Introduction
Diabetic peripheral neuropathy (DPN) is the most
common microvascular complication of diabetes, with an overall
prevalence of 50–60%, and an association with high morbidity and
mortality rates (1). DPN exhibits
characteristics of progressive, distal-to-proximal peripheral nerve
degeneration leading to pain and weakness, followed by loss of
sensation (2). Despite the
existence of various drugs that have anti-oxidative stress and
neurotrophic factor properties, which have been shown to ameliorate
DPN, the number of patients with the condition is increasing
markedly worldwide (3–7). Therefore, there is a requirement to
identify novel drugs and targets for alleviating DPN.
Glucagon-like peptide-1 receptor (GLP-1R) agonist,
liraglutide, is a type of GLP-1 derivative that has a 97%
homologous amino-acid sequence to GLP-1, with the addition of a
fatty acid (8). Liraglutide has
been established as a daily injectable treatment for type 2
diabetes mellitus, with effects including the stimulation of
insulin secretion, the suppression of glucagon and a decrease in
appetite (9). Recently, studies
have revealed the that GLP-1R agonists can exhibit beneficial
effects in the central and peripheral nervous systems, which may
have therapeutic implications for treating diabetic neuropathy
(10–12). However, the exact mechanisms
involved remain under study. Persistent hyperglycemia is believed
to activate oxidative stress and inflammatory pathways. The
activation of inflammatory pathways can upregulate the gene
expression of proinflammatory cytokines, and serve a critical role
in the initiation and perpetuation of DPN (13,14). The involvement of oxidative stress
has consistently been indicated in peripheral nerve demyelination
and degeneration, increased sensory afferent excitability and
neuropathic pain induction (1).
The present study investigated whether liraglutide alleviates the
severity of DPN by inhibiting inflammatory signaling pathways.
Materials and methods
Drugs and reagents
GLP-1R agonist liraglutide (batch no. CP51039) was
purchased from Novo Nordisk Pharmaceutical Co., Ltd., (Beijing,
China). Streptozotocin (STZ; cat. no. WXBB2432V) was obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Citric acid (cat.
no. 20100830) was from Chengdu Kelong Chemical Reagent Factory
(Chengdu, Sichuan, China) and trisodium citrate (cat. no.
2011092803) was purchased from Shanghai Real Chemical Reagent Co.,
Ltd. (Shanghai, China). Rat tumor necrosis factor-α (TNF-α; cat.
no. 100335038), interleukin-1β (IL-1β; cat. no. 101064016) and IL-6
(cat. no. 100527031) platinum enzyme-linked immunosorbent assay
(ELISA) reagents were obtained from Bender MedSystems GmbH (Vienna,
Austria). The reverse transcription kit (cat. no. R011) was
purchased from Takara Biotechnology Co., Ltd. (Dalian, China).
Antibodies against phosphorylated (p)-extracellular
signal-regulated kinases (ERK; Thr202/Tyr204), total ERK, p-c-Jun
NH2-terminal kinases (JNK; Thr183/Tyr185), total JNK, p-p38
mitogen-activated protein kinase (MAPK; Tyr322), total p38 MAPK,
p65 subunit of nuclear factor-κB (NF-κB p65), goat anti-rabbit,
rabbit anti-mouse, glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
and Histone H3 were purchased from Cell Signaling Technology, Inc.
(Danvers, MA, USA).
Experimental animals
Male Sprague-Dawley (SD) rats (n=40; 180–200 g
weight, 8 weeks old) were purchased from Shanghai Sippr-BK
Laboratory Animal Co., Ltd. (Shanghai, China). All rats were
maintained under an environmentally controlled atmosphere
(temperature, 23°C; relative humidity, 45%) and a 12 h light/12 h
dark cycle. Rats received water and food freely and were allowed to
acclimatize for 1 week before drugs injection. The specific
pathogen-free SD rat certification number was SCXK(Hu) 2013–0016.
All animal experiments were approved by the Ethics Committee of
Animal Care of the Nanjing Medical University (Huai'an, Jiangsu,
China), according to the Guidelines for Animal Experiments of the
Chinese Academy of Medical Sciences. After 12 h of fasting, the
fasting blood glucose (FPG) was monitored and selected <8.9
mmol/l FPG was selected as the rat model. Diabetic rats were then
induced by intraperitoneal injection of a single dose of STZ at 60
mg/kg body weight, in an injection volume of 1 ml/100 g. After 72
h, STZ-injected rats with a blood glucose level of >16.65 mmol/l
were selected as the diabetic animal model. The control group was
injected with equal amounts of sterile citric acid/sodium citrate
buffer. The animals were randomly divided into 4 groups (10 rats in
each): The normal control + saline group (NC), the normal control +
liraglutide group (NCL), the diabetic + saline (DM) group and the
diabetic + liraglutide (DML) group. The NCL group and the DML group
were treated with liraglutide at 200 µg/kg/day for 8 weeks.
The NC group and the DM group were treated with saline at the same
dose for 8 weeks. Blood glucose levels and body weight were
monitored each week during the experiment. Tail vein blood was used
for measuring the level of blood glucose by OneTouch®
UltraVue™ (Johnson & Johnson Medical Companies, Shanghai,
China). The diabetic animals were continuously fed for 8 weeks to
generate animals with DPN.
