Introduction
Atherosclerosis is a complex chronic inflammatory
and metabolic disease in which aberrant inflammatory responses and
dysregulation of lipid metabolism in the arterial walls at
predisposed sites plays an important role from the initiation to
progression and eventually rupture of the atherosclerotic plaque
(1). With the development of the
society, atherosclerosis and its complications considerably cause
increased morbidity and mortality worldwide and account for almost
a third of the deaths in the world (2). Many physiological mechanisms,
including metabolic, genetic, immunologic and environmental factors
have been suggested to be involved in the progression of
atherosclerosis (3). Endothelial
dysfunction plays a pivotal role in these interactions and is the
first step toward atherosclerosis, this dysfunction favors
vasospasm, thrombosis, penetration of macrophages, cellular growth
and the inflammatory reaction leading to atherosclerosis (4).
Visfatin, which was firstly found in visceral
adipose tissue and is also known as pre-B cell colony enhancing
factor (PBEF) and nicotinamide phosphoribosyl-transferase (Nampt),
plays a crucial role in a large number of metabolic and stress
responses (5). Plasma visfatin
level was negatively associated with vascular endothelial function
in patients with type 2 diabetes mellitus (6). With the discovery of the
proinflammatory role of visfatin, its potential effect in
atherosclerosis has gradually attracted much attention. Increased
expression of visfatin was detected in human unstable
atherosclerotic lesions (7,8).
In addition, visfatin has been shown to react with inflammatory
cytokines, such as interleukin-6 (IL-6) and tumor necrosis factor-α
(TNF-α), these cytokines are known to mediate pro-inflammatory and
detrimental effects in the progression of atherosclerosis (9). Moreover, our previously studies
demonstrated that ambient particulate matter inhalation accelerate
atherosclerosis in apolipoprotein E knockout (ApoE−/−)
mice, which is related to visfatin upregulation, as well as the
activation of inflammation, visfatin induced cholesterol
accumulation in macrophages and accelerated atherosclerosis through
modulating the expression of SR-A and CD36 (10,11).
Despite lipid-lowering drugs such as simvastatin and
atorvastatin are used in Clinical treatments, atherosclerosis
remains the leading cause of death in developing countries. During
long-term statin treatment, typical side effects including eczema,
increased creatine phosphokinase, dizziness, fainting and fast or
irregular heartbeat should not be ignored (12). The use of statin is limited by the
relatively frequent occurrence of serious side effects.
With increasing popularity of complementary and
alternative medicine among patients with atherosclerosis,
Traditional Chinese Medicine is becoming more and more frequently
used both in Asian and Western countries. Berberine (BBR) is a
botanical alkaloid mainly extracted from many different medicinal
herbs as Rhizoma coptidis (Huanglian) and Cortex
phellodendri (Huangbai) which are widely used in China and
other East Asian countries. Recently, increasing studies have
suggested that BBR has protective effects in cardiovascular
diseases (CVD). BBR ameliorated atherosclerosis in
hyperhomocysteinemia mice, which was related to the activation of
peroxisome proliferator-activated receptor γ (PPARγ) and subsequent
suppression of oxidative stress in endothelial cells (13). BBR inhibited the expression and
production of inflammatory cytokines IL-6, TNF-α and monocyte
chemoattractant protein 1 (MCP-1) in macrophages stimulated by
acetylated low-density lipoprotein through PPARγ activity (14). Autophagy in macrophages played a
protective role in advanced atherosclerosis, BBR inhibited
inflammation in macrophages by inducing autophagy (15). Furthermore, BBR increased
atherosclerotic plaque stability by reducing matrix
metalloproteinases-9 (MMP-9) and extracellular matrix
metalloproteinase inducer expression (16). The anti-atherogenic property of
BBR also could be linked to its preventive effect on the formation
of foam cells by suppressing cholesterol accumulation in
macrophages (17). Although
beneficial effects of BBR on atherosclerosis have been suggested,
the underlying mechanisms responsible for the amelioration of
atherosclerosis have not been fully elucidated. The effects of BBR
on visfatin expression in the development of atherosclerosis and on
visfatin-induced endothelial dysfunction remain unclear.
On the basis of these findings, we hypothesized that
BBR could prevent atherogenesis by downregulating visfatin
expression and attenuating visfatin-induced endothelial
dysfunction. ApoE−/− mouse is a genetically modified
animal model that is commonly used for spontaneous atherosclerosis
(18). Human umbilical vein
endothelial cells (HUVECs) have been essential to modern vascular
research and are considered the archetypal example of mature
endothelial cells, with a distinct and demonstrable endothelial
phenotype (19). Therefore, we
evaluated the effects of BBR on high fat diet-induced atherogenesis
in ApoE−/− mice as well as on HUVECs and investigated
the mechanisms underlying BBR-mediated modulation of
atherosclerosis.
Materials and methods
Animals and treatments
Fifty male 6-week-old ApoE−/− mice with a
genetic C57BL/6J background and 10 male 6-week-old C57BL/6J mice
were purchased from Vital River Experimental Animal Technology Co.,
Ltd. (Beijing, China) and housed in SPF grade Experimental Animal
House at Southern Medical University (Guangdong, China) in
environmentally controlled conditions (23±2°C, 55±10% relative
humidity, with a 12-h light/dark cycle) with a common 1 week
acclimatization period. All ApoE−/− mice were randomly
divided into five groups (n=10): a model group (Mod), a positive
control group (Sim), three BBR groups (BBR-L, BBR-M and BBR-H) and
were provided with unlimited access to water and Western diet (21%
fat and 0.15% cholesterol) from Medical Experimental Animal Center
of Guangdong Province for consecutive 12 weeks to establish an
animal model of atherosclerosis, while 10 C57BL/6J mice were
provided with a standard mouse chow diet as a control group (Con).
Mice in the BBR-L, BBR-M, BBR-H or Sim groups were treated with BBR
(2.5 mg/kg, purity ≥98%), BBR (5 mg/kg), BBR (10 mg/kg) or
simvastatin (5 mg/kg, purity ≥98%) (both from Sigma, St. Louis, MO,
USA), respectively. All drugs were dissolved in pure water and were
administered by gavage once a day for 12 weeks. Mice in the Mod and
Con groups were treated with the same volume of normal saline. All
animal experiments were approved by the Ethics Committee of
Southern Medical University and were conducted in accordance with
international guidelines.
Biochemical tests of serum
All mice were sacrificed by collecting whole blood
via the abdominal aorta under ether euthanasia on the last day of
the experiment after 12-h fasting. Serum was isolated from blood by
centrifuging and was stored at −80°C until required for analysis.
Serum levels of total cholesterol (TC), triglyceride (TG), high
density lipoprotein-cholesterol (HDL-C) and low density
lipoprotein-cholesterol (LDL-C) were assayed using commercially
available kits (Invitrogen, Waltham, MA, USA). The circulating
levels of serum visfatin, IL-6 and TNF-α were measured by
enzyme-linked immunosorbent assay (ELISA) according to the
manufacturer's protocols of ELISA kits (visfatin; RayBiotech,
Norcross, GA, USA) (IL-6 and TNF-α; eBioscience, San Diego, CA,
USA).
Histologic analysis
The right atrium was incised and the heart was
perfused by phosphate-buffered saline (PBS) (10 mM, pH 7.4) through
the apex of the left ventricle at a constant pressure of 100 mmHg
followed by 4% paraformaldehyde (pH 7.4) after the thoracic cavity
was opened. For each mouse, one part of aorta was used for
histological examination and the other part was used for western
blotting. The aortic root was separated from the aortic arc at the
right subclavical branching point and fixed in 10% zinc-formalin,
embedded in paraffin, sliced into 4 µm-thick sections and
stained with hematoxylin and eosin (H&E). Serial sections were
cut from the proximal 1 mm of the aortic root. Five sections were
collected at 80-µm intervals starting at a 100-µm
distance from the appearance of the aortic valves. The intima of
the aorta thickness was analyzed under a microscope (Olympus,
Tokyo, Japan). Images of the aorta were captured using a digital
camera (Olympus) and analyzed for plaque area quantification using
ImageJ software (National Institutes of Health). For each animal a
mean lesion area was calculated from five sections, reflecting the
cross-section area covered by atherosclerosis.
