Introduction
Bone remodeling involves the resorption of old bone
by osteoclasts and the formation of new bone by osteoblasts. Normal
bone physiology requires a balance between the coupled processes of
bone resorption and bone formation (1). In these processes, osteocytes
including osteoblasts and osteoclasts are involved in the
development, growth, and remodeling of bones. Disturbance in the
balance of the remodeling process results in osteopenic disorders,
including osteoporosis, rheumatoid arthritis and Paget's disease
(2).
Regulation of bone remodeling occurs through
multiple mechanisms that ultimately converge at the interaction of
osteoclasts or their precursors with osteoblasts and bone marrow
stromal cells. Osteoblasts are the bone-lining cells that are
responsible for the production of bone matrix components and
minerals during bone formation (3). They are regulated by various
transcription factors, including runt-related transcription factor
2 (Runx2), osterix and β-catenin (4).
Osteoclast precursors, such as bone marrow-derived
macrophages, express the receptors for macrophage colony
stimulating factor (M-CSF) and for receptor activator of nuclear
factor-kB ligand (RANKL), and differentiate into osteoclasts in the
presence of M-CSF and RANKL expressed by osteoblasts (5). RANKL/receptor activator of nuclear
factor-kB (RANK) signaling induces osteoclast differentiation and
activation via various transcription factors, such as nuclear
factor (NF)-κB, Fos proto-oncogene (c-Fos), and nuclear factor of
activated T-cell (NFATc1) (6,7).
c-Fos is an essential factor for the activation of NFATc1, which is
a master regulator of RANKL-induced osteoclast differentiation; it
regulates the expression of osteoclast-specific genes, including
tartrate-acid resistant acid phosphatase (TRAP), Cathepsin K,
Atp6v0d2, and osteoclast-associated receptor (OSCAR) (8).
Polygoni Multiflori Radix (PMR) is the root
of Polygonum multiflorum Thunb., and it is widely used in
East Asia. PMR exhibits a variety of pharmacological effects,
including acetylcholinesterase inhibitory activity,
neuroprotective, antioxidant, immunomodulatory, antihyperlipidemic,
anticancer, anti-inflammatory and hepatoprotective activities
(9). PMR is used in Korean
medicine to treat bone diseases by balancing the functions of
osteoblasts and osteoclasts. Specifically, water extract of PMR
exhibits antiosteoporotic efficacy in ovariectomized (OVX)-induced
osteoporosis in mice (10,11).
However, the exact signaling mechanism that leads to PMR-mediated
bone remodeling and osteoclast/osteoblast differentiation remains
unclear. In the present study, the effects of PMR on osteoclast
differentiation were investigated in RANKL-treated bone
marrow-derived macrophages (BMMs) and in ascorbic
acid/β-glycerophosphate (AA/β-GP)-induced osteoblast
differentiation. In addition, the inhibitory effect of PMR was
investigated in an animal model of lipopolysaccharide (LPS)-induced
bone loss to evaluate the in vivo efficacy of PMR.
Materials and methods
Reagents
PMR was purchased from Omniherb Corporation (Daegu,
Korea; Fig. 1). α-minimum
essential medium (α-MEM), fetal bovine serum (FBS), and antibiotics
were purchased from Gibco (Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The XTT assay kit was purchased from Roche Diagnostics
GmbH (Mannheim, Germany). Human recombinant M-CSF and RANKL were
purchased from PeproTech EC, Ltd. (London, UK). Antibodies against
c-Fos (1:1,000 dilution; cat. no. SC-7202), NFATc1 (1:1,000
dilution; cat. no. SC-7294) β-actin (1:1,000 dilution; cat. no.
SC-47778), phosphorylated (p-) SMAD family member (Smad) 1/5/8
(1:1,000 dilution; cat. no. SC-12353), and total Smad1/5/8 (1:1,000
dilution; cat. no. SC-6031-R), were purchased from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). Antibodies against
p-extracellular signal-regulated kinase (ERK; 1:1,000 dilution;
cat. no. 4370), ERK (1:1,000 dilution; cat. no. 9102), p-c-Jun
N-terminal kinase (JNK; 1:1,000 dilution; cat. no. 9251), JNK
(1:1,000 dilution; cat. no. 9252), p-p38 (1:1,000 dilution; cat.
no. 9211), p38 (1:1,000 dilution; cat. no. 9212), p-inhibitor of κB
(IκB; 1:1,000 dilution; cat. no. 2859), IκB (1:1,000 dilution; cat.
no. 4812), p-p65 NF-κB (1:1,000 dilution; cat. no. 3033), and p65
NF-κB (1:1,000 dilution; cat. no. 8242) were purchased from Cell
Signaling Technology Inc. (Beverly, MA, USA). Ascorbic acid (AA)
and β-glycerophosphate (β-GP) were purchased from Sigma-Aldrich
(Merck KGaA, Darmstadt, Germany).
Mice
Five-week-old male ICR mice were purchased from
Samtako Bio, Inc. (Osan, Korea). Mice were housed in a laminar
air-flow room maintained at a temperature of 22–24°C and a reactive
humidity of 55–60% with a 12-h light/dark cycle. All experiments
were performed in accordance with the guidelines of and approved by
the Institutional Animal Care and Use Committee of Wonkwang
University (approval no. WKU15-143).
Preparation of PMR
Preparation of PMR water extract was conducted
following the extraction protocol of Korean Plant Extract Bank
(Cheongju, Korea). PMR was dissected into small pieces, placed in
distilled water for 30 min, and then boiled using Glas-Col heating
mantle (Glas-Col LLC, Terre Haute, IN, USA) for 2 h. The extract
was filtered using a filter paper (110 mm; Advantec no. 2), and the
filtrate was concentrated at 60°C using a rotary evaporator (Buchi
Labortechnik AG, Flawil, Switzerland) and then lyophilized by a
Bondiro Freeze Dryer (IlShinBio, Dongducheon, Korea) into a dry
powder (yield, 17.9%) (12,13). The dry powder was resuspended in
distilled water, and then filtered through a 0.2 µm filter.
The PMR extract samples were deposited at the College of Pharmacy,
Wonkwang University for future use (voucher no. PMR2014).
Ultra performance liquid chromatography
(UPLC) analysis
Identification and quantification of the
constituents in the PMR extract was performed by a UPLC system that
consisted of a 1290 Infinity UPLC system (Agilent Technologies,
Inc., Santa Clara, CA, US), with Binary pump and Diode Array
Detector (DAD). The chromatographic separation was performed on C18
RP column (Halo C18 RP; 2.7 µm; 4.6×100 mm). Two solvents,
solvent A and B, were used for the mobile phases of UPLC. Solvent A
was 10 mM phosphate buffer in H2O and solvent B was 100%
acetonitrile. The gradient elution was progressed at a flow rate of
1.0 ml/min under the program: (A)/(B) = 90/10 for 0 min, followed
by 90/10 for 5 min, and 40/60 for 25 min and hold for 4 min. The UV
wavelength of the detector was fixed at 210 nm and monitored for 25
min. Column temperature was maintained constantly at 40°C. The PMR
extract and standard were dissolved in methanol and filtered using
a 0.2 µm membrane filter (Millipore; Merck KGaA, Billerica,
MA, USA).
