiboBio Co., Ltd.) was used as an internal control for
miRNA template normalization. The PCR conditions were as follows:
95°C for 20 sec, followed by 40 cycles of 95°C for 10 sec, 60°C for
20 sec and 70°C for 10 sec. The 2−ΔΔCq method was used
for analysis (21).
Statistical analysis
Values are expressed as the mean ± standard error.
Statistical analysis was performed using GraphPad Prism 5 (GraphPad
Software, Inc., La Jolla, CA, USA). Statistical significance was
determined using Student's t-test or one-way analysis of variance
with Bonferroni's post-hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-327 expression is upregulated in
fibrotic cardiac tissues
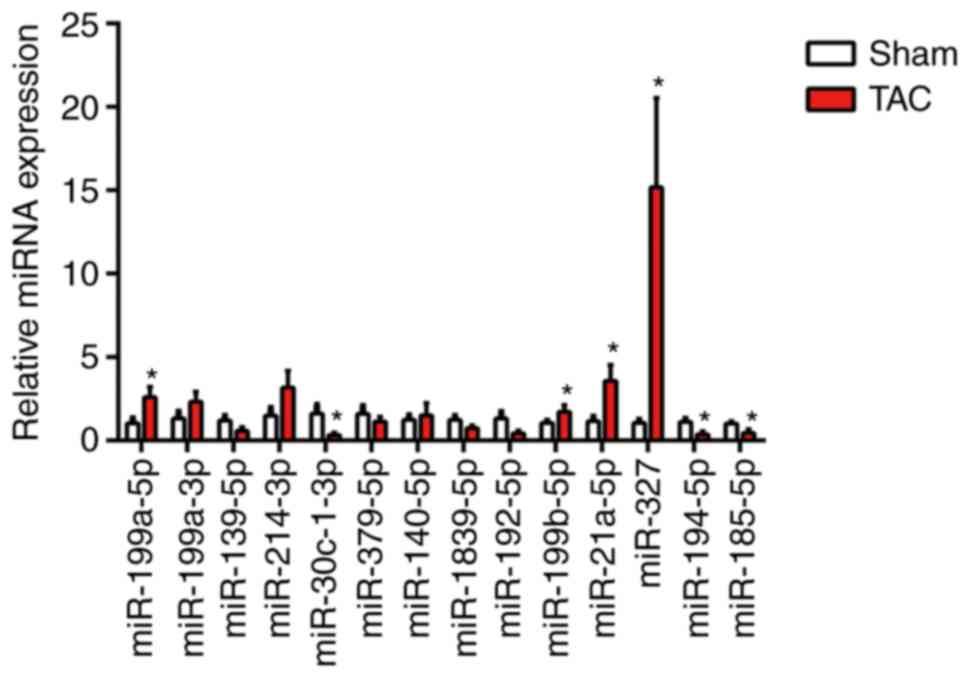
By using RT-qPCR analysis, a series of candidate
miRNAs in the heart tissue of mice at 4 weeks were measured
following TAC surgery. These candidate genes were the top 14
dysregulated miRNAs identified by miRNA arrays of post-MI
ventricles with prominent fibrosis performed in a previous study
(3). In the present study,
miR-327 was identified as the highest upregulated miRNA in fibrotic
heart tissue (Fig. 1). To confirm
the cellular expression of miR-327, its expression was examined in
isolated neonatal rat cardiomyocytes and cardiac fibroblasts. The
expression of miR-327 in cardiac fibroblasts was higher compared
with that in the unstressed cardiomyocytes (Fig. 2A).
