Introduction
Interferon (IFN) has important roles in innate
immunity to fight off viral infections and has been widely used for
the treatment of patients with hepatitis C virus (HCV) infection
(1-3). Secreted and/or exogenously
administered IFN-α/β binds to the IFN-α/β receptors and activates
the Janus kinase (Jak)/signal transducer and activator of
transcription (STAT) pathway, in which the receptor-associated
protein kinases Jak1 and tyrosine kinase 2 cause the
phosphorylation of STAT proteins on critical serine and tyrosine
residues. The activated STATs associate with IFN-stimulated gene
factor (IRF)9 to form an IRF3 transcription factor complex, and
then stimulate the expression of IFN-stimulated genes (ISGs)
through the IFN-stimulated response element (ISRE) by interacting
with its promoter/enhancer region. Hundreds of ISGs induced by IFN
act as effectors of the host defense against viruses, including
HCV.
Polyethylene glycol-conjugated IFN (PEG-IFN) and
ribavirin therapy for HCV achieves a sustained viral response (SVR)
of >70% in genotype 2/3- and 40-60% in genotype 1-infected
patients (4,5). Numerous host and viral factors have
been associated with resistance to IFN-based anti-HCV treatment,
and the presence of liver fibrosis remains an important factor
influencing the failure of HCV clearance (6-10).
Combination treatment of PEG-IFN and ribavirin with recently
developed direct-acting antivirals (DAAs) further increases the
response rate to 80-90% SVR for HCV genotype 1. However, the
response rates of patients with advanced liver fibrosis and
cirrhosis are still lower (11,12). More recently, DAAs have also made
it possible to treat HCV infection without IFN. DAA combination
therapies for HCV are well-tolerated and effective for viral
suppression, reaching a probable SVR of >90% in clinical trials
(13-15). However, it has been reported that
even with IFN-free DAA regimens, an endogenous, intrahepatic type I
IFN response may be important for achieving SVR (16). Therefore, investigating the
factors that antagonize IFN signaling in HCV treatments may be
important, irrespective of whether or not the treatment itself
involves IFN.
Excessive accumulation of extracellular matrix (ECM)
components, including fibrillar type I and III collagens,
fibronectin and laminin, is a feature of liver fibrosis (17,18), and these fibrotic ECM components
provoke diverse cellular responses, mainly through the integrin
family transmembrane receptors (19). Increased ECM stimulates integrin
receptor-mediated signaling in hepatocytes and promotes the growth
and survival of cells through the activation of several signaling
cascades, including the phosphoinositide-3 kinase (PI3K),
mitogen-activated protein kinase (MAPK) and transforming growth
factor (TGF)-β/Smad signaling pathways (20-22).
Although the presence of liver fibrosis remains an
important factor influencing the response to IFN-based anti-HCV
therapy, the molecular mechanisms by which liver fibrosis prevents
IFN from eliminating HCV have remained to be fully elucidated, and
the direct roles of ECMs in the IFN signaling pathway in hepatic
cells remain unknown. The present study investigated the effects of
ECMs on the IFN signaling cascade in hepatic cells and indicated
that the presence of ECMs, including fibrotic collagen, attenuated
IFN-mediated signaling in a β1-integrin-dependent manner and
inhibited the effects of IFN on HCV replication.
Materials and methods
Cells and cell culture
The human hepatoma-derived cell line HuH-7 was
obtained from the Japanese Cancer Research Resources Bank (Osaka,
Japan). OR6 cells derived from HuH-7 cells with the stable
transfection of the full-length genotype 1 replicon containing the
Renilla luciferase gene were selected by neomycin,
ORN/C-5B/KE (23), were used to
examine the anti-HCV effect of IFN-α. The cells were cultured and
maintained in Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) containing 10% fetal
bovine serum (FBS; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and 1% antibiotics (ampicillin/streptomycin) in 5%
CO2 at 37°C. ECM (type I collagen, laminin, type IV
collagen or fibronectin)-coated dishes (Cosmo Bio, Tokyo, Japan)
were used for cell culture to investigate the differences in cell
signaling between cells cultured on ECM-coated dishes and those
cultured on non-ECM-coated dishes, which had hydroxyl and carboxyl
groups on the surface to facilitate cell adhesion (cat. no. 150687;
Thermo Fisher Scientific, Inc.).
Reagents and antibodies
Human IFN-α was obtained from Merck KGaA. The
β1-integrin function-blocking antibody was purchased from EMD
Millipore (Billerica, MA, USA; cat. no. MABT821). The rabbit
polyclonal anti-IFN-stimulated gene (ISG) 15 (cat. no. 2743S) and
anti-protein kinase R (PKR; cat. no. 3072S) antibodies were
purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA).
The antibody against the HCV core protein (cat. no. ab2740) and HCV
nonstructural protein (NS) 5A (cat. no. ab13833) were purchased
from Abcam (Cambridge, UK). The rabbit polyclonal anti-β-actin
antibody (Cell Signaling Technology, Inc.; cat. no. 4967) was used
as a control. Anti-rabbit horseradish peroxidase (HRP) conjugated
IgG (cat. no. 7074; Cell Signaling Technology, Inc.) was used as
the secondary antibody. The integrin-linked kinase (ILK) inhibitor
Cpd 22 was purchased from EMD Millipore, and the focal adhesion
kinase (FAK) inhibitor PF 573228 was from Sigma-Aldrich (Merck
KGaA).
Plasmids and luciferase assays
The ISRE-inducible lucif-erase reporter plasmid
(p-ISRE-Luc) was from Invitrogen (Thermo Fisher Scientific, Inc.).
The ISRE-dependent transcriptions were detected by a luciferase
assay performed with the Dual-Luciferase Reporter Assay System
(Promega Corporation, Madison, WI, USA) according to the
manufacturer's protocol. Values were normalized to the luciferase
activity of the co-transfected pGL4.75 Renilla
luciferase-expressing plasmid (Promega Corp.). HCV-RNA replication
in OR6 cells was also detected with the Renilla luciferase
assay system (Promega Corp.).
HuH-7 cells or OR6 cells were seeded onto 48-well
plates with hydroxyl and carboxyl groups on the surface to
facilitate cell adhesion and 48-well type I collagen-coated plates
(cat. no. 354505; Cosmo Bio) at 1×104 cells per well.
