Introduction
Glioblastoma, namely glioblastoma multiforme (GBM),
is the most fatal cancer developed within the brain, which is
characterized by rapid progression, common therapeutic resistance
and a high probability of recurrence (1,2).
The mean survival time of patients is 12 to 15 months following
diagnosis, with a small portion of patients surviving longer than
five years (3). Disappointingly,
small improvement has been made in GBM patients' prognosis over the
years (4). Therefore, powerful
prognostic model based on molecular biomarkers are needed to
facilitate accurate prediction of GBM prognosis.
Identifying prognostic biomarkers for GBM has
attracted increasing attention. For instance, gene signature-based
survival models for prognosis prediction with high-throughput data
have been investigated in multiple studies (5,6).
Sana et al (7) have
suggested a six-microRNA (miRNA) signature-based risk score model
as an independent prognostic predictor of GBM. Based on the
observation that closely correlated genes are involved in the same
biological processes, incorporating higher-order representative
features, such as pathways, is thought to yield more stable and
robust prognosis prediction (8).
In addition, alterations in multiple pathways have important roles
in cancer initiation and progression (9,10).
Hence, characterization of pathway-level information is crucial for
improving patient survival and developing individualized cancer
therapies. Pathifier is an algorithm for pathway analysis of
high-throughput data, which could quantify deviation of each
pathway from normal behavior in a context-specific manner by using
pathway deregulation score (PDS) (11). Pathway-based transcriptomic
information of breast cancer has been used for prognosis prediction
(12). For GBM, the pathways
significantly associated with survival have been explored with
Pathifier (11). However,
prognosis stratification models based on pathway-level information
have not been studied in GBM.
In the present study, based on The Cancer Genome
Atlas (TCGA) data of GBM patients, a pathway-based prognosis
prediction model was constructed using a combination of univariate
and multivariate Cox regression analysis, PDS calculation by
Pathifier, and L1-penalized estimation-based Cox-proportional
hazards (Cox-PH) model. Additionally, the risk differentiating
power of pathway-based model was successfully validated in three
independent sets. The pathway-based model was compared to a
gene-based model for predictive robustness. Furthermore, the
pathway-based information was integrated with clinical features to
build a pathway+clinic factor-based model, with the aim of
improving the prognostic performance of the pathway-based model.
These findings may have important implications for GBM prognosis
and may hold promising potential for personalized therapeutic
intervention.
Materials and methods
Data source and preprocessing
The mRNA-seq data of 154 GBM tissue samples which
were acquired from the TCGA repository (https://gdc-portal.nci.nih.gov/, platform: Illumina
HiSeq 2000 RNA Sequencing) were considered as a training set in the
current study. An additional three validation sets were used in the
study: The gene expression profiles of 128 GBM samples numbered
'Part A' (13,14), which were downloaded from the
Chinese Glioma Genome Atlas database (CGGA, http://cgga.org.cn/); the GSE13041 dataset (platform,
GPL96; Affymetrix Human Genome U133 Array) including gene
expression data of 191 GBM samples downloaded from Gene Expression
Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/); and the GSE74187
dataset (platform, GPL6480; Agilent-014850 Whole Human Genome
Microarray 4×44K) of 60 GBM samples downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/). The clinical
characteristics of all these four sets are listed in Table I.
 | Table IClinical features of TCGA set and
three validation sets. |
Table I
Clinical features of TCGA set and
three validation sets.
