Introduction
Osteogenesis imperfecta (OI) is a clinically
heterogeneous and heritable connective tissue disorder that is
characterized by bone fragility and susceptibility to fractures
following minimal trauma (1).
Sillence et al (2)
initially categorized OI individuals into four types based on
clinical features and disease severity: Type I [(Mendelian
Inheritance in Man (MIM) 166200], type II (MIM 166210), type III
(MIM 259420), and type IV (MIM 166220). For the majority of
patients, OI is associated with mutations of COL1A1 (MIM
120150) or COL1A2 (MIM 120160), which encode type I
collagens (3). Glorieux et
al (4) described a group of
patients with OI that presented with a discrete phenotype,
including hyperplastic callus formation and calcification of the
interosseous membrane; this was designated as OI type V (MIM
610967). In 2002, Glorieux et al (5) described another novel form of OI
that presented with osteoid accumulation due to a mineralization
defect; this was designated OI type VI (MIM 613982). Patients with
OI type VI typically sustain their first fracture after the age of
6 months (6). Their sclerae are
white or faintly blue and dentinogenesis imperfecta (DI) is
uniformly absent (6). These
characteristic features of OI type VI are readily apparent in
histological bone samples, including bone biopsy specimens that
have a 'fish-scale' pattern, hyperosteoidosis, prolonged
mineralization lag time and decreased mineral apposition rate
(5). Becker et al
(7) first reported that Serpin
family F member 1 (SERPINF1) mutations cause severe
autosomal recessive OI in 2011. FK506 binding protein 10
(FKBP10) mutations were first reported by Alanay et
al (8) in 2010 in
autosomal-recessive OI. Kelley et al (9) concluded that FKBP10 mutations
also cause Bruck syndrome, which is characterized by congenital
joint contractures. Autosomal-recessive OI with FKBP10
mutations was designated as OI type XI (10). As an increasing number of
virulence genes have been discovered, the OI classification has
expanded to 15 types (10).
Previous studies have reported that the most
prevalent pathogenic genes responsible for OI in Chinese patients
were COL1A1 and COL1A2 (11,12). To the best of our knowledge, only
one report of Chinese patients with SERPINF1 mutations and
one with FKBP10 mutation have previously been reported,
respectively (13,14). In the present study, two novel
mutations in SERPINF1 and one novel compound heterozygous
mutation in FKBP10 were identified in three unrelated
Chinese families with autosomal recessive OI type VI and XI.
Materials and methods
Subjects
None of the probands belonged to consanguineous
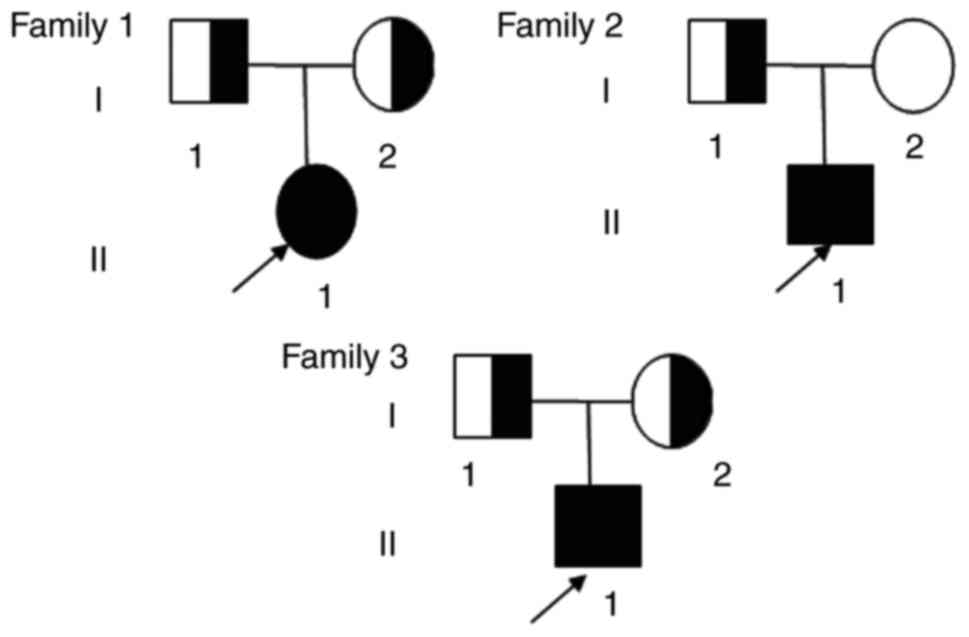
families. Three unrelated Chinese families with OI (Fig. 1) and 250 healthy control donors
were included in the present study. Patients were recruited between
May 2010 and May 2015 from our outpatient clinic, Tianlin Street
Community Centre and Fengin Street Community Centre. The control
donors included 129 females (66.6±10.0 years) and 121 males
(59.3±17.9 years). All patients and control subjects belonged to
the Han ethnic group.
Ethics statement
The present study was approved by the Ethics
Committee of the Shanghai Jiao Tong University Affiliated Sixth
People's Hospital (Shanghai, China). All adult participants
provided written informed consent prior to beginning the study. In
addition, written informed consent was provided by the parents on
behalf of the children enrolled in the present study.
Bone densitometry
The bone mineral density (BMD; g/cm2) of
the lumbar spine (L1-L4) was measured using dual-energy X-ray
absorptiometry (DXA). The probands of families 1 (proband 1) and 3
(proband 3) were assessed using Lunar Prodigy equipment (GE Lunar
Corp., Madison, WI, USA). The Lunar device was calibrated daily and
the coefficient of variability (CV) value of the DXA measurements
at L1-4 was 1.39% (15). The
proband of family 2 (proband 2) was assessed using Hologic
Discovery A equipment (Hologic, Bedford, MA, USA). The machine was
calibrated daily. The CV value of the DXA measurements at L1-4 was
0.9% (15). Lumbar spine BMD
results were converted to age- and sex-specific Z-scores according
to the reference data (16).
There are 3 DXAs in the department, including 2 GE Lunar equipment
and 1 Hologic Discovery A equipment. The patients were examined
using the DXAs depending on availability.