Immunohistochemistry
After antigen retrieval by boiling in 0.01 M sodium
citrate for 10 min, deparaffinized sections were quenched in 0.3%
hydrogen peroxide for 30 min, followed by incubated in 1% BSA in
PBS for 30 min. Sections were labeled with rabbit anti-human
visfatin antibody (1:200 dilution; Peprotech, Rocky Hill, NJ, USA)
at 37°C for 45 min and then overnight at 4°C. After washing, the
bound antibodies were conjugated with secondary antibodies at 37°C
for 1 h, and then the DAB substrate was administered and incubated
for 1 min. The sections were counterstained with hematoxylin and
the result was acquired with Image-Pro Plus 5.0 analysis software
(Media Cybernetics, Rockville, MD, USA).
Western blot analysis
The mouse aortas were homogenized in lysis buffer
containing 1% NP-40, 50 mM Tris (pH 7.4), 150 mM NaCl and 1 mM PMSF
for 30 min on ice. After centrifugation at 14,000 rpm for 20 min at
4°C, the protein concentrations were quantified by BCA protein
assay kit (Invitrogen). For immunoblots, 50 µg of protein
per sample was separated by 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
transferred to polyvinylidene difluoride (PVDF) membrane
(Millipore, Billerica, MA, USA). Membranes were blocked with 5%
skim milk in PBS with 0.1% Tween-20 (PBST) for 1 h at 37°C. Rabbit
anti-human visfatin antibody (1:1,000 dilution; Peprotech), p38
mitogen-activated protein kinase (p38 MAPK), phospho-p38 MAPK
(p-p38 MAPK), c-Jun N-terminal kinase (JNK) and phospho-JNK (p-JNK)
antibodies (1:1,000 dilution; Cell Signaling Technology, Danvers,
MA, USA) in PBST were incubated with membranes overnight at 4°C.
The membranes were washed thoroughly for 60 min with PBST before
incubation with IgG-horseradish peroxidase-conjugated secondary
antibody (1:2,000 dilution) for 1 h. Proteins were visualized with
ECL Plus (GE Healthcare, Uppsala, Sweden) on Kodak 2000MM.
Densitometric analysis was conducted by PDI ImageWare system
(Bio-Rad, Hercules, CA, USA).
Cell culture and treatments
HUVECs (Cascade Biologics, Portland, OR, USA) were
cultured in endothelial cells basal medium supplemented with 10%
fetal bovine serum (FBS; Invitrogen), penicillin (100 U/ml) and
streptomycin (100 µg/ml) at 37°C in a humidified atmosphere
containing 5% CO2 and grown to 70–80% confluence before
being treated with the indicated agents. Cells between passages 3
and 7 were used in all experiments. To further elucidate the
protective effect and the potential mechanism of BBR on
visfatin-induced HUVECs injury, HUVECs were pretreated with BBR (50
µmol/l; Sigma), p38 MAPK inhibitor SB203580 (20
µmol/l) or JNK inhibitor SP600125 (10 µmol/l) (both
from Tocris Bioscience, Ellisville, MO, USA) for 1 h and followed
by the addition of human recombinant visfatin (100 ng/ml;
Peprotech) for 24 h.
Cell viability assay
The methylthiazolyl tetrazolium (MTT) assay was
performed to investigate the cell viability according to the
manufacturer's instructions. HUVECs were plated in 96-well plates
at the density of 10,000 cells/well. Then, 20 µl MTT (5
mg/ml; Sigma) was added into cultured medium in each well for 4 h
incubation. The blue formazan crystals of viable cells were
solubilized with dimethylsulfoxide (DMSO). Absorbance was measured
at 570 nm using a microplate reader (Thermo Fisher Scientific,
Inc., Waltham, MA, USA).
Flow cytometry
HUVECs were seeded in 6-well plates after cells
reached 80% confluence. Apoptotic cells were evaluated by using an
Annexin V-FITC apoptosis detection kit (BD Biosciences, Franklin
Lakes, NJ, USA) according to the manufacturer's instructions. The
mean intensity of untreated HUVECs was considered as 100%. Changes
in the HUVECs following treatments were determined and standardized
against the untreated HUVECs.
ELISA
Supernatants of HUVECs were collected, the contents
of IL-6 and TNF-α in the cell supernatants were quantified by ELISA
according to the manufacturer's instructions as described
above.
Western blot analysis
Cultured cells were lysed in a lysis buffer, the
protein levels of p-JNK, JNK, p-p38 MAPK, p38 MAPK, Bax, Bcl-2 and
β-actin (Cell Signaling Technology) in HUVECs were subjected to
western blot analysis as mentioned above.
Statistical analysis
Data were expressed as means ± SEM from at least
three independent experiments. Statistical analyses were made
between two groups with the t-test and between multiple groups by
one-way ANOVA and a P-value <0.05 was regarded as statistically
significant.
Results
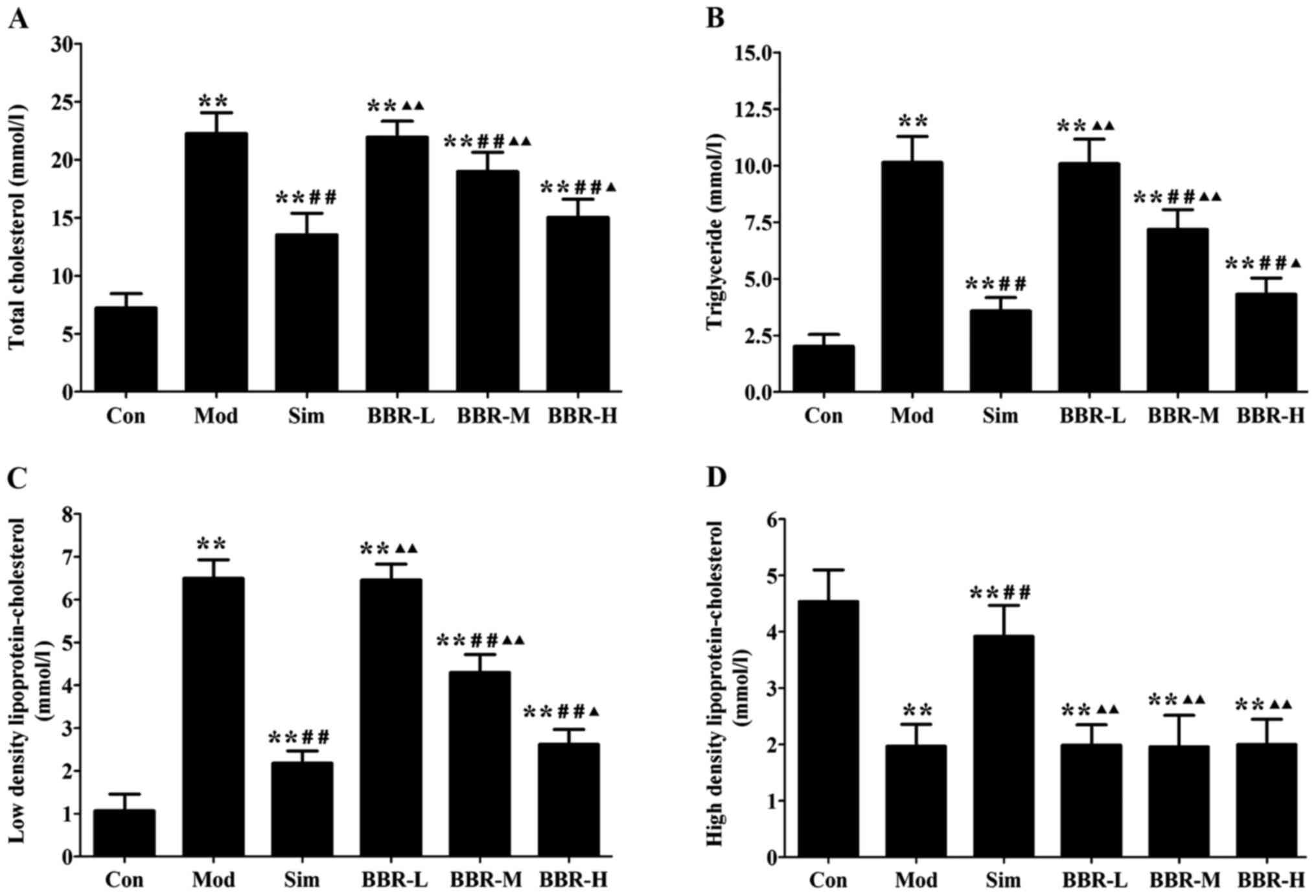
BBR decreased serum lipid profiles
Serum levels of TC, TG and LDL-C in the Mod group
were significantly increased compared with the Con group,
indicating occurrence of hyperlipidemia. After treatments of
simvastatin or BBR, serum lipid profiles were remarkably improved.
