Introduction
Increasing evidence has shown that diabetes
mellitus-induced hyperglycemic conditions cause vascular
endothelial damage (1,2) demonstrating that hyperglycemia is
associated with endothelial dysfunction and reduced blood vessel
growth, with angiogenesis often compromised in patients with
diabetes. Endothelial dysfunction has long been considered as the
first event in the development of atherosclerosis and clinical
events (3). Therefore, the
maintenance of an intact endothelial layer and improvement of its
functions are essential for the maintenance of healthy blood
vessels and the prevention of cardiovascular complications
associated with diabetes.
The E2 promoter binding factor (E2F) family of
transcription factors is involved in DNA synthesis, the cell cycle,
cell differentiation and apoptosis (4,5).
E2 promoter binding factor 1 (E2F1), which was the first member
cloned, is important in cell control, although its exact role is
controversial. It has been reported that the expression of E2F1
inhibits cell apoptosis (6),
whereas other have reported that its expression promotes cell
apoptosis (7,8). Yang et al reported that the
expression of E2F1 promoted cell apoptosis mediated by
AMP-activated protein kinase α2 (AMPKα2) (9). AMPKα2 also mediated endothelial cell
apoptosis under high glucose (HG) conditions (10), which resulted in the opposite
effect of AMPKα1 (11). E2F1 is
also important in the regulation of mammalian target of rapamycin
complex 1 (mTORC1) during autophagy (12,13). Increasing evidence has shown that
certain autophagic activations inhibit HG-induced endothelial cell
apoptosis via the mTOR signaling pathway (14); however, the regulatory mechanisms
of E2F1 in autophagy and HG-induced endothelial cell apoptosis
remain to be fully elucidated.
MicroRNAs (miRNAs) are 20–22 nucleotide non-coding
RNAs, which repress the expression of their cognate target genes by
specifically binding and cleaving mRNAs, inhibiting translation,
and/or deadenylating mRNA tails (15). miRNAs have been reported to
control various biological processes, including cell proliferation,
apoptosis, and autophagy (16–18), and miRNA (miR)-17-5p is conserved
across vertebrates and downregulated in diabetes (19–21). Bioinformatics analyses (http://www.targetscan.org/) have suggested that
miR-17-5p targets the 3′untranslated region (3′UTR) of E2F1.
However, the role of miR-17-5p in HG-induced endothelial cell
damage and the association between miR-17-5p and E2F1 remain to be
elucidated.
In the present study, it was found that HG induced
the downregulation of miR-17-5p and upregulation of E2F1 during
human umbilical vein endothelial cell (HUVEC) injury. The
downregulation of the expression of E2F1 inhibited HG-induced HUVEC
dysfunction via the suppression of mTORC1-mediated inhibition of
autophagy and AMPKα2-mediated promotion of apoptosis. These results
suggested that the inhibited expression of E2F1 protected against
HG-induced HUVEC injury through the activation of autophagy. The
overexpression of miR-17-5p inhibited E2F1-mediated HUVEC injury
under HG conditions, which was reversed following transfection with
an E2F1-overexpression vector. Finally, bifluorescein experiments
showed that miR-17-5p targeted the 3′UTR of E2F1. Taken together,
the results revealed the importance of miR-17-5p in controlling
HG-induced E2F1-mediated endothelial cell damage.
Materials and methods
Cell culture
The HUVECs, purchased from the American Type Culture
Collection (Manassas, VA, USA; CRL-1730™), were cultured in
RPMI-1640 complete medium (Invitrogen; Thermo Fisher Scientific,
Inc., USA) containing 10% fetal bovine serum (Invitrogen; Thermo
Fisher Scientific, Inc.). All cells were cultured at 37°C in a
humidified incubator containing 5% CO2, and the culture
medium was replaced every other day.
Flow cytometry
Flow cytometry was used to determine the percentage
of apoptotic HUVECs. The apoptotic cells were differentiated from
viable or necrotic cells by combined treatment with annexin V
(AV)-FITC and propidium iodide (PI). The cells were washed twice
and adjusted to a concentration of 1×106 cells/ml with
cold D-Hanks buffer. The AV-FITC (10 µl) and PI (10
µl) were then added to 100 µl of cell suspension and
incubated for 15 min at room temperature in the dark. Finally, 400
µl of binding buffer was added to each sample without
washing and analyzed using flow cytometry. Each experiment was
performed in triplicate.
Western blot analysis
Western blot analyses were performed using cell
lysates in urea buffer (8 M urea, 1 M thiourea, 0.5% CHAPS, 50 mM
dithiothreitol and 24 mM spermine). GAPDH was used as a loading
control. The samples [40 µg total protein, quantified using
the two-dimensional Quant kit (GE Healthcare Life Sciences, Little
Chalfont, UK)] were separated using 8% SDS-PAGE and transferred
onto nitrocellulose membranes (EMD Millipore, Billerica, MA, USA).
Following blocking in 5% nonfat milk for 1 h, the membranes were
incubated with primary antibodies against E2F1 (1:1,000; cat. no.
ab179445; Abcam, Cambridge, UK), AMPKα2 (1:200; cat. no. ab3760;
Abcam), mTORC1 (1:200; cat. no. ab168538; Abcam), cleaved caspase-3
(1:200; cat. no. ab2302; Abcam), B-cell lymphoma 2 (Bcl-2; 1:400;
cat. no. ab119506; Abcam), Bcl-2-associated X protein (Bax; 1:300;
cat. no. ab32503; Abcam), P62 (1:400; cat. no. ab5641; Abcam),
microtubule-associated protein 1 light chain 3 (LC3; 1:200; cat.
no. ab48394; Abcam), and GAPDH (1:2,000; cat. no. ab8245; Abcam) at
4°C overnight. Following washing with phosphate buffered saline
(PBS) three times, the membranes were incubated with horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibodies
(1:2,000; cat. no. ab172730; Abcam) for 1 h at room temperature.