| Clinical
factor | TCGA (n=154) | GSE13041
(n=191) | CGGA (n=128) | GSE74187
(n=60) |
|---|
| Age (years, mean ±
SD) | 59.84±13.54 | 53.83±13.65 | 47.41±11.83 | – |
| Sex
(male/female/−) | 99/54/1 | 116/74/1 | 62/39/27 | – |
| Chemotherapy
(yes/no/−) | 44/91/19 | – | – | – |
| Drug therapy
(yes/no/−) | 19/115/20 | – | – | – |
| Pharmaceutical
therapy (yes/no/−) | 55/84/15 | – | – | – |
| Radiation therapy
(yes/no/−) | 19/120/15 | – | – | – |
| Targeted molecular
therapy (yes/no/−) | 18/116/20 | – | – | – |
| Progression free
survival (yes/no) | – | – | – | 51/9 |
| Progression free
survival months (mean ± SD) | – | – | – | 14.94±10.56 |
| Death
(dead/alive) | 102/50 | 176/15 | 68/33 | 46/14 |
| Overall survival
(months, mean ± SD) | 12.06±10.41 | 19.37±19.41 | 14.24±7.85 | 19.15±10.58 |
Raw data (CEL files) in GSE13041 (platform, GPL96)
were processed for background correction and normalization by oligo
package (15) (http://www.bioconductor.org/packages/release/bioc/html/oligo.html)
in R language (version 3.4.1). With regard to the CGGA and GSE74187
datasets downloaded in the GPL6480 platform, probes were annotated
to genes according to platform annotation profiles. By using the
limma package (16) (https://bioconductor.org/packages/release/bioc/html/limma.html)
in R language (version 3.4.1), data was log2 transformed to achieve
normal distribution, and standardized using median
normalization.
Identification of differentially
expressed genes (DEGs)
In the TCGA set, the patients that died within 6
months following diagnosis were classified as bad prognosis, and
the patients with survival time >12 months were considered as
good prognosis. The DEGs between the bad prognosis and good
prognosis patients were screened using edgeR package in R language
(version 3.4.1) with the thresholds of |log fold change
(FC)|>0.585 and false discovery rate (FDR) <0.05.
Screening for prognosis-related genes and
clinical features
Using the survival package in R language (version
3.4.1; http://bioconductor.org/packages/survivalr/),
univariate Cox regression analysis (17) was performed to reveal the DEGs and
clinical features that are significantly associated with survival.
The genes and clinical characteristics with log-rank P-value
<0.05 were further subjected to multivariate Cox regression
analysis to identify the prognosis-related genes (17). According to the expression levels
of the prognosis-related genes, two-way hierarchical clustering
analysis based on centered Pearson correlation algorithm (18) was conducted with the pheatmap
package (19) in R language
(https://bioconductor.org/packages/release/bioc/html/pheatmap.html;
version 3.4.1).
Constructing a pathway-based prognosis
prediction model
The Gene Set Enrichment Analysis (GSEA; http://www.broadinstitute.org/gsea/) software
(20) is a freely available tool
for analysis of microarray data at the gene-level, including 217
Biocarta pathways and 186 Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathways. In order to evaluate pathway deregulation
associated with GBM, the Pathifier package (8) (http://bioconductor.org/packages/pathifier/) in R
language (version 3.4.1) was applied to calculate a PDS for each
pathway in each sample based on the expressions of the
prognosis-related genes in the TCGA set. The PDS score was
indicative of the degree of deviation in the activity of a pathway
in GBM compared to the activity in normal tissue.
The PSD matrix was inputted, and least absolute
shrinkage and selection operator (LASSO) estimation-based Cox-PH
model was then used to identify the specific predictive pathways of
prognosis by penalized package in R language (version 3.4.1). The
optimal parameter 'lambda' was determined by running 1,000
simulations through cross-validation likelihood. Consequently, a
pathway-based prognostic model was constructed with the predictive
pathways and their Cox-PH coefficients and PDS scores. The
prognosis index was calculated as follows: Prognosis Index(PI)=∑i=1nCodfpi×PDSPi;
where CoefPi stands for the Cox-PH coefficient of pathway i; and
PDSPi stands for the PDS score of pathway i.
All samples in the training set were divided into
high-risk group (above median PI) or low-risk group (below median
PI). The overall survival time of the two groups was compared using
Kaplan-Meier survival analysis (21) and log-rank test. Similarly, all
samples in each validation set (CGGA set, GSE13041 and GSE74187)
were classified by PI into two risk groups, followed by comparison
of overall survival time between the two groups. Receiver operating
characteristic (ROC) curve analysis was conducted to compare the
sensitivity and specificity of the prognosis prediction model. The
area under the curve (AUC) was calculated as well. For log-rank
test and ROC analysis, significance level was set at P<0.05.