TC, TG and LDL-C levels in the Sim, BBR-M and BBR-H groups were
significant reduced compared to the Mod group, while BBR
supplementation did not affect HDL-C levels compared to the Mod
group (Fig. 1).
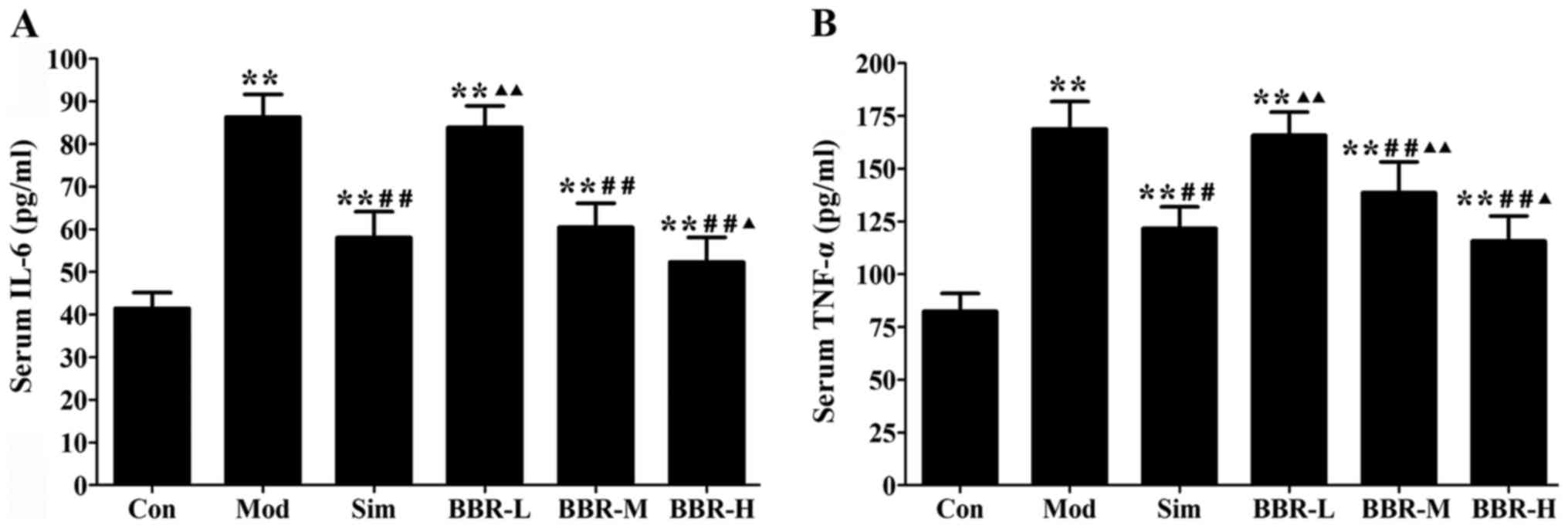
BBR decreased serum levels of
inflammatory cytokines
Western diet fed in the Mod group had remarkably
higher serum IL-6 and TNF-α levels than those of the Con group,
whereas administration with simvastatin significantly decreased the
alterations compared with the Mod group. The BBR-M and BBR-H group
showed a similar trend of simvastatin effect on serum levels of
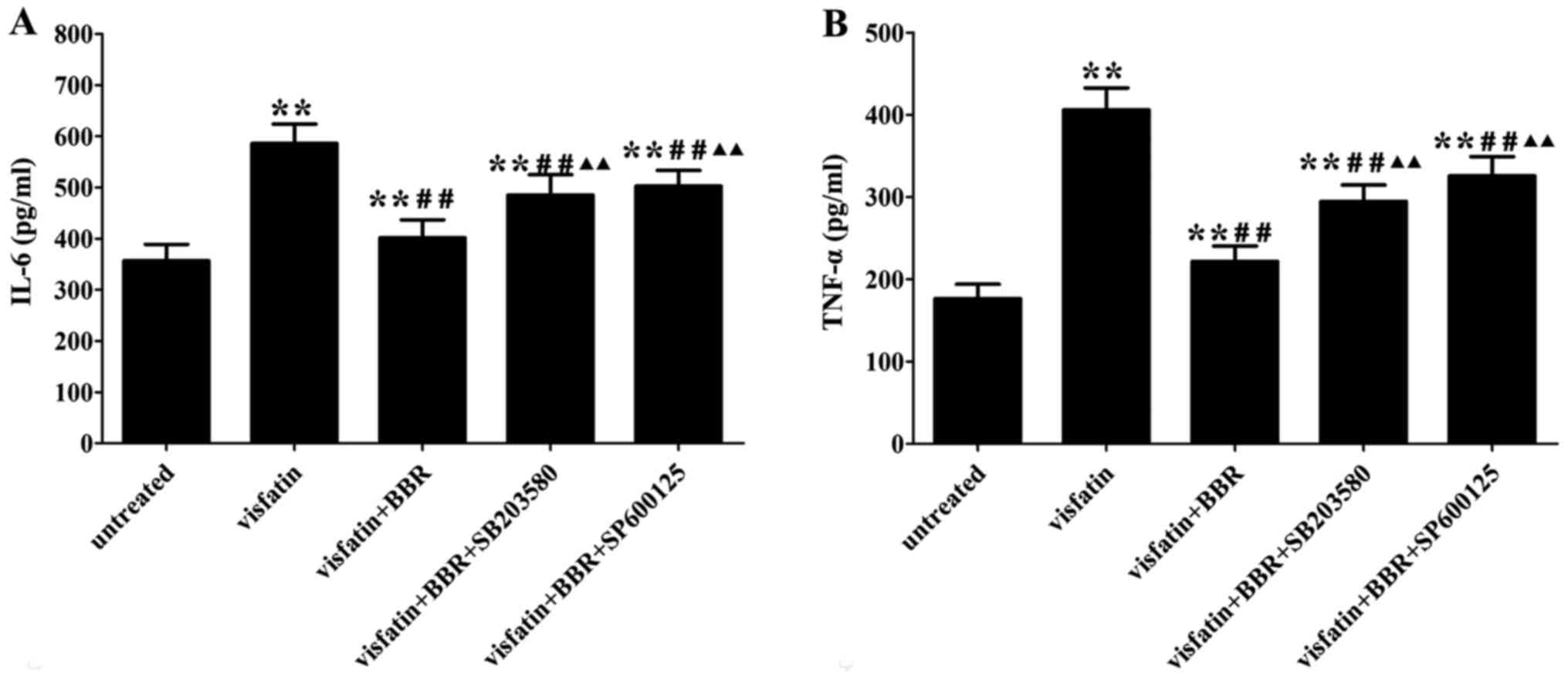
IL-6 and TNF-α (Fig. 2).