Laboratory tests
Fasting blood samples were obtained between 8 and 10
am. Blood was stored for 2-3 h at 25°C and centrifuged at 4°C for
10 min at 4,000 × g to obtain the serum. The following markers of
calcium metabolism were measured: Serum calcium (Ca), phosphorus
(P), alkaline phosphatase (ALP), intact parathyroid hormone (PTH),
and 25 hydroxy vitamin D3 [25(OH)D]. Ca, P and ALP were measured
using a HITACHI7600-020 automatic biochemistry analyser (Hitachi,
Ltd., Tokyo, Japan). All other compounds were measured using the
following kits: Intact PTH kit (Roche Diagnostics GmbH, Mannheim,
Germany) and a 25(OH)D kit (Roche Diagnostics GmbH, Mannheim,
Germany). The intra- and inter-assay CVs, respectively, were 1.5
and 2.0% for Ca, 2.1 and 2.3% for P, 2.5 and 4.5% for ALP, 5.7 and
7.3% for 25(OH)D and 1.4 and 2.9% for PTH as previously reported
(17). Pigment-epithelium-derived
factor (PEDF) was measured using an ELISA reader (R&D Systems,
Inc., Minneapolis, MN, USA). The intra- and inter-assay
coefficients of variation were 8.3 and 9.2%, respectively.
Whole exome capture and massively
parallel DNA sequencing
The exome of proband 1 and proband 3 were sequenced
to identify the pathogenic gene. Exon-enriched DNA was sequenced
using an Illumina Genome Analyser II platform according to the
manufacturer's protocol (Illumina, Inc., San Diego, CA, USA). The
raw image files were processed using Illumina Base Calling Software
v. 1.7 with default parameters and the sequence of each individual
DNA fragment was reported as 101-bp paired-end reads. The
sequencing reads were aligned to the NCBI human reference genome
(NCBI36.3; https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.12/)
using SOAPaligner 2.21 (18-20). The SOAPsnp results were filtered
using the following standards: The base quality was ≥20, the
sequencing depth was between 50-200 and the distance between two
single-nucleotide polymorphisms (SNPs) was >5 bp (21-23). Approximately 114 million reads
were quantified and mapped to the hs37d5 human reference genome DNA
sequence, giving an average read depth of 70.0-96.5 for the exome
of each proband. The percentage of the official target covered with
at least 20X was 89.06%. All 1000G and dbSNP records, the filter
region (including exonic or splicing) and the overlapping
non-coding RNA regions were filtered out. Approximately 11.4 Gb of
high-quality data were aligned to the target regions of the proband
with a per-base mismatch rate of 0.52%. As a result, the mean
coverage sequencing depth on the official target was 79. Reads that
were among the target regions for SNP identification were collected
for subsequent analysis. The consensus sequence and quality of each
allele were calculated by SOAPsnp.
Sanger sequencing analysis
As whole-exome sequencing is expensive, Sanger
sequencing of SERPINF1 was used for other OI probands
without COL1A1 and COL1A2 mutations. Mutations in
SERPINF1 for proband 2 were assessed. The results of exome
sequencing for families 1 and 2 were verified using Sanger
sequencing analysis of the SERPINF1 gene. All eight exons
and the exon-intron boundaries of SERPINF1 were amplified
from genomic DNA via polymerase chain reaction (PR). Patient
sequences were referenced to the Ensembl gene sequence
ENSG00000132386 (SERPINF1; https://www.ncbi.nlm.nih.gov/nuccore/NC_000017.11?from=1761965&to=1777565&report=genbank).
The results of next-generation Sanger sequencing
analysis of the FKBP10 gene were used to verify Family 3.
All eleven exons and the exon-intron boundaries of FKBP10
were amplified from genomic DNA via PCR. Patient sequences were
referenced to the Ensembl gene sequence ENSG00000141756
(FKBP10; https://www.ncbi.nlm.nih.gov/nuccore/NC_000017.11?from=41812262&to=41823217&report=genbank).
Direct sequencing was performed using the BigDye
Terminator Cycle Sequencing Ready Reaction kit, v. 3.1 (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA), and
the sequencing was analysed with an ABI 3730XL automated sequencer
(Applied Biosystems; Thermo Fisher Scientific, Inc.). SNPs were
identified using Polyphred (http://droog.gs.washington.edu/polyphred).
Furthermore, Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org/) and Polymorphism Phenotyping
(POLYPHEN-2; http://genetics.bwh.harvard.edu/pph2/) tools were used
to predict whether an amino acid substitution had an impact on the
biological function of the protein.
Quantitative PCR (qPCR) and detection of
the allelic copy numbers of the two families to verify
deletion
The following primers were used in the present study
and were designed using Primer 3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi):
Primers corresponding to the head site of the targeted gene
SERPINF1, forward 5′-GCC TGC TGG ACG CTG GAT TA-3′ and
reverse 5′-CACCCAGCCTAGTCCCTCTAAGC-3′; primers corresponding to the
mutagenic site of proband 2 on the targeted gene SERPINF1,
forward 5′-CCATCATTCACCGGGCTCTCT-3′ and reverse
5′-CGGGAGGCACTCTTGAGGTTC-3′; primers corresponding to the tail site
of the targeted gene SERPINF1 forward,
5′-TGGCTTTGAGTGGAACGAGGA-3′ and reverse 5′-TGATAGTCCAGCGGAAGGTG-3′.
The mutagenic site of proband 1 was near the tail of the gene
region; as such, family 1 only needed two pairs of primers
corresponding to the head and tail regions. However, family 2
required two pairs of primers corresponding to the head, tail and
mutated regions. All DNA samples from the 2 families were detected
using 2X SYBR Quant Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.) qPCR assays and the reference gene was RPP14.
Thermo cycling conditions were as follows: 95°C for 2 min followed
by 40 cycles of 95°C for 15 sec and 60°C for 15 sec followed by the
dissociation step. All reactions were performed in triplicate. The
allelic copy number of the markers in the muted region was finally
determined with the comparison of normalized data of control
samples and patients using the 2−ΔΔCq method (24).