Constructing a gene-based prognosis
prediction model
In order to establish a prognosis prediction model
based on gene expression, the prognostic genes for GBM were
identified by LASSO estimation-based Cox-PH model, with expression
matrix of the prognosis-related genes as input. Expression data of
these prognostic genes and their Cox-PH coefficients were used to
develop a gene-based prognosis prediction model as follows:
PI=∑i=1nCoefgenei×expgeni;
where CoefPi denotes the Cox-PH coefficient of gene i; and expgenei
denotes expression level of gene i.
By using this model, all samples in the TCGA set,
CGGA set, GSE13041, or GSE74187 were classified by PI into a
high-risk group and a low-risk group, separately. The overall
survival time of the two risk groups was compared by Kaplan-Meier
survival analysis and log-rank test for each set, separately,
followed by ROC analysis.
Developing a pathway+clinic factor-based
model
In order to investigate whether clinical features
could improve the predictive performance of the pathway-based
model, the significant clinical features extracted from
multivariate Cox regression analysis were combined with prognostic
pathways into the LASSO penalized step to construct a
pathway+clinic factor-based model. Similarly, samples were
separated into high-risk group and low-risk group by PI for TCGA
set, CGGA set, GSE13041, and GSE74187, separately. The model
performance was evaluated similarly to that of the pathway-based
model.
Results
DEGs in TCGA set
There were 38 bad prognosis samples and 38 good
prognosis samples in TCGA set. A total of 402 DEGs were identified
between the good and bad prognosis samples, including 84
downregulated DEGs and 318 upregulated DEGs (Fig. 1A). Two-way hierarchical clustering
analysis of these DEGs revealed that the good prognosis samples
were distinguished from the bad prognosis samples based on
expression pattern of these DEGs (Fig. 1B).
Selection of prognosis-related genes and
clinical characteristics
The 402 DEGs and clinical characteristics were
subjected to univariate Cox regression analysis. Three clinical
features, including age, chemotherapy and pharmaceutical therapy
were significantly associated with overall survival by univariate
Cox regression analysis (P<0.05, Table II). In addition, multivariate Cox
regression analysis revealed that 148 genes and pharmaceutical
therapy were independent biomarkers for prognosis in GBM (Table II).
| Table IIClinical factors significantly
associated with prognosis. |
Table II
Clinical factors significantly
associated with prognosis.
| Clinical
factor | Univariable Cox
regression
| Multivariable Cox
regression
|
|---|
| P-value | HR (95% CI) | P-value | HR (95% CI) |
|---|
| Age (≤60/>60
years) | 0.015 | 1.631
(1.096-2.427) | 0.132 | 1.409
(0.902-2.201) |
| Sex
(male/female) | 0.289 | 0.806
(0.540-1.202) | 0.210 | 0.746
(0.472-1.179) |
| Chemo therapy
(yes/no) | 0.003 | 0.503
(0.319-0.793) | 0.375 | 1.491
(0.618-3.598) |
| Drug therapy
(yes/no) | 0.104 | 0.622
(0.349-1.108) | 0.676 | 1.203
(0.506-2.864) |
| Pharmaceutical
therapy (yes/no) |
<0.001 | 0.351
(0.221-0.558) | 0.003 | 0.249
(0.010-0.626) |
| Radiation therapy
(yes/no) | 0.111 | 0.587
(0.303-1.138) | 0.168 | 0.545
(0.230-1.291) |
| Targeted molecular
therapy (yes/no) | 0.466 | 0.803
(0.444-1.451) | 0.949 | 0.975
(0.449-2.114) |
As illustrated in Fig.
