Introduction
Pulmonary hypertension (PH) caused by left heart
disease (LHD) affects over two thirds of patients with left
ventricular (LV) dysfunction (1).
PH-LHD is a common condition, contributing to the elevation of
pulmonary vascular resistance (PVR) and mean pulmonary pressure,
and eventually leading to heart failure and even mortality.
The pathogenesis of PH is complex and
multifactorial, while vascular remodeling is considered to
contribute to high PVR in PH. Endothelial dysfunction is the key
triggering factor in the pathophysiology of pulmonary arterial
hypertension (PAH). Although the etiologies are distinct, patients
with PH are characterized by the progressive remodeling of the
pulmonary vasculature and significant endothelial dysfunction
(2). Despite advances in the
therapy regimes for PAH, which is generally defined as a different
condition to PH-LHD owing to its distinct causes, no significant
progress has been made in treatment strategies for PH-LHD. However,
PH-LHD is a far more common type of PH in clinical settings.
Although current medical therapies have shown certain beneficial
effects in lowering the mortality rate of patients with PH, these
methods do not prevent the progression of remodeling of the
pulmonary artery (3).
A number of previous studies have described the
vital roles of RhoA (a small G protein) and its target and
downstream effector, Rho-kinase (ROCK), in the pathogenesis of PH
(4–9). In vascular walls, multiple cell
types, including endothelial cells, smooth muscle cells and
fibroblasts, maintain a homeostatic function and response to injury
through the RhoA/ROCK pathway (10). Other studies, including our
previous study, have demonstrated that the overexpression of ROCK
serves a critical role in the pathogenesis of PH with distinct
etiologies (4–9). In addition, ROCK signal inhibition
has been reported to have beneficial effects in a range of PH
animal models, such as in PH induced by monocrotaline, Sugen
5416/hypoxia, bleomycin or aortic banding (4–9).
Previous studies have demonstrated that the expression and activity
of ROCK are greatly increased in patients with idiopathic PH
(1,11), and that its activity is closely
correlated with the severity and duration of PH (1). Furthermore, results from several
clinical trials have indicated the efficacy of inhibition of the
ROCK signaling pathway by fasudil in patients with PH (12–14). In addition, long-term inhibition
of the ROCK pathway can improve LV geometry and LV dysfunction in
aortic-banded (15) and
hypertension-sensitive rat models (16), as well as attenuate cardiac
hypertrophy and fibrosis in mice with myocardial infarction
(17). Recently, it was reported
that fasudil attenuated PH-LHD in rats (9). However, the possible efficacy of a
later and longer-term treatment with fasudil on established PH-LHD
has yet to be fully elucidated.
In the present study, the aim was to evaluate the
effects of 4-week fasudil treatment administered from postoperative
day 35 on PH-LHD induced by supracoronary aortic banding in rats.
The study also investigated whether fasudil exerts its activity in
end-stage PH-LHD by protecting the function of endothelial
cells.
Materials and methods
Animal maintenance
Specific-pathogen-free male juvenile Sprague-Dawley
rats (90±10 g; 3–4 weeks old) were purchased from Shanghai SLAC
Laboratory Animal Co., Ltd. (Shanghai, China). The animals were
maintained in a 12-h light/dark cycle at 25°C and were provided
free access to commercial rodent food and tap water prior to the
experiments. All rats used in the experiments and all study
protocols were approved by the Institutional Animal Care and Use
Committee of Tongji University (Shanghai, China; date of
application, April 15, 2016; approval no., TJLAC-015-033).
Experimental groups
A total of 52 rats were assigned to different groups
at random. In the sham group (n=9), a titanium clip was fixed at
the mediastinal tissue beside the artery, serving as a control. As
reported previously (18), PH-LHD
was induced by banding the ascending aorta without further
treatment in the PH group (n=14). In the PH+F group (n=18), rats
with induced PH-LHD were treated with fasudil (30 mg/kg/day,
intraperitoneal; cat. no., S1573; Selleck Chemicals, Houston, TX,
USA) from day 35 after surgery, and treatment continued for 4
weeks. In the F group (n=11), healthy rats were treated with
fasudil without surgery.
Hemodynamic measurement and cardiac
hypertrophy
At 9 weeks after the surgery, hemodynamic
measurements were performed by cardiac catheterization, and the
right ventricular (RV) systolic pressure (RVSP) and pulmonary
artery pressure (PAP) were assessed by a polygraph system (Power
Lab 8/30; AD Instruments, Bella Vista, Australia) as previously
described (19,20). The animals were sacrificed on day
63 following the operation, and then the lung, RV and the combined
LV and ventricular septal (LV+S) weights were measured. The lung
tissues were also used to detect the protein expression of ROCK1,
ROCK2 and endothelin-1 (ET-1), and the mRNA expression of ROCK1,
ROCK2, RhoA, endothelin A receptor (ETAR) and endothelin B receptor
(ETBR).
Tissue histopathology and
immunofluorescence
Left lung and heart tissues were harvested and
processed for hematoxylin and eosin (H&E) staining,
immunohistochemical (IHC) and wheat germ agglutinin (WGA) staining
subsequent to being embedded in paraffin, cut into 3-µm
sections, picked up with slides and placed in an oven at 60°C for
baking, and finally removed at a normal temperature. H&E
staining was performed to assess arteriole muscularization and
median wall thickness (MWT) in the lung sections. The lung tissue
sections were stained with hematoxylin at room temperature for 5
min. The slides were incubated in water at 25°C for 30 min,
dehydrated in 100% ethanol for 10 min three times, stained with
eosin at room temperature for 5 min and then were dehydrated in
100% ethanol for 10 min three times, and mounted on cover slips.
The slides were observed under a ×200 magnification using a Nikon
positive fluorescence microscope ECLIPSE 80i (Nikon Corporation,
Tokyo, Japan). Five different fields of view were observed. The IHC
assay was performed to evaluate the pulmonary vascular structural
alterations with a monoclonal α-smooth muscle actin (α-SMA)
antibody (dilution, 1:200; cat. no. sc-32251; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). Pulmonary distal arterioles
were considered to be muscularized if >75% of the circumference
was α-SMA-positive (magnification, ×400). The MWT of the pulmonary
artery was calculated with the Image-Pro Plus 6.0 software (Media
Cybernetics, Inc., Rockville, MD, USA). IHC staining was also
performed to evaluate the expression of ROCK1 (dilution, 1:200;
cat. no. ab45171) and ROCK2 (dilution, 1:200; cat. no. ab125025;
both purchased from Abcam, Cambridge, UK) in the left lung tissue
samples.
In order to examine the cell size, WGA staining was
performed, as previously described (21). Briefly, myocardial tissues were
cut into 3-µm sections and the slides were stained for 15
min with a WGA-FITC labeled antibody (dilution, 1:200; cat. no.
W11261; Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) at room temperature subsequent to being fixed with 4%
formaldehyde for 15 min at 37°C, according to the manufacturer’s
protocol. Following labeling, the labeling solution was removed and
the cells were washed twice in Hanks’ Balanced Salt Solution. Then
the cells were observed under a ×400 magnification using a Nikon
positive fluorescence microscope ECLIPSE 80i (Nikon Corporation).
Five different fields of view were observed. Subsequently, the
myocyte cross-sectional area of the left and right ventricles was
measured using Image-Pro Plus 6.0 software.