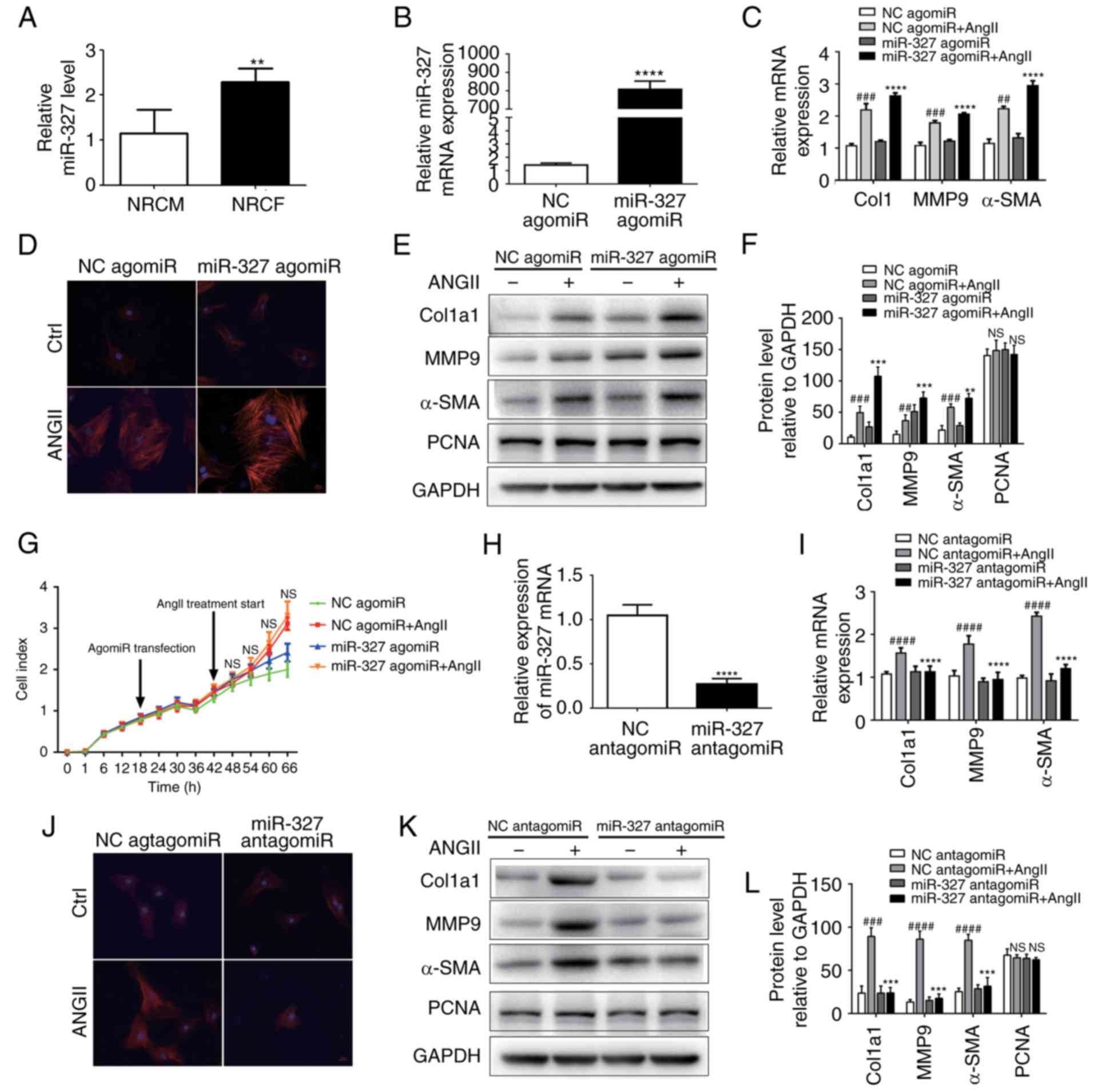 | Figure 2miR-327 promotes cardiac fibroblast
differentiation into myofibroblasts in vitro. (A) miR-327
expression in NRCF and NRCM (n=6). (B) Overexpression of miR-327
with miR-327 agomiR (n=6). (C) Effect of miR-327 agomiR treatment
on markers of cardiac fibrosis during cardiac fibroblast
differentiation as determined by RT-qPCR (n=6). (D) Fluorescence
microscopy images demonstrating the effect of miR-327
overexpression on angiotensin expression during the differentiation
of cardiac fibroblasts into myofibroblasts (n=3; scale bar, 20
µm). (E and F) Western blot analysis indicated upregulation
of fibrosis-associated markers in NRCF overexpressing miR-327
(n=3). (G) Real-time cell proliferation assay using electric
impedance as a measure of NRCF proliferation with miR-327 agomiR.
ns, P>0.05, miR321-agomiR + AngII vs. NC-agomiR + AngII. (H)
miR-327 antagomiR treatment decreased miR-327 in NRCFs (n=6). (I)
Downregulation of fibrosis-associated gene expression in NRGF
following inhibition of miR-327 (n=6). (J) Fluorescence microscopy
images demonstrating inhibition of NRCF differentiation into
myofibroblasts by inhibition of miR-327 (n=3; scale bar, 20
µm). (K and L) Downregulation of fibrosis-associated gene
expression in NRCFs following inhibition of miR-327 (n=3).