Histological examination
The rats were anesthetized by peritoneal injection
with 10% chloral hydrate (500 mg/kg) prior to sciatic nerve tissue
samples being taken. Sciatic nerve tissue from the same rat was
isolated after the NCV determination. Sciatic nerve tissue samples
(sections 4 µm thick) were fixed in 10% formalin at 4°C for
24 h, dehydrated through a graduated ethanol series and embedded in
paraffin blocks. The sciatic nerve sections were stained with
H&E at room temperature. The staining procedure was as follows:
The sections were deparaffinized, followed by 2 changes of xylene
for 10 min each, re-hydration in 2 changes of absolute alcohol for
5 min each, then 95% alcohol and 70% alcohol for 2 min, and washing
in distilled water. The sections were then stained in Harris'
hematoxylin solution for 8 min and washed under running tap water
for 5 min. This was followed by differentiation in 1% acid alcohol
for 30 sec and washing under running tap water for 1 min, and then
by bluing in 0.2% ammonia water for 30 sec, washing again for 5
min, rinsing in 95% alcohol, and counterstaining with
eosin-phloxine solution for 30 sec. The setions were then
dehydrated with 95% alcohol and absolute alcohol for 5 min each,
followed by 2 changes of xylene for 5 min each. Finally, the
setions were mounted with xylene-based mounting medium.
Morphological changes were observed under a Olympus
DX45 microscope (Camera DP72; Olympus, Tokyo, Japan). Microscopic
fields were randomly selected for observation in each paraffin
section.
Measurement of serum proinflammatory
cytokines by ELISA
The blood from the vena cava was drawn and then
centrifuged at 3,000 rpm for 5 min to measure TNF-α, IL-1β and IL-6
levels were measured by enzyme-linked immunosorbent assay (ELISA)
using a commercial kit according to the manufacturer's
protocols.
Measurement of NCV
Nerve conduction velocity (NCV) was measured in the
sciatic nerves of the rats. The rats were anesthetized by
peritoneal injection with 10% chloral hydrate (500 mg/kg).
Following disinfection of the right leg with alcohol, the
stimulating and recording electrodes were placed. The sciatic nerve
was stimulated with single supramaximal square wave pluses (1.2 V
in intensity, 1 msec in width), using the Functional Experiment
System (BL-420s; Taimeng, Sichuan, China). The distance between the
two sites of stimulation was 6 mm. The distance (D) between the
stimulating and recording electrodes, and the action potential
latency (L) of the sciatic nerve were measured to calculate the
motor NCV (MNCV) as follows: MNCV (m/sec)= D / L. For the
determination of sensory NCV (SNCV), the recording site was located
in the sciatic notch. SNCV was calculated in the same manner as the
MNCV.
Western blot analysis
The sciatic nerve tissues were washed twice with
cold PBS and lysed in an ice-cold cell lysis buffer which was
supplemented with a Protease Inhibitor Cocktail (Roche, Basel,
Switzerland) to extract the total protein. The protein
concentration was determined using the bicinchoninic acid method.
The total protein lysates were dissolved in the gel loading buffer
and boiled for 5 min. Equal amounts of protein were subjected to
SDS-PAGE electrophoresis on 10% gel and transferred to
nitrocellulose membranes (EMD Millipore, Billerica, MA, USA).
Immunodetection was performed using antibodies against NF-κBp65
(cat. no. 8242; 1:1,000), p-p38MAPK (cat. no. 8690; 1:1,000), p-ERK
(cat. no. 4370; 1:1,000) and p-JNK (cat. no. 9251; 1:1,000). After
overnight incubation with primary antibodies, the membranes were
washed in Tris buffered saline with 0.1% Tween-20 (TBST), and then
incubated in BLOTTO containing horseradish peroxidase-conjugated
rabbit anti-mouse (cat. no. 58802; 1:8,000) and goat anti-rabbit
(cat. no. 7074; 1:500) secondary antibodies (Nanjing Enogene
Biotech. Co., Ltd.) for 1 h at room temperature. The blots were
rinsed and proteins were visualized by enhanced chemiluminescence
(ECL; EMD Millipore) and quantified by densitometry (Quantity One;
Bio-Rad, Hercules, CA, USA). Membranes were stripped and reprobed
with GAPDH (cat. no. 2118; 1:2000) to evaluate the lane-loading
control. Histone-H3 (cat. no. 14269; 1:2,000), t-p38MAPK (cat. no.
2387; 1:1,000), t-ERK (cat. no. 4067; 1:1,000) and t-JNK (cat. no.
9252; 1:1,000) were used as loading controls. Results were
expressed graphically as a ratio of phosphorylated to total
protein.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The mRNA transcripts were analyzed by qPCR of the
sciatic nerve tissue. The RT-PCR reaction system (20 µl) was
as follows: 10 µl SYBR Premix Ex Taq™ (2X), 0.4 µl
PCR Forward Primer (10 µM), 0.4 µl PCR Reverse Primer
(10 µM), 0.4 µl ROX Reference Dye (50X), 2.0
µl cDNA, 6.8 µl ddH2O. Total RNA was
isolated using a standard TRIzol RNA isolation method (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) following the
manufacturer's protocols and was subjected to reverse transcription
using Promega M-MLV reverse transcriptase (cat no. 9PIM170; Promega
Corporation, Madison, WI, USA). SYBR-green qPCR was performed using
a Bio-Rad iQ5 Real-Time PCR system according to the protocols
provided with the SYBR Premix Ex Taq™ (Takara Biotechnology Co.,
Ltd.). The relative mRNA levels were normalized to the mRNA levels
of β-actin and the fold-change of each mRNA was calculated using
the 2−ΔΔCq method (15). Primer sequences for these
biomarkers are as shown in Table
I.
 | Table IPrimer sequence of different genes
for reverse transcription-quantitative polymerase chain
reaction. |
Table I
Primer sequence of different genes
for reverse transcription-quantitative polymerase chain
reaction.