Results
Clinical features Family 1
Proband 1 was an 11-year-old girl, the first
daughter of non-consanguineous parents. She was the product of a
full-term pregnancy with a normal delivery (Fig. 1), and her birth weight and length
were within normal limits. Her height and weight at the time of the
study were 132.6 cm (Z score, -1.7) and 35.4 kg (Z score, -0.3),
respectively. She experienced her first fracture in the femur at
the age of 2 years and has since experienced 5 subsequent
fractures, including both femurs between 8 and 11 years. Proband 1
has evident deformities in both lower limbs. Blue sclera, brittle
teeth and hearing loss were not observed and no family history of
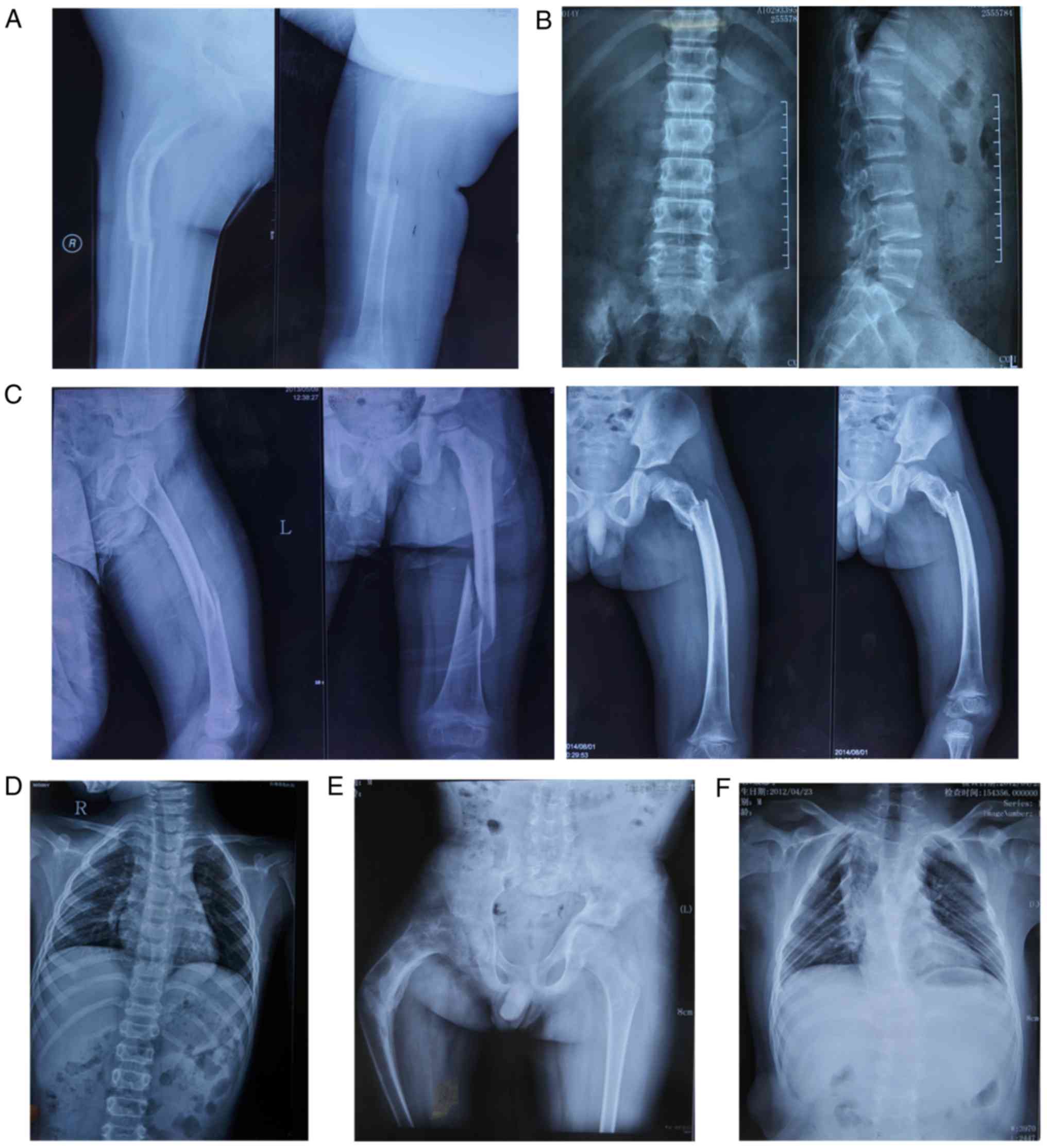
bone fragility was identified. Radiographs revealed thin cortices
and normal diaphyseal modelling with bending deformities in both
femurs (Fig. 2A). Radiographs of
the thoracic vertebra and lumbar vertebra of proband 1 revealed
multiple vertebral wedging, she did not feel any pain (Fig. 2B). Proband 1 was demonstrated to
have a lower height and lumbar spine bone mass compared with
healthy donors of the same age (Table
I). Biological test results, including serum Ca, P, ALP and PTH
were within normal ranges, whereas 25(OH)D levels were below the
normal range (Table I). Her
parents did not show any symptom of OI, except that her father was
below average height (159.9 cm; Z score, -1.9).
 | Table IGeneral features and laboratory
findings of the probands. |
Table I
General features and laboratory
findings of the probands.
| Parameter | Proband 1 | Proband 2 | Proband 3 | Normal values |
|---|
| Sex | F | M | M | |
| Age (years) | 11 | 4 | 12 | |
| Blue sclera | − | + | − | |
| Dentinogenesis
imperfecta | − | − | − | |
| Hearing loss | − | − | − | |
| Site of
fracture | B | L | R | |
| First fracture age
(years) | 2 | 3 | 10 | |
| Times of
fracture | 6 | 2 | 5 | |
| Height Z-score | −1.7a − | 0.3 | −1.9a | >–1 |
| Weight Z-score | −0.3 | −0.1 | −0.7 | >–1 |
| Lumbar spine 1-4
Z-score | −2.9a − | −2.6a | 5.9a | >–1 |
| Calcium
(mmol/l) | 2.42 | 2.49 | 2.40 | 2.08–2.60 |
| Phosphate
(mmol/l) | 1.39 | 1.27 | 1.02 | 0.80–1.60 |
| Alkaline phosphate
(U/)l | 224 | 365 | 350 | 116–380 |
| Parathyroid hormone
(pg/ml) | 58.0 | 29.3 | 54 | 15.0–65.0 |
| 25(OH)D
(ng/ml) | 16a | 34 | 18a | >30 |
Family 2
Proband 2 was a 4-year-old boy, the only son of
non-consanguineous parents (Fig.
1). He was the product of a full-term pregnancy with a normal
delivery, and his birth weight and length were within normal
limits. His height and weight at the time of the study were 102.4
cm (Z score, -0.3) and 16.1 kg (Z score, -0.1), respectively. He
experienced his first fracture in the left upper femur at the age
of 3 years. Subsequently, he experienced another fracture in the
left middle femur at the age of 4 years. Proband 2 presented with
blue sclera without brittle teeth or hearing loss. Radiographs
revealed thin cortices, normal diaphyseal modelling in the left
femur and multiple vertebral wedging (Fig. 2C and D). Proband 2 had a decreased
lumbar spine bone mass compared with healthy controls of the same
age. All biological test results were within normal ranges
(Table I). His parents did not
exhibit any symptoms of OI.