Cell culture
Human pulmonary microvascular endothelial cells
(HPMECs; cat. no., 3000) were purchased from ScienCell Research
Laboratories, Inc. (San Diego, CA, USA), while human pulmonary
artery smooth muscle cells (HPASMCs) were obtained from iCell
Bioscience, Inc. (Shanghai, China). All cells were cultured and
subcultured according to the manufacturer’s protocol. HPMECs were
incubated in Endothelial Cell medium (ScienCell Research
Laboratories, Inc.) and HPASMCs were maintained in Dulbecco’s
Modified Eagle’s Medium and high glucose medium (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% foetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc.) at 37°C under an
atmosphere of 5% CO2 and 95% air. Cells were used in the
third passage at a concentration of 5×105 cells per well
in a 12 well-plate, or 1×106 cells per well in a 6-well
plate. Cells were pre-stimulated with fasudil (10 µM) for 2
h, followed by incubation with ET-1 (10 nM; cat no. 1160; Tocris
Bioscience, Bristol, UK) for a further 6 h for the migration assay.
PBS-only treated cells were used as the control. HPMECs were used
to confirmed the nitric oxide (NO) production and migration, while
HPASMCs were used to detect the effect of fasudil on the
proliferation of SMC.
Proliferation of HPASMCs
The proliferation of HPASMCs was assessed with a
cell counting kit-8 (CCK-8; cat. no. 40203ES60; Yeasen, Shanghai,
China) assay and Ki67 detection. Briefly, HPASMCs were plated in
96-well plates at a density of 5,000 cells/well. The cells received
different treatments once they reached 50–60% confluence. CCK-8
solution (10 µl) at a 1:10 dilution with FBS-free high
glucose Dulbecco’s modified Eagle’s medium (100 µl) was
added to each well, followed by another 3 h of incubation at 37°C.
The absorbance was measured at 450 nm on a microplate reader
(SpectraMax; Molecular Devices, LLC, San Jose, CA, USA). As
previously described (22),
viability of >100% indicated cell proliferation, whereas
viability of <100% indicated cell death. In addition, cells were
collected following stimulation, stained with the
fluorophore-conjugated anti-Ki67 monoclonal antibody (0.5 mg/ml;
dilution, 1:200; cat. no. 151204; BioLegend, Inc., San Diego, CA,
USA) and then detected by FACS AriaII (BD Biosciences, Franklin
Lakes, NJ, USA).
Migration of HPMECs
HPMECs were grown to confluence on 6-well plates in
complete medium. Once confluence was reached, the cells were grown
without FBS overnight and pre-stimulated with fasudil (10
µM) for 2 h. Next, a scratch was made down the center of the
confluent monolayer with a 100-µl pipette tip. After the
scratch, 10 nM ET-1 was added, and images were captured at ×40
magnification every 2 h. The migration rate was determined by
counting the number of cells that migrated into the wound. ImageJ
software (National Institutes of Health, Bethesda, MD, USA) was
used to quantify the results.
Detection of mRNA levels by reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the tissues or cells
using TRIzol (Thermo Fisher Scientific, Inc.) and the RNA
concentration was measured using NanoDrop 2000 (Thermo Fisher
Scientific, Inc.). The PrimeScript RT reagent kit (Takara Bio,
Inc., Otsu, Japan) was then used to reverse transcribe 1 µg
RNA to cDNA. A Fast Real-Time PCR system (7900HT; Applied
Biosystems; Thermo Fisher Scientific, Inc.) with SYBR Green
MasterMix (Applied Biosystems; Thermo Fisher Scientific, Inc.) was
subsequently used for qPCR. The thermocycling conditions were as
follows: 96°C for 30 sec, 57°C for 30 sec and 72°C for 30 sec. The
experiments were performed in triplicate for each sample. Relative
quantification of mRNA was performed using the comparative
threshold cycles (Cq) method. The mRNA expression level was
measured using the 2−∆∆Cq method (23). Relative expression of each gene
normalizing to GAPDH. All qPCR primers (including for ETAR and
ETBR) used are listed in Table
I.
 | Table IQuantitative polymerase chain
reaction primers. |
Table I
Quantitative polymerase chain
reaction primers.
| Gene | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| ROCK1 |
ATTGAGCAGTTGCGTGCAAAA |
TAAGGAATGCAGGCAGAACCA |
| ROCK2 |
ACAAACCAAGCTAACTGCCT |
ACGCGCATGTGGTGTATGTA |
| Rho kinase |
CTGCGGGTACGAAGGTATCG |
AGCATCCAATCCATCCAGCA |
| RhoA |
ACCAGTTCCCAGAGGTGTATG |
TTGGGACAGAAGTGCTTGACTTC |
| Endothelin A
receptor |
CCGAGGAGCTCTAAGGGGAA |
CCAAAAGGACGCCAGAAAGC |
| Endothelin B
receptor |
AACCCGGCTAGGACTGAAAAC |
AGAAGAGATGGTGTGGCCTG |
| GAPDH |
GCCATCAACGACCCCTTCATTG |
TGCCAGTGAGCTTCCCGTTC |
NO and ET-1 serum concentration
ELISA kits were used to assess the concentration of
NO (cat. no. G-7921; Molecular Probes; Thermo Fisher Scientific,
Inc.) and ET-1 concentrations (cat. no. ADI-900-073; Enzo Life
Sciences, Inc., Farmingdale, NY, USA) in the serum at day 63
post-operation prior to euthanasia.
G-LISA assay
The protein concentration of the lung lysates was
evaluated by a bicinchoninic acid assay (cat. no. 23227; Pierce;
Thermo Fisher Scientific, Inc.). RhoA activity was also determined
in normalized lung lysates with a 96-well RhoA G-LISA Activation
Assay kit (cat. no. BK124; Cytoskeleton, Inc., Denver, CO, USA).
All assay procedures were conducted according to the manufacturer’s
protocol. The luminescence signal was quantified using a microplate
reader.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Data were analyzed using SPSS software, version 11.0
(SPSS Inc., Chicago, IL, USA). For in vivo experiments, the
Mann-Whitney U test was used for comparisons between different
groups. One-way analysis of variance with a Bonferroni post-hoc
test was used for multiple comparisons. P<0.05 was considered to
indicate a statistically significant difference.