2A, two-way hierarchical clustering analysis demonstrated that
TCGA samples were classified into two groups based on expression
data of the 148 prognosis-related genes. Group 1 included 8
patients that received pharmaceutical therapy and 64 patients that
did not receive pharmaceutical therapy. Group 2 was comprised of 47
patients that received pharmaceutical therapy and 20 patients that
did not receive pharmaceutical therapy. Chi-square test revealed
that the two groups clustered by the 148 prognosis-related genes
had a significant correlation to pharmaceutical therapy
(χ2=48.149, P=3.951×10−12). In addition,
Kaplan-Meier survival analysis revealed that group 2 had
significantly improved survival compared with group 1
(P=1.663×10−04; data not shown). According to
Kaplan-Meier survival analysis, the patients with pharmaceutical
therapy had significantly improved survival than the patients
without pharmaceutical therapy in the TCGA set
(P=4.789×10−06; Fig.
2B). These results indicate that the pharmaceutical therapy is
closely associated with prognosis.
Identification of prognostic
pathways
The expression matrix of the 148 prognosis-related
genes was transformed into PDS matrix, which was then used as
initial input for LASSO estimation-based Cox-PH model. When the
maximal cross-validation likelihood was −490.999, the optimal
lambda value reached 19.700. Consequently, 13 pathways were
selected to be prognostic pathways (Table III). These pathways involved 19
prognosis-related genes (Table
IV).
| Table IIIThirteen prognostic pathways
identified by LASSO estimation-based univariate Cox-proportional
hazards model. |
Table III
Thirteen prognostic pathways
identified by LASSO estimation-based univariate Cox-proportional
hazards model.
| Pathways | Coefficient | HR | P-value |
|---|
|
BIOCARTA_HIVNEF_PATHWAY | 0.113 | 1.002 |
<0.001 |
|
KEGG_ARACHIDONIC_ACID_METABOLISM | 3.310 | 3.168 |
<0.001 |
|
KEGG_AXON_GUIDANCE | 2.784 | 2.124 | 0.001 |
|
KEGG_CYTOKINE-CYTOKINE_RECEPTOR_INTERACTION | 0.609 | 1.021 | 0.002 |
|
KEGG_ENDOCYTOSIS | 0.647 | 1.118 | 0.003 |
|
KEGG_FOCAL_ADHESION | 1.666 | 1.460 | 0.001 |
|
KEGG_INSULIN_SIGNALING_PATHWAY |
−1.653 | 0.322 | 0.001 |
|
KEGG_NITROGEN_METABOLISM | 2.046 | 1.769 | 0.001 |
|
KEGG_O-GLYCAN_BIOSYNTHESIS | 1.133 | 1.391 | 0.001 |
|
KEGG_PATHWAYS_IN_CANCER | 2.272 | 1.889 | 0.001 |
|
KEGG_TYPE_II_DIABETES_MELLITUS | 2.394 | 2.081 | 0.001 |
|
KEGG_UBIQUITIN_MEDIATED_PROTEOLYSIS | 1.875 | 1.682 | 0.002 |
|
KEGG_CELL_ADHESION_MOLECULES | 3.288 | 2.963 | 0.002 |
| Table IVList of genes involved in the 13
prognostic pathways. |
Table IV
List of genes involved in the 13
prognostic pathways.
| Gene name | Count of involved
pathways | HR | P-value |
|---|
| MET | 12 | 0.591 | 0.049 |
| PLA2G5 | 11 | 0.944 | 0.022 |
| BIRC3 | 10 | 1.024 | 0.002 |
| HK3 | 7 | 0.662 | 0.013 |
| SOCS1 | 6 | 1.098 | 0.006 |
| NGFR | 5 | 0.906 | 0.033 |
| L1CAM | 3 | 2.716 | 0.016 |
| NKX3-1 | 2 | 3.109 |
<0.001 |
| NRXN3 | 2 | 1.953 | 0.001 |
| PTGES | 2 | 0.909 | 0.010 |
| ACAP1 | 1 | 0.326 | 0.002 |
| ALOX15B | 1 | 1.176 | 0.036 |
| CA14 | 1 | 1.236 | 0.005 |
| CA9 | 1 | 1.875 | 0.013 |
| CDH4 | 1 | 1.780 | 0.010 |
| EPHA5 | 1 | 1.336 | 0.043 |
| EPHB6 | 1 | 0.341 | 0.007 |
| GALNT12 | 1 | 0.607 | 0.003 |
| GALNT6 | 1 | 0.537 | 0.046 |
As illustrated in Fig.