The signals were detected using an ECL detection system (GE
Healthcare Life Sciences) and analyzed using ImageJ 1.42q software
(National Institutes of Health, Bethesda, MD, USA).
Cell viability assay
A Cell Counting Kit-8 assay (CCK8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) was used to assess cell
viability. The HUVECs (1×104) were seeded into 96-well
plates and incubated overnight under the conditions described
above. The cells were pretreated with 5.5, 15, 30 or 50 mM glucose
for 24–72 h. The medium was then removed and the cells were washed
three times with PBS. RPMI-1640 (90 µl) and CCK8 (10
µl) were subsequently added to each well and incubated for
1.5 h at 37°C, and a microplate reader was used to measure the
optical density at 450 nm.
Matrigel assay
In vitro neovascularization assays were
performed in human fibrin matrices. Briefly, following the
different treatments, the HUVECs were seeded onto Matrigel-coated
plates (BD Biosciences, Franklin Lakes, NJ, USA) in RPMI-1640
medium and incubated at 37°C for 12 h. The tubular structures of
the HUVECs in the Matrigel were analyzed by phase contrast
microscopy. The average numbers of tube formations from six random
phase contrast photomicrographs were used for subsequent
analyses.
Vector construction and transfection
For the overexpression of miR-17-5p, the miR-17-5p
mimic or corresponding negative control (miR-NC) were purchased
from GenePharma (Shanghai, China). The HUVECs were transfected with
either the miR-17-5p mimic or miR-NC at a final concentration of 50
nM using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The
cells were then used for miR-17-5p expression analyses or for other
experiments following transfection for 48 h.
For the downregulation of E2F1, small interfering
(si)RNA against E2F1 (5′-CGGACUCAGUGAUAAUAAUUU-3′) was synthesized
by GenePharma and transfected at a final concentration of 50 nM
using Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
For the overexpression of E2F1, AMPKα2, mTORC1,
human E2F1 and mTORC1, cDNA with the 3′UTR was cloned into the
pMSCV-hygro vector (Takara Bio, Inc., Otsu, Japan). The primers
corresponded to the National Center for Biotechnology Information
reference sequence, and included the following: E2F1 (NG_046988.1),
forward 5′-CAAAGCTTATGGCCTTGGCCGGGGCC-3′ and reverse
5′-GGCTCGAGTCAGAAATCCAGGGG-3′; AMPKα2 (BC069823.1), forward
5′-CAAAGCTTATGGCTGAGAAGCAG-3′ and reverse
5′-GGCTCGAGTCAACGGGCTAAAG-3′; and mTORC1 (AF148645.1), forward
5′-CAAAGCTTATGCCGCAGTCCAAG-3′ and reverse
5′-GGCTCGAGTCACATCACCGAGCATG-3′. The E2F1 cDNA was inserted into a
pMD18-T Simple vector (Takara Bio, Inc.) to form the pMD18-T-E2F1,
pMD18-T-AMPKα2, and pMD18-T-mTORC1 vectors. Following sequencing,
the recombinant segment of the correct clone was incised by
HindIII and XhoI (Takara Bio, Inc.). The recombinant
segment was inserted into the pMSCV-hygro vector, which was incised
by the same two restriction endonucleases. The clones were
sequenced and the correct clones were amplified and identified by
restriction enzyme digestion.
Prior to transfection, ~1×106 HUVECs were
seeded in media onto a 60-mm dish and incubated for 24 h. The
following day, the cells were transfected using the Sofast gene
transfection reagent kit (Sunma Biotechenology Co., Ltd., Xiamen,
China) according to the manufacturer's protocol. The transfected
cells were selected using hygromycin for 3–4 weeks for subsequent
experiments. The monoclonal cells were then cloned and screened for
the expression of E2F1, AMPKα2 and mTORC1.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis for the detection of
miR-17-5p
Total RNA was isolated using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Reverse
transcription was performed using the RT-qPCR system (Promega
Corporation, Madison, WI, USA). RT-qPCR analysis was performed in a
total reaction volume of 20 µl (10 µl 2xmaster mix, 4
µl cDNA, 1 µl forward primer (10 µM), 1
µl reverse primer (10 µM), and 4 µl of
double-distilled water) using SYBR® Green I Supermix
(Takara Biotechnology Co., Ltd., Dalian, China) according to the
manufacturer's protocol. The primer sequences used for PCR were as
follows: E2F1 forward, 5′-ATGGCCTTGGCCGGGGCC-3′ and reverse,
5′-TCAGAAATCCAGGGG-3′; AMPKα2 forward, 5′-ATGGCTGAGAAGCAG-3′ and
reverse, 5′-TCAACGGGCTAAAG-3′; mTORC1 forward,
5′-ATGCCGCAGTCCAAG-3′ and reverse, 5′-TCACATCACCGAGCATG-3′. All
reactions were run in triplicate on an iCycler IQ Multicolor
Detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
with the following cycling parameters: 95°C for 10 sec, followed by
40 cycles of 94°C for 15 sec, annealing at 55°C for 30 sec, and a
final extension step at 70°C for 30 sec. All quantifications were
normalized to the level of human U6 small nuclear RNA or GAPDH in
the reaction. Comparative quantification cycle (Cq)
(2−ΔΔCq) method, which compares differences in Cq values
between common reference RNA and target gene RNA, was used to
obtain the relative fold changes in gene expression (22), which compares differences in Cq
values between the common reference RNA and target gene RNAs, was
used to obtain the relative fold changes in gene expression. The
miR-17-5p, E2F1, TORC1, and AMPKα2 primers for PCR were designed by
GenePharma. The results are expressed as the mean ± standard
deviation.