Cell culture and osteoclast
differentiation
Bone marrow cells (BMCs) were obtained by flushing
the femurs and tibiae of 5-week-old ICR mice as described
previously (14). To obtain BMMs,
BMCs were seeded on culture dishes in α-MEM supplemented with 10%
FBS and M-CSF (10 ng/ml) and cultured for 1 day. Nonadherent cells
were transferred to 10 cm petri dishes and further cultured in the
presence of M-CSF (30 ng/ml) for 3 days. Floating cells were
discarded and adherent cells on dish bottoms were classified as
BMMs, which are osteoclast precursors. To induce BMMs
differentiation into osteoclasts, BMMs were cultured for 3 days
with M-CSF (30 ng/ml) and RANKL (100 ng/ml) in the presence or
absence of PMR. Then, cells were fixed with 3.7% formaldehyde for
10 min, permeabilized with 0.1% Triton X-100 for 10 min, and
stained for 30 min at 37°C with TRAP solution [1 mg/ml fast red
violet LB, 100 µg/ml naphthol AS-MX phosphate, 0.1 M sodium
acetate (pH 5.0), 50 mM sodium tartrate]. TRAP-positive cells were
counted as osteoclasts and multinuclear osteoclast cells (MNCs;
>3 nuclei/cell) under an inverted microscope (Leica
Microsystems, Bannockburn, IL, USA). A total of four fields of a
single well were selected randomly and three wells were counted for
each group. For the total TRAP activity assay, cells were lysed
with 1% Triton X-100 in TRAP assay buffer (50 mM sodium tartrate
and 0.1 M sodium acetate, pH 5.2) for 10 min and incubated in a
TRAP assay buffer with 1 mg/ml p-nitrophenyl phosphate. Following
30 min of incubation at 37°C, the reaction was stopped with 1 M
NaOH, and the absorbance was measured at 405 nm using a
spectrophotometer.
Cytotoxicity assay
The XTT assay was performed to examine the cytotoxic
effect of PMR on BMMs. Cells were seeded in a 96-well plate then
cultured for 3 days with various concentrations of PMR in the
presence of M-CSF (30 ng/ml). Following the incubation period, XTT
solution (50 µl) was added to each well and incubated for 4
h. Absorbance was measured using an ELISA reader (Thermomax;
Molecular Devices, Sunnyvale, CA, USA) at 450 nm.
Actin ring staining
Cell were fixed with 3.7% formaldehyde for 10 min
and permeabilized with 0.1% Triton X-100 for 5 min. The cells were
blocked with 1% bovine serum albumin (BSA) and then incubated with
Texas red-phalloidin (Molecular probes; Thermo Fisher Scientific,
Inc.) at room temperature for 30 min. Following washing with PBS,
nuclei were counterstained with 0.1 µg/ml DAPI for 1 min.
The images were captured using a fluorescence microscope (EVOS FL;
Thermo Fisher Scientific, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated with Isol-RNA lysis reagent
(5 Prime Inc., Gaithersburg, MA, USA), according to the
manufacturer's instructions. To obtain cDNA, equal amounts of total
RNA were reverse-transcribed using ReverTra Ace qPCR RT kit (Toyobo
Co., Ltd., Osaka, Japan). qPCR was performed in a 20 µl
reaction mixture containing 10 µl of SYBR Green Real-Time
PCR Master Mix (Toyobo Co., Ltd.), 10 pM of forward primer/reverse
primer, and 1 ng of cDNA using StepOnePlus RT-PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The primers used to
detect the genes of interest were as follows: c-Fos, forward 5′-CTG
GTG CAG CCC ACT CTG GTC-3′ and reverse 5′-CTT TCA GCA GAT TGG CAA
TCT C-3′; NFATc1, forward 5′-CAA CGC CCT GAC CAC CGA TAG-3′ and
reverse 5′-GGC TGC CTT CCG TCT CAT AGT-3′; TRAP, forward 5′-ACT TCC
CCA GCC CTT ACT AC-3′ and reverse 5′-TCA GCA CAT AGC CCA CAC CG-3′;
OSCAR, forward 5′-CTG CTG GTA ACG GAT CAG CTC CCC AGA-3′ and
reverse 5′-CCA AGG AGC CAG AAC CTT CGA AAC T-3′; Atp6v0d2, forward
5′-TCA GAT CTC TTC AAG GCT GTG CTG-3′ and reverse 5′-GTG CCA AAT
GAG TTC AGA GTG ATG-3′; Cathepsin K, forward
5′-ACGGAGGCATTGACTCTGAAGATG-3′ and reverse 5′-GTT GTT CTT ATT CCG
AGC CAA GAG-3′; Runx2, forward 5′-CCC AGC CAC CTT TAC CTA CA-3′ and
reverse 5′-CAG CGT CAA CAC CAT CAT TC-3′; alkaline phosphatase
(ALP), forward 5′-CAA GGA TAT CGA CGT GAT CAT G-3′ and reverse
5′-GTC AGT CAG GTT GTT CCG ATT C-3′; Osteocalcin, forward 5′-CTC
TCT GCT TGA GGA AGA AGC TC-3′ and reverse 5′-GTG CCC CTT AGG CAC
TAG GAG-3′; Osterix, forward 5′-CTC TCT GCT TGA GGA AGA AGC TC-3′
and reverse 5′-GTG CCC CTT AGG CAC TAG GAG-3′; Osteopontin, forward
5′-TCT GAT GAG ACC GTC ACT GC-3′ and reverse 5′-CCT CAG TCC ATA AGC
CAA GC-3′; and GAPDH, forward 5′-ACC ACA GTC CAT GCC ATC AC-3′ and
reverse 5′-TCC ACC ACC CTG TTG CTG TA-3′. The mouse GAPDH gene was
used as the internal control. The thermal cycling conditions were
as follows: 95°C for 15 min, followed by 40 cycles of 95°C for 15
sec, 58°C for 15 sec, and 72°C for 15 sec. The specificity of the
SYBR green assays was confirmed by melting-point analysis.
Expression data were calculated from the cycle threshold (Cq) value
using the 2−ΔΔCq method (15).