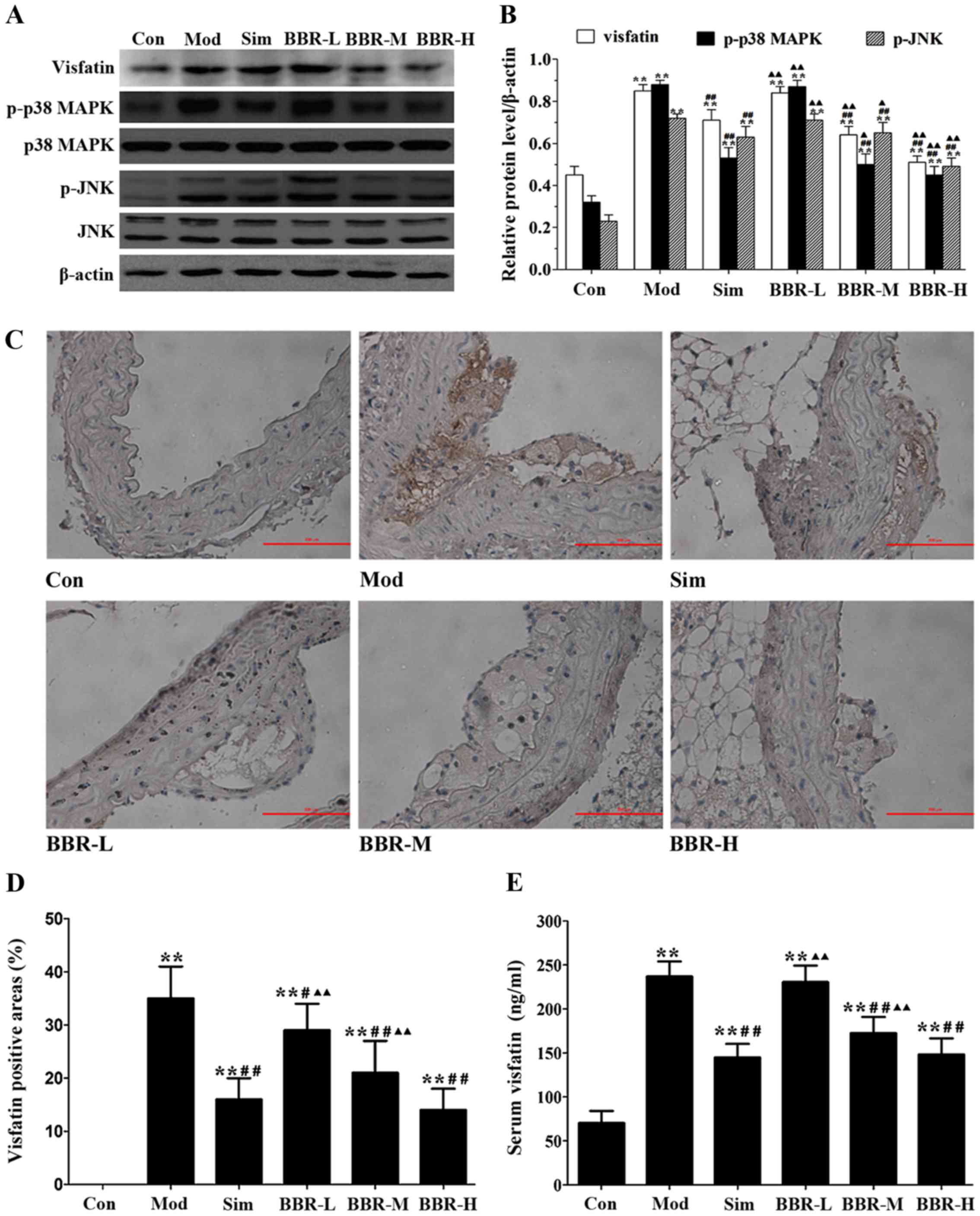
BBR downregulated the expression of
visfatin in ApoE−/− mice
The protein expression of visfatin, p-p38 MAPK and
p-JNK in the aortas of the Mod group were much higher than those of
the Con group, but were much lower than those of the Sim, BBR-M and
BBR-H groups (Fig. 3A and B).
Immunohistochemical staining results showed that significantly more
visfatin was detected in the Mod group compared with the Con group,
and less visfatin was detected in the Sim, BBR-M and BBR-H groups
compared with the Mod group (Fig. 3C
and D). Serum visfatin level in the Mod group was significantly
higher than that in the Con group, but was significantly lower than
that in the Sim, BBR-M and BBR-H groups (Fig. 3E). These results indicated that
BBR supplementation in a dose-dependent manner suppressed visfatin
expression in the progression of atherosclerosis.
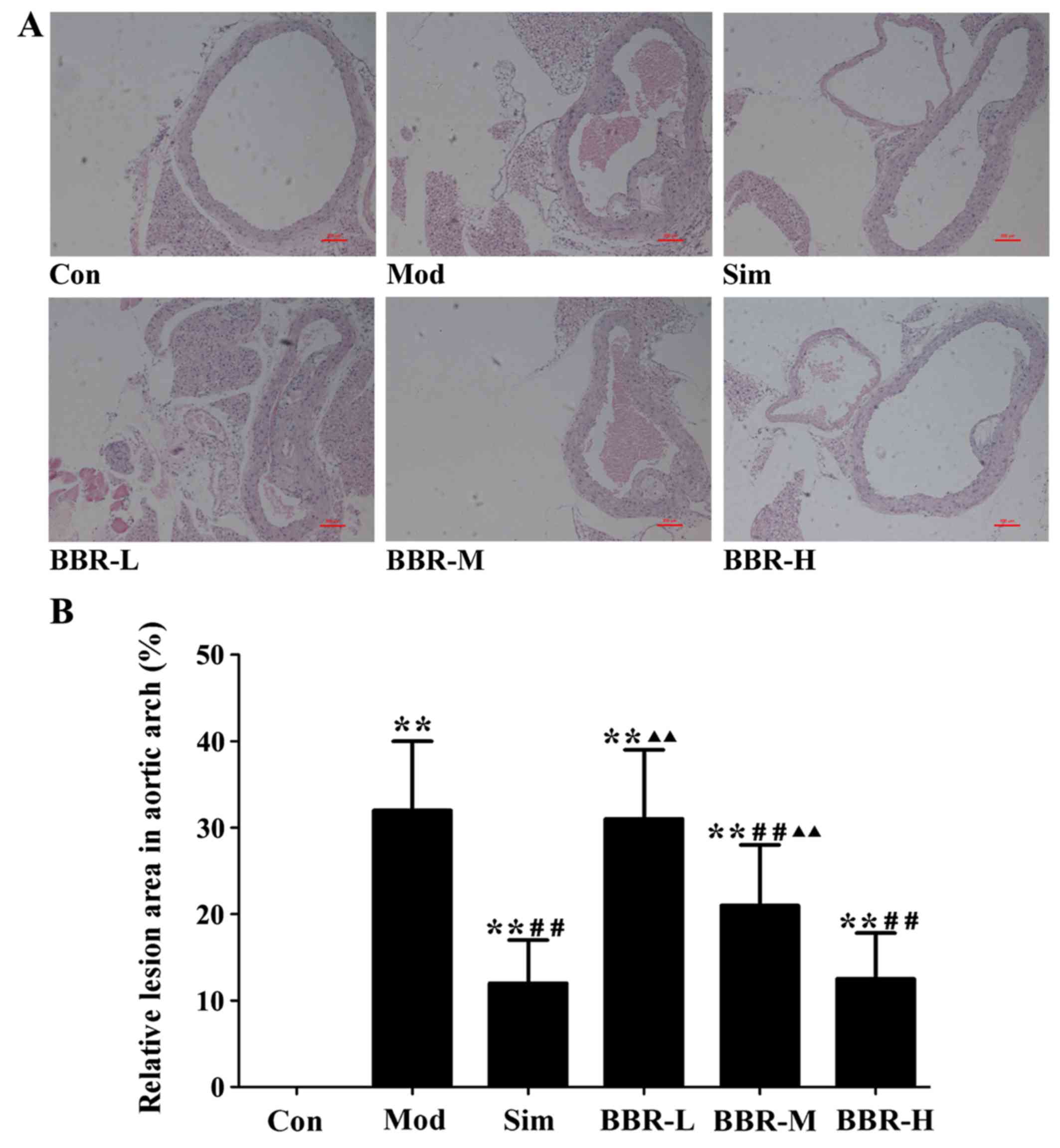
BBR suppressed the formation of
atherosclerotic lesions
The analysis of representative images from the
sections after staining with H&E (Fig. 4) showed that atherosclerotic
lesions in the Mod group were markedly larger than those of the Con
group, plaques were predominantly observed in the medial and
intimal areas of the arterial wall but less so in the adventitial
layers, whereas small, sparse plaques were observed in the Sim
group. Supplementing with BBR in the BBR-M and BBR-H groups reduced
lesion development significantly in spite of high fat diet intake
compared to the Mod group. The percentages of aortic surface area
occupied by the atherosclerotic lesions were significantly reduced
in the BBR-M and BBR-H groups compared with the Mod group. Mice fed
with the low concentration BBR showed no suppressive effect.
Cell viability following visfatin or BBR
treatment
In order to evaluate the HUVECs injury induced by
visfatin, HUVECs were stimulated with different concentrations of
human recombinant visfatin (0, 25, 50, 100, 150 and 200 ng/ml) for
24 h and HUVECs were also stimulated with 100 ng/ml visfatin for 0,
3, 6, 12, 24 and 48 h. Fig. 5A
demonstrated that a concentration of 100 ng/ml visfatin lead to
significant reduction in HUVECs viability. Fig. 5B indicated that the treatment with
100 ng/ml visfatin for 24 h lead to significant reduction in HUVECs
viability. Therefore, a treatment with 100 ng/ml visfatin for 24 h
was considered in subsequent experiments.