Family 3
Proband 3 was a 12-year-old boy, the only son of
non-consanguineous parents (Fig.
1). He was the product of a full-term pregnancy with a normal
delivery, and his birth weight and length were within normal
limits. His height and weight at the time of the study were 140.0
cm (Z score, -1.9) and 34.0 kg (Z score, -0.7), respectively. He
experienced his first fracture in the right upper femur at the age
of 10 years, with 4 subsequent fractures of the right femur.
Proband 3 was unable to walk without the aid of auxiliary equipment
following the first fracture. He did not present with blue sclera,
brittle teeth, hearing loss or congenital large joint contracture.
No family history of bone fragility was identified. Radiographs
revealed thin cortices, normal diaphyseal modelling and bending
deformities in the right femur, whilst a chest X-ray identified
light scoliosis (Fig. 2E and F).
Proband 3 had a lower lumbar spine bone mass compared with healthy
controls of the same age. Biological test results, including serum
Ca, P, ALP and PTH, were within normal ranges. However, proband 3
was identified to have a 25(OH)D deficiency (Table I). His parents did not show any
other symptoms.
Whole exome capture and massively
parallel DNA sequencing
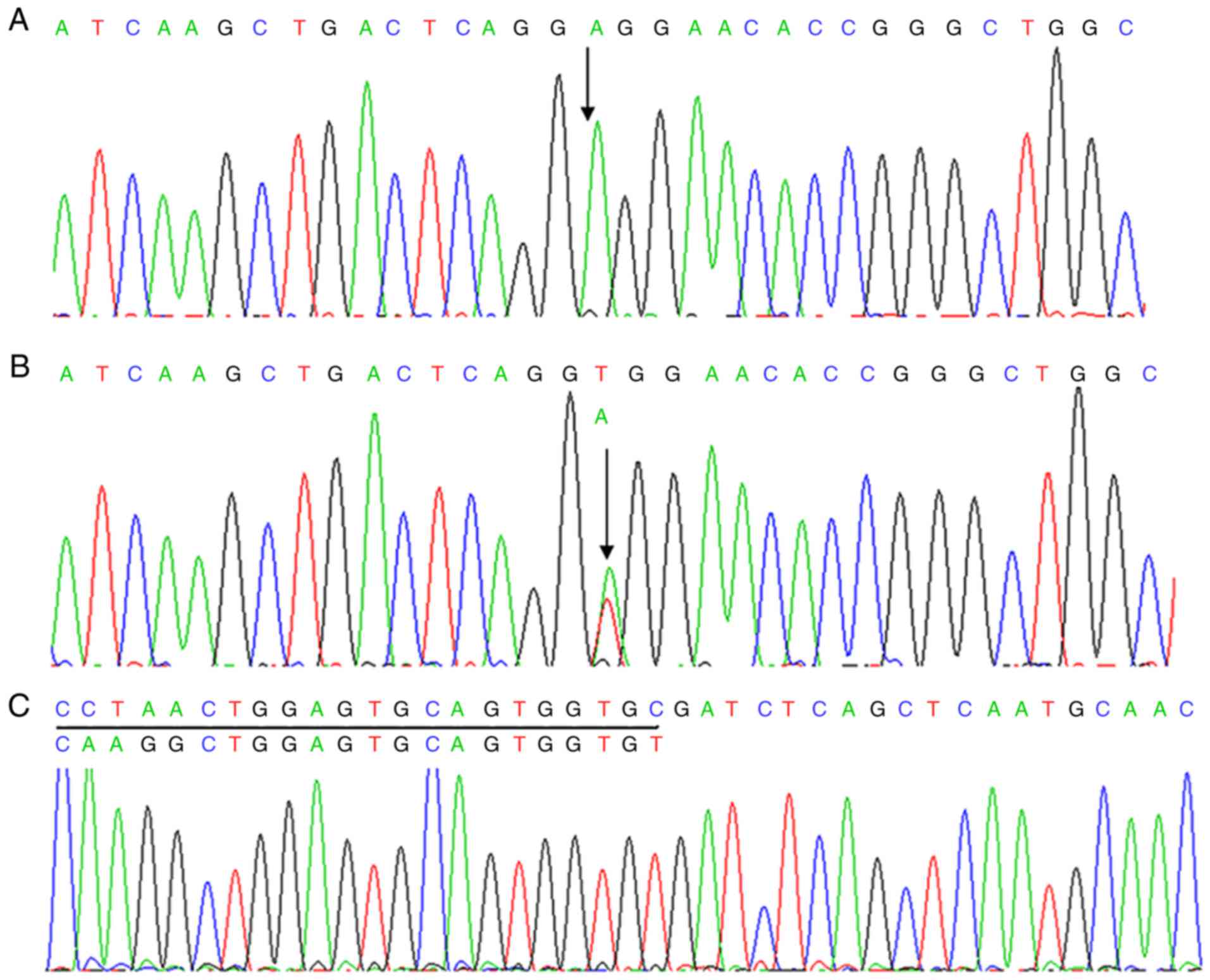
A novel missense substitution was identified in the
SERPINF1 gene of proband 1. This mutation was homozygous
according to the autosomal recessive mode of inheritance. The
homozygous T to A transition at c.1067 in exon 8 of SERPINF1
(Fig. 3) resulted in a valine to
glutamic acid substitution at p.356 (V356E). This homozygous
missense mutation occurred at a highly conserved position (Fig. 4). The results revealed that this
missense mutation was deleterious with a PROVEAN score of -4.334. A
similar result was obtained when using POLYPHEN-2.