Western blot analysis
Whole-cells were lysed in a buffer containing 50 mM
Tris-HCl, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM sodium
fluoride, 1 mM sodium vanadate, 1% deoxycholate, and protease
inhibitors. The lysates were centrifuged at 16,128 × g for 20 min
and the supernatants were collected. The protein concentration was
measured using a BCA protein assay kit (Thermo Fisher Scientific
Inc.). Equal amounts of protein (30 µg) were separated on
10% SDS-polyacrylamide gels and were transferred by
electro-blotting onto polyvinylidene difluoride membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Membranes were incubated
with blocking buffer consisting of 5% nonfat dry milk in 10 mM
Tris-HCl pH 7.5/150 mM NaCl/0.1% Tween-20 (TBST) for 1 h at room
temperature, then probed with the indicated primary antibodies
overnight at 4°C. The membraned were then washed with TBST three
times (10 min for each wash), and after washing, they were
incubated with horseradish peroxidase-conjugated secondary
antibodies [1:5,000 dilution; goat anti-mouse immunoglobulin G
(IgG)-horseradish peroxidase (HRP), cat. no. SC-2005; goat
anti-rabbit IgG-HRP, cat. no. SC-2004; rabbit anti-goat IgG-HRP,
cat. no. SC-2768] for 1 h at room temperature and washed with TBST
three times. Chemiluminescent signals were detected using the
FlourChemE system (version 1.4.1; ProteinSimple, San Jose, CA, USA)
with western enhanced chemiluminescence substrate (Bio-Rad
Laboratories, Inc.).
Transfection and luciferase reporter
assay
293T cells were seeded in a 96-well plate at a
density of 2×104 cells/well and incubated for 24 h
before transfection. The cells were transiently cotransfected with
TRAF6 and NF-κB luciferase reporter vector (pGL3-Basic Vector;
Promega Corporation, Madison, WI, USA) using the X-tremeGENE9 DNA
transfection reagent (Roche Diagnostics GmbH) for 3 h in serum-free
DMEM and then the medium were replaced by DMEM complete medium.
After 12 h of transfection, the transfected cells were incubated
with or without PMR. The cells were lysed with lysis buffer, and
luciferase activity was measured using a luciferase assay system
(Promega Corporation). Luciferase activity were normalized to the
β-galactosidase activity in each sample.
Culture of primary mouse osteoblasts and
assays
Calvaria was isolated from 1-day-old neonatal ICR
mice and was digested with 0.1% collagenase (Sigma-Aldrich; Merck
KGaA) and 0.2% dispase (Roche Diagnostics GmbH) for 5 min at 37°C
(16). After removal of the
medium, the remaining tissue was digested 4 times for 10 min at
37°C. Cell fractions were collected and used as primary mouse
osteoblasts. Cells were cultured for 3 days, and adherent cells
were used as osteo-blasts. Primary mouse osteoblasts were seeded at
a density of 1.5×104 cells/well into 48-well plates. To
induce the osteoblast differentiation, cells were induced by
osteogenic inducers: 50 µg/ml AA and 10 mM β-GP. After 24 h,
cells were cultured in the presence of 10 mM β-GP and 50
µg/ml AA with or without PMR to induce osteoblast
differentiation. On day 3, the medium was replaced with fresh
medium containing β-GP and AA with or without PMR. For ALP
staining, AA/β-GP induced osteoblasts were washed twice with PBS
and fixed in 3.7% formaldehyde. Then, they were stained with a
mixture of 0.1 mg/ml naphthol AS-MX phosphate, 0.6 mg/ml fast-blue
BB salt, 2 mM MgCl2, 5 ml N, N-dimethylformamide (MP
Biomedicals, Illkirch, France), and 100 mM Tris-HCl (pH 8.8) buffer
at 37°C for 5 to 10 min. When the cells turned blue, they were
washed twice with PBS. For alizarin red S (ARS; Sigma-Aldrich;
Merck KGaA) staining, cells were fixed in 3.7% formaldehyde in
sterile PBS and stained with 2% ARS solution.
Bone loss model
To study the effect of PMR on LPS-induced bone loss
in vivo, ICR (5-week-old males) mice were randomly divided
into 4 experimental groups (n=5 mice/group; 23–28 g body weight):
Control group, LPS-injected (disease) group, and LPS-injected and
PMR-treated group (200 and 400 mg/kg/day). After acclimatization
for 1 week, control mice were orally administered
phosphate-buffered saline (PBS), LPS-injected mice were injected
with LPS, and PMR-treated mice were orally administered with PMR.
PBS and PMR were orally administered every day for 10 days to the
treated and the control groups, respectively. LPS (5 mg/kg) and PBS
were injected intraperitoneally on days 1, 4, and 7 to the disease
and the control groups, respectively. Mice were euthanized on day
10 (17,18), the left femurs were analyzed by
high-resolution micro-computed tomography (µCT; Skyscan
1172), and the right femurs were fixed in 4% paraformaldehyde in
PBS for 1 day, decalcified for 3 weeks in 12% EDTA, and then
embedded in paraffin. Sections (5 µm thick) were prepared
using a Leica microtome RM2125RTM (Leica Microsystems, Inc.,
Bannockburn, IL, USA). The sections were stained with
hematoxylin-eosin (H&E) for histological examination, and other
sections were stained with TRAP to reveal osteoclasts on the bone
surface. The parameters for bone resorption, including the number
of osteoclasts per two fields of view per slide (n=3 slides per
group) were quantified using ImageJ 1.50i software (National
Institutes of Health, Bethesda, MD, USA). Serum mouse C-teminal
telopeptide of type I collagen (CTX-1) levels, a specific marker of
bone resorption, were measured using a CTX-1 ELISA kit (cat. no.
MBS726456; MyBioSource Inc., San Diego, CA, USA).
Micro-computed tomography analysis
µCT images were scanned with a
high-resolution SkyScan 1172 system (SkyScan/Bruker, Kontich,
Belgium) with the X-ray source at 50 kV and 201 µA with a
0.5-mm aluminum filter. Images were captured every 0.7° over an
angular range of 180°. Raw images were reconstructed from a stack
of 2-dimensional images using commercial software (NRecon, version
1.6.2.0; SkyScan). The trabecular bones between 6.889 and 3.608 mm
away from the epiphyseal plate of distal femurs were manually
selected as a region of interest (ROI) and the contouring of images
was performed every 50 axial slices. The bone morphometric
parameters of ROI calculated using proprietary software (CTAn;
Skyscan): Trabecular bone volume as a fraction of total tissue
volume (BV/TV, %), trabecular thickness (Tb.Th, µm),
trabecular seperation (Tb.Sp, µm) and trabecular number
(Tb.N, 1/mm). The three-dimensional visualization images were
obtained by using the 3D-creator software (Ant; SkyScan).
Statistical analysis
Experiments were conducted independently at least
three times and all data were presented as mean ± standard
deviation. All statistical analyses were performed with SPSS
version 12.0 Software (SPSS, Inc., Chicago, IL, USA). Statistical
differences were analyzed using one-way analysis of variance with
Fisher's Least Significant Difference test. P<0.05 was
considered to indicate a statistically significant difference.
Results
PMR inhibits RANKL-induced osteoclast
differentiation
To identify the main constituents of PMR, UPLC
analysis was performed. By comparing with emodin and
2,3,5,4′-tetrahy-droxystilbene 2-O-β-D-glucoside (THS), the two
standard marker compounds, emodin and THS were identified in the
extract of PMR (Fig. 2).