After culture for 48 h, HuH-7 cells were transfected with
p-ISRE-Luc, a luciferase reporter plasmid driven by the promoter
region of ISRE (Clontech Laboratories, Inc., Mountainview, CA, USA)
and co-transfected with pGL4.75, a plasmid that encodes the
Renilla luciferase reporter gene (Promega Corporation),
using Lipofectamine™ LTX and PLUS ligand (Thermo Fisher Scientific,
Inc.) in accordance with the manufacturer's protocols. Following
incubation for 6 h, the medium was changed to serum- and
antibiotic-free medium. The cells were then treated with IFN-α at
the indicated concentrations for 12 h. OR6 cells were cultured for
48 h and subsequently treated with IFN-α at the indicated
concentrations for 12 h. Following the IFN-α treatment, HuH-7 and
OR6 cells were washed twice with PBS and lysed. The cell extracts
were immediately assayed for luciferase activity using a
Multi-label plate reader (Wallac 1420 ARVOsx; PerkinElmer, Inc.,
Waltham, MA, USA).
Semi-quantitative reverse transcription
polymerase chain reaction (RT-PCR)
The total RNA was extracted from the cultured
hepatocellular carcinoma cells using ISOGEN (Nippon Gene, Tokyo,
Japan) in accordance with the manufacturer's protocol. The
concentration of RNA was determined with a spectrophotometer, and
the integrity of the samples was confirmed by visualizing 28S and
18S ribosomal RNA bands under ultraviolet light after gel
electrophoresis. RT-PCR was performed as described previously
(24). The primers used in the
experiments were as follows: ISG15 sense, 5′-GCAGCGAACTCATCTTTG-3′
and antisense, 5′-GCCCTTGTTATTCCTCACC-3′; PKR sense,
5′-GTCCTCTGGTTCTTTTGCTAC-3′ and antisense,
5′-TCCCAACAGCCATTGTAG-3′; GAPDH sense, 5′-ACGCATTTGGTCGTATTGGG-3′
and antisense 5′-TGATTTTGGAGGGATCTCGC-3′.
Western blot analysis
HuH-7 or OR6 cells were seeded on 35-mm plastic
dishes or 35-mm type I collagen-coated dishes at 2×105
cells per dish and cultured under various conditions for 48 h. They
were collected and lysed with extraction buffer containing 50 mM
Tris (pH 7.5), 0.1% SDS and 1 mM phenylmethylsulfonylfluoride. The
lysate was sonicated for 5 min (sonication for 10 sec, pause for 20
sec, repeated 10 times) at 4°C and clarified by centrifugation at
12,000 × g for 10 min, and the supernatant was then collected.
After measuring the protein concentration using a protein assay kit
(cat. no. 5000113-5000115; Bio-Rad Laboratories, Hercules, CA,
USA), 30 µg of protein was mixed with SDS sample buffer,
separated by SDS-PAGE (6% acrylamide for β1-integrin, 10% for PKR,
HCV-NS5A, HCV core protein and β-actin, 15% for ISG15), transferred
to a polyvinylidene difluoride membrane (Bio-Rad Laboratories) and
blocked with 0.1% Tween-20 and 5% skimmed milk overnight at 4°C.
The membranes were incubated with the primary antibodies described
above in Tris-buffered saline with 1% skimmed milk at a dilution of
1:1,000 overnight at 4°C. The specific bands were visualized by
further incubation with anti-rabbit HRP-conjugated secondary
antibodies at 1:1,000 dilution in Tris-buffered saline with 1%
skimmed milk for 1 h at room temperature, followed by a
chemiluminescence reaction using Amersham ECL Prime (GE Healthcare,
Little Chalfont, UK) in accordance with the manufacturer's
protocol.
Treatment of cells with anti-β1-integrin
antibody
To investigate whether the ECM affects IFN signaling
via β1-integrin, cells were treated with β1-integrin
function-blocking antibody. HuH-7 cells were cultured as described
above and treated with β1-integrin function-blocking antibody at 1
µg/l in DMEM for 6 h at 37°C prior to IFN-α treatment.
Treatment of cells with inhibitors
To investigate whether the effect of ECM on IFN
signaling proceeds via ILK or FAK, cells were treated with ILK
inhibitor or FAK inhibitor. HuH-7 cells were cultured as described
above and treated with FAK or ILK inhibitor at 0.1 and 1 µM,
respectively, prior to IFN-α treatment.
Statistical analyses
Differences between two groups were analyzed using
Student's t-test, and P<0.05 was considered to indicate a
statistically significant difference. All experiments were
performed at least three times. Values are expressed as the mean ±
standard deviation. Analysis of variance followed by a post-hoc
multiple comparisons test was performed to compare multiple groups.
Tukey's test was used when all pairwise comparisons were performed
and Dunnett's test was used when one control was compared to all
other experiment groups. We used JMP® version 12 (SAS
Institute, Inc., Cary, NC, USA) software for statistical
analysis.
Results
Impairment of IFN-α signaling in HuH-7 on
ECM-coated dishes
With the progression of liver fibrosis, the levels
of ECM components are increased in the liver. To evaluate the
effects of ECM on IFN signaling, HuH-7 cells were cultured on ECM
(type I collagen, fibronectin, type IV collagen and laminin)-coated
dishes and compared to cells cultured on non-coated plastic dishes.
The cells were transfected with ISRE-luciferase plasmids, and the
luciferase activities were measured after IFN-α treatment. As
presented in Fig. 1A, the ISRE
luciferase activities of HuH-7 cells induced by IFN-α were
significantly reduced when they were cultured on plates coated with
ECM components, except for laminin, compared with those cultured on
normal plastic plates. The IFN-α-induced mRNA or protein expression
of ISGs, including ISG15 and PKR, in HuH-7 cells cultured on type I
collagen-coated dishes was compared with those in cells cultured on
non-coated plastic dishes. As displayed in Fig. 1B and C, the expression of ISG15
and PKR was decreased at the mRNA and protein level, indicating
that IFN-α signaling was attenuated in cells grown on the type I
collagen-coated dishes.