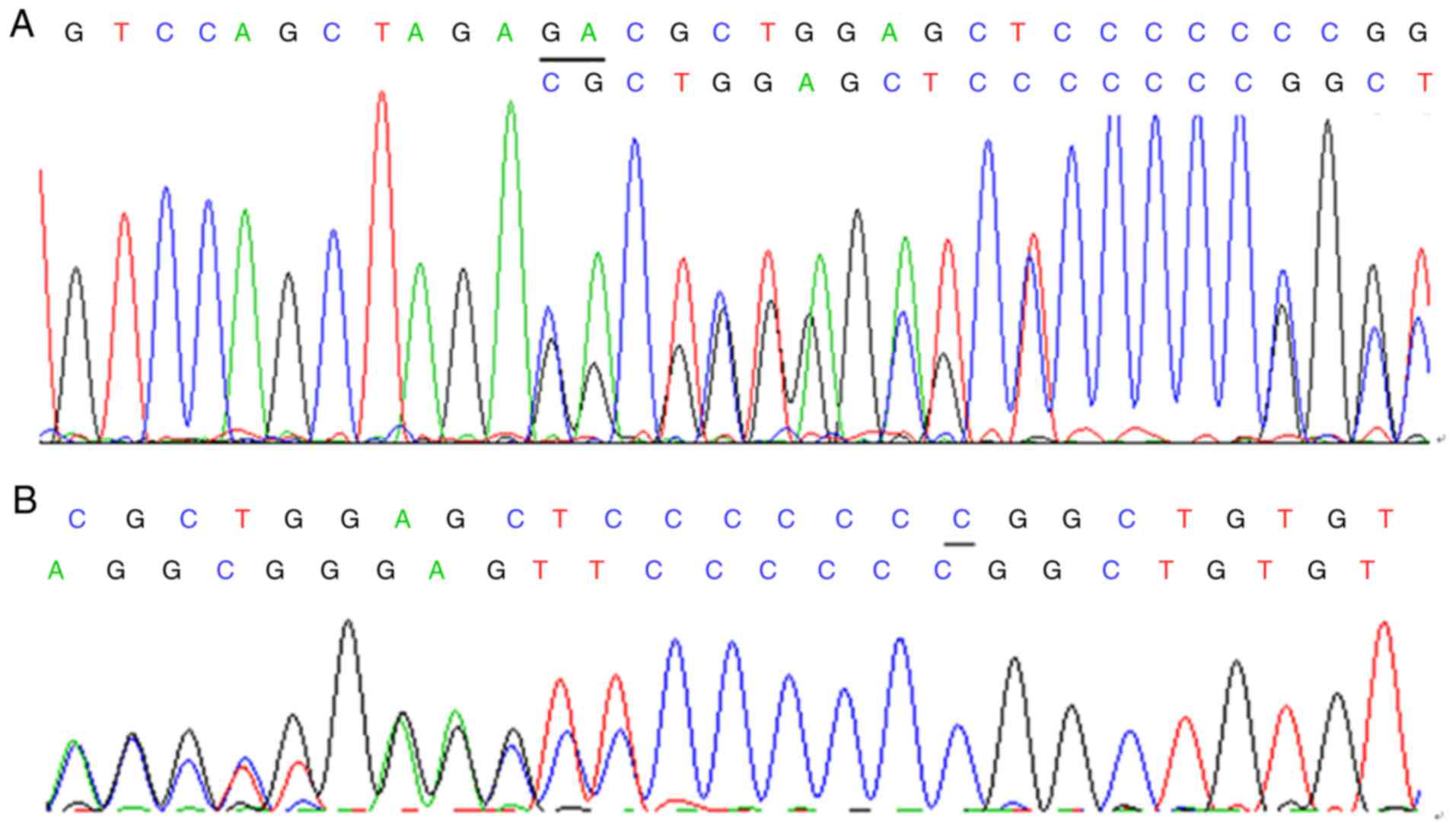
A novel compound heterozygous mutation that
consisted of a nonsense mutation (c.813_814delGA,
p.Glu271AspfsX101) and another nonsense mutation (c.831delC,
p.Gly278AlafsX20) was identified in exon 5 of the FKBP10 gene in
proband 3 (Fig. 5A and B).
Validation of the SERPINF1 and FKBP10
germline mutation
The Sanger sequencing results for the
SERPINF1 gene were consistent with the whole exome
sequencing results. A homozygous V356E (c.1067T>A) mutation was
identified in proband 1. The heterozygous V356E (c.1067T>A)
mutation was found in proband 1's father (Fig. 3B). However, no mutation in
SERPINF1 was observed in the proband's mother, and no V356E
mutation was identified in the control subjects. According to OI
variant databases (https://oi.gene.le.ac.uk/variants.php?select_db=SERPINF1&action=view_all),
the identified mutation is novel.
A homozygous p.Ala96_Gly215del (c.283+473_643+
104del) mutation of SERPINF1 was identified in proband 2
(Fig. 3C). The same heterozygous
mutation was identified in the proband's father, whereas no
mutation was observed in the proband's mother. The same compound
heterozygous mutation was identified in proband 3 by further Sanger
sequencing. The heterozygous mutation (c.831delC, p.Gly278AlafsX20)
of FKBP10 was present in proband 3's father, whereas the
heterozygous mutation (c.813_814delGA, p.Glu271AspfsX101) was
identified in the proband's mother.
In the present study, qPCR was repeatedly performed
to detect allelic copy numbers of both families and verify whether
there were deletions. The results for family 1 revealed a deletion
of SERPINF1 in the proband and her mother. However, the
results for family 2 demonstrated that there was no SERPINF1
deletion in the proband or her mother. Furthermore, the genotypes
of all SNPs of proband 2 were homozygous. This suggests that, due
to the uniparental disomy, the region of the chromosome of the
proband of family 2 was derived from the father although the total
copy number was unchanged.
Detection of serum PEDF
PEDF was undetectable in serum samples from proband
1. Serum samples were not obtained from her parents as initially
only the whole blood of the parents was reserved and they did not
return to have the serum extracted. Serum PEDF was also
undetectable in proband 2. However, for proband 2's father and
mother, the PEDF serum concentration was 13.8 and 37.8
μg/ml, respectively. PEDF serum concentrations were
determined for 10 normal subjects. The median value was 22.4
μg/ml, the minimum was 12.0 μg/ml and the maximum was
39.5 μg/ml.
Discussion
The first causal gene of recessive OI, CRTAP
(MIM 605497), was reported in 2006 (25,26). A number of additional genes,
including LEPRE1 (MIM 610339), PPIB (MIM 123841),
FKBP10 (MIM 607063), SERPINH1 (MIM 600943), and
SP7 (MIM 606633) mutations, have since been reported to
cause severe or lethal autosomal recessive OI (8,27-30). Becker et al (7) identified a lack of homozygosity of
the SERPINF1 gene product, PEDF, which caused severe OI in
four patients. Alanay et al (8) first discovered homozygosity for
mutations in FKBP10 in a cohort of five consanguineous
Turkish families and a Mexican-American family, causing
autosomal-recessive OI.