Treatment of BMMs with M-CSF and RANKL for 4 days induced
TRAP-positive multinucleated osteoclasts (Fig. 3A). As shown in Fig. 3A, PMR treatment reduced the number
of TRAP-positive cells and inhibited RANKL-induced TRAP activity in
a dose-dependent manner. In addition, PMR treatment (25, 50 and 100
µg/ml) suppressed the formation of TRAP-positive MNCs
(Fig. 3A), without affecting the
viability of osteoclast precursor cells (Fig. 3C), suggesting that the inhibitory
effect of PMR on osteoclast differentiation is not due to
cytotoxicity. Based on the results of morphological analysis and
MNC formation, the dose of 50 µg/ml PMR was used for
subsequent experiments to investigate its antiosteoclastogenic
effect. The formation of F-actin rings is necessary for
osteoclastic bone resorption, therefore the effect of PMR on
F-actin ring formation was examined next. Characteristic F-actin
ring formation was observed in the untreated control, whereas
treatment with PMR strongly inhibited the F-actin ring formation
and morphology (Fig. 3B). These
findings suggested that PMR distinctly inhibited TRAP-positive
multinucleated osteoclasts and F-actin ring formation.
PMR inhibits RANKL-mediated induction of
c-Fos and NFATc1
Since PMR inhibited RANKL-induced osteoclast
differentiation, the effect of PMR on the expression of c-Fos and
NFATc1, transcription factors essential for osteoclast
differentiation, was investigated. RT-qPCR analysis indicated that
PMR treatment significantly inhibited the transcriptional levels of
c-Fos and NFATc1 at 6, 12 and 48 h (Fig. 4A and B). In addition,
RANKL-induced expression of c-Fos and NFATc1 mRNA was suppressed by
PMR in a dose-dependent manner (Fig.
4A and B). Western blot analysis confirmed that PMR treatment
significantly decreased RANKL-induced expression levels of c-Fos
and NFATcl at the protein level (Fig.
4C). These results suggested that PMR inhibited RANKL-induced
osteoclast differentiation through down-regulation of c-Fos and
NFATcl.
PMR inhibits RANKL-induced mRNA
expression of TRAP, OSCAR, ATP6v0d2 and Cathepsin
K. NFATc1 induces the expression of various
osteoclast-specific genes, including TRAP, OSCAR, ATP6v0d2 and
Cathepsin K, during RANKL-induced osteoclast differentiation
(8). To characterize the
PMR-inhibitory effect on osteoclast differentiation through
regulation of osteoclast-specific gene expression, the mRNA levels
of genes associated with osteoclast differentiation were analyzed
using RT-qPCR. PMR treatment downregulated the expression of TRAP
and OSCAR (Fig. 5A), which are
associated with osteoclast differentiation, as well as suppressed
the expression of ATPv0d2 (Fig.
5A), which affects cell-to-cell fusion and cell migration. The
expression of Cathepsin K, which is related to bone-resorbing
activity, was also inhibited by PMR treatment at 48 h (Fig. 5A). Furthermore, PMR treatment
significantly reduced the mRNA expression of TRAP, OSCAR, Atp6v0d2
and Cathepsin K in a dose-dependent manner (Fig. 5B). Based on these observations,
PMR may have inhibited osteoclast differentiation by controlling
the expression of several genes that are necessary for this
process.
 | Figure 5(A and B) PMR inhibits the expression
of osteoclast-specific genes. BMMs were stimulated with M-CSF (30
ng/ml) and RANKL (100 ng/ml) in the presence or absence of PMR for
the indicated time periods and indicated concentrations. The mRNA
expression levels of TRAP, OSCAR, ATP6vod2 and Cathepsin K were
analyzed by reverse transcription-quantitative polymerase chain
reaction. **P<0.01 and ***P<0.001 vs.
control group; #P<0.05, ##P<0.01 and
###P<0.001 vs. RANKL-treated group at the
corresponding time and concentration. PMR, Polygoni
Multiflori Radix; BMMs, bone marrow macrophages; M-CSF,
macrophage colony stimulating factor; RANKL, receptor activator of
nuclear factor-kB ligand; TRAP, tartrate-acid resistant acid
phosphatase; OSCAR, osteoclast-associated receptor; ATP6vod2,
ATPase H+ transporting lysosomal 38 kDa V0 subunit
d2. |
PMR suppresses the RANKL-induced p38 and
ERK/NF-κB signaling pathways
A key signaling event induced by the binding of
RANKL to it receptor, RANK, is the activation of mitogen-activated
protein kinase (MAPKs), Akt and NF-κB signaling (6). To identify the molecular mechanism
of inhibition and the pathways influenced by PMR, BMMs were treated
with RANKL in the absence or presence of PMR for 0-30 min.
RANKL-induced activation of p38 and ERK was inhibited by PMR
treatment within 5 min, whereas phosphorylation of JNK was not
affected by treatment with PMR (Fig.
6A). These findings suggest that the inhibitory effect of PMR
on osteo-clast differentiation is primarily mediated by the
suppression of the p38 and ERK signaling pathways. NF-kB is
downstream of MAPK signaling and is a major transcription factor
for RANKL-activated osteoclastogenesis (19). Thus, degradation of IκB and
activation of NF-κB were evaluated by western blot analysis.
RANKL-induced degradation of IκB and activation of NF-κB were
inhibited by PMR treatment within 5 min (Fig. 6B). Furthermore, a luciferase
reporter assay was performed to examine the effect of PMR on
RANKL-induced NF-κB activation. PMR treatment significantly
suppressed the NF-κB transcription activity (Fig. 6C). These results showed PMR
inhibits RANKL-induced NF-κB transcriptional activity through
increased phosphorylation of IκB.