##P<0.01, ###P<0.001,
####P<0.0001 vs. respective NC agomiR or NC
antagomiR; **P<0.01, ***P<0.001,
****P<0.0001 vs. respective NC agomiR + AngII or NC
antagomiR + Ang II. Ctrl, control; NC, negative control; ns, no
significance; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction; Ang, angiotensin; col, collagen; α-SMA,
α-smooth muscle actin; MMP, matrix metalloproteinase; miR,
microRNA; PCNA, proliferating cell nuclear antigen; TGF,
transforming growth factor; TAC, transverse aortic constriction;
NRCF, neonatal cardiac fibroblasts; NRCM, neonatal
cardiomyocytes. |
Differentiation of cardiac fibroblasts in
vitro is attenuated by miR-327 inhibition
The development of myofibroblasts from fibroblasts
is a critical step in the pathogenesis of cardiac fibrosis. In
vitro experiments were used to improve the current
understanding of the role that miR-327 serves in regulating
fibrosis. AgomiR-stimulated overexpression of miR-327 promoted
angiotensin II-induced cardiac fibroblast differentiation, as
indicated by the increase in α-SMA expression and staining
(Fig. 2B–D). The protein
expression of MMP-9 and Col1a1 was increased (Fig. 2E and F); however, cardiac
fibroblast proliferation was not affected (Fig. 2G). By contrast, antagomiR-induced
downregulation of miR-327 decreased cardiac fibroblast
differentiation, although the proliferation remained unaffected
(Fig. 2G–J). These results
suggested that, in vitro, cardiac fibroblast differentiation
into myofibroblasts was attenuated by miR-327 inhibition.
Cardiac hypertrophy and pressure
overload-induced fibrosis are prevented by miR-327 inhibition
To investigate the effect of miR-327 on stressed
hearts, miR-327 antagomiR was administered daily by tail-vein
injection to mice for 3 days prior to TAC surgery. At 4 weeks
following surgery, the loss of miR-327 in the heart was confirmed
by RT-qPCR (Fig. 3A). Examination
of the appearance of the hearts and the heart weight normalized to
either body weight or tibia length demonstrated that miR-327
antagomiR administration prevented TAC-induced increases in LV
volume and mass (Fig. 3B and C).
Echocardiography revealed significantly improved EF, LV mass and LV
posterior wall thickness in miR-327 antagomiR-treated mice compared
with those in the control mice (Fig.
3D and E).
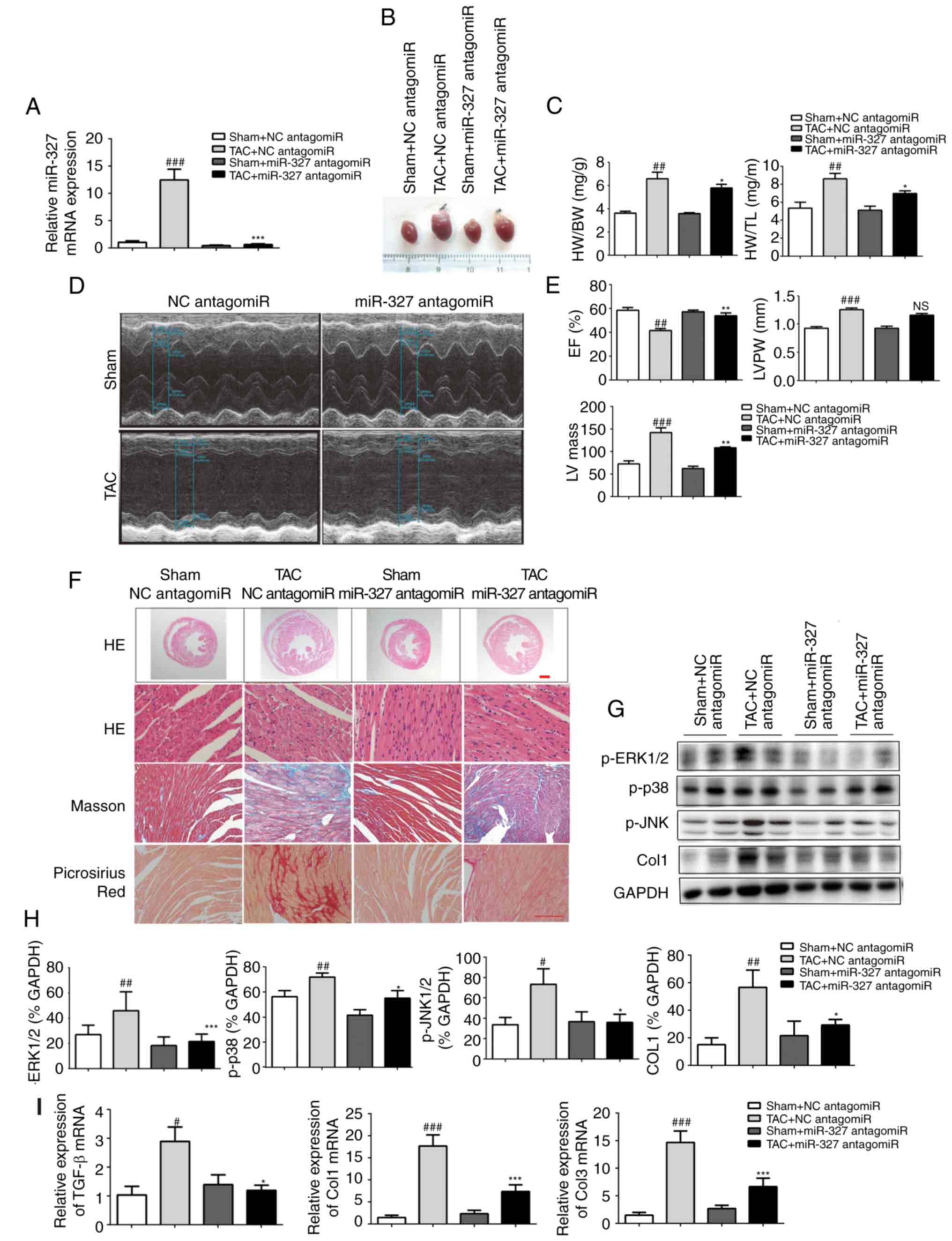 | Figure 3Cardiac hypertrophy is attenuated and