The SERPINF1 gene is located on chromosome
17p13.3 and encodes PEDF, which is a glycoprotein of the Serpin
superfamily secreted by retinal pigment epithelial cells (7). In the musculoskeletal system,
collagen-1 is bound by PEDF, which may act as an inhibitor of bone
resorption by inhibiting osteoclast maturation via osteoprotegerin
and receptor activator of nuclear factor-kB ligand (7). Li et al (31) observed that PEDF suppressed the
expression of genes that inhibit mineralization, leading to
enhanced osteoblastic differentiation and increased matrix
mineralization. In the present study, probands 1 and 2 experienced
their first fracture when they were older than 1 year. Both
probands suffered recurrent fractures of the femur, with proband 1
more severely affected than proband 2. Proband 1 was 11 years old
and proband 2 was 4 years old at the time of the present study;
this discrepancy in age may be responsible for the difference in
phenotypic severity of OI. The sclera of proband 1 was white,
whereas that of proband 2 was faintly blue. Both probands lacked
dentinogenesis imperfecta. The phenotype of Chinese patients in the
present study was similar to that of other patients reported in
previous studies (5,7,13,32-35). Both probands were able to walk
independently during the intermittent period between fractures,
although they fractured their femurs many times. However, Caucasian
patients with OI typically experience severe limb deformity and are
mostly unable to walk independently (5,7,32-35). Phenotypes of Korean patients with
type VI OI and Inuit patients have also been reported to be more
severe compared with those presented in this study (5,35).
Probands 1 and 2 presented with multiple vertebral wedging, however
they did not report experiencing backache or lumbago. Moderate to
severe bone fragility was observed in Chinese patients by Wang
et al (13), further
supporting the hypothesis that the phenotype of Chinese patients
with OI is less severe compared with Caucasian and Korean
patients.
To date, a total of 82 mutations in the
SERPINF1 gene, including 37 substitutions, 23 deletions, 20
duplications and 2 insertions, have been described (https://oi.gene.le.ac.uk/variants.php?select_db=SERPINF1&action=view_unique).
The novel missense mutation identified in proband 1 consisted of a
homozygous T to A transition at c.1067 in exon 8 (V356E) of
SERPINF1. In proband 2, the novel deletion mutation
identified comprised a homozygous p.Ala96_Gly215del
(c.283+473_643+104del) deletion of SERPINF1, including the
loss of intron 4, exon 4, exon 5 and part of introns 3 and 5. At
present, this mutation is the only large deletion identified that
has been identified among mutations of the SERPINF1 gene.
The father of proband 2 was demonstrated to have a heterozygous
deletion of SERPINF1 with no symptoms of OI and normal
stature. This indicates that carriers are asymptomatic despite the
large fragment deletion. Proband 2 had comparatively mild symptoms;
he was able to walk, jump and play with other children during the
intermittent period between fractures. Furthermore, his height is
within the normal range. The association between the severe
mutation type and relatively mild phenotype of this proband should
be further studied. Rauch et al (36) reported that the absence of
circulating PEDF was specific to OI type VI. In the present study,
probands 1 and 2 had undetectable serum concentrations of PEDF,
which indicates that their mutations were pathogenic. In addition,
the mutations occurred at a highly conserved position and were
predicted as deleterious by PROVEAN and polyphen software.
Interestingly, the families of probands 1 and 2 in
the present study exhibited nonpaternity. Both probands had
homozygous mutations in the SERPINF1 gene, whilst the
fathers from both families showed heterozygous mutations. However,
both mothers had no mutation on this allele. The mothers may
therefore have a deleted SERPINF1 allele. The results of
allelic copy number detection revealed that proband 1 and her
mother exhibited a SERPINF1 deletion, meaning that family 1
did not deviate from Mendelian inheritance. However, no deletion of
SERPINF1 was observed in family 2. Furthermore, the
genotypes of all SNPs of proband 2 were homozygous. It was
therefore assumed that the uniparental disomy of proband 2 was in
part because his chromosome originated from his father, although
the total copy number was unchanged. Wang et al (13) also reported this phenomenon in a
family that included a proband with a homozygous in-frame
duplication, a heterozygous carrier mother and a father without
mutations. It was suggested that Sanger sequencing of this
phenomenon may reveal nonpaternity or partial de novo
mutation.
The first FKBP10 mutations were identified in
families with a moderate to severe form of OI without contractures
or webbing (8). Steinlein et
al (37) also described
patients with FKBP10 mutations affected by severe autosomal
recessive OI without contractures. However, Shaheen et al
(38) discovered two brothers
with Bruck syndrome, in which multiple joint contracture was caused
by a mutation in the FKBP10 gene. Kelley et al
(9) concluded that FKBP10
mutations are the cause of recessive OI and Bruck syndrome. Zhou
et al (14) also reported
a Chinese patient with OI and Bruck syndrome resulting from
mutations in the FKBP10 gene. In the present study, the
patients presented with moderate OI without contractures or
webbing. The majority of patients with OI experience their first
fractures soon after birth or in infancy (8,14,37). However, in the present study,
proband 3 suffered his first fracture when he was 10 years old,
consistent with the phenotype described by Schwarze et al
(39). Proband 3 did not have
blue sclerae or dentinogenesis imperfecta and presented with
scoliosis that developed during adolescence. His fracture was
restricted on the right femur and resulted in deformity in this
area. The results of the present study verified that FKBP10
may cause Bruck syndrome or isolated OI. In addition, Schwarze
et al (39) reported that
isolated OI and Bruck syndrome may occur in siblings of the same
family.
A total of 119 variants in the FKBP10 gene
have been identified, including 32 substitutions, 17 deletions, 62
duplications and 8 insertion/deletions and mutations primarily
located in exon 5 (32.04%) and exon 6 (22.33%) (https://oi.gene.le.ac.uk/variants_statistics.php).
The novel compound heterozygous mutation in proband 3 described
herein was located in exon 5. Of these mutations, c.831dupC and
c.948dupT occur most frequently, having been reported 21 and 19
times, respectively (https://oi.gene.le.ac.uk/variants.php?select_db=FKBP10&action=view_unique).
In particular, the mutation c.831dupC has been reported in
different origins, including the families of Turkish, Mexican,
South African and Caucasian origin (8,9,39).