PMR stimulates AA/β-GP-induced osteoblast
differentiation and mRNA expression of osteoblast differentiation
markers
The effect of PMR on osteoblast differentiation was
assessed by measuring the ALP activity and staining in the cells,
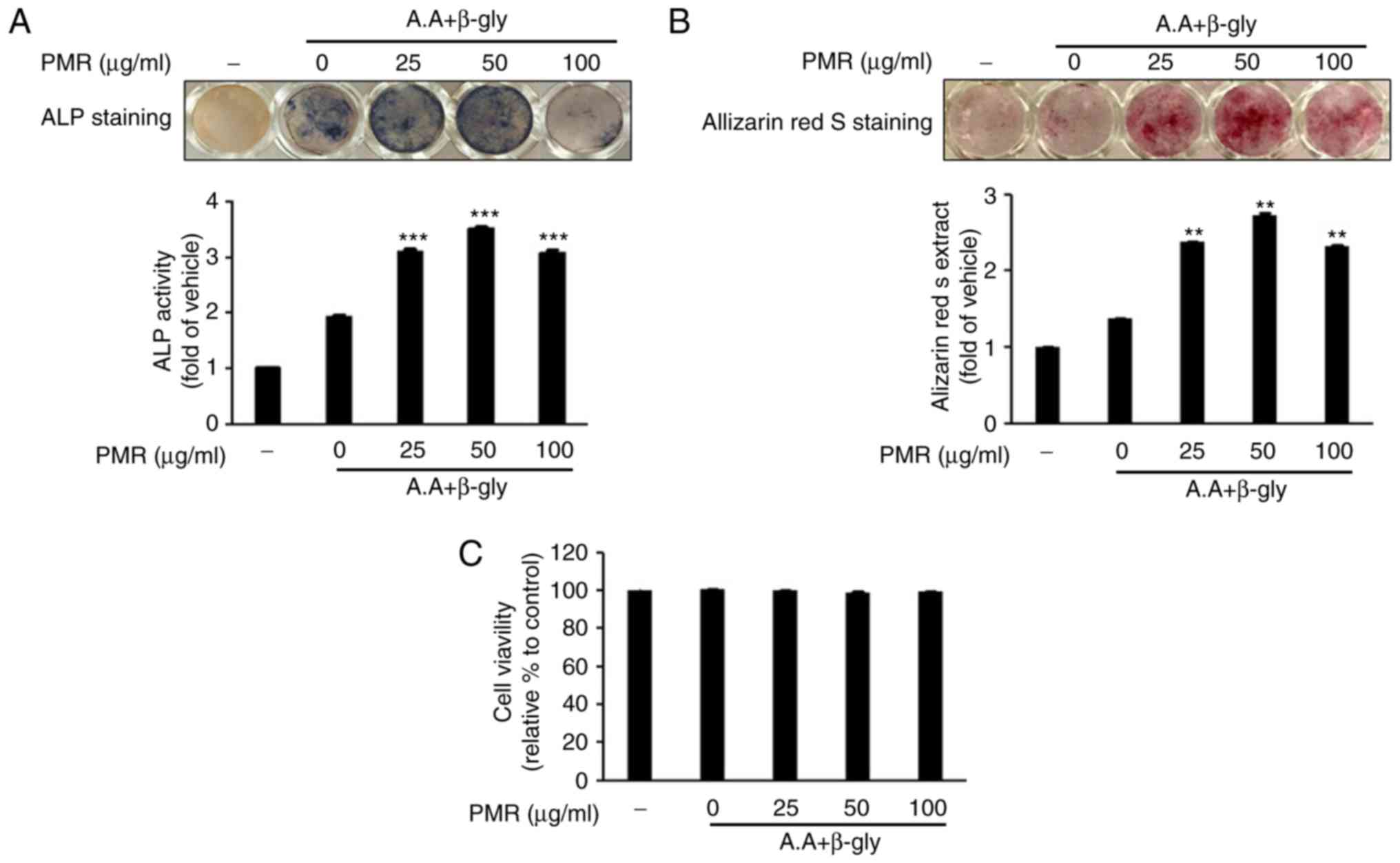
as an early marker of osteoblastogenesis. As illustrated in
Fig. 7A, expression and activity
of ALP were significantly increased following 25 and 50
µg/ml PMR treatment. These results demonstrated that PMR
enhanced osteoblast differentiation by increasing ALP activity. To
elucidate the effect of PMR-induced mineralization of extracellular
matrix (ECM) during osteoblastogenesis, osteoblasts were stained
with ARS 21 days following induction of differentiation. The ECM of
the differentiated osteoblasts that were not treated with PMR
exhibited only slight mineralization (Fig. 7B). However, increased ECM
mineralization was observed in the cells treated with 25 and 50
µg/ml PMR for 21 days (Fig.
7B). The quantified optical density of Alizarin red dissolved
in cetylpyridinum chloride solution was significantly higher in the
PMR-treated osteoblasts compared with the untreated osteoblasts
(Fig. 7B). Of note, PMR treatment
had no effect in cell viability of the differentiated osteoblasts
(Fig. 7C).
The mRNA expression of genes related to osteoblast
differentiation was investigated next by RT-qPCR. The mRNA
expression levels of Runx2, ALP, osteocalcin and osterix were
demonstrated to be significantly upregulated in the PMR-treated
group compared with the untreated osteoblasts (Fig. 8A). These findings suggested that
PMR treatment enhanced osteoblast differentiation and mRNA
expression of osteoblast differentiation markers.
 | Figure 8Effect of PMR on osteoblast-specific
markers and on the p38 and Smad pathways. (A) Primary mouse
osteoblasts were seeded in 6-well plates at a density
2×105 cells/well and cultured in the absence or presence
of β-GP (10 mM) and AA (50 µg/ml) for the indicated days.
Levels of mRNA expression of Runx2, ALP, osterix and osteocalcin
were analyzed by reverse transcription-quantitative polymerase
chain reaction. (B) Primary mouse osteoblasts were serum-starved
for 2 h, and treated with PMR (50 µg/ml) for the indicated
time. The cell lysates were analyzed by western blotting with the
indicated antibodies. β-actin was used as an internal control.
*P<0.05, **P<0.01 and
***P<0.001 vs. control group; #P<0.05,
##P<0.01 and ###P<0.001 vs.
AA/β-GP-induced group at the corresponding time. PMR, Polygoni
Multiflori Radix; AA, ascorbic acid; β-GP, β-glycerophosphate;
Runx2, runt-related transcription factor 2; ALP, alkaline
phosphatase; p-, phosphorylated; ERK, extracellular
signal-regulated kinase. |
PMR promotes AA/β-GP-induced osteoblast
differentiation via the p38 and Smad pathway
Since the expression of osteoblast-related genes,
including Runx2, ALP, osteocalcin and osterix, is modulated via the
p38 and Smad signaling pathway, western blot analysis was performed
to determine the signaling pathway involved in the PMR-regulated
osteoblast differentiation. PMR increased the phosphorylation of
p38 after 5 min of treatment and activated ERK and Smad 1/5/8 after
15 min of treatment (Fig. 8B).
These results indicate that PMR might upregulate osteoblast-related
genes via the p38 and Smad signaling pathways.
PMR prevented LPS-induced bone loss in a
mouse model
The efficacy of PMR in inhibiting in vivo
bone destruction was investigated in a mouse model of LPS-induced
osteolysis. PMR was administered orally to ICR mice for 10 days. In
the µCT analyses, a 2- or 3-dimensional visualization of the
femoral area revealed a loss in trabecular bone density following
LPS treatment (Fig. 9A).
LPS-induced bone loss clearly decreased in the femurs of the
PMR-treated and LPS-injected group (Fig. 9A). Morphometric analyses of the
femurs revealed significant reductions in the BV/TV and an increase
in the Tb.Sp. in the LPS-injected group (Fig. 9B). The Tb.N of the LPS-injected
group exhibited a slight reduction, even though usually Tb.N
expresses significant decrease (Fig.