| Genes | Primer sequences
(5′–3′) |
|---|
| TNF-α | F:
GTCTGTGCCTCAGCCTCTTC |
| R:
TGGAACTGATGAGAGGGAGC |
| IL-1β | F:
CACCTCTCAAGCAGAGCACAGA |
| R:
ACGGGTTCCATGGTGAAGTC |
| IL-6 | F:
TCCAGTTGCCTTCTTGGGAC |
| R:
GTACTCCAGAAGACCAGAGG |
| ICAM-1 | F:
AGGTATCCATCCATCCCACA |
| R:
GCCACAGTTCTCAAAGCACA |
| MCP-1 | F:
GTGCTGACCCCAATAAGGAA |
| R:
TGAGGTGGTTGTGGAAAAGA |
| Neuritin | F:
GGGCGAAAGATATGTGGGAT |
| R:
CGAGAGAGACACCAGGAGCA |
| NGF | F:
CTGGACCCAAGCTCACCTCA |
| R:
GTGGATGAGCGCTTGCTCCT |
| NSE | F:
GAACTATCCTGTGGTCTCC |
| R:
CGACATTGGCTGTGAACTTG |
| NOX4 | F:
GAAGCCCATTTGAGGAGTCA |
| R:
GGGTCCACAGCAGAAAACTC |
| β-actin | F:
ACGGGGTCACCCACACTGTGC |
| R:
CTAGAAGCATTTGCGGTGGACGATG |
Statistical analysis
All statistical analyses were conducted using
statistical analysis software (SPSS 18.0; IBMM, Armonk, NY, USA).
Comparisons between groups were performed using either paired
Student's t-tests or one-way analysis of variance, where indicated.
Data are presented as the mean ± standard deviation. Differences
were considered significant at values of P<0.05.
Results
Body weight and blood glucose level
At 8 weeks post-liraglutide treatment, the rats in
the DM group exhibited an increased blood glucose level and
decreased body weight compared with the rats in the NC group.
Treatment with liraglutide did not alter body weight or blood sugar
levels in the groups (Fig.
1).
Histological analysis of DPN among
groups
The myelin of the myelinated nerve fibers in the NC
group and the NCL group appeared dense, round and uniform, with an
ordered lamellar structure presenting with neither axonal shrinkage
nor swelling (Fig. 2A and B).
However, in the DM group, the myelin sheath was thin, loose and
disorganized, with vacuolar-like defects (Fig. 2C). Following treatment with
liraglutide for 8 weeks, the density of the myelin nerve fibers was
partially restored in the DML group (Fig. 2D).
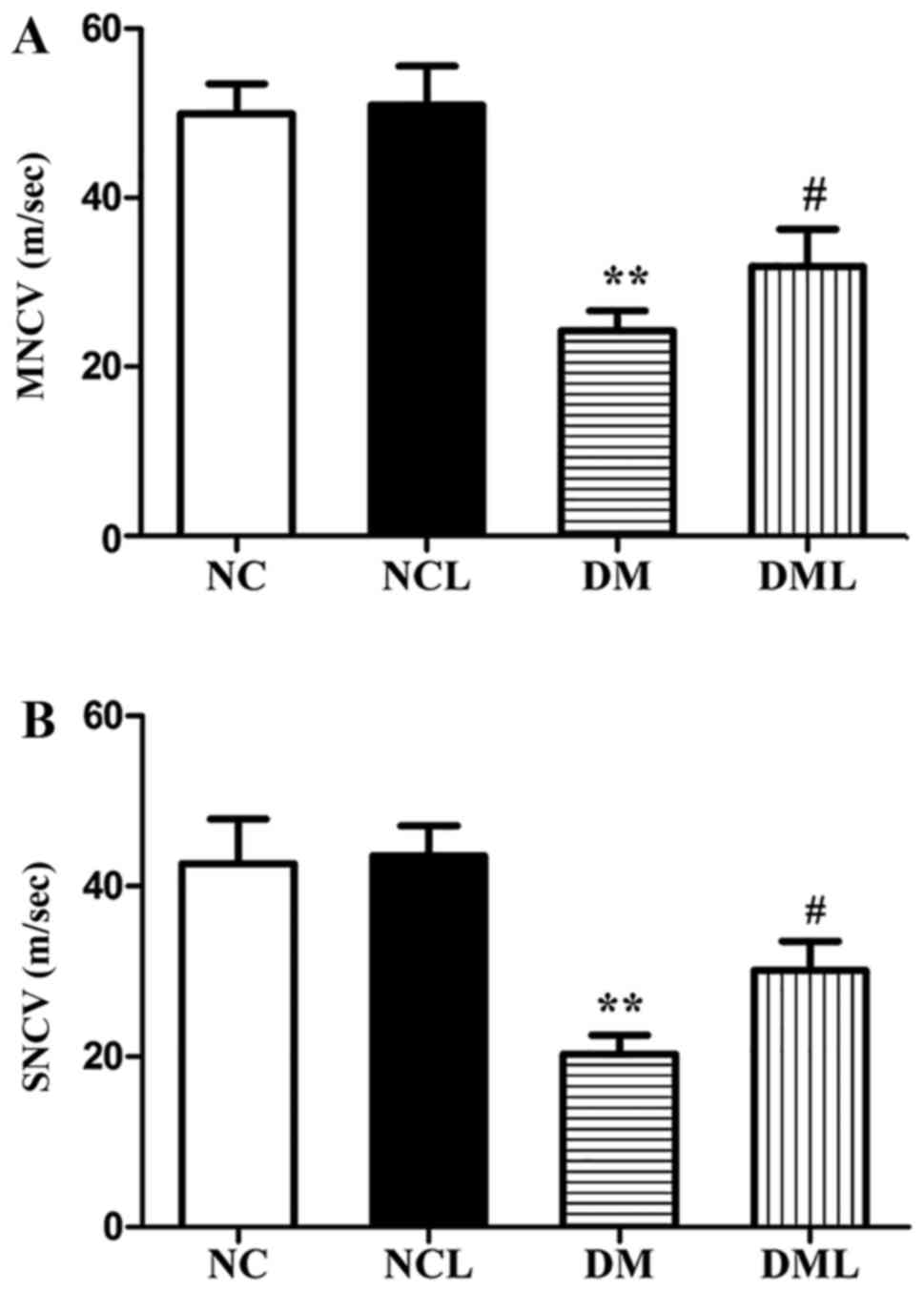
Liraglutide improves sciatic NCV
Sciatic nerve MNCV and SNCV of the 4 groups are
presented in Fig. 3. The DM group
exhibited significantly reduced MNCV and SNCV compared with the NC
group (P<0.01). MNCV and SNCV were each significantly increased
(P<0.01) in the DML compared with those in the DM group
(Fig. 3).