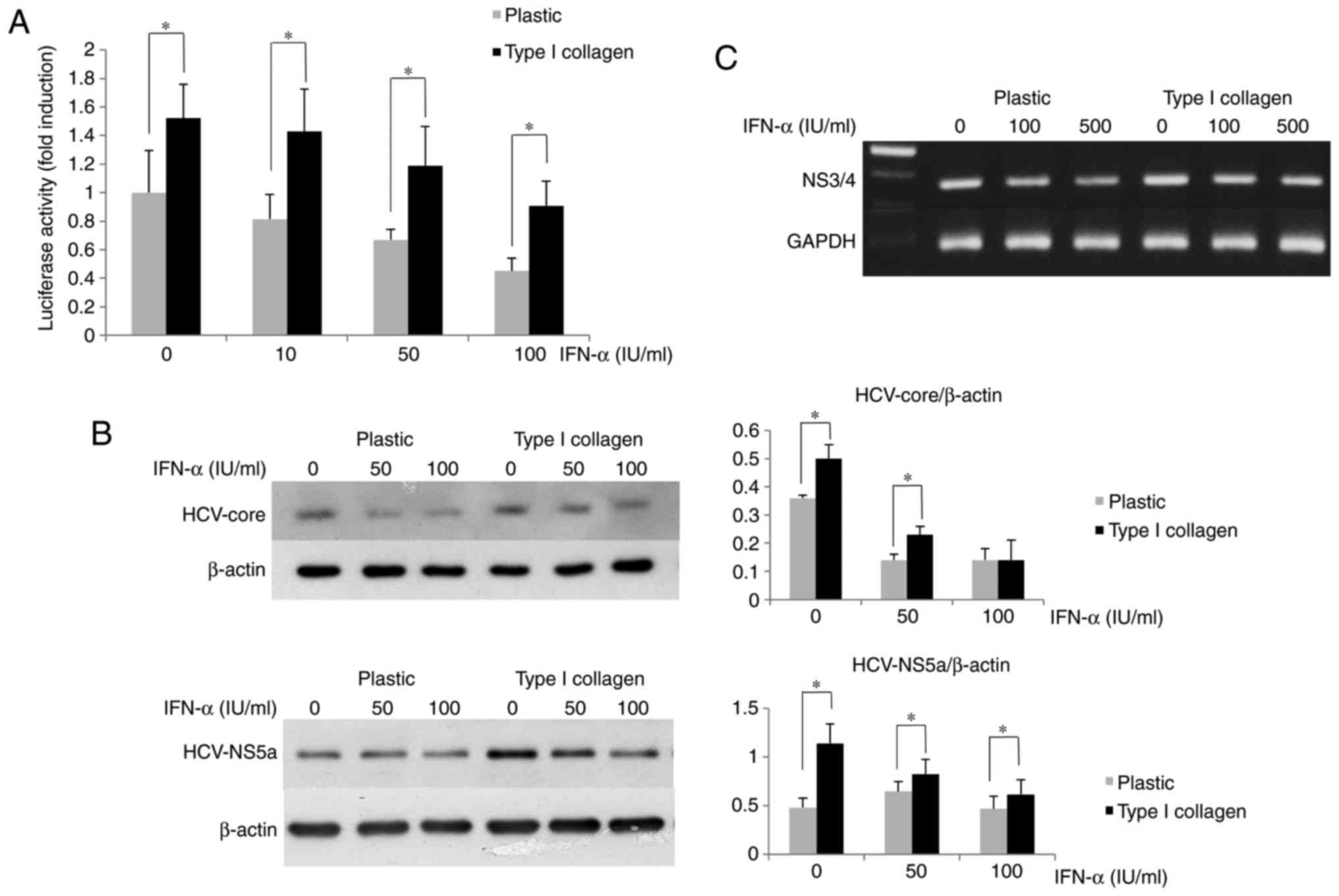
Type I collagen inhibits the
IFN-α-associated suppression of HCV-RNA replication in OR6
cells
The suppressive effect of IFN-α on HCV replication
was assessed using OR6 cells cultured on type I collagen-coated
dishes to determine the effects of ECM on HCV replication. In the
type I collagen-coated plates, the luciferase activity of OR6 cells
was higher than that in the cells cultured on normal plates
(Fig. 2A). The amounts of HCV
core and NS5A protein expression in OR6 cells grown on type I
collagen-coated dishes were measured by western blot analysis,
revealing that their expression in the cells cultured on the coated
dishes was higher than that in the cells cultured on the normal
plastic dishes (Fig. 2B). The
HCV-RNA expression in OR6 cells grown on type I collagen-coated
dishes measured by RT-PCR was higher than that in cells cultured on
normal dishes (Fig. 2C). These
results indicated that the inhibitory effect of IFN-α on HCV
replication was impaired in OR6 cells cultured on type I
collagen-coated dishes compared with the effect in those cultured
on normal plastic dishes.
Inhibition of β1-integrin function
restores IFN-α-induced signaling
The mechanisms of the ECM-mediated inhibition of
IFN-α signaling were then examined. Since β1-integrin is a major
subunit of ECM receptors and is known to influence diverse
signaling pathways in adherent cells, the expression and role of
β1-integrin in HuH-7 cells was examined. As presented in Fig. 3A, the β1-integrin expression of
cells on type I collagen-coated dishes was increased compared with
that in cells cultured on plastic dishes. It was then investigated
whether β1-integrin, which was highly expressed in HuH-7 cells
cultured on type I collagen-coated dishes, affected IFN-α
signaling. The cells were optionally pre-treated with β1-integrin
function-blocking antibody, and after optional IFN-α treatment for
12 h, the ISRE luciferase activity was measured. The ISRE
luciferase activity of HuH-7 activated by IFN-α was clearly higher
in the plastic dishes compared with the collagen-coated dishes.
When β1-integrin function blocking antibody was administered, there
was a significant elevation of ISRE luciferase activity observed in
the collagen coated dish, but not in the plastic dish (Fig. 3B). In HuH-7 cells treated with
β1-integrin function-blocking antibody, the IFN-α-induced ISG
protein expression was also increased compared with that in cells
without blocking treatment (Fig.