9B). However, these reductions were recovered in the
PMR-treated and LPS-injected group (Fig. 9B). Histological analysis of
sections from the femurs also demonstrated the preventive effect of
PMR on trabecular bone loss induced by LPS (Fig. 9C). In addition, the increased
number of TRAP-positive osteoclasts induced by LPS was
significantly reduced in the PMR-treated and LPS-injected group
(Fig. 9C). CTX-1 is a new marker
of bone resorption, which can be used to assess the antiresorptive
activity and to evaluate increases in osteoclast numbers (20). Serum levels of CTX-1 in mice
receiving PMR were lower compared with the PMR-untreated and
LPS-injected group (Fig. 9D).
These findings demonstrated that PMR treatment prevented
LPS-induced bone loss in mice by inhibiting bone resorption and
stimulating bone formation.
 | Figure 9Effect of PMR on LPS-induced bone
loss in vivo. (A) Representative three-dimensional
reconstruction images of femurs from high-resolution µCT
analysis. (B) The BV/TV, Tb.Sp, Tb.Th, and Tb.N of the femurs were
determined using the µCT data, analyzed by Skyscan 1172
software. (C) Dissected femora were fixed, decalcified, embedded in
paraffin and sectioned. Sections were stained with H&E (upper
images) and with TRAP (lower images). The number of osteoclasts per
field was counted using the histomorphometric results. (D) Serum
levels of CTX-1 were determined using a mouse ELISA kit.
*P<0.05, **P<0.01,
***P<0.001 vs. control group; #P<0.05
and ##P<0.01 vs. LPS-treated group. PMR, Polygoni
Multiflori Radix; LPS, lipopolysaccharide; µCT,
micro-computed tomography; BV/TV, trabecular bone volume/total
tissue volume; Tb.Sp, trabecular separation; Tb.Th, trabecular
thickness; Tb.N, trabecular number; H&E, hematoxylin and eosin;
TRAP, tartrate-acid resistant acid phosphatase. CTX-1, C-teminal
telopeptide of type I collagen. |
Discussion
Osteoporosis is a common disease characterized by
low bone mass and microarchitectural deterioration of bone tissue,
which result in fragility fractures (21). It is widely recognized as a
serious public health problem afflicting more than 200 million
people worldwide. Risk factors of osteoporosis include
endocrinological, nutritional and genetic factors, such as
hyperparathyroidism and deficiency of estrogen, vitamin D, or
calcium (22). Most agents used
for treatment and prevention of osteoporosis, such as
bisphosphonates, selective estrogen receptor modulators and
estrogen, are inhibitors of bone resorption (23). Although these agents are
effective, they have several side effects. Therefore, many
scientists have been searching for alternative approaches without
side effects for the prevention and treatment of osteoporosis
through inhibition of osteoclasts and promotion of osteoblasts. The
present study demonstrated that PMR inhibited RANKL-induced
osteoclast differentiation and stimulated the mineralization
activity of osteoblasts.
RANKL is expressed by osteoblastic cells/bone marrow
stromal cells; it binds to the RANK receptor on osteoclast
precursor cells, and stimulates their differentiation into mature
osteoclasts. The RANKL-RANK interaction promotes osteoclast
differentiation, which is involved in the formation and survival of
osteoclasts (24). In addition,
RANKL induces the expression of transcription factors, including
c-Fos, and NFATc1, which are essential for osteoclast
differentiation (25,26). Previous studies have demonstrated
that NFATc1 is a master regulator of osteoclastogenesis, which
autoamplifies and induces the expression of osteoclast-specific
genes, such as activator protein 1 (AP-1), TRAP, calcitonin
receptor, Cathepsin K and OSCAR, through cooperation with c-Fos
(27,28). The present data indicated that PMR
significantly inhibited RANKL-induced expression of c-Fos and
NFATc1, and consequently, suppressed the mRNA expression of
osteoclast-specific gene markers TRAP, OSCAR, Cathepsin K, and
Atp6v0d2.
NFATc1 induction is a downstream event to RANKL
signaling, which is induced by p38 activation. It may be postulated
that this event involves the increase in the transactivation
ability of p65 NF-κB upon Ser-536 phosphorylation by the p38 MAPK
(29,30). Because of RANKL stimulation,
subsequent ubiquitination and proteasomal degradation of IκB occur,
and IκB releases NF-κB, which translocates into the nucleus and
initiates the transcription of target genes (31). In the present study, PMR inhibited
p38 and NF-κB activation induced by RANKL treatment. Specifically,
NF-κB transcriptional activity was strongly suppressed following
PMR treatment. Thus, the downregulation of NF-κB-dependent
transcription might be a mechanism by which PMR inhibits
RANKL-induced c-Fos and NFATc1 expression during osteoclast
differentiation. The mechanism underlying PMR-mediated inhibition
of osteoclast differentiation may be related to the reduced
expression of osteoclast-specific genes.
Furthermore, PMR treatment stimulated osteoblast
differentiation and increased the expression of genes associated
with bone formation in primary mouse osteoblasts. ALP activity
reflects the early stages of AA/β-GP-induced osteogenic
differentiation and has a key role in bone mineralization by
initiating and/or promoting the formation of hydroxyapatite
crystals in the matrix vesicles of osteoblasts (32). Several transcription factors,
including ALP, Runx2, osterix and osteocalcin, are prominent
osteoblast-specific markers (33,34). Runx2, a key transcription factor
for osteogenesis and bone formation, is exclusively expressed in
mineralized tissues and osteoblasts. It serves a crucial role in
osteoblastogenesis through the induction of major
osteoblast-specific genes, including ALP, osteocalcin, and type I
collagen (35,36). Specifically, both Runx2 and
osterix have been known as master transcription factors for
osteoblast differentiation and for controlling the expression of
bone-related genes (35,37). It was reported that
Runx2-deficient mice completely lack osteoblasts and bone formation
(38,39). Osteocalcin gene expression is
initiated during late stages of osteoblast differentiation at the
onset of ECM mineralization (40). Thus, the present results
demonstrated that PMR enhanced early and late stages of osteoblast
differentiation by increasing ALP activity and increasing
mineralization, respectively. In addition, PMR treatment increased
the expression of osteoblast differentiation markers ALP, Runx2,
osterix and osteocalcin. Signaling transduction of osteoblast
differentiation specifically occurs through both a canonical
Smad-dependent pathway and a non-canonical Smad-independent
signaling pathway (such as the p38 MAPK pathway). Both the Smad
(Smad 1, 5, and 8) and the p38 MAPK pathways converge at the Runx2
gene to control mesenchymal precursor cell differentiation
(41,42). In the present study, PMR treatment
induced the phosphorylation of p38 and Smad 1/5/8 and subsequently,
stimulated Runx2 expression. Through these actions, PMR enhanced
osteoblast differentiation.