Liraglutide decreases the level of
proinflammatory cytokines in serum and sciatic nerves
The results data showed a significant increase in
serum proinflammatory cytokine (TNF-α, IL-6 and IL-1β) level in the
DM group compared with that in rats of the NC and NCL groups.
Following 8 weeks of treatment with liraglutide, the level of
TNF-α, IL-1β and IL-6 in the peripheral serum of the DML group was
significantly suppressed compared with that of the DM group
(Table II). In addition, a
similar phenomenon was observed at the gene level, where the mRNA
expression of TNF-α, IL-6 and IL-1β was increased in the sciatic
nerves of the rats in the DM group compared with that in the NC and
NCL group. The mRNA expression levels of TNF-α, IL-6 and IL-1β in
the sciatic nerves were attenuated in the DML group compared with
that in the DM group (Fig. 4A–C).
In the present study, it was also observed that the mRNA expression
levels of MCP-1 and ICAM-1 were increased in the sciatic nerves of
the DM group compared with that in the NC and NCL groups.
Subsequent to liraglutide treatment, the mRNA expression levels of
MCP-1 and ICAM-1 were decreased in the sciatic nerves of the DML
group compared with that in the DM group (Fig. 4D and E).
 | Figure 4mRNA expression levels of TNF-α,
IL-6, IL-1β, ICAM-1 and MCP-1 were quantified in the sciatic nerve
by quantitative polymerase chain reaction. The relative mRNA levels
were normalized to the mRNA levels of β-actin and the fold-change
of each mRNA was calculated using the 2−ΔΔCq method. (A)
TNF-α; (B) IL-6; (C) IL-1β; (D) ICAM-1; and (E) MCP-1.
**P<0.01 vs. the NC group; ##P<0.01 vs.
the DM group. NC, normal control + saline group; NCL, normal
control + liraglutide group; DM, diabetic + saline group; DML,
diabetic + liraglutide group; TNF-α, tumor necrosis factor-α; IL,
interleukin; ICAM-1, intercellular adhesion molecule 1; MCP-1,
monocyte chemoattractant protein-1. |
| Table IISerum proinflammatory cytokine levels
of each group of rats treated with saline or liraglutide for 8
weeks. |
Table II
Serum proinflammatory cytokine levels
of each group of rats treated with saline or liraglutide for 8
weeks.
| Groups | TNF-α, pg/ml | IL-6, pg/ml | IL-1β, pg/ml |
|---|
| NC | 86.10±6.61 | 145.58±16.54 | 248.65±120.27 |
| NCL | 90.49±13.42 | 151.10±25.81 | 250.11±50.08 |
| DM |
134.40±27.87a |
254.62±25.46a |
530.68±65.78a |
| DML | 96.12±7.72b |
166.80±28.15c |
258.41±20.32c |
Liraglutide inhibits the expression of
NOX4 in the rat sciatic nerve tissue
The IOD value of NOX4 staining in the NC group was
13.60±3.51; the IOD value of NOX4 staining in the NCL group was
14.02±4.22; the IOD value of NOX4 staining in the DM group was
56.60±6.91; the IOD value of NOX4 staining in the DML group was
24.43±9.01. There was a significant increase in the IOD value of
NOX4 staining in diabetic rat sciatic nerves compared with that in
the NC group (Fig. 5A and B).
Following liraglutide treatment for 8 weeks, the IOD value of NOX4
staining in the DML group was reduced compared with that in the DM
group (Fig. 5A and B). In
addition, it was also observed that the mRNA expression of NOX4 was
increased significantly (P<0.01) in the DM group compared with
that of the NC group (Fig. 5C).
Following treatment with liraglutide for 8 weeks, the mRNA
expression of NOX4 was significantly lower (P<0.01) in the DML
group compared with that in the DM group (Fig. 5C).
mRNA expression levels of neurotrophic
factor in the sciatic nerve
In this study, the role of neurotrophic factor in
DPN was also observed. Gene expression of neuritin, nerve growth
factor (NGF) and neuron-specific enolase (NSE) was quantified by
qPCR in the sciatic nerves of 4 groups (Fig. 6). It was observed that the mRNA
expression levels of neuritin and NGF were reduced in the sciatic
nerves of the DM group compared with those of the NC and NCL
groups. However, NSE was significantly increased in the sciatic
nerves of the DM group compared with those of the NC and NCL
groups. Subsequent to 8 weeks of treatment with liraglutide, the
mRNA expression of neuritin and NGF was significantly increased
(P<0.01) in the DML group compared with that of the DM group,
NSE was reduced in the sciatic nerves of the DML group compared
with that of the DM group (Fig.