ventricular function is preserved by miR-327 inhibition after TAC
surgery. (A) Decreased miR-327 in hearts of mice treated with
miR-327 antagomiR as determined by RT-qPCR (n=6). (B)
Representative images of isolated mouse hearts (scale in mm). (C)
miR-327 inhibition attenuated the HW/BW and HW/TL ratios (n=6). (D
and E) Echocardiograms indicated that miR-327 inhibition preserved
EF and attenuated LVPW and LV mass (n=6). (F) Histological
examination indicated that miR-327 inhibition reduced cardiac
hypertrophy and fibrosis (n=3; scale bar, 500 µm in the top
panel and 50 µm in the others). (G and H) Western blot
analysis revealed that miR-327 inhibition prevented the
phosphorylation of MAPK proteins (n=3). (I) miR-327 inhibition
caused a downregulation of fibrosis-associated genes in mice (n=6).
##P<0.01, ###P<0.001 vs. respective
Sham + NC antagomiR; *P<0.05, **P<0.01,
***P<0.001 vs. respective TAC + NC antagomiR. ns, no
significance; nc, negative control; col, collagen; ERK,
extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase; miR, microRNA; TGF, transforming growth factor; TAC,
transverse aortic constriction; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; EF, ejection
fraction; HW, heart weight; BW, body weight; TL, tibia length;
LVPW, left ventricular posterior wall; MAPK, mitogen-activated
protein kinase; TGF, transforming growth factor. |
miR-327 antagomiR-treated mice also possessed
decreased cardiac chamber sizes and cardiomyocyte cross-sectional
areas (Fig. 3F). The level of
cardiac fibrosis was then assessed, given that this factor is
critical in the progression of pathological cardiac remodeling.
Masson trichrome and picrosirius red staining were used to
visualize collagen deposition in LV sections. miR-327 antagomiR
treatment attenuated TAC-induced fibrosis (Fig. 3F).
The mitogen-activated protein kinase (MAPK) cascade
is a central signaling cascade known to promote the development of
cardiac hypertrophy (22). To
investigate the antihyper-trophic effects of miR-327 antagomiR, the
protein levels of activated MAPK members, including p-JNK, p-p38,
and p-ERK1/2 were analyzed. Downregulation of miR-327 by antagomiR
prevented phosphorylation of the MAPK proteins, which is typically
caused by TAC (Fig. 3G and H).
Finally, the mRNA levels of known pathological cardiac fibrosis
markers, including TGF-β, Col1a1 and Col3a1, were observed to be
decreased in miR-327 antagomiR-treated mice (Fig. 3I).