3C). The effects of β1-integrin-blocking antibody on HCV-RNA
replication in OR6 cells were then evaluated. As presented in
Fig. 3D, when OR6 cells were
treated with β1-integrin blocking antibody prior to IFN-α
treatment, the Renilla luciferase activity was reduced
compared with that in cells without β1-integrin blocking. As
displayed in Fig. 3E, when cells
were cultured on type I collagen-coated dishes, β1-integrin
function-blocking antibody reduced the expression of HCV-NS5a,
although no clear differences were seen between the groups treated
with or without β1-blocking antibody when cells were cultured on
plastic. It appeared that β1-integrin blocking affects endogenous
IFN-α signaling and improves the effect of IFN treatment on HCV-RNA
replication. These results suggest that type I collagen may support
HCV replication via attenuation of IFN signaling in a
β1-integrin-dependent manner, and that the ECM-stimulated integrin
signal may promote HCV replication in an IFN-dependent and
-independent manner.
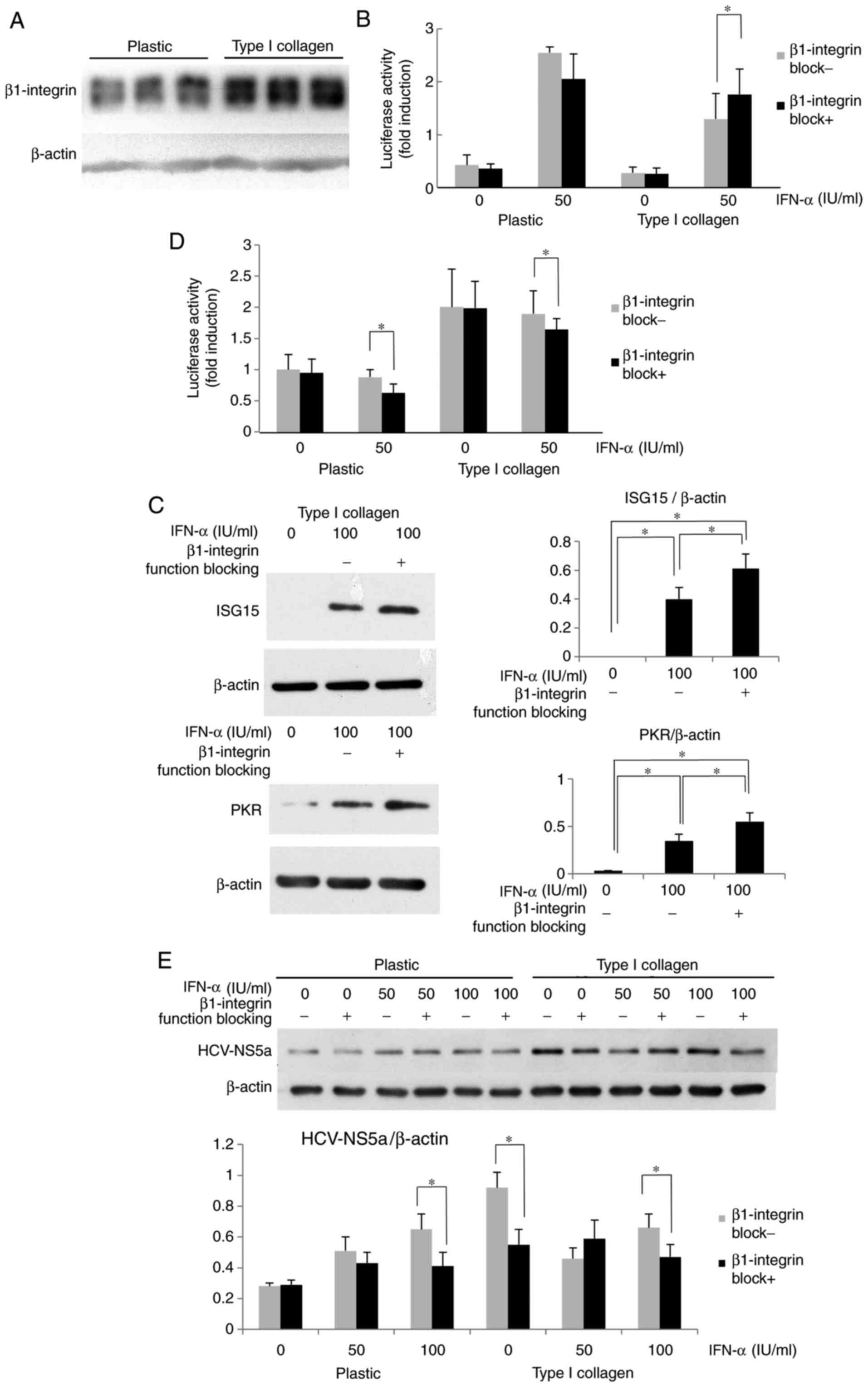 | Figure 3Attenuation of IFN-α signaling by
type I collagen is β1-integrin-dependent. (A) β1-integrin was
overexpressed in Huh7 cells grown on type I collagen-coated dishes.
(B) Improvement in the ISRE luciferase activity after treatment
with β1-integrin function-blocking antibody in HuH-7 cells cultured
on type I collagen-coated dishes. Cells were treated with
β1-integrin function-blocking antibody (1 µg/ml) for 6 h.
The ISRE-luciferase activity was measured after IFN-α treatment for
12 h. The results are presented as the mean fold induction of the
controls. (C) Improvement in the ISG protein expression by
treatment with β1-integrin function-blocking antibody in HuH-7
cells grown on type I collagen-coated dishes. The cells were
cultured on type I collagen-coated dishes for 3 days and then
treated with β1-integrin function-blocking antibody for 6 h,
followed by treatment with IFN-α for 12 h. The ISG15 and PKR
expression was measured by western blot analysis with β-actin used
as a control. (D) Improvement in the suppressive effect of IFN-α on
HCV replication in OR6 cells cultured on type I collagen-coated
dishes. After 6-h treatment with β1-integrin function-blocking
antibody (1 µg/ml), the cells were treated with IFN-α for 12
h, and the Renilla luciferase activity was then measured.