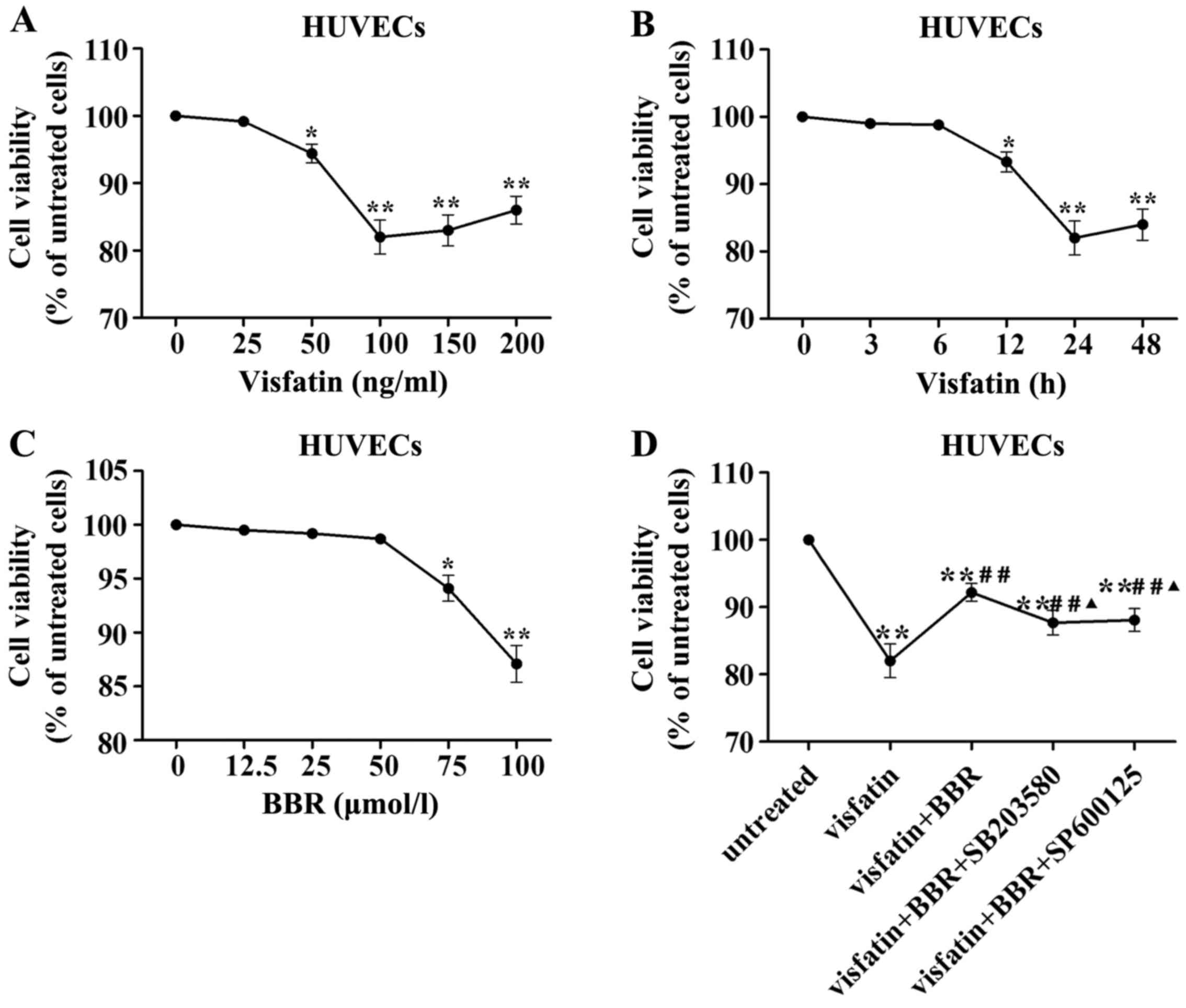 | Figure 5Effects of visfatin and berberine on
human umbilical vein endothelial cell (HUVEC) viability. (A) HUVECs
were stimulated with different concentrations of visfatin (0, 25,
50, 100, 150 and 200 ng/ml) for 24 h. (B) HUVECs were stimulated
with 100 ng/ml visfatin for 0, 3, 6, 12, 24 and 48 h. (C) HUVECs
were stimulated with berberine at 0, 12.5, 25, 50, 75 and 100
µmol/l for 24 h. (D) HUVECs were pretreated with 50
µmol/l berberine, 20 µmol/l SB203580 or 10
µmol/l SP600125 for 1 h and followed by the addition of 100
ng/ml visfatin for 24 h. *P<0.05 and
**P<0.01 vs. untreated cells; #P<0.05
and ##P<0.01 vs. visfatin group;
▲P<0.05 and ▲▲P<0.01 vs. visfatin +
berberine group. |
To exclude the possible effect of BBR on viability
of HUVECs, we evaluated the viability of HUVECs treated with BBR at
0, 12.5, 25, 50, 75 and 100 µmol/l for 24 h by MTT assay.
There was no significant difference in the viability between the
BBR treatment at 0–50 µmol/l for 24 h (Fig. 5C). Thus, BBR pretreatment at
concentration of 50 µmol/l was used in subsequent
experiments.
Visfatin treatment significantly reduced HUVEC
viability compared to the untreated cells, which was reversed by
BBR administration. This effect of BBR was diminished by SB203580
and SP600125 compared to the BBR-treated cells (Fig. 5D).
BBR inhibits the inflammatory response in
visfatin-treated HUVECs
Visfatin treatment significantly increased the
contents of IL-6 and TNF-α in the cell supernatants compared to the
untreated cells, which was reversed by BBR administration. The
anti-inflammatory property of BBR was markedly diminished by
SB203580 and SP600125 compared to the BBR-treated cells (Fig. 6).
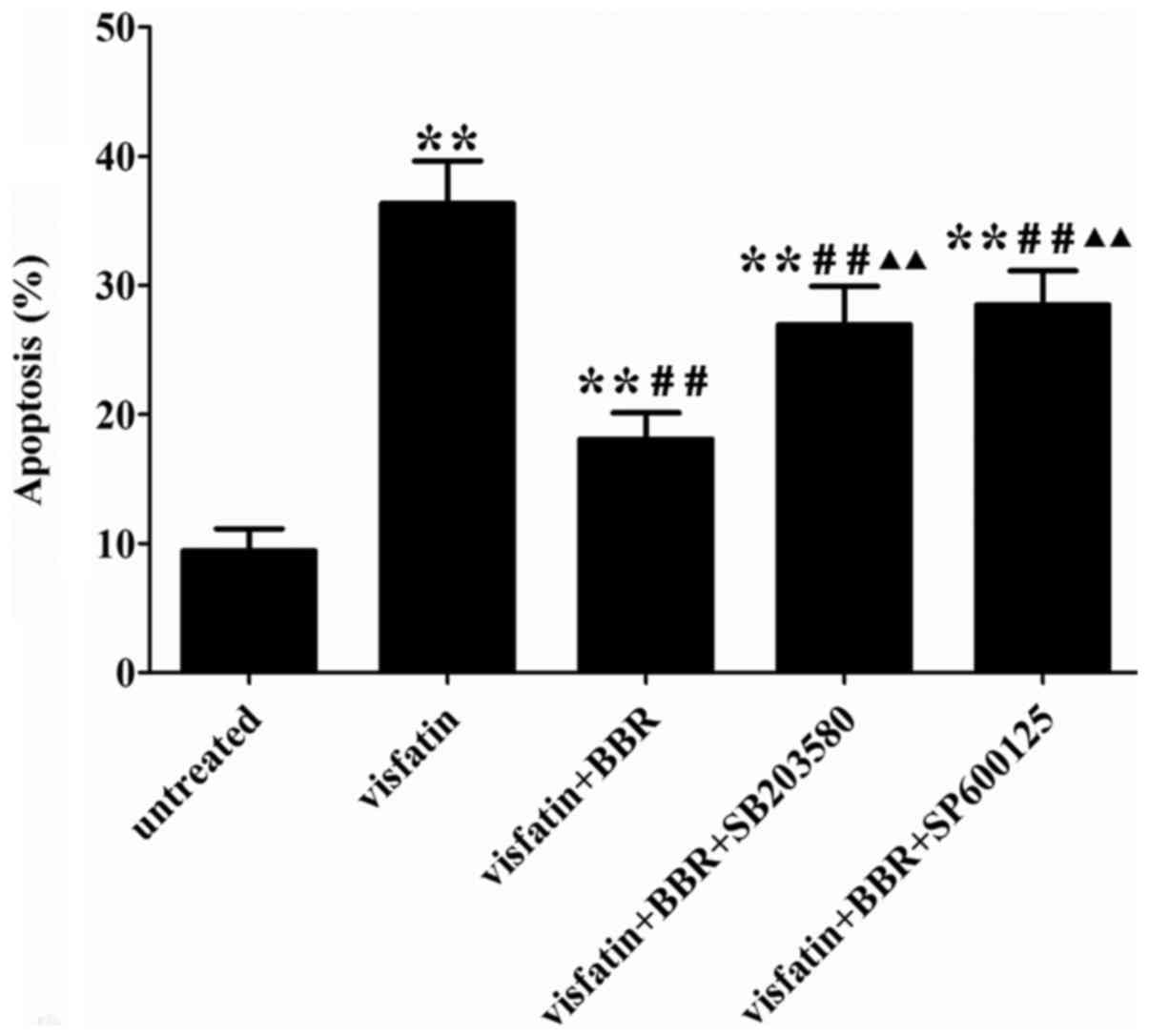
BBR suppressed visfatin-induced apoptosis
in HUVECs
Visfatin treatment significantly increased HUVECs
apoptosis compared to the untreated cells, which was reversed by
BBR administration. The anti-apoptotic effect of BBR was markedly
diminished by SB203580 and SP600125 compared to the BBR-treated
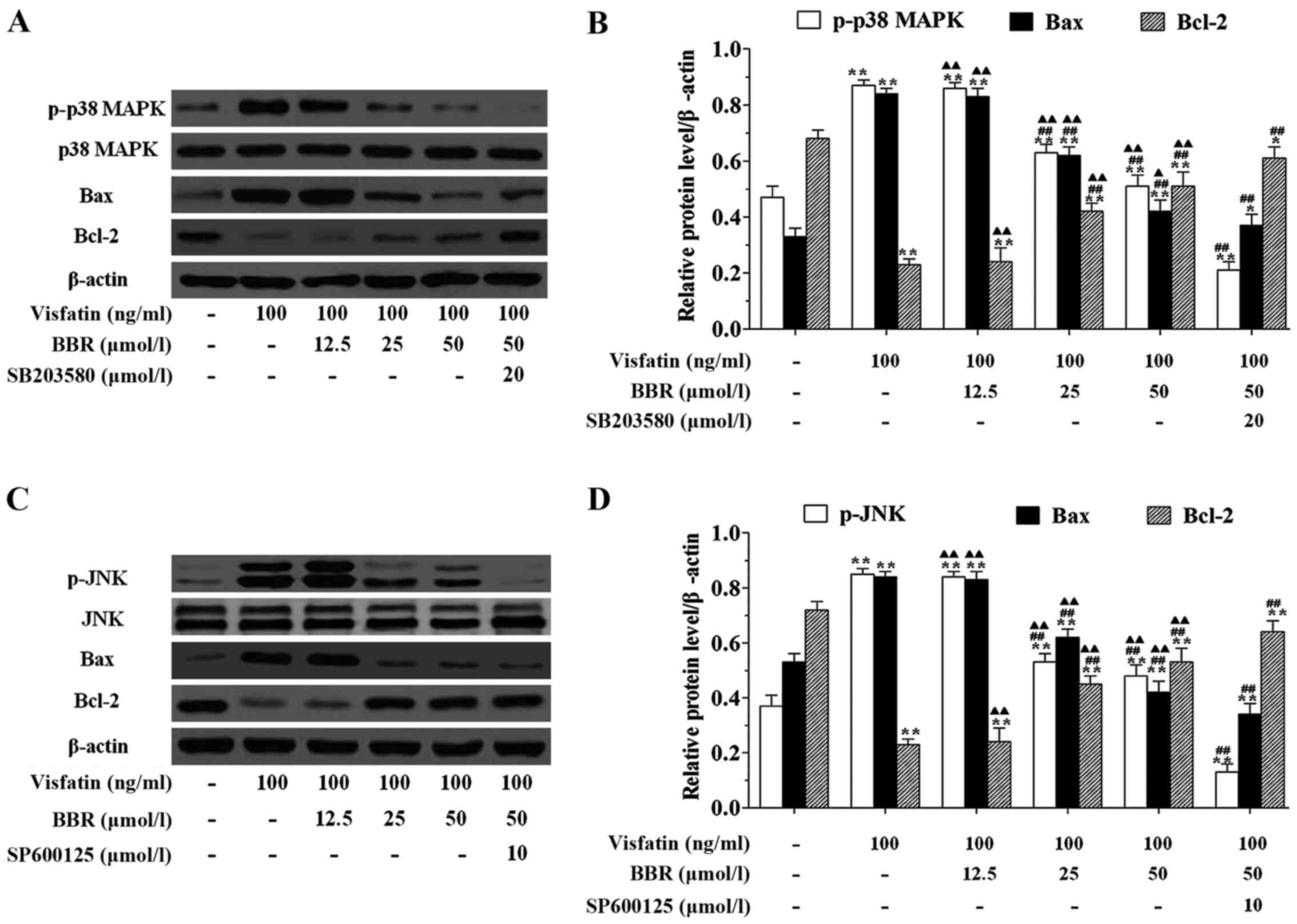
cells (Fig. 7). In order to
explore the mechanism for the anti-apoptotic effect of BBR on
HUVECs, we examined the effects of BBR treatment on the expression
of the pro-apoptotic protein Bax and the anti-apoptotic protein
Bcl-2. Fig. 8 showed that
visfatin markedly upregulated the protein expression of p-p38 MAPK,
p-JNK and Bax, but downregulated the protein expression of Bcl-2 in
HUVECs compared to the untreated cells. Twenty-five and 50
µmol/l BBR pretreatments significantly downregulated the