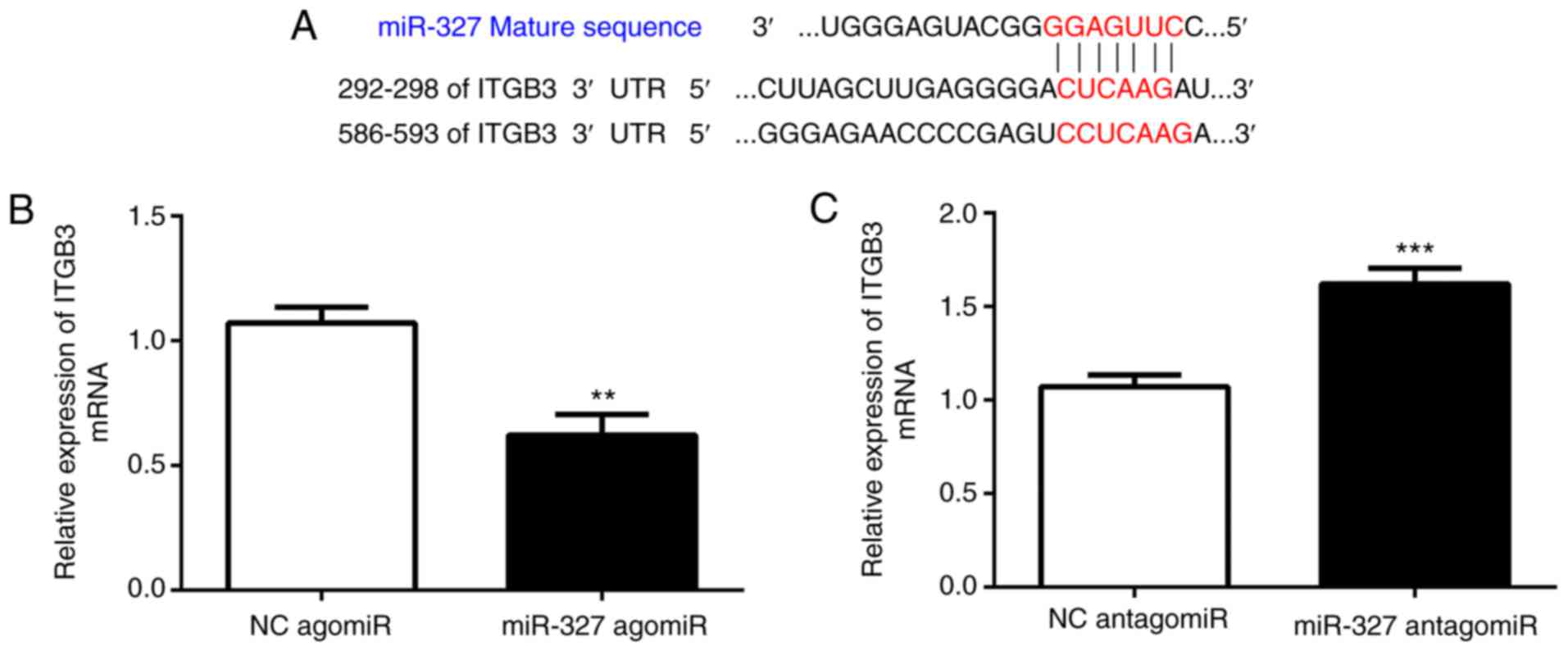
miR-327 targets integrin (ITG)B3,
contributing to its effect on cardiac fibrosis
A bioinformatics analysis with TargetScan
(http://www.targetscan.org) suggested
that ITGB3 is a direct target of miR-327. Cardiac fibroblasts were
transfected with miR-327 agomiR, antagomiR or the scrambled control
to determine whether miR-327 regulates ITGB3. As assessed via
RT-qPCR, ITGB3 expression was increased by miR-327 antagomiR and
decreased by miR-327 agomiR, suggesting the ability of miR-327 to
regulate expression of endogenous ITGB3 in cardiac fibroblasts
(Fig. 4B and C).
Discussion
Myocardial fibrosis is a frequent hallmark of
cardiomyopathy (4). Although its
original cause is known to be part of a physiological process,
sustained stress induces pathological hypertrophy. In addition,
interstitial fibrosis increases the stiffness of the myocardium.
Subsequently, diastolic dysfunction-induced global cardiac
remodeling leads to dilated cardiomyopathy and heart failure
(23,24). Despite the devastating nature of
cardiac fibrosis, effective treatment strategies currently are not
available. Thus, with regard to the treatment of cardiac
dysfunction and heart failure, identification of therapeutic
targets is of importance. Aberrant miRNA expression has been linked
to cardiac fibrosis and heart failure (9,25);
Therefore, miRNAs have emerged as novel therapeutic targets for the
treatment of fibrotic changes (6,26–28).
miRNAs are endogenous, non-coding small RNA
molecules. miRs are recruited to the RNA-induced silencing complex
and regulate target genes through diverse mechanisms (29). The importance of miRNAs in
numerous cellular processes has been well documented. However, the
significance of miRNAs in heart function, particularly with regard
to cardiac fibrosis, has remained elusive. miR-122, miR-145,
miR-134, miR-133a and miR-370 were reported to be associated with
coronary artery disease, even following adjustment for other
cardiovascular risk factors (30–33). Dysregulated miRNAs, particularly
miR-21 and miR-29b, were previously indicated to contribute to
cardiac fibrosis (34,35). Increased expression of miR-21 in
fibroblasts promotes their proliferation, suggesting the important
role of miR-21 in cardiac remodeling (36,37). By miRNA arrays, it was previously
demonstrated that high levels miR-327 are associated with
hypertrophic cardiac tissues (3).
In the present study investigated whether miR-327 serves a role in
the development of pathological cardiac hypertrophy and fibrosis.