(E) Improvement in the suppressive effect of IFN-α on HCV protein
expression. HCV-NS5a expression was suppressed by co-treatment with
β1-integrin function-blocking antibody in OR6 cells grown on type I
collagen-coated dishes. The OR6 cells were cultured on type I
collagen-coated dishes for 3 days and then treated with β1-integrin
function-blocking antibody for 6 h, followed by treatment with
IFN-α for 12 h. The HCV-NS5a expression was measured by western
blot analysis with β-actin used as a control. Values are expressed
as the mean ± standard deviation (n=3). *P<0.05. IFN,
interferon; OR6 cells, HuH-7 cells stably transfected with
full-length HCV-RNA fused with Renilla luciferase HCV,
hepatitis C virus; NS, nonstructural protein; ISRE, IFN-stimulated
response element; ISG, IFN-stimulated gene; PKR, protein kinase
R. |
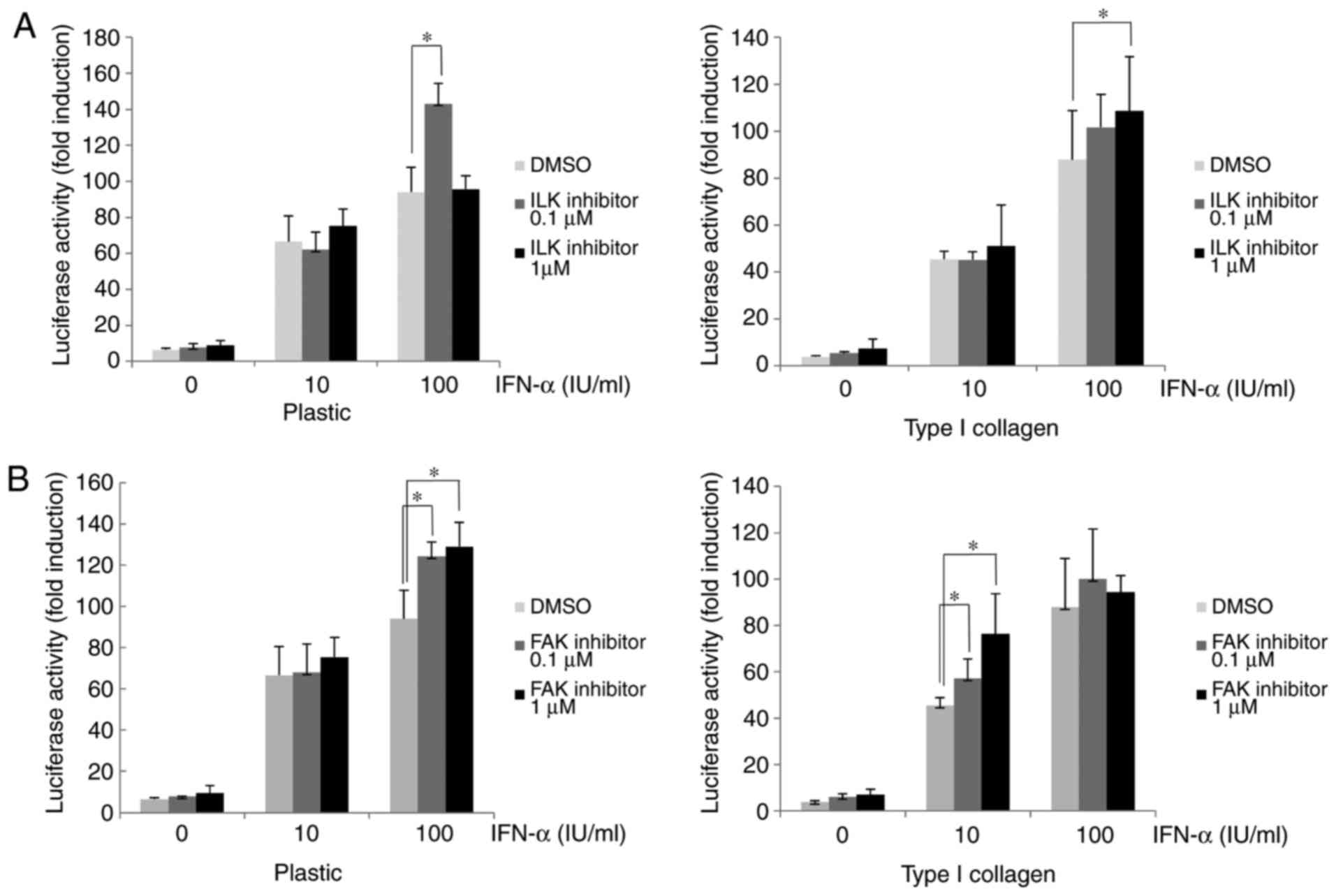
Attenuation of IFN-α signaling via
β1-integrin involves ILK and FAK
Various proteins associated with integrin α/β
heterodimers, including FAK and ILK, are known be involved in the
activation of diverse cellular signaling pathways (19-21). In the present study, cells were
treated with ILK or FAK inhibitor to determine the roles of ILK and
FAK in the β1-integrin-mediated attenuation of IFN-α signaling.
After treatment with ILK or FAK inhibitor, the IFN-α-induced
ISRE-luciferase activity in Huh-7 cells cultured on type I
collagen-coated dishes was higher than that in
dimethyl-sulfoxide-treated control cells (Fig. 4A and B). In the plastic dishes in
the ILK 0.1 µM with IFN-α 100 IU/ml group, the
ISRE-luciferase activity was significantly increased compared with
the DMSO control, while in the ILK 1 µM with IFN-α 100 IU/ml
group an increase in ISRE-luciferase activity was not observed. In
the collagen-coated dishes in the ILK 1 µM with IFN-α 100
IU/ml group the ISRE-luciferase activity was significantly
increased compared with the DMSO group, whereas it was not
significantly increased in the ILK 0.1 µM with IFN-α 100
IU/ml group. Although these results suggested that ISRE-luciferase
activity was increased when ILK was inhibited, the effect of the
ILK inhibitor was matrix-dependent and an excessive inhibition of
ILK in a plastic dish may cancel the effects and reverse the
increase in ISRE-luciferase activity. The ISG15 and PKR expression
in HuH-7 cells treated with ILK or FAK inhibitor was then examined.
As presented in Fig. 4C, the
cells cultured on type I collagen-coated dishes had a lower
expression of PKR and ISG15 after IFN-α treatment compared with the
plastic dishes, and subsequent treatment with ILK inhibitor
restored their expression. Similarly, treatment with FAK inhibitor
restored the reduced PKR and ISG15 expression induced by IFN-α in
cells cultured on type I collagen-coated dishes (Fig. 4D). These results suggested that
the IFN-α-induced attenuation of ISRE luciferase activity and ISG
expression in cells cultured on type I collagen-coated dishes were
mediated by ILK and FAK downstream of β1-integrin.