3, two-way hierarchical clustering analysis based on the 13
prognostic pathways PDS scores categorized all samples of TCGA set
into two groups: Group I (n=69) and group II (n=85). There were 35
samples with pharmaceutical therapy and 25 samples without
pharmaceutical therapy in Group I. Group II had 30 samples with
pharmaceutical therapy and 49 samples without pharmaceutical
therapy. The proportion of pharmaceutical therapy was significantly
different between the two groups (χ2=4.899, P=0.027),
indicating that this grouping approach had a significant
association with pharmaceutical therapy. These results indicate
that pharmaceutical therapy is an important prognostic clinical
feature for GBM.
Prognostic performance of the
pathway-based model
The pathway-based model calculated a PI for each
sample with Cox-PH coefficients of the 13 prognostic pathways, and
the TCGA samples were then separated by PI into the high-risk group
(n=77) and the low-risk group (n=77). According to Kaplan-Meier
survival curves (Fig. 4A), the
high-risk group had significantly shorter overall survival time
compared with the low-risk group (10.38±8.68 vs. 13.74±11.71
months, respectively; P= 0.005). The AUC was 0.987 (Fig. 4F), suggesting that the
pathway-based model could predict the survival outcome of GBM
patients.
The robustness of this pathway-based model was
validated in the CGGA, GSE13041 and GSE74187 sets. All samples in
each set were classified into high-risk group or low-risk group
with the threshold of median PI. For CGGA set, improved survival
was observed in the low-risk group compared with the high-risk
group (16.69±8.35 vs. 12.28±7.16 months, respectively; P=0.007;
Fig. 4B), with an AUC of 0.969
(Fig. 4F). The pathway-based
model also exhibited good predictive power with P=0.004
(22.56±21.47 vs. 16.21±16.65 months, respectively; Fig. 4C) and AUC of 0.929 (Fig. 4F) in the GSE13041 set. In the
GSE74187 set, the low-risk patients (n=30) had markedly longer
overall survival time (23.40±11.56 vs. 14.89±7.54 months;
P=1.635×10−04; Fig.
4D) and progress-free survival (PFS) time (19.23±12.01 month
vs. 10.66±6.68; P=0.0003841; Fig.
4E), compared with the high-risk patients (n=30). Furthermore,
the ROC curve demonstrated an AUC value of 0.984 for overall
survival, and 0.961 for PFS (Fig.
4F). These results indicate that the prognostic power of the
pathway-based model is successfully validated in all the three
independent sets.
Prognostic performance of the gene-based
model
Based on expression data of the 148
prognosis-related genes, LASSO estimation-based Cox-PH model
uncovered 22 genes that were significantly associated with survival
(Table V). The 22-gene
signature-based model calculated a PI for each sample as described
above. All patients of the TCGA set were divided by PI into
high-risk group (n=77) or low-risk group (n=77). According to
Kaplan-Meier survival analysis, overall survival time was
significantly different between the high-risk group and the
low-risk group (8.007±6.43 vs. 16.12±11.98 months, respectively;
P=6.166×10−11; Fig.
5A). The ROC value was 0.989 (Fig. 5F), consistent with the results of
the Kaplan-Meier curve analysis.
| Table VTwenty-two prognostic genes
identified by LASSO-based univariate Cox-proportional hazards
model. |
Table V
Twenty-two prognostic genes
identified by LASSO-based univariate Cox-proportional hazards
model.