LPS promotes osteoclast differentiation, fusion,
survival, and activation through the release of RANKL, interleukin
(IL)-1, and tumor nexrosis factor (TNF)-α (43,44). LPS was reported to potently
stimulate bone resorption in both in vitro and in
vivo studies (45,46). Therefore, the LPS-induced bone
loss model has become one of the well-established
inflammation-mediated osteoporosis models in mice. In a previous
study, PMR prevented osteoporosis in an ovariectomized-induced
osteoporosis model by increasing bone weight and bone
thickness/length ratio, and modulating levels of bone mineral
contents, including ALP, phosphorus and femoral calcium (10,11). In the present study, PMR treatment
attenuated the LPS-induced trabecular bone loss compared with the
LPS-injected group, in BV/TV, Tb.Sp. Despite Tb.N not showing
significant differences between the control and LPS-injected
groups, PMR treatment resulted in an increased Tb.N in the 400
mg/kg treatment group. Serum concentration of CTX-1, a bone
resorption marker, was lower in the PMR-treated and LPS-injected
group compared with the LPS-injected group alone. These findings
demonstrated that PMR treatment results in ameliorated LPS-induced
bone loss in an in vivo model.
Several phytochemicals including various stilbenes,
quinones, flavonoid, phospholipids and other compounds have been
identified in PMR. Phytochemicals emodin (a quinone) and THS (a
stilbene) are main characteristic components extracted from the PMR
(9). Recently, a study reported
that emodin modulated bone remodeling by inhibiting RANKL-induced
osteoclast differentiation in BMMs and stimulated osteoblast
formation on primary osteoblast cells (47). THS has been extracted from the
roots of Polygonum multiflorum Thunb. and has been reported
to promote bone mineral density and bone strength in the femoral
bones of rats. The present study identified emodin and THS as PMR
components. These results suggested that the complementary effect
of these components of PMR may contribute to the inhibitory effect
of PMR on RANKL-induced osteoclast differentiation and its
stimulatory effect on osteoblast formation.
In conclusion, PMR treatment inhibited RANKL-induced
NF-κB transcriptional activity via the p38 and ERK signaling
pathways, and this downregulation of NF-κB was involved in the
inhibitory effect of PMR on RANKL-induced c-Fos and NFATc1 during
osteoclast differentiation. Furthermore, PMR treatment increased
ALP and mineralization activity, and elevated markers of osteoblast
differentiation Runx2, ALP, osterix and osteocalcin, in mouse
calvarial primary osteo-blasts. Finally, PMR reduced the
LPS-mediated bone loss in an in vivo model. PMR may modulate
bone metabolism via dual actions of reducing osteoclast
differentiation and stimulating osteoblast formation. Taken
together, PMR might be an effective natural product for the
treatment of bone diseases.
Acknowledgments
Not applicable.
References
|
1
|
Shaw AT and Gravallese EM: Mediators of
inflammation and bone remodeling in rheumatic disease. Semin Cell
Dev Biol. 49:2–10. 2016. View Article : Google Scholar :
|
|
2
|
Nagao M, Feinstein TN, Ezura Y, Hayata T,
Notomi T, Saita Y, Hanyu R, Hemmi H, Izu Y, Takeda S, et al:
Sympathetic control of bone mass regulated by osteopontin. Proc
Natl Acad Sci USA. 108:17767–17772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Florencio-Silva R, Sasso GR, Sasso-Cerri
E, Simoes MJ and Cerri PS: Biology of bone tissue: Structure,
function, and factors that influence bone cells. Biomed Res Int.
2015:4217462015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Komori T: Regulation of osteoblast
differentiation by transcription factors. J Cell Biochem.
99:1233–1239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Suda T, Takahashi N, Udagawa N, Jimi E,
Gillespie MT and Martin TJ: Modulation of osteoclast
differentiation and function by the new members of the tumor
necrosis factor receptor and ligand families. Endocr Rev.
20:345–357. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Takayanagi H: Osteoimmunology: Shared
mechanisms and crosstalk between the immune and bone systems. Nat
Rev Immunol. 7:292–304. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takayanagi H, Kim S, Koga T, Nishina H,
Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al:
Induction and activation of the transcription factor NFATc1 (NFAT2)
integrate RANKL signaling in terminal differentiation of
osteoclasts. Dev Cell. 3:889–901. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim K, Kim JH, Lee J, Jin HM, Lee SH,
Fisher DE, Kook H, Kim KK, Choi Y and Kim N: Nuclear factor of
activated T cells c1 induces osteoclast-associated receptor gene
expression during tumor necrosis factor-related activation-induced
cytokine-mediated osteoclastogenesis. J Biol Chem. 280:35209–35216.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin L, Ni B, Lin H, Zhang M, Li X, Yin X,
Qu C and Ni J: Traditional usages, botany, phytochemistry,
pharmacology and toxicology of Polygonum multiflorum Thunb: A
review. J Ethnopharmacol. 159:158–183. 2015. View Article : Google Scholar
|
|
10
|
Kim MJ, Seo BI, Shin SS and Park JH:
Effect of Polygoni multiflori Radix and cynanchi wilfordii Radix on
prevention of osteoporosis in ovariectomized rats. Kor J Herbol.
19:23–34. 2004.
|
|
11
|
Do YJ, Ku SK, Kim HT, Oh T, Cho YM, Kim
SW, Ryu IS and Lee KW: Antiosteoporotic effects of Polygoni
Multiflori Radix (PMR) in ovariectomized (OVX)-induced osteoporosis
in ddY mice. J Vet Clin. 28:375–386. 2011.
|
|
12
|
Kim JY, Baek JM, Ahn SJ, Cheon YH, Park
SH, Yang M, Choi MK and Oh J: Ethanolic extract of Schizonepeta
tenuifolia attenuates osteoclast formation and activation in vitro
and protects against lipopolysaccharide-induced bone loss in vivo.
BMC Complement Altern Med. 16:3012016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ha H, Shim KS, Kim T, An H, Lee CJ, Lee KJ
and Ma JY: Water extract of Acer tegmentosum reduces bone
destruction by inhibiting osteoclast differentiation and function.
Molecules. 19:3940–3954. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rho TW, Lee SY, Han SY, Kim JH, Lee KH,
Kim DS, Kwak HB and Kim YK: Glycyrrhizae Radix inhibits osteoclast
differentiation by inhibiting c-Fos-dependent NFATc1 expression. Am
J Chin Med. 45:283–298. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar
|
|
16
|
Sharma-Bhandari A, Park SH, Kim JY, Oh J
and Kim Y: Lysyl oxidase modulates the osteoblast differentiation
of primary mouse calvaria cells. Int J Mol Med. 36:1664–1670. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song J, Jing Z, Hu W, Yu J and Cui X:
α-Linolenic acid inhibits receptor activator of NF-κB ligand
induced (RANKL-induced) osteoclastogenesis and prevents
inflammatory bone loss via downregulation of nuclear
Factor-KappaB-inducible nitric oxide synthases (NF-κB-iNOS)
signaling pathways. Med Sci Monit. 23:5056–5069. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim JY, Park SH, Baek JM, Erkhembaatar M,
Kim MS, Yoon KH, Oh J and Lee MS: Harpagoside inhibits
RANKL-induced osteoclastogenesis via Syk-Btk-PLCγ2-Ca2+
signaling pathway and prevents Inflammation-mediated bone loss. J
Nat Prod. 78:2167–2174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Feng X: RANKing intracellular signaling in
osteoclasts. IUBMB Life. 57:389–395. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Naylor K and Eastell R: Bone turnover
markers: Use in osteoporosis. Nat Rev Rheumatol. 8:379–389. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hofbauer LC: From bone cell biology to
novel therapies of osteoporosis. Drug Res. 65(Suppl 1): S14–S15.