6).
Liraglutide inhibits the p-p38 MAPK/NF-κB
pathway
As is known, there are three distinct subfamilies of
MAPKs: ERK, JNK and p38 kinases. In the present study, the levels
of MAPK were examined in the sciatic nerve tissue of each group. At
8 weeks post-liraglutide treatment, it was found that p-p38 MAPK
was inhibited in the DML group compared with that in the DM group
(Fig. 7). However, there was no
significant difference in the protein expression levels of p-ERK
and p-JNK among the groups (Fig.
7). The protein expression levels of NF-κB p65 were also
examined in the nucleus and cytoplasm in the sciatic nerve tissue
of each group. Cytoplasmic NF-κB p65 expression in the DM group was
significantly lower (P<0.05) than that in the NC and NCL groups
(Fig. 7). However, nuclear NF-κB
p65 expression in the DM group was significantly increased
(P<0.01) compared with that in the NC and NCL groups (Fig. 7). Following treatment with
liraglutide for 8 weeks, the NF-κB pathway was markedly inhibited
in the DML group (Fig. 7).
 | Figure 7(A) Representative western blotting
of p- and t-ERK, p- and t-JNK, p- and t-p38 MAPK, and nuclear and
cytoplasmic NF-κB p65 expression in NC, NCL, DM and DML groups. (B)
Semi-quantitative densitometric analysis was used to summarize the
fold-change in p- to t-ERK, p- to t-JNK, p- to t-p38 MAPK, nuclear
NF-κB p65 to histone H3 and cytoplasmic NF-κB p65 to GAPDH in each
group. *P<0.05 vs. the NC group;
**P<0.01 vs. the NC group; #P<0.05 vs.
the DM group; ##P<0.01 vs. the DM group. NC, normal
control + saline group; NCL, normal control + liraglutide group;
DM, diabetic + saline group; DML, diabetic + liraglutide group; p-,
phosphorylated; t-, total; ERK, extracellular signal-regulated
kinase; JNK, c-Jun NH2-terminal kinases; MAPK, mitogen-activated
protein kinase; GAPDH, glyceraldehyde 3-phosphate
dehydrogenase. |
Discussion
DPN is the most frequently occurring chronic
complication of diabetes and is associated with significant
morbidity and mortality in diabetes patients (16). Various theories have been proposed
concerning the pathogenesis of DPN, including oxidative stress,
polyol pathway activation, increased amounts of advanced glycation
end products, protein kinase C and MAPK activation, increased
levels of inflammatory cytokines, including TNF-α and IL-6, and
neurotrophic factor deficiency (1,17–22). Among the various theories
concerning the pathogenesis of DPN, the pathway most vital for the
development and progression of DPN appears to be that of
inflammation (1). The role of
inflammation in DPN has previously been reported (23,24). NF-κB and Nrf2 pathways are 2
important pathways mediating cellular homeostasis through
controlling nueroinflammation (1). Current theories that are focused on
inflammation cytokine milieu and signaling pathways, such as the
nuclear factor erythroid 2-related factor 2 and NK-κB pathways,
discuss the role of inflammation in the development and progression
of DPN, with an emphasis on therapeutic strategies.
The glucose-dependent insulinotropic hormone GLP-1
is produced by cleaving pro-glucagon in intestinal L cells
(25–28). GLP-1R agonists are approved to
improve glycemic control in diabetes patients, based on their
abilities to slow gastric emptying, to suppress glucagon secretion
and to stimulate insulin secretion from pancreatic β-cells in a
glucose-dependent manner (29).
In addition to their insulinotropic actions, certain studies have
demonstrated that GLP-1R agonists exhibit neurotrophic and
neuroprotective properties in certain neurons and neural cells
(16,30–34). GLP-1 receptors are present on
sciatic nerve axons, Schwann cells and dorsal root ganglia (DRG)
sensory neurons of normal and diabetic rats (11). Previous studies have suggested
that GLP-1 can enhance neuronal survival and plasticity in the
brain (29). GLP-1 could thus be
considered to possess the ability for the enhancement of
neuroprotection within the peripheral nervous system, but not in
the central nervous system. A previous study also demonstrated that
GLP-1 receptor agonism could be neuroprotective in an experimental
model of sensory neuropathy (35). Based on these findings, Liu et
al (12) showed that
synthetic exendin-4 may ameliorate DPN in skin and sciatic nerves
through activating the GLP-1 receptor, antiapoptotic effects and
restoration of cyclic adenosine monophosphate content. The present
study investigated how liraglutide alleviates the severity of DPN
by inhibiting inflammatory signaling pathways.
Previous studies demonstrated that persistent
hyperglycemia is a major nerve damaging factor (36,37); therefore, the normalization of the
blood glucose level is vital for DPN therapy. However, such
intensive therapies in DPN patients may increase the mortality risk
and are likely associated with frequent severe hypoglycemia.
Therefore, to prevent DPN, therapeutic strategies other than those
those that target blood glucose normalization are required. In the
present study, liraglutide did not alter body weight or blood sugar
levels in any of the groups. Based on this, whether liraglutide
alleviates the severity of DPN independent of glycemic control was
investigated. It was observed that the myelin sheath of the DM
group was thin, loose, disorganized and exhibited vacuolar-like
defects, and MNCV and SNCV were significantly reduced. Subsequent
to 8 weeks of treatment with liraglutide, morphological and
functional abnormalities were ameliorated in DML group rats. The
present study therefore aimed to investigate the mechanism of
GLP-1R agonists in the progression of DPN.