In the present study, proband 3 also had a nonsense mutation
located at c.831, with a deleted C instead of a duplicate C. These
results suggest that codon 831 of the FKBP10 gene may
represent a mutation hotspot for human OI.
In conclusion, the results of the present study
indicate that a novel missense mutation, c.1067T>A, in exon 8
and a large fragment deletion mutation, c.283+473_643+104del, in
the SERPINF1 gene cause type VI autosomal recessive OI in
Chinese patients. The present study also demonstrated that a
patient may experience mild symptoms of OI despite having a large
fragment deletion in the SERPINF1 gene. Furthermore, the
phenotype of Chinese patients with type VI OI appears to be less
severe compared with Caucasian and Korean patients. Deletion of the
SERPINF1 gene, as observed in one of the proband's parents,
or the uniparental disomy of the proband may result from one
heterozygous parent carrier and one parent without identified
mutation producing a child with a homozygous mutation. The novel
compound heterozygous mutations c.831delC and c.813_814delGA in
FKBP10 are responsible for moderate OI without Bruck
syndrome in Chinese patients. Codon 831 of the FKBP10 gene
may represent a mutation hotspot for OI in humans. The results of
the present study may improve the clinical and pathogenic gene
spectrum of recessively inherited forms of OI in China.
Acknowledgments
The authors would like to thank the Center for
Genetic & Genomic Analysis, Genesky Biotechnologies Inc.
(Shanghai, China) for their assistance with gene
identification.
Notes
[1]
Funding
The present study was supported by the National
Basic Research Program of China (grant no. 2014CB942903), National
Natural Science Foundation of China (NSFC; grant nos. 81370978 and
30800387), and Chongqing City Fundamental And Advanced Research
Projects (grant no. CSTC2013jcyjC00009) and the Science and
Technology Commission of Shanghai municipality (grant nos.
14JC1405000 and 14ZR1431900).
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
ZZ recruited patients, designed the study, revised
and approved the final version of the manuscript. HZ recruited
patients, performed experiments, and wrote the manuscript. YX, HY
and CW performed experiments and revised the manuscript. JG, JH, WF
and WH performed experiments and analyzed the data. All authors
read and approved the final manuscript.
[4] Ethics
approval and consent to participate
The present study was approved by the Ethics
Committee of the Shanghai Jiao Tong University Affiliated Sixth
People's Hospital (Shanghai, China). All adult participants
provided written informed consent prior to beginning the study. In
addition, written informed consent was provided by the parents on
behalf of the children enrolled in the present study.
[5] Consent for
publication
All the probands and their parents consented to
publication of this study.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Rauch F and Glorieux FH: Osteogenesis
imperfecta. Lancet. 363:1377–1385. 2004. View Article : Google Scholar
|
|
2
|
Sillence DO and Rimoin DL: Classification
of osteogenesis imperfect. Lancet. 1:1041–1042. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cheung MS and Glorieux FH: Osteogenesis
imperfecta: Update on presentation and management. Rev Endoc Metab
Disord. 9:153–160. 2008. View Article : Google Scholar
|
|
4
|
Glorieux FH, Rauch F, Plotkin H, Ward L,
Travers R, Roughley P, Lalic L, Glorieux DF, Fassier F and Bishop
NJ: Type V osteogenesis imperfecta: A new form of brittle bone
disease. J Bone Miner Res. 15:1650–1658. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Glorieux FH, Ward LM, Rauch F, Lalic L,
Roughley PJ and Travers R: Osteogenesis imperfecta type VI: A form
of brittle bone disease with a mineralization defect. J Bone Miner
Res. 17:30–38. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tucker T, Nelson T, Sirrs S, Roughley P,
Glorieux FH, Moffatt P, Schlade-Bartusiak K, Brown L and Rauch F: A
co-occurrence of osteogenesis imperfecta type VI and cystinosis. Am
J Med Genet A. 158A. pp. 1422–1426. 2012, View Article : Google Scholar
|
|
7
|
Becker J, Semler O, Gilissen C, Li Y, Bolz
HJ, Giunta C, Bergmann C, Rohrbach M, Koerber F, Zimmermann K, et
al: Exome sequencing identifies truncating mutations in human
SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum
Genet. 88:362–371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alanay Y, Avaygan H, Camacho N, Utine GE,
Boduroglu K, Aktas D, Alikasifoglu M, Tuncbilek E, Orhan D, Bakar
FT, et al: Mutations in the gene encoding the RER protein FKBP65
cause autosomal-recessive osteogenesis imperfecta. Am J Hum Genet.
86:551–559. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kelley BP, Malfait F, Bonafe L, Baldridge
D, Homan E, Symoens S, Willaert A, Elcioglu N, Van Maldergem L,
Verellen-Dumoulin C, et al: Mutations in FKBP10 cause recessive
osteogenesis imperfecta and Bruck syndrome. J Bone Miner Res.
26:666–672. 2011. View Article : Google Scholar
|
|
10
|
Valadares ER, Carneiro TB, Santos PM,
Oliveira AC and Zabel B: What is new in genetics and osteogenesis
imperfecta classification? J Pediatr. 90:536–541. 2014. View Article : Google Scholar
|
|
11
|
Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ,
Yu JB, Gu JM, Hu WW, Wang C, He JW and Fu WZ: The identification of
novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese
patients with osteogenesis imperfecta. J Bone Miner Metab.
30:69–77. 2012. View Article : Google Scholar
|
|
12
|
Zhang H, Yue H, Wang C, Hu W, Gu J, He J,
Fu W, Hu Y, Li M and Zhang Z: Clinical characteristics and the
identification of novel mutations of COL1A1 and COL1A2 in 61
Chinese patients with osteogenesis imperfecta. Mol Med Rep.
14:4918–4926. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang JY, Liu Y, Song LJ, Lv F, Xu XJ, San
A, Wang J, Yang HM, Yang ZY, Jiang Y, et al: Novel mutations in
SERPINF1 result in rare osteogenesis imperfecta type VI. Calcif
Tissue Int. 100:55–66. 2017. View Article : Google Scholar
|
|
14
|
Zhou P, Liu Y, Lv F, Nie M, Jiang Y, Wang
O, Xia W, Xing X and Li M: Novel mutations in FKBP10 and PLOD2
cause rare Bruck syndrome in Chinese patients. PloS One.