Luciferase reporter assay
To construct the luciferase reporter vectors, the
3′UTR of E2F1 cDNA fragments containing the predicted potential
miR-17-5p binding sites [predicted using TargetScan (http://www.targetscan.org/) for bioinformatics
analyses on September 25, 2017 using the search term 'E2F1'] were
amplified by PCR and subcloned downstream of the luciferase gene in
the PYr-MirTarget luciferase vector (Ambion; Thermo Fisher
Scientific, Inc.). The 3′UTR of E2F1 (containing the binding sites
for miR-17-5p) was amplified from a cDNA library with the following
primers: Forward 5′-CTCGAGGCGCGTGGGGGGGCTCTAACTGCACTTTCGGCC-3′ and
reverse 5′-GCGGCCGCCAGGGACCCCTGCCCTTG-3′. The mutant 3′UTR of E2F1
(in which five nucleotides were mutated in the binding sites) was
amplified using the following primer sequences: Forward
5′-CTCGAGGCGCGTGGGGGGGCTCTAACTGGTGAATCGGCC-3′ and reverse
5′-GCGGCCGCTGCCACAGTTTTGGCAGTGAAC-3′.
For the luciferase assays, 2×105 293T
cells (American Type Culture Collection, Manassas, VA, USA) were
cultured in 24-well plates and co-transfected with 50 ng of the
corresponding vectors containing firefly luciferase together with
25 ng of miR-17-5p or the control. Transfection was performed using
Lipofectamine® 2000 reagent. At 48 h post-transfection,
the relative luciferase activity was calculated by normalizing the
firefly luminescence to the Renilla luminescence using the
Dual-Luciferase Reporter assay (Promega Corporation) according to
the manufacturer's protocol.
Immunofluorescence
The cells were incubated with LC3 antibodies (cat.
no. 3215; 1:500; Cell Signaling Technology, Inc., Danvers, MA, USA)
at 4°C overnight, and were then incubated with FITC-conjugated goat
anti-mouse secondary antibody (cat. no. 3654; 1:1,000; Cell
Signaling Technology, Inc.) for 1 h at room temperature in the
dark. Following several washes with PBS, the slides were incubated
with DAPI for 3 min and then mounted in glycerol. The fluorescence
was assessed using a fluorescence microscope.
Statistical analysis
Continuous variables are expressed as the mean ±
standard deviation. One-way analysis of variance was performed for
multiple comparisons using GraphPad Prism software, version 5.0
(GraphPad Software, Inc., La Jolla, CA, USA). P≤0.05 was considered
to indicate a statistically significant difference.
Results
miR-17-5p and E2F1 are involved in
HG-induced HUVEC injury
To determine the effect of HG on endothelial cells,
the HUVECs were treated with different concentrations of glucose
(5.5–50 mM) for different durations (0–72 h). The cell viability
was then determined using a CCK8 kit assay. The results showed that
HG (15–50 mM) treatment significantly suppressed the cell
viability, compared with that under normal glucose levels (5.5 mM).
This effect was concentration- and time-dependent. Treatment with
HG (30 mM) for 48 h resulted in an almost 50% loss of viability
(Fig. 1A), therefore, 30 mM
glucose and an induction duration of 48 h were we selected for the
subsequent experiments.
 | Figure 1miR-17-5p and E2F1 are involved in
HG-induced functional disruption of HUVECs. (A) HUVECs were
incubated with increasing concentrations of glucose (5.5, 15, 30
and 50 mM) for 0–72 h. Cell viability was measured using a Cell
Counting Kit-8 assay. The percentage of viable cells was analyzed.
**P<0.01 and ***P<0.001 vs. normal group. (B)
Apoptosis was determined using annexin V/propidium iodide staining
following induction with NG or HG for 48 h. (C) Percentages of
apoptotic cells. ***P<0.001, vs. normal group. (D)
Expression of miR-17-5p in HUVECs detected by reverse
transcription-quantitative polymerase chain reaction analysis
following induction with normal glucose (5.5 mM) or HG for 48 h.
***P<0.001, vs. normal group. (E) Expression of E2F1
was measured by western blot analysis and (F) relative protein
expression was analyzed. ***P<0.001, vs. normal
group. (G) Tube formation capability of HUVECs from different
treatment groups was measured (magnification, ×400); tube formation
capability was decreased following treatment with HG. (H) Average
numbers of tube formations for each field were statistically
analyzed. ***P<0.001, vs. NG group. All data are
expressed as the mean ± standard deviation (n=3). HUVECs, human
umbilical vein endothelial cells; HG, high glucose (30 mM); miR,
microRNA; E2F1, E2 promoter binding factor 1. |
Following induction for 48 h with HG (30 mM), the
HUVECs were collected for apoptosis analyses using flow cytometry.
The results showed that the percentage of apoptotic HUVECs reached
almost 35%, compared with the normal groups (Fig. 1B and C). The RT-qPCR analysis
showed that the expression of miR-17-5p was decreased by HG
induction (Fig. 1D) and the
results of the western blot analysis showed that HG treatment
promoted the expression of E2F1 (Fig.
1E and F). A tube formation assay was then performed. As shown
in Fig. 1G and H, there was a
significant decrease in endothelial tube formation following
exposure to HG conditions. The number of endothelial branch points
decreased by ~60%, compared with that in the control group,
suggesting that HG induced HUVEC apoptosis and inhibited
differentiation into blood vessels. Therefore, miR-17-5p and E2F1
may be involved in these processes.
Inhibition of the expression of E2F1
attenuates HG-induced HUVEC injury via the activation of
autophagy
To further determine whether the expression of E2F1
was involved in HG-induced endothelial injury, siRNA against E2F1
was constructed and transfected into HUVECs. Following culture for
48 h, the protein (Fig. 2A) and
mRNA (Fig. 2B) expression levels
of E2F1 were measured using western blot and RT-qPCR analyses,
respectively. The results showed that the mRNA and protein
expression levels of E2F1 were significantly decreased following
transfection with siRNA against E2F1. HUVECs with or without
downregulated E2F1 were then treated with either normal or HG
conditions for 48 h. The flow cytometry showed that the
downregulation of E2F1 reversed HG-induced HUVEC apoptosis
(Fig. 2C and D). The western blot
analyses showed that inhibition of the expression of E2F1 reversed
HG-induced apoptosis in relation to protein expression levels,
including those of caspase-3 and Bax, but promoted the expression
of Bcl-2 (Fig. 2E–H), suggesting
that the expression of E2F1 affected HG-induced HUVEC apoptosis.