Discussion
Although the presence of advanced liver fibrosis or
cirrhosis in patients with chronic HCV is a major predictive factor
for the failure of IFN-based antiviral therapy (6-10),
the molecular mechanisms of the resistance to IFN action by
fibrosis remain elusive. The recent development of IFN-free oral
DAA regimens has markedly increased the rate of SVR in HCV-infected
patients (13-15). However, even in patients treated
with DAA without IFN, an altered IFN response in the liver tissue
was observed to be associated with the failure of DAA treatment,
suggesting the importance of an adequate host IFN response for the
eradication of HCV (16). The
present study investigated whether ECM components that are
increased in the fibrotic liver directly affect IFN signaling in
vitro. The results indicated that ECM components, e.g. type I
collagen, attenuate the IFN-α-induced ISRE-mediated transcriptional
activity and the expression of a subset of ISGs in a
β1-integrin-dependent manner, and that the presence of ECM
components decreased the inhibitory effects of IFN-α on HCV
replication in HCV replicon cells.
Hepatic stellate cells (HSCs) are a major
ECM-producing cell type that are activated after exposure to
liver-injurious stimuli, including HCV, and have central roles in
the development of liver fibrosis (17,18). Several studies have investigated
the roles of HSCs on HCV replication in hepatocytes and revealed
that the activation of HSCs via the innate immune system produced
antiviral cytokines, including IFN-β and IFN-λ, and inhibited the
replication of HCV in hepatocytes. Therefore, activated HSCs appear
to have an anti-viral phenotype against HCV infection, while
conversely producing various ECM components to thereby contribute
to the development of liver fibrosis (25-27).
The present results indicated that culture of cells
on type I collagen-coated plates resulted in an increased
expression of β1-integrin and the attenuation of IFN-stimulated
ISRE activity. Integrins, a heterodimer complex consisting of an α
and β subunit, serve as major receptors for ECM (19-21). Integrins have been reported to
interact with a wide variety of cytokine receptor-mediated signals,
including the MAPK (28-30), PI3K/mammalian target of rapamycin
(mTOR) (29,31,32) and TGF-β/Smad pathways (30,33,34). However, the interaction between
integrins and IFN receptor-mediated signals has remained to be
fully elucidated. TGF-β/Smad signaling was reported to be involved
in IFN resistance in HCV patients with advanced liver fibrosis
(35). TGF-β is a potent
stimulator of the production of various ECM components, including
collagen (17,18), and is also known to increase the
expression of integrins (36).
Shirasaki et al (35)
demonstrated that TGF-β1 inhibited the IFN-induced expression of
ISG and the IFN-mediated suppression of HCV replication in
HCV-transfected HuH-7 cells. They also reported that TGF-β1
impaired the mTOR activation required for IFN-induced ISG
expression (37,38) by downregulating the expression of
Ras homolog enriched in brain and increasing that of suppresser of
cytokine signaling 3 under conditions of reduced amino acid levels.
Furthermore, replacement of branched-chain amino acids, which are
reduced in patients with liver cirrhosis and known to activate mTOR
signaling, restored the effects of DAAs. Since integrins activate
the PI3K/mTOR pathway (29,31,32,39), the ECM-induced inhibition of IFN
signaling does not appear to be due to the inhibition of the mTOR
pathway that was observed under conditions of reduced amino acid
concentrations.
Among various proteins associated with integrin-α/β
heterodimers, kinases including FAK and ILK are involved in the
activation of diverse cellular signaling pathways, including the
MAPK (extracellular signal-regulated kinase, p38 and c-Jun
N-terminal kinase), PI3K/Akt/mTOR/ribosomal protein S6 kinase β-1
pathway, TGF-β/Smad pathway and PKCs (19-21,40,41). The present study demonstrated that
the IFN-stimulated ISRE activity was attenuated via β1-integrin,
indicating the presence of an interaction between integrins and IFN
signaling. The results also suggested a possible involvement of ILK
and FAK in the ECM-mediated attenuation of IFN signaling and ISG
expression, since the inhibition of ILK or FAK partially restored
the ISRE-luciferase activity and ISG expression. Since
integrin-associated proteins, including ILK and FAK, have
significant roles in the modulation of growth factor/cytokine
receptor-mediated signaling, further investigation is required to
identify the key molecules involved in the integrin-mediated
inhibition of IFN signaling.
Another possible role of IFN resistance in liver
fibrosis may be linked to hepatocarcinogenesis. Liver cancer
develops more often from the fibrotic liver than the normal liver,
even after the achievement of SVR in patients with HCV treated with
IFN-based therapy (17,18,42,43). Accumulated ECM in the fibrotic
liver has been suggested to contribute to hepatocarcinogenesis via
a wide variety of signaling pathways that promote hepatocyte
proliferation and survival, including integrin-mediated signaling
(34,44). While IFNs have been reported to
exert diverse antiviral effects, type I IFNs, including IFN-α, have
been used for the treatment of numerous human cancer types, and the
type I IFN status is associated with the outcome of anti-cancer
therapy (45). Indeed, IFN is
known to suppress liver cancer growth via the induction of
apoptosis in vitro and in vivo (46,47). Therefore, the attenuation of
IFN-signaling by ECM may contribute to the frequent development of
liver cancer from the fibrotic liver.
In conclusion, the present study indicated that ECM
inhibited IFN signaling in a β1-integrin-dependent manner, possibly
involving FAK and ILK, and impairment of antiviral activity by
IFN-α in HCV-replicon cells. These results may provide a mechanism
for the role of fibrosis in IFN resistance, which may be harnessed
for the treatment of cirrhotic patients with HCV infection.
Acknowledgments
Not applicable.
Funding
This study was supported by a Grant-in-Aid for
Scientific Research from the Ministry of Education, Culture,
Sports, Science and Technology of Japan (grant no. 16590606 to TM
and IO).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
IO and TM devised the study and designed the
experiments. TK, SI, XJ, SM and IO performed the experiments. NS,
MI and NK established and provided the replicon cells. TK, SM, NK,
YE, KA, KF and IO analyzed the data. TK and IO drafted the
manuscript. All authors have read and approved the final
manuscript.