protein expression of p-p38 MAPK, p-JNK and Bax, but upregulated
the protein expression of Bcl-2 in HUVECs compared to the
visfatin-treated cells, however, the effects of BBR on regulating
the expression of this protein were significantly diminished by
SB203580 and SP600125 pretreatments.
Discussion
ApoE−/− mice are unable to produce the
key glycoprotein ApoE essential for transport and metabolism of
lipids. Fed with normal chow, ApoE−/− mice start to
develop atherosclerosis at the age of 1 to 2 months (20). Therefore, we chose 6-week-old
ApoE−/− mice on a high-fat diet for 12 weeks in this
study to examine the development of atherosclerosis at initial
stages. As expected, the sizes of atherosclerotic lesions within
aorta in the Mod group were remarkably increased as compared with
the Con group.
Adipocytes produce a variety of bioactive peptides,
termed adipokines, which play a major role in whole body glucose
and lipid metabolism, as well as in the pathogenesis of
atherosclerosis. Visfatin, a 52 kDa pro-inflammatory and
potentially insulin-mimetic adipokine, is expressed by the
macrophages infiltrating adipose tissue and is produced in response
to inflammatory signals (21).
Recently visfatin has emerged as one of the most reliable and
attractive biomarkers for the diagnosis and risk stratification of
patients suffering from CVD. Visfatin was regarded as an
independent risk factor for greater carotid intima-media thickness
(IMT), a positive association between visfatin and metabolic
syndrome was noted, mainly among individuals with carotid
atherosclerosis (22). A clinical
study indicated that serum visfatin is increased in hemodialysis
patients with and without diabetes, this association with IMT may
be involved in the pathogenesis of atherosclerosis in chronic renal
failure patients (23). FK866, as
a visfatin antagonist, significantly decreased the visfatin-induced
expression of inflammatory mediators including IL-6 and IL-8 via
the upregulation of nuclear factor-κB (NF-κB) activation in human
coronary artery endothelial cells, which may contribute to a
potential therapy for atherosclerosis (24). In this study, serum visfatin
level, visfatin protein in the aorta and the distribution of
visfatin in the athero-sclerotic plaque in the Mod group were much
higher than those in the Con group, these results indicated that
visfatin may play a significant role in the pathogenesis of
atherosclerosis. This is in agreement with another study suggesting
that visfatin significantly destabilized atherosclerotic plaques in
ApoE−/− mice (25).