Taken together, the results showed that downregulation of the
expression of E2F1 significantly suppressed HG-induced endothelial
cell injury.
 | Figure 2Downregulation of E2F1 inhibits
HG-induced HUVEC dysfunction by promoting the activation of
autophagy. HUVECs were transfected with siRNA against E2F1 for 48 h
prior to induction with normal glucose (5.5 mM) or HG (30 mM) for
48 h. Expression of E2F1 was detection by (A) western blot analysis
and (B) reverse transcription-quantitative polymerase chain
reaction analysis. ***P<0.001, vs. control. (C)
Apoptosis was determined by annexin V/propidium iodide staining
following transfection with siRNA against E2F1 prior to induction
with normal glucose (5.5 mM) or HG (30 mM) for 48 h. (D) Percentage
of apoptotic cells was measured. **P<0.01 and
***P<0.001, vs. normal group;
###P<0.001, vs. HG group. (E) Western blot analyses
showed the expression of apoptosis-related proteins AMPKα2, (F)
cleaved caspase-3, (G) Bcl-2 and (H) Bax. Relative protein levels
were analyzed. *P<0.05, **P<0.01 and
***P<0.001, vs. normal group; #P<0.05,
##P<0.01 and ###P<0.001, vs. HG group.
(I) Autophagy plaques were analyzed by immunofluorescence
(magnification, ×400). (J) Western blot analyses showed the
expression of autophagy-related proteins, (K) mTORC1, (L) LC3 and
(M) P62. The relative protein levels were analyzed.
*P<0.05 and ***P<0.001, vs. normal
group; ##P<0.01 and ###P<0.001, vs. HG
group. (N) Tube formation capability of HUVECs from different
treatment groups was measured (magnification, ×400). (O) Average
number of tube formations for each field was statistically
analyzed. ***P<0.001, vs. normal group;
##P<0.01, vs. HG group. All data are expressed as the
mean ± standard deviation (n=3). HUVECs, human umbilical vein
endothelial cells; HG, HG, high glucose; E2F1, E2 promoter binding
factor 1; AMPKα2, AMP-activated protein kinase α2; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X protein; mTORC1, mammalian
target of rapamycin complex 1; LC3, microtubule-associated protein
1 light chain 3; NC, negative control; si, small interfering
RNA. |
The immunofluorescence results showed that the
downregulation of E2F1 promoted the autophagy of HUVECs under HG
conditions (Fig. 2I). The western
blot analysis showed that the protein expression of LC3-II was
increased and that of P62 was decreased following the downregulated
expression of E2F1 (Fig. 2J–M),
suggesting that the downregulation of E2F1 inhibited HG-induced
endothelial cell injury relative to the activation of autophagy. A
previous study reported that certain types of autophagy inhibited
cell damage under stress conditions (14). The tube formation assay showed
that the downregulation of E2F1 restored angiogenesis, even under
HG conditions (Fig. 2N and
O).
AMPKα2 and mTORC1 are involved in
E2F-mediated regulation of autophagy and apoptosis
Increasing evidence has shown that E2F1 regulates
AMPKα2 and mTORC1, which are associated with the regulation of
autophagy and apoptosis, respectively (9,12,23). To further determine whether the
regulation of autophagy and apoptosis by E2F1 involves AMPKα2 and
mTORC1, AMPKα2- and mTORC1-over-expression vectors were constructed
and transfection into HUVECs for 48 h. The mRNA and protein
expression levels of AMPKα2 and mTORC1 were measured by RT-qPCR and
western blot analyses, respectively (Fig. 3A–D). The results of the flow
cytometry showed that the overexpression of AMPKα2 or mTORC1
recovered HG-induced HUVEC apoptosis, even following the
downregulation of E2F1, suggesting the involvement of AMPKα2 and
mTORC1 in the regulation of apoptosis. The immunofluorescence
showed that the overexpression of mTORC1 suppressed autophagy under
HG conditions, even following the downregulation of E2F1, however,
the overexpression of AMPKα2 had no effect under exposure to HG
following the downregulation of E2F1 (Fig. 3G), suggesting that the
downregulation of E2F1 promoted autophagy by suppressing the
expression of mTORC1, but not AMPKα2. The tube formation assay
showed that the overexpression of AMPKα2 and mTORC1 suppressed
angiogenesis under exposure to HG conditions, even following the
downregulation of E2F1 (Fig. 3H and
I).
 | Figure 3AMPKα2 and mTORC1 are involved in
E2F1-mediated autophagy and apoptosis regulation. (A) RT-qPCR and
(B) western blot analyses showing the expression of AMPKα2 in
HUVECs transfected with the AMPKα2-overexpression vector for 48 h.
***P<0.001l, vs. control group. (C) RT-qPCR and (D)
western blot analyses showing the expression of mTORC1 in HUVECs
transfected with the mTORC1-overexpression vector for 48 h.
***P<0.001, vs. control group. (E) Apoptosis was
determined by annexin V/propidium iodide staining following
transfection with siRNA against E2F1 and the AMPKα2- or
mTORC1-overexpression vector alone or in combination prior to
induction with normal glucose (5.5 mM) or HG (30 mM) for 48 h. (F)
Percentage of apoptotic cells was measured. **P<0.01
and ***P<0.001, vs. normal group;
###P<0.001, vs. HG + siE2F1 group. (G) Autophagy
plaques were analyzed by immunofluorescence (magnification, ×400).
(H) Tube formation capability of HUVECs from different treatment
groups was measured (magnification, ×400). (I) Average numbers of
tube formations for each field were statistically analyzed.