Ethical approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gale M Jr and Foy EM: Evasion of
intracellular host defense by hepatitis C virus. Nature.
436:939–945. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Borden EC, Sen GC, Uze G, Silverman RH,
Ransohoff RM, Foster GR and Stark G: Interferons at age 50: Past,
current and future impact on biomedicine. Nat Rev Drug Discov.
6:975–990. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weber F: Interaction of hepatitis C virus
with type I interferon system. World J Gastroenterol. 13:4818–4823.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fried MW, Shiffman ML, Reddy KR, Smith C,
Marinos G, Gonçales FL Jr, Häussinger D, Diago M, Carosi G,
Dhumeaux D, et al: Peginterferon alfa-2a plus ribavirin for chronic
hepatitis C virus infection. N Engl J Med. 347:975–982. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Manns MP, McHutchison JG, Gordon SC,
Rustgi VK, Shiffman M, Reindollar R, Goodman ZD, Koury K, Ling M
and Albrecht JK: Peginterferon alfa-2b plus ribavirin compared with
interferon alfa-2b plus ribavirin for initial treatment of chronic
hepatitis C: A randomised trial. Lancet. 358:958–965. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marrache F, Consigny Y, Ripault MP,
Cazals-Hatem D, Martinot M, Boyer N, Degott C, Valla D and
Marcellin P: Safety and efficacy of peginterferon plus ribavirin in
patients with chronic hepatitis C and bridging fibrosis or
cirrhosis. J Viral Hepat. 12:421–428. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Everson GT, Hoefs JC, Seeff LB, Bonkovsky
HL, Naishadham D, Schiffman ML, Kahn JA, Lok ASF, Di Bisceglie AM,
Lee WM, et al: Impact of disease severity on outcome of antiviral
therapy for chronic hepatitis C: Lessons from the HALT-C trial.
Hepatology. 44:1675–1684. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bruno S, Shiffman ML, Roberts SK, Gane EJ,
Messinger D, Hadziyannis SJ and Marcellin P: Efficacy and safety of
peginterferon alfa-2a (40KD) plus ribavirin in hepatitis C patients
with advanced fibrosis and cirrhosis. Hepatology. 51:388–397. 2010.
View Article : Google Scholar
|
|
9
|
Cheng WS, Roberts SK, McCaughan G, Sievert
W, Weltman M, Crawford D, Rawlinson W, Marks PS, Thommes J,
Rizkalla B, et al: Low virological response and high relapse rates
in hepatitis C genotype 1 patients with advanced fibrosis despite
adequate therapeutic dosing. J Hepatol. 53:616–623. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asselah T, Estrabaud E, Bieche I, Lapalus
M, De Muynck S, Vidaud M, Saadoun D, Soumelis V and Marcellin P:
Hepatitis C: Viral and host factors associated with non-response to
pegylated interferon plus ribavirin. Liver Int. 30:1259–1269. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jacobson IM, McHutchison JG, Dusheiko G,
Di Bisceglie AM, Reddy KR, Bzowej NH, Marcellin P, Muir AJ, Ferenci
P, Flisiak R, et al: Telaprevir for previously untreated chronic
hepatitis C virus infection. N Engl J Med. 364:2405–2416. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bruno S, Vierling JM, Esteban R, Nyberg
LM, Tanno H, Goodman Z, Poordad F, Bacon B, Gottesdiener K,
Pedicone LD, et al: Efficacy and safety of boceprevir plus
peginterferon-ribavirin in patients with HCV G1 infection and
advanced fibrosis/cirrhosis. J Hepatol. 58:479–487. 2013.
View Article : Google Scholar
|
|
13
|
Liang TJ and Ghany MG: Current and future
therapies for hepatitis C virus infection. N Engl J Med.
368:1907–1917. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Casey LC and Lee WM: Hepatitis C virus
therapy update 2013. Curr Opin Gastroenterol. 29:243–249.
2013.PubMed/NCBI
|
|
15
|
Shah N, Pierce T and Kowdley KV: Review of
directacting antiviral agents for the treatment of chronic
hepatitis C. Expert Opin Investig Drugs. 22:1107–1121. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meissner EG, Wu D, Osinusi A, Bon D,
Virtaneva K, Sturdevant D, Porcella S, Wang H, Herrmann E,
McHutchison J, et al: Endogenous intrahepatic IFNs and association
with IFN-free HCV treatment outcome. J Clin Invest. 124:3352–3363.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Friedman SL: Mechanisms of hepatic
fibrosis. Gastroenterology. 134:1655–1669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Inagaki Y and Okazaki I: Emerging insights
into transforming growth factor beta Smad signal in hepatic
fibrogenesis. Gut. 56:284–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hynes RO: Integrins: Bidirectional,
allosteric signaling machines. Cell. 110:673–687. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hehlgans S, Hasse M and Cordes N:
Signalling via integrins: Implications for cell survival and
anticancer strategies. Biochem Biophys Acta. 1775:163–180.
2017.
|
|
21
|
Millard M, Odde S and Neamati N: Integrin
targeted therapeutics. Thearanostics. 1:154–188. 2011. View Article : Google Scholar
|
|
22
|
Hayashida T: Integrins modulate cellular
fibrogenesis at multiple levels; Regulation of TGF-β signaling.
Endocr Metab Immune Disord Drug Targets. 10:302–319. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ikeda M, Abe K, Dansako H, Nakamura T,
Naka K and Kato N: Efficient replication of a full-length hepatitis
C virus genome, strain O, in cell culture, and development of a
luciferase reporter system. Biochem Biophys Res Commun.
329:1350–1359. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ozaki I, Zhang H, Mizuta T, Ide Y, Eguchi
Y, Yasutake T, Sakamaki T, Pestell RG and Yamamoto K:
Menatetrenone, a vitamin K2 analogue, inhibits hepatocellular
carcinoma cell growth by suppressing cyclin D1 expression through
inhibition of nuclear factor kappaB activation. Clin Cancer Res.