The expression of pro-inflammatory cytokines were regulated by a
variety of intracellular signaling pathways, including MAPKs. In
previous findings, p38 MAPK and JNK signaling pathways have been
shown to be potentially important mediators in promoting
atherosclerosis. In vivo experiments showed that IL-4
intervention attenuated ox-LDL-induced atherosclerotic lesions in
ApoE−/− mice via inhibition of JNK signaling pathway
(26). Notably, irisin
significantly reduced the severity of aortic atherosclerosis by
blocking the activation of p-p38 MAPK in ApoE−/− mice
fed on a high-cholesterol diet (27). In addition, in vitro
experiments indicated that norepinephrine enhanced
lipopolysaccharide-induced MMP-9 expression as well as MMP-9
activity in human THP-1 cells by promoting the activation of p-JNK
(28). Furthermore, BBR protected
against lipopolysaccharide-induced apoptosis by suppressing
JNK-mediated signaling (29). Our
results showed that BBR intervention markedly decreased
visfatin-induced expression of p-p38 MAPK and p-JNK, suggesting
that BBR may protect against atherosclerosis via inhibition of p38
MAPK and JNK signaling pathways.
Atherosclerosis is a chronic inflammatory disease,
activation of pro-inflammatory cytokines play a central role in the
etiology of atherosclerosis by increasing monocyte adhesion, smooth
muscle cell proliferation, endothelial dysfunction, oxidative
stress, and vascular calcification. Elevated circulating levels of
IL-6 and TNF-α were observed in atherosclerosis patients. IL-6
induces oxidative stress and endothelial dysfunction by
overexpression of the angiotensin II type 1 (AT1) receptor in the
atherosclerotic process (30).
TNF-α may play an atherogenic role by upregulating the expression
of MCP-1, vascular cell adhesion molecule-1 (VCAM-1) and
intracellular cell adhesion molecule-1 (ICAM-1) in the vascular
wall, and by inducing oxidized LDL (Ox-LDL) uptake and scavenger
receptor class A (SR-A) expression in macrophages (31). In the present study, decreased
atherosclerotic plaque area, lower serum levels of visfatin, IL-6
and TNF-α, lower expression of visfatin protein in the aorta and
lower distribution of visfatin in the atherosclerotic plaque were
detected in the BBR administered ApoE−/− mice, moreover,
decreased contents of IL-6 and TNF-α were measured in the
supernatants of BBR pretreated HUVECs compared to the
visfatin-treated cells, such interactions may help to explain the
anti-inflammatory properties of BBR in the formation of
atherosclerosis.
Endothelial dysfunction is the initial step in the
progression of atherosclerosis (4). Increasing evidence clearly indicates
that the endothelium may play a vital role in the regulation of
vascular inflammation, the key initiating step of the earliest
stage of atherosclerosis is sub-endothelial accumulation of
cholesterol and monocyte-derived macrophages, leading to chronic
inflammation (32). Apoptosis is
a form of cell death which may occur in response to a wide range of
stimuli such as inflammatory cytokines, bacterial toxins and
chemotherapeutic drugs (33).
Endothelial dysfunction induced by endothelial cell apoptosis plays
an essential role in contributing to the pathogenesis of
atherosclerosis. Vascular endothelial cell apoptosis may result in
increased permeability of the endothelial monolayer through loss of
endothelial cells. This loss of integrity could facilitate the
migration and deposition of lipids, monocytes and smooth muscle
cells into the intima, further damaging the vasculature,
propagating atherosclerotic plaque erosion and enhancing thrombus
formation (34). The Bcl-2
protein family and related cytoplasmic proteins are key regulators
of apoptosis. Bcl-2 is an anti-apoptotic protein, its survival
function is opposed by close relatives such as Bax. As a
pro-apoptotic protein, Bax is a mutant of Bcl-2 at the BH1 or BH2
domain, with the property of abrogating the death suppressor
activity of Bcl-2 (35). In our
current study, visfatin treatment significantly increased HUVECs
apoptosis and markedly upregulated the protein expression of Bax,
but downregulated the protein expression of Bcl-2 in HUVECs
compared to the control group, however, the pro-apoptotic effect of
visfatin was reversed by BBR administration in HUVECs. Results also
showed that BBR pretreatment significantly downregulated the
protein expression of p-p38 MAPK, p-JNK and Bax, but upregulated
the protein expression of Bcl-2 in HUVECs compared to the
visfatin-treated cells, however, which were significantly
diminished by SB203580 and SP600125 pretreatments. Taken together,
these results indicated that BBR suppressed visfatin-induced HUVECs
apoptosis via the inhibition of p38 MAPK and JNK signaling
pathways.
Hyperlipidemia, as a result of accumulation of
lipids in blood has come to issue in therapy for the
atherosclerosis. Oxidized phospholipids contribute to inflammation
within the artery wall, initiating atherogenic chemokine expression
that leads to monocyte adhesion (36). Therefore, lipids can be regarded
as triggers of the inflammatory process in atherosclerosis.
Clinical and epidemiologic observations have consistently
documented that LDL-C concentration is positively correlated with
atherosclerosis (37). In this
study, we found that simvastatin or BBR treatment partly recovered
high serum lipid profile induced by Western diet and their
anti-hyperlipidemia effects were comparable.
In conclusion, the findings of our study indicate
that BBR significantly ameliorates the incidence of Western
diet-induced atherosclerosis in ApoE−/− mice, the
protective effect of BBR likely resulted from the reduced
inflammatory response, lowered serum lipid profiles, and attenuated
visfatin-induced endothelial dysfunction. The mechanisms underlying
these therapeutic effects involved inhibition of p38 MAPK and JNK
signaling pathways. Thus, our study presented that BBR could be
used for the protection of atherosclerosis. Further investigation
is required to focus on the emerging issues from this study.
Abbreviations:
|
BBR
|
berberine
|
|
IL-6
|
interleukin-6
|
|
TNF-α
|
tumor necrosis factor-α
|
|
MMPs
|
matrix metalloproteinase
|
|
HDL-C
|
high density
lipoprotein-cholesterol
|
|
LDL-C
|
low density
lipoprotein-cholesterol
|
|
TC
|
total cholesterol
|
|
TG
|
triglyceride
|
|
NF-κB
|
nuclear factor-κB
|
|
ApoE−/−
|
apolipoprotein E knockout
|
|
HUVECs
|
human umbilical vein endothelial
cells
|
|
CVD
|
cardiovascular diseases
|
|
MCP-1
|
monocyte chemoattractant protein 1
|
|
H&E
|
hematoxylin and eosin
|
|
IMT
|
intima-media thickness
|
|
p38 MAPK
|
p38 mitogen-activated protein
kinase
|
|
JNK
|
c-Jun N-terminal kinase
|
Acknowledgments
Not applicable.
Notes
[1]
Funding
The present study was supported by grants from the
National Nature Science Foundation of China (no. 81660770), the
Natural Science Foundation of Jiangxi Province (no.
20161BAB215256), the Science and Technology Planning Project of
Guangdong Province (no. 2016A020226023) and the China Postdoctoral
Science Foundation (no. 2016M592476).
[2] Availability
of data and material
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
QW performed the histologic analysis of aorta,
biochemical tests of serum, and was a major contributor in writing
the manuscript. ZL analyzed the immunohistochemistry and ELISA. YY
performed the western blot analysis. XC performed the cell
viability assay and flow cytometry. All authors read and approved
the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Ross R: Atherosclerosis - an inflammatory
disease. N Engl J Med. 340:115–126. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee GY, Kim JH, Oh GT, Lee BH, Kwon IC and
Kim IS: Molecular targeting of atherosclerotic plaques by a
stabilin-2-specific peptide ligand. J Control Release. 155:211–217.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lu H and Daugherty A: Atherosclerosis.
Arterioscler Thromb Vasc Biol. 35:485–491. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vanhoutte PM: Endothelial dysfunction: The
first step toward coronary arteriosclerosis. Circ J. 73:595–601.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fukuhara A, Matsuda M, Nishizawa M, Segawa
K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T,
Murakami H, et al: Visfatin: A protein secreted by visceral fat
that mimics the effects of insulin. Science. 307:426–430. 2005.