***P<0.001, vs. normal group;
###P<0.001, vs. HG + siE2F1 group. All data are
expressed as the mean ± standard deviation (n=3). HUVECs, human
umbilical vein endothelial cells; HG, HG, high glucose; E2F1, E2
promoter binding factor 1; AMPKα2, AMP-activated protein kinase α2;
mTORC1, mammalian target of rapamycin complex 1; LC3,
microtubule-associated protein 1 light chain 3; NC, negative
control; si, small interfering RNA; RT-qPCR, reverse
transcription-quantitative polymerase chain reaction analysis. |
Overexpression of miR-17-5p protects
against HG-induced endothelial cell injury by targeting
E2F1-mediated autophagy suppression and apoptosis promotion
Boinformatics analyses (http://www.targetscan.org/) suggested that miR-17-5p
targets the 3′UTR of E2F1. To determine whether miR-17-5p-regulated
HG-induced endothelial injury was associated with E2F1, an
E2F1-overexpression vector was constructed and transfected into
HUVECs. Following 48-h transfection, the mRNA and protein
expression levels were detected by RT-qPCR (Fig. 4A) and western blot (Fig. 4B) analyses, respectively. The
results showed that the expression of E2F1 was significantly
increased. The RT-qPCR analysis also showed that the expression of
E2F1 had no effect on miR-17-5p under exposure to HG conditions
(Fig. 4C). To determine whether
the overexpression of miR-17-5p reversed HG-induced endothelial
injury, the HUVECs were transfected with a miR-17-5p mimic and the
expression of miR-17-5p was detected by RT-qPCR analysis following
transfection for 48 h. The results showed that the expression of
miR-17-5p was significantly increased, compared with that in the
negative control (Fig. 4D). The
flow cytometry showed that the over-expression of miR-17-5p
significantly suppressed HG-induced HUVEC apoptosis, however, the
overexpression of E2F1 recovered the HG-induced HUVEC apoptosis,
even when miR-17-5p was overexpressed (Fig. 4E and F). As miR-17-5p was unable
to silence the transfected E2F1 without the 3′UTR region, it was
possible that miR-17-5p suppressed HG-induced HUVEC apoptosis by
targeting E2F1. The immunofluorescence showed that the
overexpression of mTORC1 suppressed autophagy under HG conditions,
even when E2F1 was downregulated. The western blot analysis showed
that the overexpression of miR-17-5p inhibited HG-induced HUVEC
apoptosis by repression of the E2F1/AMPKα2 signaling pathway, but
restoration of the expression of E2F1 reversed the
miR-17-5p-induced inhibition of apoptosis under HG conditions
(Fig. 4G–K).
 | Figure 4Overexpression of miR-17-5p protects
against HG-induced endothelial cell injury by targeting
E2F1-mediated suppression of autophagy and promotion of apoptosis.
HUVECs were transfection with an E2F1-overexpression vector or an
miR-17-5p mimic for 48 h prior to induction with normal glucose
levels (5.5 mM) or HG (30 mM) for 48 h. The expression of E2F1 was
detected by (A) RT-qPCR and (B) western blot analyses following
transfection with an E2F1-overexpression vector or the NC vector.
***P<0.001, vs. control. (C) Effects of E2F1 and HG
on the expression of miR-17-5p were determined using RT-qPCR
analysis. ***P<0.001, vs. normal group. (D)
Expression of miR-17-5p was detected using RT-qPCR following
transfection with the miR-17-5p mimic or NC.
***P<0.001, vs. control. (E) Apoptosis was determined
using annexin V/PI staining. (F) Percentage of apoptotic cells was
measured. *P<0.05 and ***P<0.001, vs.
normal group. ###P<0.001, vs. HG group;
$$$P<0.001, vs. the HG + miR-17-5p mimic group. (G)
Western blot analysis showing the expression of apoptosis-related
proteins, (H) AMPKα2, (I) cleaved caspase-3, (J) Bcl-2 and (K) Bax.
The relative protein levels were analyzed.
***P<0.001, vs. normal group; ##P<0.01 and
###P<0.001, vs. HG group; $$$P<0.001,
vs. HG + miR-17-5p mimic group. (L) Autophagy plaques were analyzed
by immunofluorescence (magnification, ×400). (M) Western blot
analyses showing the expression of autophagy-related proteins, (N)
mTORC1, (O) LC3 and (P) p62. The relative protein levels were
analyzed. ***P<0.001, vs. normal group;
##P<0.01 and ###P<0.001, vs. HG group;
$$$P<0.001, vs. HG + miR-17-5p mimic group. (Q) Tube
formation capability of HUVECs from different treatment groups was
measured (magnification, ×400). The results showed that the tube
formation capability of HUVECs decreased following treatment with
HG. All data are expressed as the mean ± standard deviation (n=3).
HUVECs, human umbilical vein endothelial cells; miR, microRNA; HG,
HG, high glucose; E2F1, E2 promoter binding factor 1; AMPKα2,
AMP-activated protein kinase α2; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; mTORC1, mammalian target of rapamycin
complex 1; LC3, microtubule-associated protein 1 light chain 3; NC,
negative control; RT-qPCR, reverse transcription-quantitative
polymerase chain reaction analysis. |
The results of the immunofluorescence showed that
the overexpression of miR-17-5p promoted autophagy under HG
conditions, however, the overexpression of E2F1 inhibited the
miR-17-5p-mediated promotion of autophagy (Fig. 4L). The western blot analysis
showed that the overexpression of miR-17-5p promoted HUVEC
autophagy under HG conditions by repression of the E2F1/mTORC1
signaling pathway, but the restored expression of E2F1 reversed the
miR-17-5p-induced promotion of autophagy under HG conditions
(Fig. 4M–P). The tube formation
assay showed that miR-17-5p overexpression promoted angiogenesis
even under exposure to HG conditions, which was reversed when E2F1
was overexpressed (Fig. 4Q).