13:2236–2245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang B, Trippler M, Pei R, Lu M, Broering
R, Gerken G and Schlaak JF: Toll-like receptor activated human and
murine hepatic stellate cells are potent regulators of hepatitis C
virus replication. J Hepatol. 51:1037–1045. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Li J, Wang X, Ye L, Zhou Y and Ho
W: Induction of interferon-λ contributes to Toll-like
receptor-3-activated hepatic stellate cell-mediated hepatitis C
virus inhibition in hepatocytes. J Viral Hepat. 20:385–394. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Alisi A, Arciello M, Petrini S, Conti B,
Missale G and Balsano C: Focal adhesion kinase (FAK) mediates the
induction of pro-oncogenic and fibrogenic phenotypes in hepatitis C
virus (HCV)-infected cells. PLoS One. 7:e441472012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu X and Assoian RK: Integrin-dependent
activation of MAP kinase: A link to shape-dependent cell
proliferation. Mol Biol Cell. 6:273–282. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Khwaja A, Rodriguez-Viciana P, Wennström
S, Warne PH and Downward J: Matrix adhesion and Ras transformation
both activate a phosphoinositide 3-OH-kinase and protein kinase
B/Akt cellular survival pathway. EMBO J. 16:2783–2793. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang H, Ozaki I, Mizuta T, Yoshimura T,
Matsuhashi S, Eguchi Y, Yasutake T, Hisatomi A, Sakai T and
Yamamoto K: Transforming growth factor-beta 1-induced apoptosis is
blocked by beta 1-integrin-mediated mitogen-activated protein
kinase activation in human hepatoma cells. Cancer Sci. 95:878–886.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yau CY, Wheeler JJ, Sutton KL and Hedley
DW: Inhibition of integrin-linked kinase by a selective small
molecule inhibitor, QLT0254, inhibits the PI3K/PKB/mTOR, Stat3, and
FKHR pathways and tumor growth, and enhances gemcitabine-induced
apoptosis in human orthotopic primary pancreatic cancer xenografts.
Cancer Res. 65:1497–1504. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Riaz A, Ilan N, Vlodavsky I, Li JP and
Johansson S: Characterization of heparanase-induced
phosphatidylinositol 3-kinase-AKT activation and its integrin
dependence. J Biol Chem. 288:12366–12375. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Garamszegi N, Garamszegi SP,
Samavarchi-Tehrani P, Walford E, Schneiderbauer MM, Wrana JL and
Scully SP: Extracellular matrix-induced transforming growth
factor-beta receptor signaling dynamics. Oncogene. 29:2368–2380.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ozaki I, Hamajima H, Matsuhashi S and
Mizuta T: Regulation of TGF-β1-induced pro-apoptotic signaling by
growth factor receptors and extracellular matrix receptor integrins
in the liver. Front Physiol. 2:782011. View Article : Google Scholar
|
|
35
|
Shirasaki T, Honda M, Shimakami T, Murai
K, Shiomoto T, Okada H, Takabatake A, Tokumaru A, Sakai Y,
Yamashita T, et al: Impaired interferon signaling in chronic
hepatitis C patients with advanced fibrosis via the transforming
growth factor beta signaling pathway. Hepatology. 60:1519–1530.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang D, Zhou GH, Birkenmeier TM, Gong J,
Sun L and Brattain MG: Autocrine transforming growth factor beta-1
modulates the expression of integrin alpha 5 beta1 in human colon
carcinoma FET cells. J Biol Chem. 270:14154–14159. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kaur S, Sassano A, Dolniak B, Joshi S,
Majchrzak-Kita B, Baker DP, Hay N, Fish EN and Platanias LC: Role
of the Akt pathway in mRNA translation of interferon-stimulated
genes. Proc Natl Acad Sci USA. 105:4808–4813. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lekmine F, Uddin S, Sassano A, Parmar S,
Brachmann SM, Majchrzak B, Sonenberg N, Hay N, Fish EN and
Platanias LC: Activation of the p70 S6 kinase and phosphorylation
of the 4E-BP1 repressor of mRNA translation by type I interferons.
J Biol Chem. 278:27772–27780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zeller KS, Idevall-Hagren O, Stefansson A,
Velling T, Jackson SP, Downward J, Tengholm A and Johansson S:
PI3-kinase 110α mediates β1 integrin-induced Akt activation and
membrane protrusion during cell attachment and initial spreading.
Cell Signal. 22:1838–1848. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tang Q, Zhao S, Wu J, Zheng F, Yang L, Hu
J and Hann SS: Inhibition of integrin-linked kinase expression by
emodin through crosstalk of AMPKα and ERK1/2 signaling and
reciprocal interplay of Sp1 and c-Jun. Cell signaling.
27:1469–1477. 2015. View Article : Google Scholar
|
|
41
|
Hirata E, Girotti MR, Viros A, Hooper S,
Spencer-Dene B, Matsuda M, Larkin J, Marais R and Sahai E:
Intravital imaging reveals how BRAF inhibition generates
drug-tolerant microenvironments with high integrin β1/FAK
signaling. Cancer Cell. 27:574–588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Asahina Y, Tsuchiya K, Tamaki N, Hirayama
I, Tanaka T, Sato M, Yasui Y, Hosokawa T, Ueda K, Kuzuya T, et al:
Effect of aging on risk for hepatocellular carcinoma in chronic
hepatitis C virus infection. Hepatology. 52:518–527. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Morgan RL, Baack B, Smith BD, Yartel A,
Pitasi M and Falck-Ytter Y: Eradication of hepatitis C virus
infection and the development of hepatocellular carcinoma: A
meta-analysis of observational studies. Ann Intern Med.
158:329–337. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang DY and Friedman SL:
Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology.
56:769–775. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zitvogel L, Galluzzi L, Kepp O, Smyth MJ
and Kroemer G: Type I interferons in anticancer immunity. Nat Rev
Immunol. 15:405–414. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yano H, Iemura A, Haramaki M, Ogasawara S,
Takayama A, Akiba J and Kojiro M: Interferon alfa receptor
expression and growth inhibition by interferon alfa in human liver
cancer cell lines. Hepatology. 29:1708–1717. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kusano H, Akiba J, Ogasawara S, Sanada S,
Yasumoto M, Nakayama M, Ueda K, Ueda K, Kurita T, Todoroki K, et
al: Pegylated interferon-α2a inhibits proliferation of human liver
cancer cells in vitro and in vivo. PLoS One. 8:e831952013.
View Article : Google Scholar
|