Results
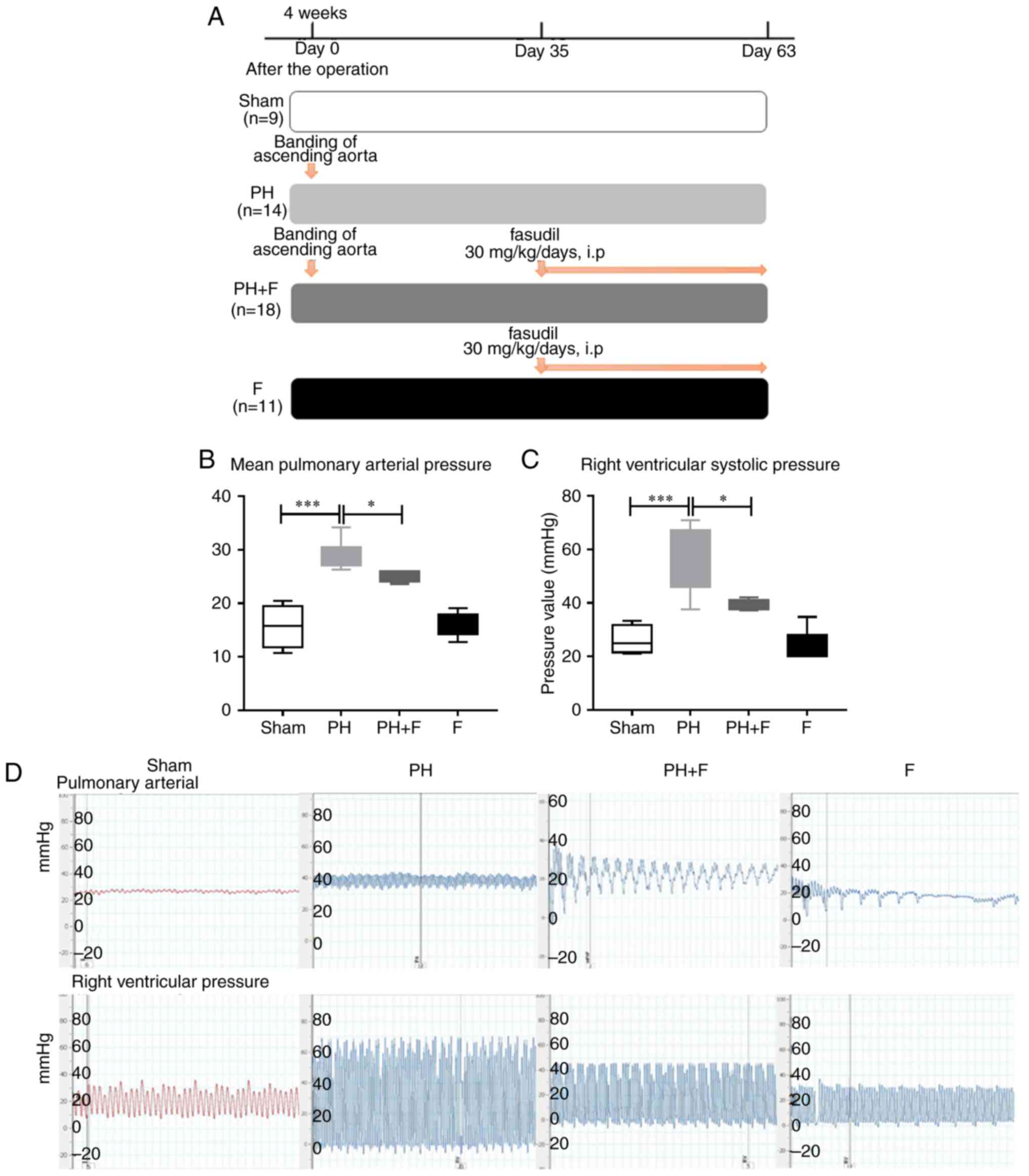
Fasudil downregulates the mean PAP in
rats following supracoronary aortic banding
As reported previously (24), supracoronary aortic banding for 9
weeks led to a marked elevation in the mean PAP and RVSP in rats
compared with that in the sham group (P<0.001; Fig. 1). To investigate whether fasudil
treatment was able to reverse this elevation, rats were treated
with fasudil (30 mg/kg/day, intraperitoneal) for 4 weeks,
commencing at day 35 after supracoronary aortic banding (Fig. 1A). The data revealed that fasudil
treatment decreased the mean pulmonary pressure and the RVSP
compared with those in the PH group (Fig. 1B–D; P<0.05) at postoperative
day 63. Compared with the sham group, there were no differences in
either the mean PAP or RVSP when treated with fasudil alone in the
F group.
Fasudil attenuates the ventricular
hypertrophy in a rat PH-LHD model
The body weight (BW) of the rats in each group were
measured. After 9 weeks of aortic banding, the BW was significantly
decreased as compared with that in the sham group (P<0.01;
Fig. 2A). Rats in the PH+F group
had significantly higher BW compared with those in the PH group
(P<0.01). Furthermore, the RV and LV+S weights were assessed to
evaluate the hypertrophy of the left and right ventricles. As shown
in Fig. 2B and C, there were no
differences in the RV or LV+S weight between the sham and PH
groups. Notably, at day 63, the ratio of (LV+S)/RV weight increased
significantly (P<0.01; Fig.
2D), while the RV/(LV+S) ratio was markedly decreased
(P<0.005; Fig. 2E in rats with
PH-LHD as compared with the ratio in sham rats. Rats treated with
fasudil also exhibited a reduction in (LV+S)/RV ratio (Fig. 2D). The ratios of (LV+S)/BW and
RV/BW were further used to assess the extent of ventricular
hypertrophy. A total of 9 weeks of supracoronary aortic banding