E2F1 is the direct target of
miR-17-5p
To determine the possible interaction between
miR-17-5p and E2F1, bioinformatics screening for its possible
target genes was performed using an online 3′UTR binding site
prediction database (http://www.targetscan.org/). The overlap analyses
showed that miR-17-5p had a broadly conserved binding site
(Fig. 5A). A mutated version of
the E2F1 3′UTR was constructed in which five complementary
nucleotides in the binding site were altered (Fig. 5B). This mutated construct was
fused to the luciferase coding region (PYr-E3F1 3′UTR) and
co-transfected into 293T cells with the miR-17-5p mimic (Fig. 5C). The relative luciferase
activity showed that when the wild-type E2F1 3′UTR was
co-transfected with the miR-17-5p mimic, the expression of E2F1 was
significantly decreased (P<0.001), compared with the cells
co-transfected with the control miRNA. However, this effect was not
observed following treatment with the mutant 3′UTR of E2F1,
indicating that miR-17-5p can specifically target and suppress the
3′UTR of E2F1. Western blot analyses further confirmed that the
expression of miR-17-5p significantly inhibited the protein
expression of E2F1 in vitro (Fig. 5D).
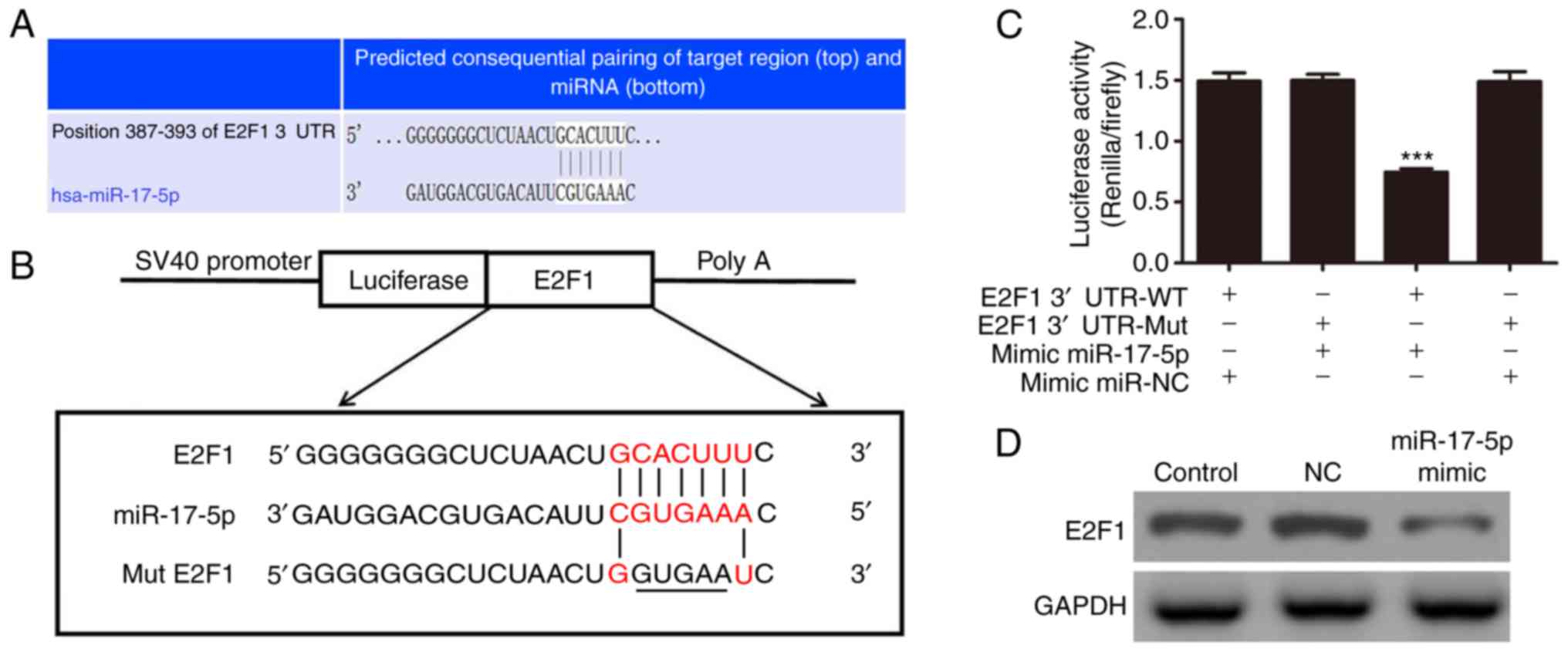 | Figure 5E2F1 is a potential target of
miR-17-5p. (A) Complementary sequences between miR-17-5p and the
3′UTR of E2F1 mRNA were obtained using publicly available
algorithms. (B) Mutated version of the E2F1 3′UTR is shown. (C)
3′UTR of E2F1 was fused to the luciferase coding region (PYr-E2F1
3′UTR) and co-transfected into HUVECs with the miR-17-5p mimic to
confirm that E2F1 was the target of miR-17-5p. The PYr-E2F1 3′UTR
and miR-17-5p mimic constructs were co-transfected into 293T cells
with a control vector, and the relative luciferase activity was
determined at 48 h post-transfection. The data are expressed as the
mean ± standard deviation. ***P<0.001, vs. control.
(D) Western blot analysis of the effect of the expression of E2F1
in HUVECs transfected with the miR-17-5p mimic (n=5). Expression
levels of GAPDH were detected as an endogenous control. The data
are expressed as the mean ± standard deviation.
***P<0.001, vs. control. HUVECs, human umbilical vein
endothelial cells; E2F1, E2 promoter binding factor 1; 3′UTR,
3′untranslated region; miR, microRNA; WT, wild-type; Mut, mutant;
NC, negative control. |
Discussion
Increasing evidence has shown that exposing cultured
HUVECs to high concentrations of glucose induces
oxidative/nitrosative stresses, including the generation of
intracellular reactive oxygen species that cause cellular death.