| Gene name | Coefficient | HR | P-value |
|---|
| AZGP1 | 0.138 | 1.375 | 0.049 |
| CA9 | 0.019 | 1.875 | 0.013 |
| COL22A1 | 0.844 | 1.592 | 0.002 |
| CPNE6 | 0.249 | 1.006 | 0.030 |
| EN2 | 0.063 | 0.838 | 0.016 |
| FERMT1 |
−0.649 | 0.355 | 0.003 |
| GPC5 | 0.162 | 2.420 | 0.002 |
| HES5 |
−0.098 | 0.776 | 0.019 |
| HIST3H2A |
−0.865 | 0.997 | 0.002 |
| HOXB2 | 0.365 | 1.519 | 0.012 |
| HOXC10 | 0.229 | 1.250 | 0.024 |
| IGFBP6 | 0.216 | 1.415 | 0.000 |
| L1CAM | 0.336 | 2.716 | 0.016 |
| LRRC61 | 0.429 | 2.325 |
<0.001 |
| MSTN |
−0.659 | 1.389 | 0.001 |
| NEUROD1 |
−0.254 | 1.066 | 0.011 |
| NRXN3 | 0.236 | 1.953 | 0.001 |
| OLFM1 | 0.842 | 2.143 | 0.005 |
| PTPRN | 0.594 | 0.723 |
<0.001 |
| PYROXD2 | 0.226 | 0.920 | 0.002 |
| RGS7 | 0.012 | 0.915 | 0.011 |
| RPL39L | 1.051 | 2.100 | 0.000 |
The CGGA, GSE13041 and GSE74187 sets were then used
to validate the pathway-based model. The gene-based model gave a
P-value of 0.071 (13.49±7.41 vs. 15.48±8.59 months; Fig. 5B) and an AUC value of 0.94
(Fig. 5F) for the CGGA set, and a
P-value of 0.011 (16.59±18.08 vs. 22.18±20.38 months; Fig. 5C) and an AUC value of 0.911
(Fig. 5F) for GSE13041.
Additionally, there were obvious differences in both overall
survival time (14.94±9.08 vs. 23.36±10.42 months; P=0.001; Fig. 5D) and PFS time (9.41±7.16 vs.
20.06±11.90 months; P=4.831×10−06; Fig. 5E) between the different risk
groups in GSE74187, with an AUC value of 0.987 for overall survival
and 0.963 for PFS time (Fig. 5F).
These results demonstrated that the gene-based model could
successfully classify patients into two risk groups with
significantly different overall survival time in GSE13041 and
GSE74187. However, poor risk differentiation power was observed for
the gene-based model in the CGGA set (P=0.071; Fig. 5B). Thus, the gene-based model was
inferior to the pathway-based model for prognosis prediction in
GBM.
Prognostic performance of the
pathway+clinic factor-based model
In order to improve the prognostic performance of
the pathway-based model, a prognosis prediction model of the 13
prognostic pathways combined with pharmaceutical therapy was
constructed in the present study. Based on the PI calculated by
this pathway+clinic factor-based model, all patients in the TCGA
set were classified into high-risk group (n=77) and low-risk group
(n=77). Significantly different overall survival time was observed
between the high-risk and low-risk group (6.54±4.62 months vs.
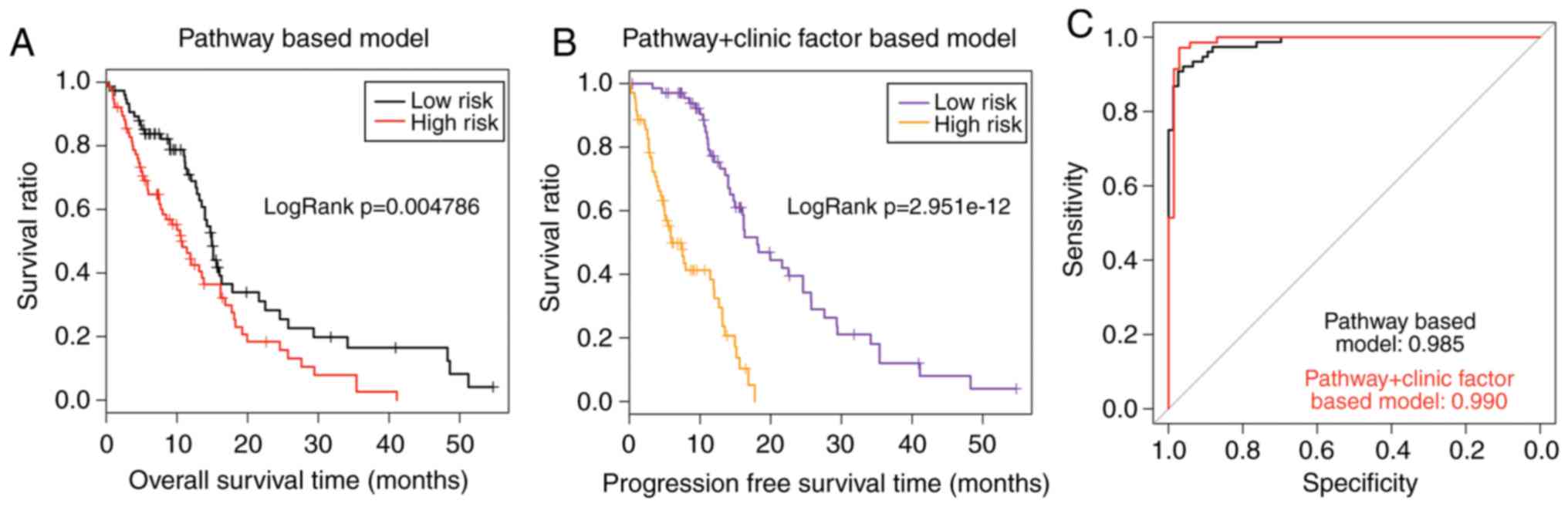
15.79±10.89 months; P=2.951×10−12; Fig. 6B). The AUC value was 0.990 for the
TCGA set (Fig. 6C). As presented
in Fig. 6A and B, the P-value of
the pathway+clinic factor-based model was markedly more significant
compared with the pathway-based model (2.951×10−12 vs.
0.004786). In addition, the AUC value of the pathway+clinic