Certain studies have shown the association between
inflammation and DPN (1,23,24). However, the mechanism has not been
fully elucidated. Therefore, the present study aimed to investigate
whether inflammatory mediators are central to the pathogenesis of
DPN. The serum levels of TNF-α, IL-6 and IL-1β were found to be
significantly higher in the DM group than the control groups.
Following 8 weeks of treatment with liraglutide, the level of
TNF-α, IL-1β, and IL-6 in the peripheral serum of the DML group was
significantly suppressed compared with that of the DM group. It was
also observed that the mRNA expression levels of TNF-α, IL-6,
IL-1β, MCP-1 and ICAM-1 in the rat sciatic nerves were attenuated
in the DML group. Hence, it was shown that GLP-1R agonists reduced
the serum levels of TNF-α, IL-1β and IL-6, and inhibited the mRNA
expression levels of proinflammataory cytokines in the rat sciatic
nerve.
The MAPK pathway is a classical pathway that can
indirectly or directly initiate the production of inflammatory
mediators and the activation of NF-κB (1). A previous study showed that high
glucose activated JNK and p38 MAPK which did not result in cell
damage. However, oxidative stress activated ERK and p38 MAPKs which
resulted in cellular damage. In the dorsal root ganglia of type 1
diabetic rats, ERK and p38 MAPK were activated at 8 weeks, and then
JNK was activated at 12 weeks (38). The present study examined the
activation of p38 MAPK in the sciatic nerve from 8-week
streptozotocin-induced diabetic rats; however, there was no
activation of ERK and JNK in the sciatic nerves. Following 8 weeks
of treatment with liraglutide, GLP-1R agonists inhibited the p38
MAPK activation. The activation of transcription factor NF-κB p65
was also observed in the sciatic nerves of the
streptozotocin-induced diabetic rats. Following treatment with
liraglutide for 8 weeks, the activation of NF-κB p65 was markedly
inhibited in the DML group. We hypothesize that persistent
hyperglycemia in the DM group ould induce the inflammatory cascade
through the activation of the p38 MAPK/NF-κB pathway. p38
MAPK/NF-κB is a transcription modulator that upregulates the gene
expression of proinflammatory cytokines. The anti-inflammatory
effects of GLP-1R agonists may be mediated through the inhibition
of the activation of the p38 MAPK/NF-κB pathway, as activated p38
MAPK/NF-κB promotes the gene expression of TNF-α, IL-6 and IL-1β.
The present study demonstrates that the p38 MAPK/NF-κB pathway can
provide a targeted approach for the prevention of inflammatory
changes in DPN.
Previous studies have demonstrated that oxidative
stress has a significant impact on the pathogenesis of diabetes,
including the associated complications such as DPN (4–6). A
previous study demonstrated that oxidative stress serves a key
contributory role in the progressive nature of DPN (1). Our present study also suggested that
NOX4 is an important source of reactive oxygen species (ROS)
production. Upon activation, it induces the production of
superoxide via oxidative stress and inflammatory responses in the
mitochondria' such ROS are indicated to be involved in the
pathogenesis of diabetic complications, such as neuropathy. NOX4 is
a subunit of NADPH, so it was as a marker of oxidative stress in
the present study. There was a significant increase in the IOD
value of NOX4 staining in the diabetic rat sciatic nerves.
Following 8 weeks of treatment with liraglutide, the IOD value of
NOX4 staining in the DML group was significantly lower than that in
the DM group. It was also observed that the mRNA expression of NOX4
was significantly lower in the DML group, thus indicating DPN
amelioration in liraglutide-treated rats.
A number of studies have suggested that neuritin and
NGF serve key roles in the progressive nature of DPN (39). Karamoysoyli et al (22) showed that neuritin levels could be
manipulated in diabetes to provide a potential therapeutic target
in the management of neuropathy via mitogen-activated protein
kinase activation. In the present study, it was observed that the
mRNA expression levels of neuritin and NGF were reduced in the DM
group. Following treatment with liraglutide for 8 weeks, the mRNA
expression of neuritin and NGF was significantly increased in the
DML group, thus indicating that liraglutide may ameliorate DPN.
NSE, which is a highly soluble intracellular enzyme normally
located in the cytoplasm in neuroendocrine cells, may be a novel
biomarker of peripheral neuropathy in diabetes. Li et al
(40) showed that serum NSE
levels were increased in diabetic neuropathy subjects and that the
elevated NSE levels were closely associated with DPN. In the
present study, to the best of our knowledge, it was observed for
the first time that the mRNA expression levels of NSE increased
significantly in the DM group compared with the controls. The mRNA
expression of NSE was significantly reduced in the DML group
compared with that in the DM group, thus indicating that
liraglutide may ameliorate DPN.
In conclusion, these data demonstrate that
inflammation and oxidative stress serve key roles in the
pathogenesis of DPN, and that GLP-1R agonists may prevent nerve
dysfunction in the sciatic nerves of diabetic rats via the p38
MAPK/NF-κB signaling pathways, independent of glycemic control. Use
of GLP-1R agonists may be a therapeutic strategy for slowing the
progression of DPN.
Acknowledgments
The authors wish to thank Professor Dawei Gong from
the School of Medicine, University of Maryland (Baltimore, MA, USA)
for helping in revising the paper and the experimental methods. The
authors also wish to thank Dr Wei Li from the Affiliated Hospital
of Xuzhou Medical College for providing help with the study
design.
Notes
[1]
Funding
This study was supported by the National Natural
Science Foundation of China Grant Award (grant nos. 81200595,
81400807 and 81700723), the Project of Six High-Peak Talents of
Jiangsu Province (grant no. WSN-101) and the Research Project of
Jiangsu Provincial Commission of Health and Family Planning (grant
nos. H201253, H201554, F201549 and H201667).