Using in vivo and in vitro models, it was
demonstrated that miR-327 antagomiR inhibited cardiac hypertrophy
and fibrosis. Cardiac fibrosis occurs in numerous types of heart
disease, including diabetic cardiomyopathy, MI, dilated
cardiomyopathy, aortic stenosis and hypertrophic cardiomyopathy
(38,39). In cardiac fibrosis, cardiac
fibroblasts proliferate and transform into myofibroblasts (40). The present study demonstrated that
cardiac fibroblast differentiation, which was enhanced by miR-327
overexpression, was attenuated by miR-327 inhibition. In addition,
miR-327 levels were enhanced in cardiac fibroblasts compared with
those in cardiomyocytes. Esposito et al (41) indicated that TAC-induced pressure
overload is associated with the activation of MAPKs (ERK1/2, p38
MAPK and JNK) in mice. Consistent with numerous animal studies,
clinical studies reported that in failing human hypertrophic
hearts, ERK1/2, JNK and p38 MAPK were significantly activated
(42). In line with these
results, MAPK activation has been demonstrated to be a key element
directly enhancing cardiac remodeling (43,44). Through a bioinformatics analysis
performed in the present study, ITGB3 was identified as a target
gene for miR-327 in cardiac fibroblasts. ITGB3, which is an
ITGB-chain subunit encoded by a serotonin-associated gene on
chromosome 17, was reported to be linked with the risk of cancer,
including colorectal cancer and acute myeloid leukemia (45,46). The ITGB3 family includes the
subtypes αIIb β3 and αvβ3. The ITGB3 gene encodes the integrin β3
subunit and has been consistently identified as a quantitative
locus for regulating serum levels of 5-hydroxytryptamin levels
(47). In the present study,
ITGB3 was inhibited by miR-327 antagomiR, suggesting that ITGB3 may
be a target gene of miR-327 inducing differentiation of cardiac
fibroblasts. However, the direct association between ITGB3 and MAPK
and their functional roles in the regulation of cardiac fibrosis
require further confirmation.
In summary, the present study suggested that cardiac
fibrosis may increase miR-327 expression and that miR-327 antagomiR
improves LV mass and attenuates cardiac fibrosis during cardiac
hypertrophy. The protective effect of miR-327 inhibition against
cardiac fibrosis was mediated through inhibition of TAC-induced
increases in ERK1/2, p38 and JNK phosphorylation. ITGB3 may be a
target gene of miR-327 that induces differentiation of cardiac
fibroblasts. Thus, a previously unknown role for miR-327 in cardiac
hypertrophy and fibrosis was identified in the present study, but
this requires to be confirmed in future studies.
Glossary
Abbreviations
Abbreviations:
|
BW
|
body weight
|
|
EF
|
ejection fraction
|
|
FBS
|
fetal bovine serum
|
|
FGF
|
fibroblast growth factor
|
|
HW
|
heart weight
|
|
LV
|
left ventricle
|
|
LVPW
|
left ventricular posterior wall
|
|
MAPK
|
mitogen-activated protein kinase
|
|
NRCF
|
neonatal rat cardiac fibroblasts
|
|
TAC
|
transverse aortic constriction
|
|
TL
|
tibia length
|
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 81627802
and 81570247), the Natural Science Foundation of Jiangsu Province
for Youth (grant no. BK20141024), the Six Talent Peaks project in
Jiangsu Province (grant no. 2015-WSN-29) and the Priority Academic
Program Development of Jiangsu Higher Education Institutions.
Notes
[1] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Huang S, Zou X, Zhu JN, Fu YH, Lin QX,
Liang YY, Deng CY, Kuang SJ, Zhang MZ, Liao YL, et al: Attenuation
of microrna-16 derepresses the cyclins d1, d2 and e1 to provoke
cardiomyocyte hypertrophy. J Cell Mol Med. 19:608–619. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chien KR, Shimizu M, Hoshijima M,
Minamisawa S and Grace AA: Toward molecular strategies for heart
disease-past, present, future. Jpn Circ J. 61:91–118. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang
H, Xu J, Che L, Chen X, Sluijter JP, et al: Crucial role of mir-433
in regulating cardiac fibrosis. Theranostics. 6:2068–2083. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thannickal VJ, Zhou Y, Gaggar A and Duncan
SR: Fibrosis: Ultimate and proximate causes. J Clin Invest.
124:4673–4677. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thum T and Lorenzen JM: Cardiac fibrosis
revisited by microRNA therapeutics. Circulation. 126:800–802. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olson EN: Micrornas as therapeutic targets
and biomarkers of cardiovascular disease. Sci Transl Med.
6:239ps32014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang X, Wu D, Du H, Nie F, Pang X and Xu
Y: MicroRNA-135a is involved in podocyte injury in a transient
receptor potential channel 1-dependent manner. Int J Mol Med.