2015. View Article : Google Scholar
|
|
22
|
Chau DL, Edelman SV and Chandran M:
Osteoporosis and diabetes. Curr Diab Rep. 3:37–42. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nardone V, D'Asta F and Brandi ML:
Pharmacological management of osteogenesis. Clinics. 69:438–446.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boyce BF and Xing L: Functions of
RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem
Biophys. 473:139–146. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kang MR, Jo SA, Yoon YD, Park KH, Oh SJ,
Yun J, Lee CW, Nam KH, Kim Y, Han SB, et al: Agelasine D suppresses
RANKL-induced osteoclastogenesis via down-regulation of c-Fos,
NFATc1 and NF-kB. Mar Drugs. 12:5643–5656. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kong L, Zhao Q, Wang X, Zhu J, Hao D and
Yang C: Angelica sinensis extract inhibits RANKL-mediated
osteoclastogenesis by down-regulated the expression of NFATc1 in
mouse bone marrow cells. BMC Complement Altern Med. 14:4812014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Asagiri M, Sato K, Usami T, Ochi S,
Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, et
al: Autoamplification of NFATc1 expression determines its essential
role in bone homeostasis. J Exp Med. 202:1261–1269. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ikeda F, Nishimura R, Matsubara T, Tanaka
S, Inoue J, Reddy SV, Hata K, Yamashita K, Hiraga T, Watanabe T, et
al: Critical roles of c-Jun signaling in regulation of NFAT family
and RANKL-regulated osteoclast differentiation. J Clin Invest.
114:475–484. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang H, Ryu J, Ha J, Chang EJ, Kim HJ,
Kim HM, Kitamura T, Lee ZH and Kim HH: Osteoclast differentiation
requires TAK1 and MKK6 for NFATc1 induction and NF-kappaB
transactivation by RANKL. Cell Death Differ. 13:1879–1891. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iotsova V, Caamaño J, Loy J, Yang Y, Lewin
A and Bravo R: Osteopetrosis in mice lacking NF-kappaB1 and
NF-kappaB2. Nat Med. 3:1285–1289. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hayden MS and Ghosh S: Signaling to
NF-kappaB. Genes Dev. 18:2195–2224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Orimo H and Shimada T: The role of
tissue-nonspecific alkaline phosphatase in the phosphate-induced
activation of alkaline phosphatase and mineralization in SaOS-2
human osteoblast-like cells. Mol Cell Biochem. 315:51–60. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karsenty G: Transcriptional control of
skeletogenesis. Annu Rev Genomics Hum Genet. 9:183–196. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lian JB, Stein GS, Javed A, van Wijnen AJ,
Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ and Young DW:
Networks and hubs for the transcriptional control of
osteoblasto-genesis. Rev Endocr Metab Disord. 7:1–16. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ducy P, Zhang R, Geoffroy V, Ridall AL and
Karsenty G: Osf2/Cbfa1: A transcriptional activator of osteoblast
differentiation. Cell. 89:747–754. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Franceschi RT and Xiao G: Regulation of
the osteoblast-specific transcription factor, Runx2: Responsiveness
to multiple signal transduction pathways. J Cell Biochem.
88:446–454. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cancedda R, Castagnola P, Cancedda FD,
Dozin B and Quarto R: Developmental control of chondrogenesis and
osteogenesis. Int J Dev Biol. 44:707–714. 2000.PubMed/NCBI
|
|
38
|
Komori T, Yagi H, Nomura S, Yamaguchi A,
Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, et al:
Targeted disruption of Cbfa1 results in a complete lack of bone
formation owing to maturational arrest of osteoblasts. Cell.
89:755–764. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Otto F, Thornell AP, Crompton T, Denzel A,
Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen
BR, et al: Cbfa1, a candidate gene for cleidocranial dysplasia
syndrome, is essential for osteoblast differentiation and bone
development. Cell. 89:765–771. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lian JB, Stein GS, Stein JL and van Wijnen
AJ: Osteocalcin gene promoter: Unlocking the secrets for regulation
of osteoblast growth and differentiation. J Cell Biochem Suppl.
30–31:62–72. 1998. View Article : Google Scholar
|
|
41
|
Chen G, Deng C and Li YP: TGF-β and BMP
signaling in osteoblast differentiation and bone formation. Int J
Biol Sci. 8:272–288. 2012. View Article : Google Scholar :
|
|
42
|
Lee KS, Kim HJ, Li QL, Chi XZ, Ueta C,
Komori T, Wozney JM, Kim EG, Choi JY, Ryoo HM and Bae SC: Runx2 is
a common target of transforming growth factor beta1 and bone
morphogenetic protein 2, and cooperation between Runx2 and Smad5
induces osteoblast-specific gene expression in the pluripotent
mesenchymal precursor cell line C2C12. Mol Cell Biol. 20:8783–8792.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Islam S, Hassan F, Tumurkhuu G, Dagvadorj
J, Koide N, Naiki Y, Mori I, Yoshida T and Yokochi T: Bacterial
lipopolysaccharide induces osteoclast formation in RAW 264.7
macrophage cells. Biochem Biophys Res Commun. 360:346–351. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mörmann M, Thederan M, Nackchbandi I,
Giese T, Wagner C and Hänsch GM: Lipopolysaccharides (LPS) induce
the differentiation of human monocytes to osteoclasts in a tumour
necrosis factor (TNF) alpha-dependent manner: A link between
infection and pathological bone resorption. Mol Immunol.
45:3330–3337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ishihara Y, Nishihara T, Maki E, Noguchi T
and Koga T: Role of interleukin-1 and prostaglandin in in vitro
bone resorption induced by Actinobacillus actinomycetemcomitans
lipopolysaccharide. J Periodontal Res. 26:155–160. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Orcel P, Feuga M, Bielakoff J and De
Vernejoul MC: Local bone injections of LPS and M-CSF increase bone
resorption by different pathways in vivo in rats. Am J Physiol.
264:E391–E397. 1993.PubMed/NCBI
|
|
47
|
Kim JY, Cheon YH, Kwak SC, Baek JM, Yoon
KH, Lee MS and Oh J: Emodin regulates bone remodeling by inhibiting
osteoclas-togenesis and stimulating osteoblast formation. J Bone
Miner Res. 29:1541–1553. 2014. View Article : Google Scholar
|