[2] Availability
of data and materials
All data generated or analyzed during this study are
included in this published article.
[3] Authors'
contributions
JM and HZ coordinated the study and contributed to
the acquisition of funding, study design, data collection,
statistical analysis, draft and revision of the paper. MS, XZ and
XL contributed to the study design, acquisition of data, and
statistical analysis. JC, RZ, and XW contributed to acquisition of
data, statistical analysis and revision of important intellectual
content. All authors read and approved the final manuscript.
[4] Ethics
approval and consent to participate
All animal experiments were approved by the Ethics
Committee of Animal Care of the Nanjing Medical University,
according to the Guidelines for Animal Experiments of the Chinese
Academy of Medical Sciences.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Sandireddy R, Yerra VG, Areti A,
Komirishetty P and Kumar A: Neuroinflammation and oxidative stress
in diabetic neuropathy: Futuristic strategies based on these
targets. Int J Endocrinol. 2014:6749872014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
O'Brien PD, Sakowski SA and Feldman EL:
Mouse models of diabetic neuropathy. ILAR J. 54:259–272. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bae SM, Bae MN, Kim EY, Kim IK, Seo MW,
Shin JK, Cho SR and Jeong GH: Recurrent insulin autoimmune syndrome
caused by α-Lipoic acid in type 2 diabetes. Endocrinol Metab
(Seoul). 28:326–330. 2013. View Article : Google Scholar
|
|
4
|
Oyenihi AB, Ayeleso AO, Mukwevho E and
Masola B: Antioxidant strategies in the management of diabetic
neuropathy. BioMed Res Int. 2015:5150422015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Verdile G, Keane KN, Cruzat VF, Medic S,
Sabale M, Rowles J, Wijesekara N, Martins RN, Fraser PE and
Newsholme P: Inflammation and oxidative stress: The molecular
connectivity between insulin resistance, obesity, and Alzheimer's
disease. Mediators Inflamm. 2015:1058282015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiao Q, Yang YA, Zhao XY, He LS, Qin Y, He
YH, Zhang GP and Luo JD: Oxidative stress contributes to the
impaired sonic hedgehog pathway in type 1 diabetic mice with
myocardial infarction. Exp Ther Med. 10:1750–1758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang XW, Liu FQ, Guo JJ, Yao WJ, Li QQ,
Liu TH and Xu LP: Antioxidation and anti-inflammatory activity of
Tang Bi Kang in rats with diabetic peripheral neuropathy. BMC
Complement Altern Med. 15:662015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sato K, Kameda M, Yasuhara T, Agari T,
Baba T, Wang F, Shinko A, Wakamori T, Toyoshima A, Takeuchi H, et
al: Neuroprotective effects of liraglutide for stroke model of
rats. Int J Mol Sci. 14:21513–21524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Inoue K, Maeda N, Kashine S, Fujishima Y,
Kozawa J, Hiuge-Shimizu A, Okita K, Imagawa A, Funahashi T and
Shimomura I: Short-term effects of liraglutide on visceral fat
adiposity, appetite, and food preference: A pilot study of obese
Japanese patients with type 2 diabetes. Cardiovasc Diabetol.
10:1092011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jaiswal M, Martin CL, Brown MB, Callaghan
B, Albers JW, Feldman EL and Pop-Busui R: Effects of exenatide on
measures of diabetic neuropathy in subjects with type 2 diabetes:
Results from an 18-month proof-of-concept open-label randomized
study. J Diabetes Complications. 29:1287–1294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jolivalt CG, Fineman M, Deacon CF, Carr RD
and Calcutt NA: GLP-1 signals via ERK in peripheral nerve and
prevents nerve dysfunction in diabetic mice. Diabetes Obes Metab.
13:990–1000. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu WJ, Jin HY, Lee KA, Xie SH, Baek HS
and Park TS: Neuroprotective effect of the glucagon-like peptide-1
receptor agonist, synthetic exendin-4, in streptozotocin-induced
diabetic rats. Br J Pharmacol. 164:1410–1420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ganesh Yerra V, Negi G, Sharma SS and
Kumar A: Potential therapeutic effects of the simultaneous
targeting of the Nrf2 and NF-κB pathways in diabetic neuropathy.
Redox Biol. 1:394–397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ramesh G, MacLean AG and Philipp MT:
Cytokines and chemokines at the crossroads of neuroinflammation,
neurodegeneration, and neuropathic pain. Mediators Inflamm.
2013:4807392013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak and Schmittgen: Analysis of relative
gene expression data using real-time quantitative PCR and the
2-ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
16
|
Höke A: Animal models of peripheral
neuropathies. Neurotherapeutics. 9:262–269. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chapter MC, White CM, DeRidder A, Chadwick
W, Martin B and Maudsley S: Chemical modification of class II G
protein-coupled receptor ligands: Frontiers in the development of
peptide analogs as neuroendocrine pharmacological therapies.
Pharmacol Ther. 125:39–54. 2010. View Article : Google Scholar
|
|
18
|
Douglas DS and Popko B: Mouse forward
genetics in the study of the peripheral nervous system and human
peripheral neuropathy. Neurochem Res. 34:124–137. 2009. View Article : Google Scholar :
|
|
19
|
Edwards JL, Vincent AM, Cheng HT and
Feldman EL: Diabetic neuropathy: Mechanisms to management.