40:1511–1519. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou B, Li H and Shi J: miR-27 inhibits
the NF-κB signaling pathway by targetingleptin in osteoarthritic
chondrocytes. Int J Mol Med. 40:523–530. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Thum T: Noncoding RNAs and myocardial
fibrosis. Nat Rev Cardiol. 11:655–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tao H, Yang JJ and Shi KH: Non-coding RNAs
as direct and indirect modulators of epigenetic mechanism
regulation of cardiac fibrosis. Expert Opin Ther Targets.
19:707–716. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumarswamy R and Thum T: Non-coding RNAs
in cardiac remodeling and heart failure. Circ Res. 113:676–689.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Espinoza-Lewis RA: Wang DZ. MicroRNAs in
heart development. Curr Top Dev Biol. 100:279–317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi J, Wang L, Lu Y, Ji Y, Wang Y, Dong K,
Kong X and Sun W: Protective effects of seabuckthorn pulp and seed
oils against radiation-induced acute intestinal injury. J Radiat
Res. 58:24–32. 2017. View Article : Google Scholar :
|
|
15
|
Bourajjaj M, Armand AS, da Costa Martins
PA, Weijts B, van der Nagel R, Heeneman S, Wehrens XH and De Windt
LJ: Nfatc2 is a necessary mediator of calcineurin-dependent cardiac
hypertrophy and heart failure. J Biol Chem. 283:22295–22303. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu Y, Zhu X, Li J, Fang R, Wang Z, Zhang
J, Li K, Li X, Bai H, Yang Q, et al: Glycine prevents pressure
overload induced cardiac hypertrophy mediated by glycine receptor.
Biochem Pharmacol. 123:40–51. 2017. View Article : Google Scholar
|
|
17
|
Zaid TM, Yeung TL, Thompson MS, Leung CS,
Harding T, Co NN, Schmandt RS, Kwan SY, Rodriguez-Aguay C,
Lopez-Berestein G, et al: Identification of FGFR4 as a potential
therapeutic target for advanced-stage, high-grade serous ovarian
cancer. Clin Cancer Res. 19:809–820. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng Y, Zhu Y, Zhang J, Duan X and Zhang
Y: Large accumulation of collagen and increased activation of mast
cells in hearts of mice with hyperlipidemia. Arq Bras Cardiol.
109:404–409. 2017.In English, Portuguese. PubMed/NCBI
|
|
19
|
Irelan JT, Wu MJ, Morgan J, Ke N, Xi B,
Wang X, Xu X and Abassi YA: Rapid and quantitative assessment of
cell quality, identity, and functionality for cell-based assays
using real-time cellular analysis. J Biomol Screen. 16:313–322.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xing D and Orsulic S: A genetically
defined mouse ovarian carcinoma model for the molecular
characterization of pathway-targeted therapy and tumor resistance.
Proc Natl Acad Sci USA. 102:6936–6941. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔC T method. Methods. 25:402–408.
2001. View Article : Google Scholar
|
|
22
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maillet M, van Berlo JH and Molkentin JD:
Molecular basis of physiological heart growth: Fundamental concepts
and new players. Nat Rev Mol Cell Biol. 14:38–48. 2013. View Article : Google Scholar
|
|
24
|
Dirkx E, Gladka MM, Philippen LE, Armand
AS, Kinet V, Leptidis S, El Azzouzi H, Salic K, Bourajjaj M, da
Silva GJ, et al: Nfat and mir-25 cooperate to reactivate the
transcription factor hand2 in heart failure. Nat Cell Biol.
15:1282–1293. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Melman YF, Shah R and Das S: Micrornas in
heart failure: Is the picture becoming less mirky. Circ Heart Fail.
7:203–214. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roncarati R, Viviani Anselmi C, Losi MA,
Papa L, Cavarretta E, Da Costa Martins P, Contaldi C, Saccani Jotti
G, Franzone A, Galastri L, et al: Circulating miR-29a, among other
up-regulated microRNAs, is the only biomarker for both hypertrophy
and fibrosis in patients with hypertrophic cardiomyopathy. J Am
Coll Cardiol. 63:920–927. 2014. View Article : Google Scholar
|
|
27
|
Wang J, Liew OW, Richards AM and Chen YT:
Overview of MicroRNAs in cardiac hypertrophy, fibrosis, and
apoptosis. Int J Mol Sci. 17:pii: E7492016. View Article : Google Scholar
|
|
28
|
Wu C, Dong S and Li Y: Effects of
miRNA-455 on cardiac hypertrophy induced by pressure overload. Int
J Mol Med. 35:893–900. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ling H, Fabbri M and Calin GA: Micrornas
and other non-coding RNAs as targets for anticancer drug
development. Nat Rev Drug Discov. 12:847–865. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gacoń J, Kabłak-Ziembicka A, Stępień E,
Enguita FJ, Karch I, Derlaga B, Żmudka K and Przewłocki T:
Decision-making microRNAs (miR-124, -133a/b, -34a and -134) in
patients with occluded target vessel in acute coronary syndrome.