increased the ratios of (LV+S)/BW and RV/BW, whereas treatment with
fasudil reversed these elevated values (Fig. 2F and G). These results suggested
that hypertrophy occurred in the left and right ventricles at day
63 post-surgery, while LV hypertrophy was more pronounced than RV
hypertrophy. WGA staining further confirmed that fasudil attenuated
the myocyte hypertrophy of both the left and right ventricles in
PH-LHD rats (Fig. 2H). As
expected, there were no differences in the above indexes between
the sham and F groups.
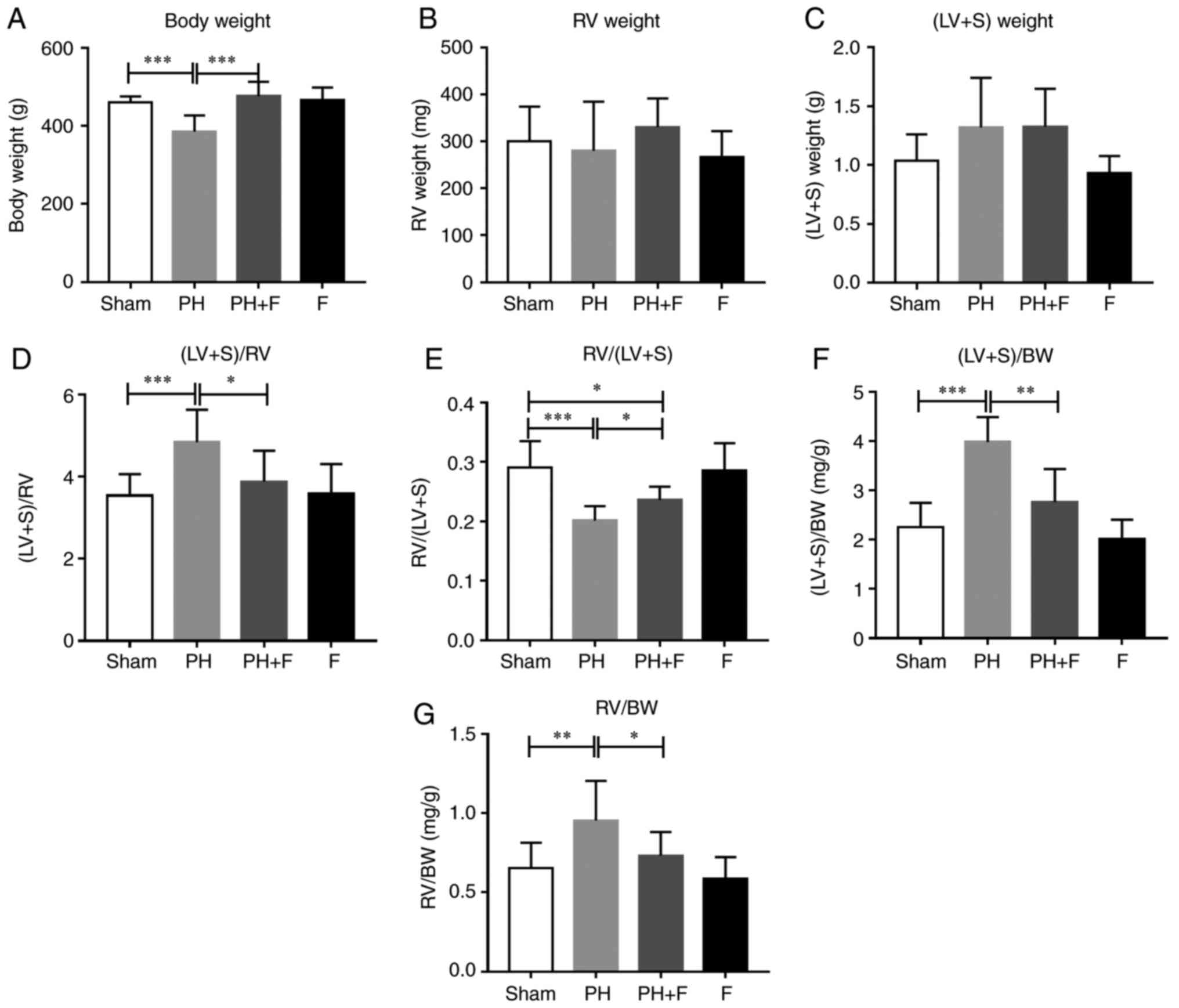 | Figure 2Effect of fasudil on BW and
ventricular hypertrophy. (A) BW; (B) RV weight; and (C) LV+S
weight. (D) (LV+S)/RV indicating LV hypertrophy. (E) RV/(LV+S). (F)
(LV+S)/BW ratios. (G) RV/BW ratios, indicating RV hypertrophy. (H)
Wheat germ agglutinin staining conducted to evaluate the CSA of the
left and right ventricles in the heart sections. Representative
results from six different sections are shown (n=9 in Sham group,
n=14 in PH group, n=18 in PH+F group, and n=11 in F group)
(magnification, ×400). *P<0.05,
**P<0.01 and ***P<0.001. PH, pulmonary
hypertension; F, fasudil; RV, right ventricular; LV, left
ventricular; S, ventricular septal; CSA, myocyte cross-sectional
area; BW, body weight. |
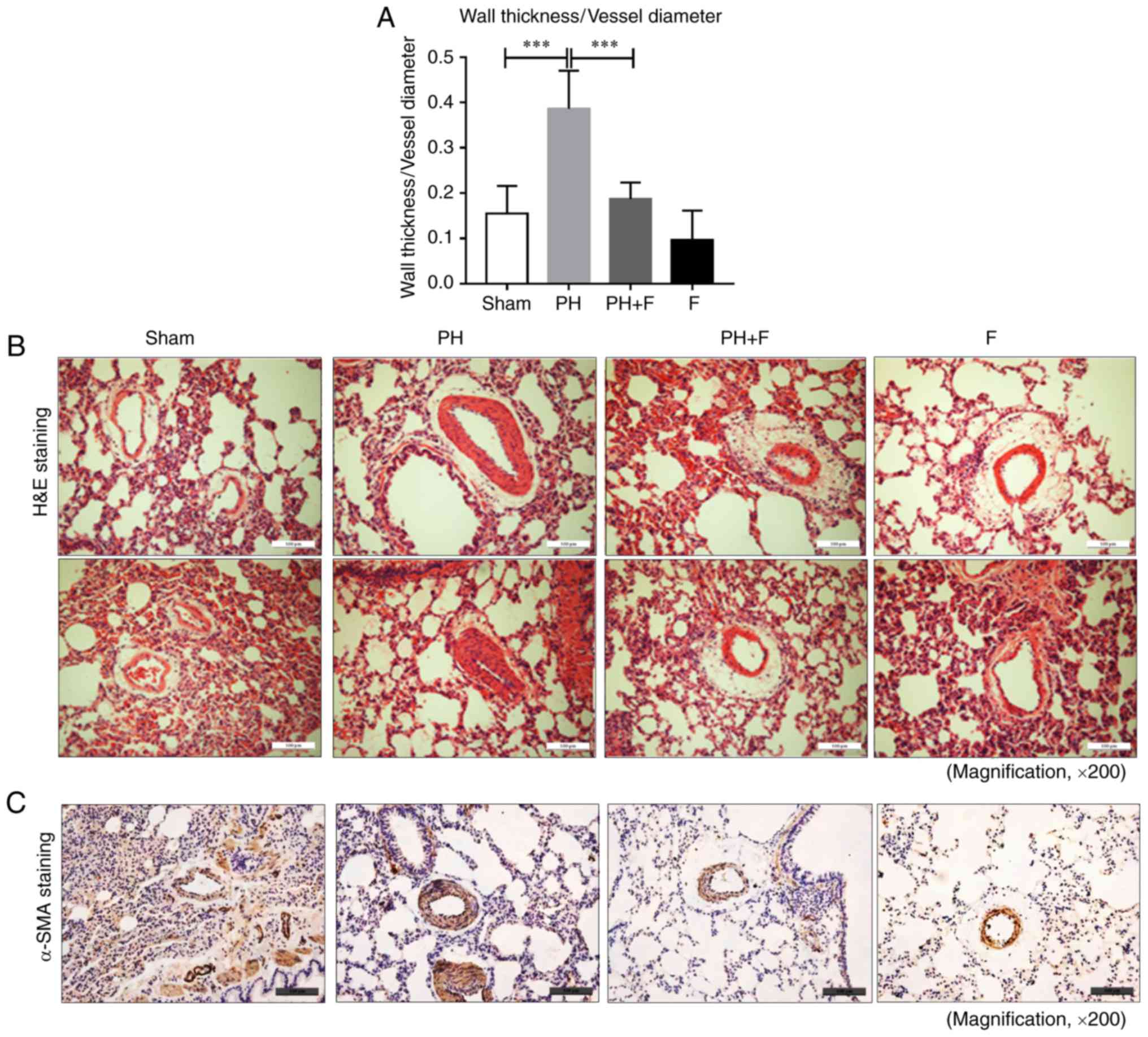
Fasudil prevents pulmonary vascular
remodeling in rats with PH-LHD
To determine whether fasudil administration impeded
pulmonary remodeling in rats with PH-LHD, arteriole muscularization
and MWT were assessed in the lung sections. As displayed by H&E
staining in Fig. 3A and B, 9
weeks of supracoronary aortic banding resulted in an increase in
MWT (P<0.01), whereas administration of fasudil markedly reduced
MWT and muscularization. Compared with the sham group, there were
no differences in MWT when treated with fasudil alone in the F
group. Representative IHC images for α-SMA are presented in
Fig. 3C.