View Article : Google Scholar
|
|
6
|
Takebayashi K, Suetsugu M, Wakabayashi S,
Aso Y and Inukai T: Association between plasma visfatin and
vascular endothelial function in patients with type 2 diabetes
mellitus. Metabolism. 56:451–458. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dahl TB, Yndestad A, Skjelland M, Øie E,
Dahl A, Michelsen A, Damås JK, Tunheim SH, Ueland T, Smith C, et
al: Increased expression of visfatin in macrophages of human
unstable carotid and coronary atherosclerosis: Possible role in
inflammation and plaque destabilization. Circulation. 115:972–980.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Auguet T, Aragonès G, Guiu-Jurado E,
Berlanga A, Curriu M, Martinez S, Alibalic A, Aguilar C, Camara ML,
Hernández E, et al: Adipo/cytokines in atherosclerotic secretomes:
Increased visfatin levels in unstable carotid plaque. BMC
Cardiovasc Disord. 16:1492016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Moschen AR, Kaser A, Enrich B, Mosheimer
B, Theurl M, Niederegger H and Tilg H: Visfatin, an adipocytokine
with proinflammatory and immunomodulating properties. J Immunol.
178:1748–1758. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wan Q, Cui X, Shao J, Zhou F, Jia Y, Sun
X, Zhao X, Chen Y, Diao J and Zhang L: Beijing ambient particle
exposure accelerates atherosclerosis in ApoE knockout mice by
upregulating visfatin expression. Cell Stress Chaperones.
19:715–724. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou F, Pan Y, Huang Z, Jia Y, Zhao X,
Chen Y, Diao J, Wan Q and Cui X: Visfatin induces cholesterol
accumulation in macrophages through up-regulation of scavenger
receptor-A and CD36. Cell Stress Chaperones. 18:643–652. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin CF, Chang YH, Liu JC, Chuang MT and
Chien LN: Statin use associated with a reduced risk of pneumonia
requiring hospitalization in patients with myocardial infarction: A
nested case-control study. BMC Cardiovasc Disord. 16:242016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li H, He C, Wang J, Li X, Yang Z, Sun X,
Fang L and Liu N: Berberine activates peroxisome
proliferator-activated receptor gamma to increase atherosclerotic
plaque stability in Apoe(−/−) mice with hyperhomocysteinemia. J
Diabetes Investig. 7:824–832. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen FL, Yang ZH, Liu Y, Li LX, Liang WC,
Wang XC, Zhou WB, Yang YH and Hu RM: Berberine inhibits the
expression of TNFalpha, MCP-1, and IL-6 in AcLDL-stimulated
macrophages through PPARgamma pathway. Endocrine. 33:331–337. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fan X, Wang J, Hou J, Lin C, Bensoussan A,
Chang D, Liu J and Wang B: Berberine alleviates ox-LDL induced
inflammatory factors by up-regulation of autophagy via AMPK/mTOR
signaling pathway. J Transl Med. 13:922015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang Z, Wang L, Meng S, Wang Y, Chen T
and Wang C: Berberine reduces both MMP-9 and EMMPRIN expression
through prevention of p38 pathway activation in PMA-induced
macrophages. Int J Cardiol. 146:153–158. 2011. View Article : Google Scholar
|
|
17
|
Zimetti F, Adorni MP, Ronda N, Gatti R,
Bernini F and Favari E: The natural compound berberine positively
affects macrophage functions involved in atherogenesis. Nutr Metab
Cardiovasc Dis. 25:195–201. 2015. View Article : Google Scholar
|
|
18
|
Zhang SH, Reddick RL, Piedrahita JA and
Maeda N: Spontaneous hypercholesterolemia and arterial lesions in
mice lacking apolipoprotein E. Science. 258:468–471. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Goon PK, Watson T, Shantsila E, Boos CJ
and Lip GY: Standardization of circulating endothelial cell
enumeration by the use of human umbilical vein endothelial cells. J
Thromb Haemost. 5:870–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Whitman SC: A practical approach to using
mice in atherosclerosis research. Clin Biochem Rev. 25:81–93.
2004.
|
|
21
|
Varma V, Yao-Borengasser A, Rasouli N,
Bodles AM, Phanavanh B, Lee MJ, Starks T, Kern LM, Spencer HJ III,
McGehee RE Jr, et al: Human visfatin expression: Relationship to
insulin sensitivity, intramyocellular lipids, and inflammation. J
Clin Endocrinol Metab. 92:666–672. 2007. View Article : Google Scholar
|
|
22
|
Zhong M, Tan HW, Gong HP, Wang SF, Zhang Y
and Zhang W: Increased serum visfatin in patients with metabolic
syndrome and carotid atherosclerosis. Clin Endocrinol (Oxf).
69:878–884. 2008. View Article : Google Scholar
|
|
23
|
El-Shishtawy SH, Mosbah O, Sherif N,
Metwaly A, Hanafy A and Kamel L: Association between serum visfatin
and carotid atherosclerosis in diabetic and non-diabetic patients
on maintenance hemodialysis. Electron Physician. 8:1966–1972. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shi KL, Qian JY, Qi L, Mao DB, Chen Y, Zhu
Y and Guo XG: Atorvastatin antagonizes the visfatin-induced
expression of inflammatory mediators via the upregulation of NF-κB
activation in HCAECs. Oncol Lett. 12:1438–1444. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao Li B, Liu Y, Meng H, Wang B, Qi J,
Zhang T, Li H, Zhao T, Sun PH, et al: Visfatin destabilizes
atherosclerotic plaques in apolipoprotein E-deficient mice. PLoS
One. 11:e01482732016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao XN, Li YN and Wang YT: Interleukin-4
regulates macrophage polarization via the MAPK signaling pathway to
protect against atherosclerosis. Genet Mol Res. 15:2016.
|
|
27
|
Zhang Y, Mu Q, Zhou Z, Song H, Zhang Y, Wu
F, Jiang M, Wang F, Zhang W, Li L, et al: Protective effect of
irisin on atherosclerosis via suppressing oxidized low density
lipoprotein induced vascular inflammation and endothelial
dysfunction. PLoS One. 11:e01580382016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yin X, Zhou L, Han F, Han J, Zhang Y, Sun
Z, Zhao W, Wang Z and Zheng L: Beta-adrenoceptor activation by
norepinephrine enhances lipopolysaccharide-induced matrix
metallopro-teinase-9 expression through the ERK/JNK-c-Fos pathway
in human THP-1 cells. J Atheroscler Thromb. 24:55–67. 2016.
View Article : Google Scholar
|
|
29
|
Guo J, Wang L, Wang L, Qian S, Zhang D,
Fang J and Pan J: Berberine protects human umbilical vein
endothelial cells against LPS-induced apoptosis by blocking
JNK-mediated signaling. Evid Based Complement Alternat Med.
2016:69839562016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wassmann S, Stumpf M, Strehlow K, Schmid
A, Schieffer B, Böhm M and Nickenig G: Interleukin-6 induces
oxidative stress and endothelial dysfunction by overexpression of
the angiotensin II type 1 receptor. Circ Res. 94:534–541. 2004.
View Article : Google Scholar
|
|
31
|
Ohta H, Wada H, Niwa T, Kirii H, Iwamoto
N, Fujii H, Saito K, Sekikawa K and Seishima M: Disruption of tumor
necrosis factor-alpha gene diminishes the development of
atherosclerosis in ApoE-deficient mice. Atherosclerosis. 180:11–17.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kumar S, Kim CW, Simmons RD and Jo H: Role
of flow-sensitive microRNAs in endothelial dysfunction and
atherosclerosis: Mechanosensitive athero-miRs. Arterioscler Thromb
Vasc Biol. 34:2206–2216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X, Guo Y, Wang C and Yu H, Yu X and
Yu H: MicroRNA-142 3p Inhibits chondrocyte apoptosis and
inflammation in osteoarthritis by targeting HMGB1. Inflammation.
39:1718–1728. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choy JC, Granville DJ, Hunt DW and McManus
BM: Endothelial cell apoptosis: Biochemical characteristics and
potential implications for atherosclerosis. J Mol Cell Cardiol.
33:1673–1690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Furnkranz A, Schober A, Bochkov VN,
Bashtrykov P, Kronke G, Kadl A, Binder BR, Weber C and Leitinger N:
Oxidized phospholipids trigger atherogenic inflammation in murine
arteries. Arterioscler Thromb Vasc Biol. 25:633–638. 2005.
View Article : Google Scholar
|
|
37
|
Stein O and Stein Y: Atheroprotective
mechanisms of HDL. Atherosclerosis. 144:285–301. 1999. View Article : Google Scholar : PubMed/NCBI
|