Kardiol Pol. 74:280–288. 2016. View Article : Google Scholar
|
|
31
|
Gao H, Guddeti RR, Matsuzawa Y, Liu LP, Su
LX, Guo D, Nie SP, Du J and Zhang M: Plasma levels of microRNA-145
are associated with severity of coronary artery disease. PLoS One.
10:e01234772015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gao W, He HW, Wang ZM, Zhao H, Lian XQ,
Wang YS, Zhu J, Yan JJ, Zhang DG, Yang ZJ and Wang LS: Plasma
levels of lipometabolism-related miR-122 and mir-370 are increased
in patients with hyperlipidemia and associated with coronary artery
disease. Lipids Health Dis. 11:552012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang F, Long G, Zhao C, Li H, Chaugai S,
Wang Y, Chen C and Wang DW: Plasma microRNA-133a is a new marker
for both acute myocardial infarction and underlying coronary artery
stenosis. J Transl Med. 11:2222013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Thum T, Gross C, Fiedler J, Fischer T,
Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et
al: MicroRNA-21 contributes to myocardial disease by stimulating
map kinase signalling in fibroblasts. Nature. 456:980–984. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang J, Huang W, Xu R, Nie Y, Cao X, Meng
J, Xu X, Hu S and Zheng Z: MicroRNA-24 regulates cardiac fibrosis
after myocardial infarction. J Cell Mol Med. 16:2150–2160. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ye Y, Perez-Polo JR, Qian J and Birnbaum
Y: The role of microRNA in modulating myocardial
ischemia-reperfusion injury. Physiol Genomics. 43:534–542. 2011.
View Article : Google Scholar
|
|
37
|
Dong S, Cheng Y, Yang J, Li J, Liu X, Wang
X, Wang D, Krall TJ, Delphin ES and Zhang C: MicroRNA expression
signature and the role of microRNA-21 in the early phase of acute
myocardial infarction. J Biol Chem. 284:29514–29525. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar
|
|
39
|
Gyöngyösi M, Winkler J, Ramos I, Do QT,
Firat H, McDonald K, González A, Thum T, Diez J, Jaisser F, et al:
Myocardial fibrosis: Biomedical research from bench to bedside. Eur
J Heart Fail. 19:177–191. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Weber KT, Sun Y, Bhattacharya SK, Ahokas
RA and Gerling IC: Myofibroblast-mediated mechanisms of
pathological remodelling of the heart. Nat Rev Cardiol. 10:15–26.
2013. View Article : Google Scholar
|
|
41
|
Esposito G, Perrino C, Schiattarella GG,
Belardo L, di Pietro E, Franzone A, Capretti G, Gargiulo G, Pironti
G, Cannavo A, et al: Induction of mitogen-activated protein kinases
is proportional to the amount of pressure overload. Hypertension.
55:137–143. 2010. View Article : Google Scholar
|
|
42
|
Haq S, Choukroun G, Lim H, Tymitz KM, del
Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T and
Molkentin JD: Differential activation of signal transduction
pathways in human hearts with hypertrophy versus advanced heart
failure. Circulation. 103:670–677. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Peterson KL: Pressure overload hypertrophy
and congestive heart failure. Where is the 'Achilles' heel'. J Am
Coll Cardiol. 39:672–675. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rose BA, Force T and Wang Y:
Mitogen-activated protein kinase signaling in the heart: Angels
versus demons in a heart-breaking tale. Physiol Rev. 90:1507–1546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lei Y, Huang K, Gao C, Lau QC, Pan H, Xie
K, Li J, Liu R, Zhang T, Xie N, et al: Proteomics identification of
ITGB3 as a key regulator in reactive oxygen species-induced
migration and invasion of colorectal cancer cells. Mol Cell
Proteomics. 10:M110 0053972011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Miller PG, Al-Shahrour F, Hartwell KA, Chu
LP, Järås M, Puram RV, Puissant A, Callahan KP, Ashton J, McConkey
ME, et al: In vivo RNAi screening identifies a leukemia-specific
dependence on integrin beta 3 signaling. Cancer Cell. 24:45–58.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mazalouskas M, Jessen T, Varney S,
Sutcliffe JS, Veenstra-VanderWeele J, Cook EH Jr and Carneiro AM:
Integrin β3 haploinsufficiency modulates serotonin transport and
antidepressant-sensitive behavior in mice. Neuropsychopharmacology.
40:2015–2024. 2015. View Article : Google Scholar : PubMed/NCBI
|