Inhibition of ROCK signaling by fasudil
in rats with PH-LHD
To determine the efficacy of fasudil in vivo,
the expression levels of ROCK1 and ROCK2 was assessed in the lungs
of rats in different groups. As shown in Fig. 4A and B, ROCK1 and ROCK2 mRNA
levels were upregulated in the lungs of the PH group compared with
the sham group, and fasudil treatment decreased the extent of this
elevation. The RhoA mRNA level exhibited similar results (Fig. 4C). RhoA activity in the lung
homogenates was also increased in the PH-LHD rats, and fasudil
administration decreased the RhoA activity (Fig. 4D). There were no significant
differences in the mRNA levels of ROCK1, ROCK2 and RhoA and the
activity of RhoA between the sham and F groups. The IHC staining
results further demonstrated that fasudil inhibited the
upregulation of ROCK1 and ROCK2 protein levels by PH-LHD in the rat
lungs (Fig. 4E).
 | Figure 4Fasudil inhibited ROCK signaling
in vivo. (A) ROCK1, (B) ROCK2 and (C) RhoA mRNA expression
levels in lung tissues are shown. (D) RhoA activity in lung
homogenates, as measured by a G-LISA RhoA activation assay. (E)
Immunohistochemical staining results of ROCK1 and ROCK2 protein
levels in lung tissues (magnification, ×400; n=9 rats in Sham
group, n=14 rats in PH group, n=18 rats in PH+F group, and n=11 in
F group). *P<0.05, **P<0.01 and
***P<0.001. PH, pulmonary hypertension; F, fasudil;
ROCK, Rho-kinase. |
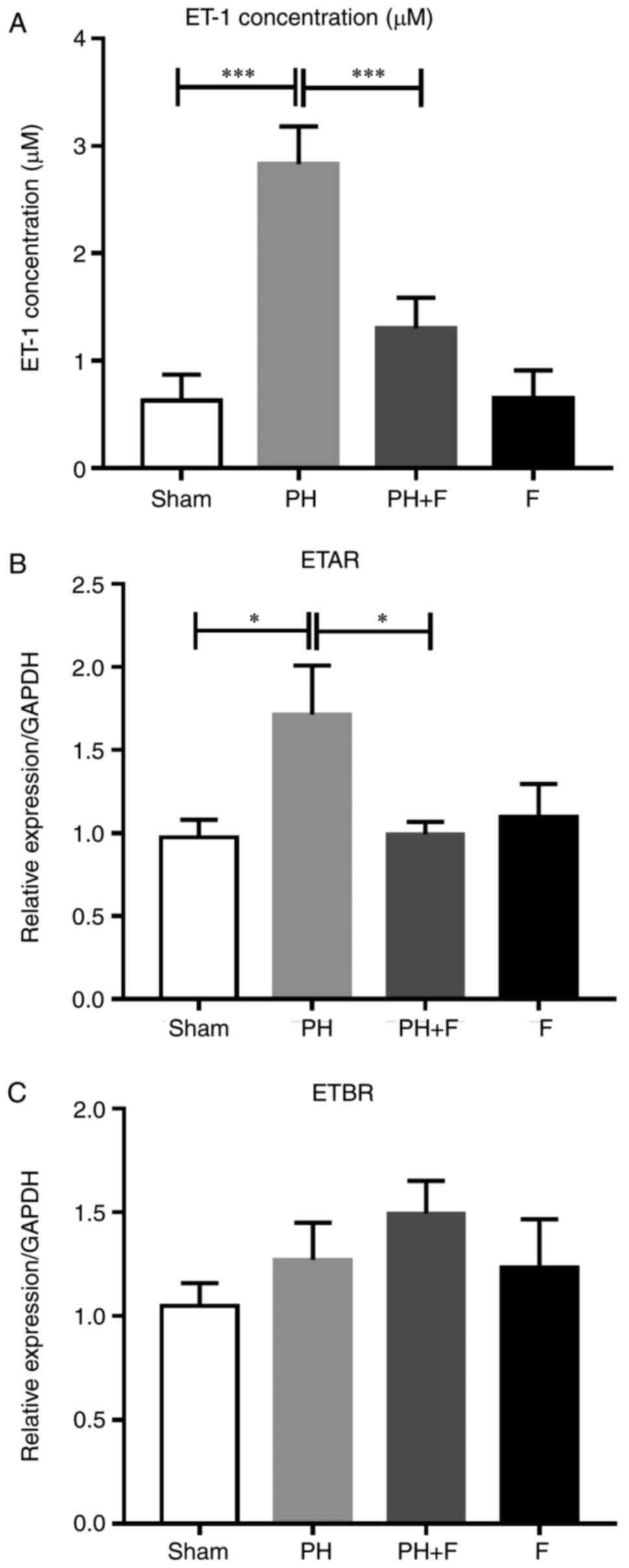
Expression levels of ET-1 and ET-1
receptors in the rat model of PH-LHD
Previous studies have demonstrated that the severity
of PH is associated with high ET-1 levels (25,26). Thus, ET-1 levels were evaluated in
the serum of rats. As shown in Fig.
5A, the ET-1 levels were significantly higher in the PH-LHD
rats compared with that in the sham group, and the inhibition of
ROCK signaling by fasudil reversed this elevation. Next, the mRNA
levels of two ET-1 receptors (ETAR and ETBR) were detected. As
shown in Fig. 5B, ETAR expression
decreased in the PH+F group compared with that in the PH group. By
contrast, no significant differences in ETBR mRNA expression level
were detected between groups (Fig.
5C).
Fasudil inhibits the migration of HPMECs
and preserves lung endothelial function
Endothelial dysfunction is considered to be the key
initiating factor in the pathophysiology of PAH (2). Therefore, the present study sought
to determine how the administration of fasudil affects the lung
endothelial cells in rats with PH-LHD. As shown in Fig. 6A, the mean number of endothelial
cells in each lung arteriole increased in the PH group and
decreased in PH-LHD rats receiving fasudil treatment. However,
there was no difference in endothelial cell apoptosis in the lung
arterioles (data not shown). Previously, data confirmed that
activation of ROCK signaling inhibited eNOS activity and NO
production (27–29). To define whether administration of
fasudil was able to attenuate PH-LHD via promoting NO production
in vivo, the NO concentration was detected in the serum of
rats. Compared with the sham group, the NO levels were reduced in
the PH group, whereas treatment with fasudil for 4 weeks restored
the NO levels in the serum (Fig.
6B). Furthermore, ET-1 treatment decreased the NO production by
HPMECs in vitro, whereas fasudil treatment reversed this
reduction (Fig. 6C). Fig. 6D and E indicate that ET-1 also
promoted the migration of HPMECs, whereas fasudil treatment
inhibited this promotion.
Since the administration of fasudil markedly reduced
MWT and muscularization, the study next determined whether the
administration of fasudil suppressed the proliferation of HPASMCs
in vitro. As shown in Fig.
6F–H, the addition of fasudil reversed the elevation in the
viability and proliferation of HPASMCs that was induced by
ET-1.
Discussion
There is no effective cure for the most common
subset of PH, namely PH-LHD (30). Previous studies and our previous
data have demonstrated that inhibition of ROCK signaling had
beneficial effects in different PH animal models, including
decreasing the PVR (4–9). The administration of fasudil between
postoperative day 29 and 42 induced PH-LHD regression (9). Furthermore, the efficacy and safety
of the short-term administration of fasudil in treating human
patients with PH has been demonstrated in clinical trials (12–14,31). In the present study, fasudil (30
mg/kg/day) was administrated to rats commencing on day 35 after
ascending aortic-banding, and treatment was maintained for 4 weeks.
The treatment starting date was later, and the overall period was
longer in comparison with those previously reported by Dai et
al (9). The current data
revealed that this late, long-term regime significantly decreased
MWT and RVSP, suggesting the potential efficacy of a later and
longer-term treatment with fasudil in preventing hemodynamic
disorders in patients with end-stage established PH-LHD. This
evidence further expands the data regarding the effective
time-frame for the administration of fasudil in the treatment of
PH-LHD.