Pharmacol Ther. 120:1–34. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lupachyk S, Stavniichuk R, Komissarenko
JI, Drel VR, Obrosov AA, El-Remessy AB, Pacher P and Obrosova IG:
Na+/H+-exchanger-1 inhibition counteracts
diabetic cataract formation and retinal oxidative-nitrative stress
and apoptosis. Int J Mol Med. 29:989–998. 2012.PubMed/NCBI
|
|
21
|
Tesfaye S, Boulton AJ and Dickenson AH:
Mechanisms and management of diabetic painful distal symmetrical
polyneuropathy. Diabetes Care. 36:2456–2465. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karamoysoyli E, Burnand RC, Tomlinson DR
and Gardiner NJ: Neuritin mediates nerve growth factor-induced
axonal regeneration and is deficient in experimental diabetic
neuropathy. Diabetes. 57:181–189. 2008. View Article : Google Scholar
|
|
23
|
Gorska-Ciebiada M, Saryusz-Wolska M,
Borkowska A, Ciebiada M and Loba J: Serum levels of inflammatory
markers in depressed elderly patients with diabetes and mild
cognitive impairment. PLoS One. 10:e01204332015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zong H, Ward M, Madden A, Yong PH, Limb
GA, Curtis TM and Stitt AW: Hyperglycaemia-induced pro-inflammatory
responses by retinal Müller glia are regulated by the receptor for
advanced glycation end-products (RAGE). Diabetologia. 53:2656–2666.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Heni M, Kullmann S, Gallwitz B, Häring HU,
Preissl H and Fritsche A: Dissociation of GLP-1 and insulin
association with food processing in the brain: GLP-1 sensitivity
despite insulin resistance in obese humans. Mol Metab. 4:971–976.
2015. View Article : Google Scholar
|
|
26
|
May AA and Woods SC: Is endogenous GLP-1 a
major influence on the orbitofrontal cortex? Mol Metab. 4:977–978.
2015. View Article : Google Scholar
|
|
27
|
Meng J, Ma X, Wang N, Jia M, Bi L, Wang Y,
Li M, Zhang H, Xue X, Hou Z, et al: Activation of GLP-1 receptor
promotes bone marrow stromal cell osteogenic differentiation
through beta-catenin. Stem Cell Reports. 6:6332016. View Article : Google Scholar
|
|
28
|
Yamamoto T, Nakade Y, Yamauchi T,
Kobayashi Y, Ishii N, Ohashi T, Ito K, Sato K, Fukuzawa Y and
Yoneda M: Glucagon-like peptide-1 analogue prevents nonalcoholic
steatohepatitis in non-obese mice. World J gastroenterol.
22:2512–2523. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Seino Y and Yabe D: Glucose-dependent
insulinotropic polypeptide and glucagon-like peptide-1: Incretin
actions beyond the pancreas. J Diabetes Investig. 4:108–130. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Greig NH, Tweedie D, Rachmany L, Li Y,
Rubovitch V, Schreiber S, Chiang YH, Hoffer BJ, Miller J, Lahiri
DK, et al: Incretin mimetics as pharmacologic tools to elucidate
and as a new drug strategy to treat traumatic brain injury.
Alzheimers Dement. 10(Suppl): S62–S75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Harkavyi A and Whitton PS: Glucagon-like
peptide 1 receptor stimulation as a means of neuroprotection. Br J
Pharmacol. 159:495–501. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Janssens J, Etienne H, Idriss S, Azmi A,
Martin B, Maudsley S and Systems-Level G: Systems-level G
protein-coupled receptor therapy across a neurodegenerative
continuum by the GLP-1 receptor system. Front Endocrinol
(Lausanne). 5:1422014.
|
|
33
|
Li Y, Perry T, Kindy MS, Harvey BK,
Tweedie D, Holloway HW, Powers K, Shen H, Egan JM, Sambamurti K, et
al: GLP-1 receptor stimulation preserves primary cortical and
dopaminergic neurons in cellular and rodent models of stroke and
Parkinsonism. Proc Natl Acad Sci USA. 106:1285–1290. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Salcedo I, Tweedie D, Li Y and Greig NH:
Neuroprotective and neurotrophic actions of glucagon-like
peptide-1: An emerging opportunity to treat neurodegenerative and
cerebrovascular disorders. Br J Pharmacol. 166:1586–1599. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Perry T, Holloway HW, Weerasuriya A,
Mouton PR, Duffy K, Mattison JA and Greig NH: Evidence of
GLP-1-mediated neuroprotection in an animal model of
pyridoxine-induced peripheral sensory neuropathy. Exp Neurol.
203:293–301. 2007. View Article : Google Scholar :
|
|
36
|
Katagi M, Terashima T, Okano J, Urabe H,
Nakae Y, Ogawa N, Udagawa J, Maegawa H, Matsumura K, Chan L, et al:
Hyperglycemia induces abnormal gene expression in hematopoietic
stem cells and their progeny in diabetic neuropathy. FEBS Lett.
588:1080–1086. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yan LJ: Pathogenesis of chronic
hyperglycemia: From reductive stress to oxidative stress. J
Diabetes Res. 2014:1379192014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Purves T, Middlemas A, Agthong S, Jude EB,
Boulton AJ, Fernyhough P and Tomlinson DR: A role for
mitogen-activated protein kinases in the etiology of diabetic
neuropathy. FASEB J. 15:2508–2514. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang C and Salton SR: The role of
neurotrophins in major depressive disorder. Transl Neurosci.
4:46–58. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li J, Zhang H, Xie M, Yan L, Chen J and
Wang H: NSE, a potential biomarker, is closely connected to
diabetic peripheral neuropathy. Diabetes Care. 36:3405–3410. 2013.
View Article : Google Scholar : PubMed/NCBI
|