These injuries are associated with impaired endothelial cell tube
formation (24). Several studies
have reported that the dysregulation of miRNA in diabetic
endothelial cells influences their functions (25,26). Therefore, targeting and regulating
miRNA in an advanced pathway is a strategy for overcoming
HG-induced endothelial injury.
In 1986, Kovesdi et al identified the cell
cycle-related factor, E2F1 (27).
Since its discovery, increasing evidence has shown that E2F is not
only involved in cell cycle regulation and apoptotic signal
transduction, but is also closely associated with growth,
apoptosis, metastasis and autophagy (28–30). The results of the present study
showed that the expression of E2F1 was upregulated following
exposure to HG conditions, and showed that the downregulation of
E2F1 improved HG-induced endothelial injury. The present study also
demonstrated that HG-induced endothelial injury was associated with
E2F1/AMPKα2 and E2F1/mTORC1. Fox et al reported that the
expression of AMPKα2 mediated cell apoptosis (31), whereas the expression of mTORC1
inhibited autophagy (32,33). The present study also showed that
down-regulation of the expression of E2F1 significantly suppressed
the expression of AMPKα2 and mTORC1, suggesting that the
downregulation of E2F1 inhibited HG-induced apoptosis, and reversed
the inhibition of mTORC1-mediated autophagy. Taken together, these
results confirmed that increased autophagy within certain limits
suppressed HG-induced cellular damage (34,35).
The present study investigated miRNAs as molecular
targets of E2F1. The in vitro results showed that the
overexpression of miR-17-5p was important for enhanced HUVEC
survival under HG conditions. miR-17 has been shown to control
cellular proliferation and apoptosis by targeting the E2F family of
transcription factors (36,37), indicating that the overexpression
of miR-17-5p significantly reverses HG-induced cellular damage by
targeting the E2F1/AMPKα2- and E2F1/mTORC1-l-mediated promotion of
apoptosis and inhibition of autophagy, respectively. Bifluorescein
experiments confirmed that miR-17-5p bound predominantly to the
3′UTR of E2F1, resulting in downregulation of its expression.
In conclusion, the present study showed that E2F1
was expressed at high levels in HUVECs under HG conditions, leading
to endothelial cell damage. The study also showed that the
overexpression of miR-17-5p increased cell survival and functional
recovery by targeting the E2F1/AMPKα2- and E2F1/mTORC1-mediated
promotion of apoptosis and inhibition of autophagy, respectively.
These findings characterized a unique mechanism involved in the
HG-induced activation of E2F1 and cell apoptosis, in which the
overexpression of miR-17-5p induced the activation of
autophagy.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
YY and ML generated and analyzed the data. XL
designed the experiments and drafted the manuscript. All authors
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chao CL, Chuang CP, Cheng YF, Lee KR,
Chang Y, Cheng SP, Chan WK and Ho FM: The protective role of
autophagy in matrix metalloproteinase-mediated cell transmigration
and cell death in high-glucose-treated endothelial cells.
Inflammation. 39:830–838. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Weikel KA, Cacicedo JM, Ruderman NB and
Ido Y: Knockdown of GSK3β increases basal autophagy and AMPK
signalling in nutrient-laden human aortic endothelial cells. Biosci
Rep. 36:pii: e00382. 2016. View Article : Google Scholar
|
|
3
|
Kang H, Ma X, Liu J, Fan Y and Deng X:
High glucose-induced endothelial progenitor cell dysfunction. Diab
Vasc Dis Res. 14:381–394. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cam H and Dynlacht BD: Emerging roles for
E2F: Beyond the G1/S transition and DNA replication. Cancer Cell.
3:311–316. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin Z, Ren N, Jiang Y, Xu W, Shi Y and Liu
G: Adenovirus-mediated E2F-1 gene transfer augments
gemcitabine-induced apoptosis in human colon cancer cells. Clin
Lab. 61:1435–1444. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang X, Liu G, Qiu J, Zhang N, Ding J and
Hua K: E2F1-regulated long non-coding RNA RAD51-AS1 promotes cell
cycle progression, inhibits apoptosis and predicts poor prognosis
in epithelial ovarian cancer. Sci Rep. 7:44692017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Y, Zhou Y, Xiao L, Zheng S, Yan N and
Chen D: E2f1 mediates high glucose-induced neuronal death in
cultured mouse retinal explants. Cell Cycle. 16:1824–1834. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu Y, Ma S, Xia Y, Lu Y, Xiao S, Cao Y,
Zhuang S, Tan X, Fu Q, Xie L, et al: Loss of GCN5 leads to
increased neuronal apoptosis by upregulating E2F1- and
Egr-1-dependent BH3-only protein Bim. Cell Death Dis. 8:e25702017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang W, Park IJ, Yun H, Im DU, Ock S, Kim
J, Seo SM, Shin HY, Viollet B, Kang I, et al: AMP-activated protein
kinase α2 and E2F1 transcription factor mediate doxorubicin-induced
cytotoxicity by forming a positive signal loop in mouse embryonic
fibroblasts and non-carcinoma cells. J Biol Chem. 289:4839–4852.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abdel Malik R, Zippel N, Frömel T, Heidler
J, Zukunft S, Walzog B, Ansari N, Pampaloni F, Wingert S, Rieger
MA, et al: AMP-activated protein kinase α2 in neutrophils regulates
vascular repair via Hypoxia-inducible factor-1α and a network of
proteins affecting metabolism and apoptosis. Circ Res. 120:99–109.
2017. View Article : Google Scholar :
|
|
11
|
Liu C, Liang B, Wang Q, Wu J and Zou MH:
Activation of AMP-activated protein kinase alpha1 alleviates
endothelial cell apoptosis by increasing the expression of
anti-apoptotic proteins Bcl-2 and survivin. J Biol Chem.