PH-LHD is primarily caused by LV failure; thus, it
is crucial to improve LV function and reverse the increase in PVR
for patients with LV dysfunction. However, therapies using anti-PH
vasodilators are associated with exacerbated LV hypertrophy in
animal models subjected to HF (32), and deteriorated pathology in
patients with congestive HF (33). In addition, fasudil has been
demonstrated to markedly attenuate LV dysfunction and LV
hypertrophy induced by partial abdominal aortic constriction
(34). It has also been reported
that the long-term suppression of ROCK signaling with fasudil
contributes to LV geometry and dysfunction in aortic-banded rats
(15), and represses the
remodeling of the infarcted heart in a murine model (17). The findings of the present study
suggested that hypertrophy occurred in the left and right
ventricles at day 63 post-supracoronary aortic banding surgery,
while LV hypertrophy was much more pronounced in comparison with RV
hypertrophy. Administration of fasudil significantly attenuated the
myocyte hypertrophy of both the left and right ventricles in PH-LHD
rats. This evidence indicated that starting fasudil treatment at a
late stage is effective in preventing ventricular dysfunction and
the progression of PH-LHD.
Although there are different etiologies, patients
with PH often present with similar pulmonary vascular remodeling to
those with PAH. Consistent with previous findings reporting that
fasudil can attenuate high flow-induced (35) and hypoxic pulmonary vascular
remodeling in rats (36), the
present study demonstrated that blocking ROCK with fasudil can
effectively reverse pulmonary vascular remodeling in PH-LHD rat
models. Additionally, the current study results further
demonstrated the effective time frame of fasudil treatment in a rat
PH-LHD model. Further investigations in patients are required to
verify the efficacy of long-term fasudil treatment in end-stage
PH-LHD.
Recent studies have demonstrated the critical roles
of Rho/ROCK in the pathogenesis of PH (35,37,38). The expression and activity of ROCK
were identified to be markedly increased in the lungs of PH
patients compared with the controls, and its activity level was
closely associated with the severity and duration of PH (1). ROCKs are serine-threonine kinases
with two known isoforms, ROCK1 (p160ROCK or ROKβ) and ROCK2 (ROKα).
The results of the present study similarly demonstrated the
enhancement of ROCK1, ROCK2 and RhoA mRNA expression levels, and of
RhoA activity in a rat PH-LHD model. In addition, the protective
function of fasudil against PH-LHD progression was demonstrated,
which was attributed to the suppression of RhoA/ROCK activation in
the lungs. Previously, Dai et al (9) reported the upregulation of ROCK 2,
but not ROCK 1 in the lungs of AOB28 and AOB42 rats, and that
treatment with fasudil significantly decreased the expression of
ROCK 2, but not ROCK 1. In the current study, all detections were
performed at day 63 after supracoronary aortic banding surgery. The
IHC results demonstrated that fasudil inhibited the upregulation of
both ROCK1 and ROCK2 protein levels induced by PH-LHD in rat lungs
at day 63. Furthermore, treatment with fasudil inhibited the up
regulation of ROCK1 and ROCK2 levels induced by PH-LHD in pulmonary
distal arteriole endothelial cells in rats. A recent study revealed
that ROCK1 and ROCK2 contribute to the profibrotic responses of
endothelial cells (39).
Therefore, our results suggested that the efficacy of fasudil in
blunting the progression of end-stage LHD-PH in rat models may
partly contribute to the inhibition of ROCK1, as well as ROCK2, in
pulmonary endothelial cells.
ET-1 is highly expressed in the lung, compared with
other organs (40), and the
severity of PH is positively associated with the ET-1 level
(25,26). ET-1 can activate ROCK via binding
to ETAR in endothelial cells (41–43). Data from the present study
demonstrated that ascending aortic banding for 63 days resulted in
a significant increase in the mean PAP and up regulation of ET-1 in
the serum of rats, which is consistent with the observations of
previous studies (44,45). Treatment with fasudil reduced the
expression levels of ET-1 and ETAR in the lungs of rats with
PH-LHD. This indicated that blockade of ROCK signaling by fasudil
may inhibit ET-1 production and activity. The observation that ET-1
can activate Rho signaling supports the hypothesis that decreased
RhoA activity in the lungs of rats with PH-LHD may be attributed to
a combination of direct inhibition of ROCK signaling and reduction
of ET-1 expression.
Pulmonary vascular remodeling is caused primarily by
endothelial cell proliferation, apoptosis and dysfunction (46). The impairment of endothelial
function serves a key role in the progression of PH (47). The present study results indicated
that fasudil treatment may prevent cell proliferation in
small-to-moderate size pulmonary arteries in the PH group. Although
the increase in endothelial cell apoptosis is considered to be the
driving factor of PAH onset (48), the endothelial cell apoptosis
levels in this rat PH-LHD model were similar to those in the sham
rats, suggesting that an effect on endothelial cell apoptosis may
not mediate the protective effect of fasudil. By contrast, it was
also identified that treatment with ET-1 contributed to the
migration of HPMECs, and the addition of fasudil inhibited this
process. This is consistent with a previous study reporting that
the dysfunctional angiogenesis of endothelial cells serves a
critical role in the initiation and development of PAH (49). The findings of the present study
support the hypothesis that fasudil can inhibit dysfunctional
angiogenesis in a rat PH-LHD model.
A previous study revealed that the imbalanced
release of endothelial vasoactive mediators, such as NO,
prostacyclin or ET-1, serves a key role in controlling the
pulmonary vascular tone, hypertrophy and remodeling in PH (47). In addition, the inhibition of ROCK
signaling increases eNOS expression, thus serving a protective role
in the vasculature (28,29,38). A later study reported that ET-1
regulates the phosphorylation state of eNOS via activating the ROCK
signaling pathway (41). In the
present study, it was observed that the NO levels significantly
decreased in the serum of the PH group, and that the administration
of fasudil upregulated the NO level. Further data confirmed that
exogenous ET-1 decreased NO production in cultured HPMECs, while
the inhibition of ROCK signaling by fasudil reversed this
reduction. This is consistent with the recent finding that
increased ET-1 levels regulate the endothelial NO production via
ROCK, therefore modulating cerebral circulation (41). Previous studies revealed that
fasudil treatment inhibited the proliferation of PASMCs induced by
5-hydroxytryptamine (10) or
platelet-derived growth factor (50). In the current study, the results
demonstrated that the addition of fasudil also prevented the
elevation of viability and proliferation induced by ET-1 in
HPASMCs. Therefore, fasudil exerted its role on both pulmonary
endothelial cells and PASMCs.
In conclusion, the inhibition of the ROCK signaling
pathway by the late, long-term administration of fasudil is
sufficient to ameliorate the development of established PH-LHD. In
addition, fasudil decreased the expression levels of ET-1 and ETAR,
depressed the proliferation of endothelial cells in pulmonary
arterioles, and promoted NO production in rats with PH-LHD.
Furthermore, fasudil inhibited the migration of HPMECs and the
proliferation of HPASMCs induced by ET-1. These results suggest
that late, long-term treatment with fasudil may be a promising
strategy to prevent and attenuate the development of PH-LHD in
middle- to end-stage patients through the inhibition of the
RhoA/ROCK signaling pathway.
Acknowledgments
The authors would like to thank Dr Ping Yuan from
the Department of Pulmonary Function of the Shanghai Pulmonary
Hospital (Shanghai, China) for hemodynamic measurements and advice
on analyzing the hemodynamic data.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81370434, 81670458,
81470393 and 81302545), the Shanghai Leading Talents (no. 2012053),
the Innovative Project of Pudong Science and Technology (no.
Pkj2013-z03), the Health Industry Project of Pudong Health Bureau
of Shanghai (no. PW2013E-1) and the Program for Young Excellent
Talents in Tongji University (no. 2015KJ073).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article, and more detailed data during
the current study are available from the corresponding author on
reasonable request.