9:e1075942014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, He JW, Gao G, Yue H, Yu JB, Hu
WW, Gu JM, Hu YQ, Li M, Fu WZ, et al: Polymorphisms in the HOXD4
gene are not associated with peak bone mineral density in Chinese
nuclear families. Acta Pharmacol Sin. 31:977–983. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maynard LM, Guo SS, Chumlea WC, Roche AF,
Wisemandle WA, Zeller CM, Towne B and Siervogel RM: Total-body and
regional bone mineral content and areal bone mineral density in
children aged 8-18 y: The Fels Longitudinal study. Am J Clin Nutr.
68:1111–1117. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu HK, Zhang Z, Ke YH, He JW, Fu WZ, Zhang
CQ and Zhang ZL: High prevalence of vitamin D insufficiency in
China: Relationship with the levels of parathyroid hormone and
markers of bone turnover. PloS One. 7:e472642012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li R, Li Y, Kristiansen K and Wang J:
SOAP: Short oligonucleotide alignment program. Bioinformatics.
24:713–714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Z, Xia W, He J, Zhang Z, Ke Y, Yue
H, Wang C, Zhang H, Gu J, Hu W, et al: Exome sequencing identifies
SLCO2A1 mutations as a cause of primary hypertrophic
osteoarthropathy. Am J Hum Genet. 90:125–132. 2012. View Article : Google Scholar :
|
|
20
|
Li Y, Vinckenbosch N, Tian G,
Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao
H, Korneliussen T, et al: Resequencing of 200 human exomes
identifies an excess of low-frequency non-synonymous coding
variants. Nat Genet. 42:969–972. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shi Y, Li Y, Zhang D, Zhang H, Li Y, Lu F,
Liu X, He F, Gong B, Cai L, et al: Exome sequencing identifies
ZNF644 mutations in high myopia. PLoS Genet. 7:e10020842011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Morello R, Bertin TK, Chen Y, Hicks J,
Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH,
Vranka J, et al: CRTAP is required for prolyl 3-hydroxylation and
mutations cause recessive osteogenesis imperfecta. Cell.
127:291–304. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barnes AM, Chang W, Morello R, Cabral WA,
Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, et
al: Deficiency of cartilage-associated protein in recessive lethal
osteogenesis imperfecta. N Engl J Med. 355:2757–2764. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Willaert A, Malfait F, Symoens S, Gevaert
K, Kayserili H, Megarbane A, Mortier G, Leroy JG, Coucke PJ and De
Paepe A: Recessive osteogenesis imperfecta caused by LEPRE1
mutations: Clinical documentation and identification of the splice
form responsible for prolyl 3-hydroxylation. J Med Genet.
46:233–241. 2009. View Article : Google Scholar
|
|
28
|
van Dijk FS, Nesbitt IM, Zwikstra EH,
Nikkels PG, Piersma SR, Fratantoni SA, Jimenez CR, Huizer M,
Morsman AC, Cobben JM, et al: PPIB mutations cause severe
osteogenesis imperfecta. Am J Hum Genet. 85:521–527. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Christiansen HE, Schwarze U, Pyott SM,
AlSwaid A, Al Balwi M, Alrasheed S, Pepin MG, Weis MA, Eyre DR and
Byers PH: Homozygosity for a missense mutation in SERPINH1, which
encodes the collagen chaperone protein HSP47, results in severe
recessive osteogenesis imperfecta. Am J Hum Genet. 86:389–398.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lapunzina P, Aglan M, Temtamy S,
Caparrós-Martín JA, Valencia M, Letón R, Martínez-Glez V, Elhossini
R, Amr K, Vilaboa N and Ruiz-Perez VL: Identification of a
frameshift mutation in Osterix in a patient with recessive
osteogenesis imperfecta. Am J Hum Genet. 87:110–114. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li F, Song N, Tombran-Tink J and Niyibizi
C: Pigment epithelium derived factor suppresses expression of
Sost/Sclerostin by osteocytes: Implication for its role in bone
matrix mineralization. J Cell Physiol. 230:1243–1249. 2015.
View Article : Google Scholar
|
|
32
|
Homan EP, Rauch F, Grafe I, Lietman C,
Doll JA, Dawson B, Bertin T, Napierala D, Morello R, Gibbs R, et
al: Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J
Bone Miner Res. 26:2798–2803. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Venturi G, Gandini A, Monti E, Dalle
Carbonare L, Corradi M, Vincenzi M, Valenti MT, Valli M, Pelilli E,
Boner A, et al: Lack of expression of SERPINF1, the gene coding for
pigment epithelium-derived factor, causes progressively deforming
osteogenesis imperfecta with normal type I collagen. J Bone Miner
Res. 27:723–728. 2012. View Article : Google Scholar
|
|
34
|
Caparrós-Martin JA, Valencia M, Pulido V,
Martínez-Glez V, Rueda-Arenas I, Amr K, Farra C, Lapunzina P,
Ruiz-Perez VL, Temtamy S and Aglan M: Clinical and molecular
analysis in families with autosomal recessive osteogenesis
imperfecta identifies mutations in five genes and suggests
genotype-phenotype correlations. Am J Med Genet A. 161A:1354–1369.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cho SY, Ki CS, Sohn YB, Kim SJ, Maeng SH
and Jin DK: Osteogenesis imperfecta Type VI with severe bony
deformities caused by novel compound heterozygous mutations in
SERPINF1. J Korean Med Sci. 28:1107–1110. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rauch F, Husseini A, Roughley P, Glorieux
FH and Moffatt P: Lack of circulating pigment epithelium-derived
factor is a marker of osteogenesis imperfecta type VI. J Clin
Endocrinol Metab. 97:E1550–E1556. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Steinlein OK, Aichinger E, Trucks H and
Sander T: Mutations in FKBP10 can cause a severe form of isolated
Osteogenesis imperfecta. BMC Med Genet. 12:1522011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shaheen R, Al-Owain M, Sakati N, Alzayed
ZS and Alkuraya FS: FKBP10 and Bruck syndrome: Phenotypic
heterogeneity or call for reclassification? Am J Hum Genet.
87:306–307; author reply 308. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schwarze U, Cundy T, Pyott SM,
Christiansen HE, Hegde MR, Bank RA, Pals G, Ankala A, Conneely K,
Seaver L, et al: Mutations in FKBP10, which result in Bruck
syndrome and recessive forms of osteogenesis imperfecta, inhibit
the hydroxylation of telopeptide lysines in bone collagen. Hum Mol
Genet. 22:1–17. 2013. View Article : Google Scholar :
|