285:15346–15355. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Real S, Meo-Evoli N, Espada L and Tauler
A: E2F1 regulates cellular growth by mTORC1 signaling. PLoS One.
6:e161632011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meo-Evoli N, Almacellas E, Massucci FA,
Gentilella A, Ambrosio S, Kozma SC, Thomas G and Tauler A:
V-ATPase: A master effector of E2F1-mediated lysosomal trafficking,
mTORC1 activation and autophagy. Oncotarget. 6:28057–28070. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, Zhang S, Wang Y, Yang M, Zhang N,
Jin Z, Ding L, Jiang W, Yang J, Sun Z, et al: Autophagy inhibits
high glucose induced cardiac microvascular endothelial cells
apoptosis by mTOR signal pathway. Apoptosis. 22:1510–1523. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xu J, Wang Y, Tan X and Jing H: MicroRNAs
in autophagy and their emerging roles in crosstalk with apoptosis.
Autophagy. 8:873–882. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhai H, Fesler A and Ju J: MicroRNA: A
third dimension in autophagy. Cell Cycle. 12:246–250. 2013.
View Article : Google Scholar :
|
|
18
|
Han W, Fu X, Xie J, Meng Z, Gu Y, Wang X,
Li L, Pan H and Huang W: MiR-26a enhances autophagy to protect
against ethanol-induced acute liver injury. J Mol Med.
93:1045–1055. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dong D, Fu N and Yang P: MiR-17
downregulation by high glucose stabilizes Thioredoxin-interacting
protein and removes thioredoxin inhibition on ASK1 leading to
apoptosis. Toxicol Sci. 150:84–96. 2016. View Article : Google Scholar :
|
|
20
|
Hajarnis S, Lakhia R, Yheskel M, Williams
D, Sorourian M, Liu X, Aboudehen K, Zhang S, Kersjes K, Galasso R,
et al: microRNA-17 family promotes polycystic kidney disease
progression through modulation of mitochondrial metabolism. Nat
Commun. 8:143952017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nandakumar P, Tin A, Grove ML, Ma J,
Boerwinkle E, Coresh J and Chakravarti A: MicroRNAs in the miR-17
and miR-15 families are downregulated in chronic kidney disease
with hypertension. PLoS One. 12:e01767342017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods.
25:402–408. 2001. View Article : Google Scholar
|
|
23
|
Ladu S, Calvisi DF, Conner EA, Farina M,
Factor VM and Thorgeirsson SS: E2F1 inhibits c-Myc-driven apoptosis
via PIK3CA/Akt/mTOR and COX-2 in a mouse model of human liver
cancer. Gastroenterology. 135:1322–1332. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rezabakhsh A, Ahmadi M, Khaksar M,
Montaseri A, Malekinejad H, Rahbarghazi R and Garjani A: Rapamycin
inhibits oxidative/nitrosative stress and enhances angiogenesis in
high glucose-treated human umbilical vein endothelial cells: Role
of autophagy. Biomed Pharmacother. 93:885–894. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhu L, Wang G, Fischbach S and Xiao X:
Suppression of microRNA-205-5p in human mesenchymal stem cells
improves their therapeutic potential in treating diabetic foot
disease. Oncotarget. 8:52294–52303. 2017.PubMed/NCBI
|
|
26
|
Reddy MA and Natarajan R: Targeting
miR-200c to ameliorate diabetes-induced endothelial dysfunction.
Diabetes. 65:1152–1154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kovesdi I, Reichel R and Nevins JR: E1A
transcription induction: Enhanced binding of a factor to upstream
promoter sequences. Science. 231:719–722. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Engelmann D and Pützer BM: The dark side
of E2F1: In transit beyond apoptosis. Cancer Res. 72:571–575. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang P, Long M, Zhang S, Cheng Z, Zhao X,
He F, Liu H and Ming L: Hypoxia inducible factor-1α regulates
autophagy via the p27-E2F1 signaling pathway. Mol Med Rep.
16:2107–2112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Korah J, Canaff L and Lebrun JJ: The
retinoblastoma tumor suppressor protein (pRb)/E2 promoter binding
factor 1 (E2F1) pathway as a novel mediator of TGFβ-induced
autophagy. J Biol Chem. 291:2043–2054. 2016. View Article : Google Scholar
|
|
31
|
Fox MM, Phoenix KN, Kopsiaftis SG and
Claffey KP: AMP-activated protein kinase α2 isoform suppression in
primary breast cancer alters AMPK growth control and apoptotic
signaling. Genes Cancer. 4:3–14. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bartolomeo R, Cinque L, De Leonibus C,
Forrester A, Salzano AC, Monfregola J, De Gennaro E, Nusco E,
Azario I, Lanzara C, et al: mTORC1 hyperactivation arrests bone
growth in lysosomal storage disorders by suppressing autophagy. J
Clin Invest. 127:3717–3729. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tan HW, Sim AY and Long YC: Glutamine
metabolism regulates autophagy-dependent mTORC1 reactivation during
amino acid starvation. Nat Commun. 8:3382017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin C, Zhang M, Zhang Y, Yang K, Hu J, Si
R, Zhang G, Gao B, Li X, Xu C, et al: Helix B surface peptide
attenuates diabetic cardiomyopathy via AMPK-dependent autophagy.
Biochem Biophys Res Commun. 482:665–671. 2017. View Article : Google Scholar
|
|
35
|
Zhang C, Hou B, Yu S, Chen Q, Zhang N and
Li H: HGF alleviates high glucose-induced injury in podocytes by
GSK3β inhibition and autophagy restoration. Biochim Biophys Acta.
1863:2690–2699. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pickering MT, Stadler BM and Kowalik TF:
miR-17 and miR-20a temper an E2F1-induced G1 checkpoint to regulate
cell cycle progression. Oncogene. 28:140–145. 2009. View Article : Google Scholar :
|