Authors’ contributions
RZ, JW and FL contributed equally to the present
study, and participated in the research, acquisition and analysis
of the data. LH, XL, QM, YJ, ZW and AY participated in data
acquisition. XZ, JW and RZ contributed to the conception and design
of the study. YG, HF, XZ and ZL participated in the data analysis
and interpretation. XZ and RZ participated in the article drafting,
revising and final approval of the version to be submitted. XZ, HF
and ZL contributed to the funding application.
Ethics approval and consent to
participate
All rats used in the experiments and all study
protocols were approved by the Institutional Animal Care and Use
Committee of Tongji University (Shanghai, China; date of
application, April 15, 2016; approval no., TJLAC-015-033).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Doe Z, Fukumoto Y, Takaki A, Tawara S,
Ohashi J, Nakano M, Tada T, Saji K, Sugimura K, Fujita H, et al:
Evidence for Rho-kinase activation in patients with pulmonary
arterial hypertension. Circ J. 73:1731–1739. 2009. View Article : Google Scholar
|
|
2
|
Delgado JF, Conde E, Sánchez V, López-Ríos
F, Gómez-Sánchez MA, Escribano P, Sotelo T, Gómez de la Cámara A,
Cortina J and de la Calzada CS: Pulmonary vascular remodeling in
pulmonary hypertension due to chronic heart failure. Eur J Heart
Fail. 7:1011–1016. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Galie N, Hoeper MM, Humbert M, Torbicki A,
Vachiery JL, Barbera JA, Beghetti M, Corris P, Gaine S, Gibbs JS,
et al: Guidelines for the diagnosis and treatment of pulmonary
hypertension: The task force for the diagnosis and treatment of
pulmonary hypertension of the European Society of Cardiology (ESC)
and the European Respiratory Society (ERS), endorsed by the
International Society of Heart and Lung Transplantation (ISHLT).
Eur Heart J. 30:2493–2537. 2009. View Article : Google Scholar
|
|
4
|
Abe K, Shimokawa H, Morikawa K, Uwatoku T,
Oi K, Matsumoto Y, Hattori T, Nakashima Y, Kaibuchi K, Sueishi K
and Takeshit A: Long-term treatment with a Rho-kinase inhibitor
improves monocrotaline-induced fatal pulmonary hypertension in
rats. Circ Res. 94:385–393. 2004. View Article : Google Scholar
|
|
5
|
Bei Y, Hua-Huy T, Duong-Quy S, Nguyen VH,
Chen W, Nicco C, Batteux F and Dinh-Xuan AT: Long-term treatment
with fasudil improves bleomycin-induced pulmonary fibrosis and
pulmonary hypertension via inhibition of Smad2/3 phosphorylation.
Pulm Pharmacol Ther. 26:635–643. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elias-Al-Mamun M, Satoh K, Tanaka S,
Shimizu T, Nergui S, Miyata S, Fukumoto Y and Shimokawa H:
Combination therapy with fasudil and sildenafil ameliorates
monocrotaline-induced pulmonary hypertension and survival in rats.
Circ J. 78:967–976. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mouchaers KT, Schalij I, de Boer MA,
Postmus PE, van Hinsbergh VW, van Nieuw Amerongen GP, Vonk
Noordegraaf A and van der Laarse WJ: Fasudil reduces
monocrotaline-induced pulmonary arterial hypertension: Comparison
with bosentan and sildenafil. Eur Respir J. 36:800–807. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Oka M, Homma N, Taraseviciene-Stewart L,
Morris KG, Kraskauskas D, Burns N, Voelkel NF and McMurtry IF: Rho
kinase-mediated vasoconstriction is important in severe occlusive
pulmonary arterial hypertension in rats. Circ Res. 100:923–929.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dai ZK, Wu BN, Chen IC, Chai CY, Wu JR,
Chou SH, Yeh JL, Chen IJ and Tan MS: Attenuation of pulmonary
hypertension secondary to left ventricular dysfunction in the rat
by Rho-kinase inhibitor fasudil. Pediatr Pulmonol. 46:45–59. 2011.
View Article : Google Scholar
|
|
10
|
Raja SG: Evaluation of clinical efficacy
of fasudil for the treatment of pulmonary arterial hypertension.
Recent Pat Cardiovas Drug Discov. 7:100–104. 2012. View Article : Google Scholar
|
|
11
|
Guilluy C, Eddahibi S, Agard C, Guignabert
C, Izikki M, Tu L, Savale L, Humbert M, Fadel E, Adnot S, et al:
RhoA and Rho kinase activation in human pulmonary hypertension:
Role of 5-HT signaling. Am J Respir Crit Care Med. 179:1151–1158.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li F, Xia W, Yuan S and Sun R: Acute
inhibition of Rho-kinase attenuates pulmonary hypertension in
patients with congenital heart disease. Pediatr Cardiol.
30:363–366. 2009. View Article : Google Scholar
|
|
13
|
Kojonazarov B, Myrzaakhmatova A,
Sooronbaev T, Ishizaki T and Aldashev A: Effects of fasudil in
patients with high-altitude pulmonary hypertension. Eur Respir J.
39:496–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang X, Wang YF, Zhao QH, Jiang R, Wu Y,
Peng FH, Xu XQ, Wang L, He J and Jing ZC: Acute hemodynamic
response of infused fasudil in patients with pulmonary arterial
hypertension: A randomized, controlled, crossover study. Int J
Cardiol. 177:61–65. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Phrommintikul A, Tran L, Kompa A, Wang B,
Adrahtas A, Cantwell D, Kelly DJ and Krum H: Effects of a Rho
kinase inhibitor on pressure overload induced cardiac hypertrophy
and associated diastolic dysfunction. Am J Physiol Heart Circ
Physiol. 294:H1804–H1814. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Satoh S, Ueda Y, Koyanagi M, Kadokami T,
Sugano M, Yoshikawa Y and Makino N: Chronic inhibition of Rho
kinase blunts the process of left ventricular hypertrophy leading
to cardiac contractile dysfunction in hypertension-induced heart
failure. J Mol Cell Cardiol. 35:59–70. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hattori T, Shimokawa H, Higashi M, Hiroki
J, Mukai Y, Tsutsui H, Kaibuchi K and Takeshita A: Long-term
inhibition of Rho-kinase suppresses left ventricular remodeling
after myocardial infarction in mice. Circulation. 109:2234–2239.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yin N, Kaestle S, Yin J, Hentschel T,
Pries AR, Kuppe H and Kuebler WM: Inhaled nitric oxide versus
aerosolized iloprost for the treatment of pulmonary hypertension
with left heart disease. Criti Care Med. 37:980–986. 2009.
View Article : Google Scholar
|
|
19
|
Yuan P, Wu WH, Gao L, Zheng ZQ, Liu D, Mei
HY, Zhang ZL and Jing ZC: Oestradiol ameliorates monocrotaline
pulmonary hypertension via NO, prostacyclin and endothelin-1
pathways. Eur Respir J. 41:1116–1125. 2013. View Article : Google Scholar
|
|
20
|
Fan YF, Zhang R, Jiang X, Wen L, Wu DC,
Liu D, Yuan P, Wang YL and Jing ZC: The phosphodiesterase-5
inhibitor vardenafil reduces oxidative stress while reversing
pulmonary arterial hypertension. Cardiovasc Res. 99:395–403. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dolber PC, Beyer EC, Junker JL and Spach
MS: Distribution of gap junctions in dog and rat ventricle studied
with a double-label technique. J Mol Cell Cardiol. 24:1443–1457.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu C, Liu F, Zhou X, Cheng Z, Yang X, Xiao
H, Chen Q and Cai K: Effect of protein kinase C on proliferation
and apoptosis of T lymphocytes in idiopathic thrombocytopenic
purpura children. Cell Mol Immunol. 2:197–203. 2005.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Hentschel T, Yin N, Riad A, Habbazettl H,
Weimann J, Koster A, Tschope C, Kuppe H and Kuebler WM: Inhalation
of the phosphodiesterase-3 inhibitor milrinone attenuates pulmonary
hypertension in a rat model of congestive heart failure.
Anesthesiology. 106:124–131. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Keller RL, Tacy TA, Hendricks-Munoz K, Xu
J, Moon-Grady AJ, Neuhaus J, Moore P, Nobuhara KK, Hawgood S and
Fineman JR: Congenital diaphragmatic hernia: Endothelin-1,
pulmonary hypertension, and disease severity. Am J Respir Crit Care
Med. 182:555–561. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kobayashi H and Puri P: Plasma endothelin
levels in congenital diaphragmatic hernia. J Pediatr Surg.
29:1258–1261. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sugimoto M, Nakayama M, Goto TM, Amano M,
Komori K and Kaibuchi K: Rho-kinase phosphorylates eNOS at
threonine 495 in endothelial cells. Biochem Biophys Res Commun.
361:462–467. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wolfrum S, Dendorfer A, Rikitake Y,
Stalker TJ, Gong Y, Scalia R, Dominiak P and Liao JK: Inhibition of
Rho-kinase leads to rapid activation of phosphatidylinositol
3-kinase/protein kinase Akt and cardiovascular protection.
Arterioscler Thromb Vasc Biol. 24:1842–1847. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ming XF, Viswambharan H, Barandier C,
Ruffieux J, Kaibuchi K, Rusconi S and Yang Z: Rho GTPase/Rho kinase
negatively regulates endothelial nitric oxide synthase
phosphorylation through the inhibition of protein kinase B/Akt in
human endothelial cells. Mol Cell Biol. 22:8467–8477. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gerges C, Gerges M, Lang MB, Zhang Y,
Jakowitsch J, Probst P, Maurer G and Lang IM: Diastolic pulmonary
vascular pressure gradient: A predictor of prognosis in
̔out-of-proportion̓ pulmonary hypertension. Chest. 143:758–766.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Y and Wu S: Effects of fasudil on
pulmonary hypertension in clinical practice. Pulm Pharmacol Ther.
46:54–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Oie E, Yndestad A, Robins SP, Bornerheim
R, Asberg A and Attramadal H: Early intervention with a potent
endothelin-A/endothelin-B receptor antagonist aggravates left
ventricular remodeling after myocardial infarction in rats. Basic
Res Cardiol. 97:239–247. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kaluski E, Cotter G, Leitman M,
Milo-Cotter O, Krakover R, Kobrin I, Moriconi T, Rainisio M, Caspi
A, Reizin L, et al: Clinical and hemodynamic effects of bosentan
dose optimization in symptomatic heart failure patients with severe
systolic dysfunction, associated with secondary pulmonary
hypertension-a multi-center randomized study. Cardiology.
109:273–280. 2008. View Article : Google Scholar
|
|
34
|
Balakumar P and Singh M: Differential role
of rho-kinase in pathological and physiological cardiac hypertrophy
in rats. Pharmacology. 78:91–97. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li F, Xia W, Li A, Zhao C and Sun R:
Long-term inhibition of Rho kinase with fasudil attenuates high
flow induced pulmonary artery remodeling in rats. Pharmacol Res.
55:64–71. 2007. View Article : Google Scholar
|
|
36
|
Sun XZ, Tian XY, Wang DW and Li J: Effects
of fasudil on hypoxic pulmonary hypertension and pulmonary vascular
remodeling in rats. Eur Rev Med Pharmacol Sci. 18:959–964.
2014.PubMed/NCBI
|
|
37
|
Fukumoto Y, Matoba T, Ito A, Tanaka H,
Kishi T, Hayashidani S, Abe K, Takeshita A and Shimokawa H: Acute
vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients
with severe pulmonary hypertension. Heart. 91:391–392. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nunes KP, Rigsby CS and Webb RC:
RhoA/Rho-kinase and vascular diseases: What is the link. Cell Mol
Life Sci. 67:3823–3836. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Knipe RS, Probst CK, Lagares D, Franklin
A, Spinney JJ, Brazee PL, Grasberger P, Zhang L, Black KE, Sakai N,
et al: The Rho Kinase Isoforms ROCK1 and ROCK2 each contribute to
the development of experimental pulmonary fibrosis. Am J Respir
Cell Mol Biol. 58:471–481. 2018. View Article : Google Scholar
|
|
40
|
Matsumoto H, Suzuki N, Onda H and Fujino
M: Abundance of endothelin-3 in rat intestine, pituitary gland and
brain. Biochem Biophys Res Commun. 164:74–80. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Faraco G, Moraga A, Moore J, Anrather J,
Pickel VM and Iadecola C: Circulating endothelin-1 alters critical
mechanisms regulating cerebral microcirculation. Hypertension.
62:759–766. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Miao L, Dai Y and Zhang J: Mechanism of
RhoA/Rho kinase activation in endothelin-1-induced contraction in
rabbit basilar artery. Am J Physiol Heart Circ Physiol.
283:H983–H989. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ma MM, Li SY, Wang M and Guan YY:
Simvastatin attenuated cerebrovascular cell proliferation in the
development of hypertension through Rho/Rho-kinase pathway. J
Cardiovasc Pharmacol. 59:576–582. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tan MS, Chai CY, Wu JR, Yeh JL, Chen IJ,
Kwan AL, Jeng AY, Yang HY, Lee MH and Dai ZK: Differential change
in expression of pulmonary ET-1 and eNOS in rats after chronic left
ventricular pressure overload. Exp Biol Med (Maywood). 231:948–953.
2006.
|
|
45
|
Chou SH, Chai CY, Wu JR, Tan MS, Chiu CC,
Chen IJ, Jeng AY, Chang CI, Kwan AL and Dai ZK: The effects of
debanding on the lung expression of ET-1, eNOS, and cGMP in rats
with left ventricular pressure overload. Exp Biol Med (Maywood).
231:954–959. 2006.
|
|
46
|
Sakao S, Tatsumi K and Voelkel NF:
Endothelial cells and pulmonary arterial hypertension: Apoptosis,
proliferation, interaction and transdifferentiation. Respir Res.
10:952009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Budhiraja R, Tuder RM and Hassoun PM:
Endothelial dysfunction in pulmonary hypertension. Circulation.
109:159–165. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Long L, Ormiston ML, Yang X, Southwood M,
Gräf S, Machado RD, Mueller M, Kinzel B, Yung LM, Wilkinson JM, et
al: Selective enhancement of endothelial BMPR-II with BMP9 reverses
pulmonary arterial hypertension. Nat Med. 21:777–785. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tuder RM, Cool CD, Yeager M,
Taraseviciene-Stewart L, Bull TM and Voelkel NF: The pathobiology
of pulmonary hypertension. Endothelium Clin Chest Med. 22:405–418.
2001. View Article : Google Scholar
|
|
50
|
Liu AJ, Ling F, Wang D, Wang Q, Lu XD and
Liu YL: Fasudil inhibits platelet-derived growth factor-induced
human pulmonary artery smooth muscle cell proliferation by
up-regulation of p27kip(1) via the ERK signal pathway. Chin Med J
(Engl). 124:3098–3104. 2011.
|