factor-based model was larger compared with the pathway-based model
(0.990 vs. 0.985; Fig. 6C). These
results demonstrated that the pathway+clinic factor-based model had
better performance than the pathway-based model for risk assessment
of GBM.
Discussion
GBM, the most common malignancy in brain, which is
characterized by inevitable recurrence and dismal prognosis results
in decreased health-related quality of life (22). It has been hypothesized that the
prognosis models based on higher-order representative relationships
with genes, such as pathways and network modules, have more stable
and accurate results (23–25).
Therefore, the present study focused on exploring prognosis models
based on the degree of pathway dysregulation caused by GBM, in
combination with Cox-PH model and LASSO estimation. A total of 148
genes were identified to be significantly associated with prognosis
by Cox regression analysis. Based on the expression matrix of the
148 prognosis-related genes, LASSO estimation-based Cox-PH model
identified 13 prognostic pathways. A pathway-based model was
constructed with the Cox-PH coefficients and the PDS scores of
these pathways. The pathway-based model was trained on the TCGA
set, and tested on three independent sets (CGGA, GSE13041, and
GSE74187) of different sample sizes, which were downloaded from
different platforms. The results demonstrated that the
pathway-based model could successfully classify patients in each
set into two risk groups with significantly different survival
outcome. In addition, the present study also constructed a
gene-based prognosis prediction model with the expression matrix
and the Cox-PH coefficients of the 22 prognostic genes. The
prognostic power of this gene-based model was validated in only two
of the three validation sets, suggesting that the pathway-based
model performed better than the gene-based model in terms of
outcome prediction. Therefore, the prognostic performance of the
pathway+clinic factor-based model was evaluated next, since the
pathway-based model was superior to the gene-based model for
prognosis prediction in GBM.
It has been demonstrated that models using genomic
data combined with clinical data exhibit more accurate prognosis
prediction compared with models using genomic data or clinical data
alone (26). Cheng et al
(27) have demonstrated that
combined clinical and genomic model is superior over models based
on either data type in terms of prognostic accuracy. Additionally,
Huang et al (12) have
provided strong evidence that integration of clinical and genomic
information could greatly improve prognosis prediction compared
with using only one type of information. In order to further
improve the prediction power of this pathway-based model, a
pathway+clinic factor-based prognostic model was developed with the
13 prognostic pathways and a significant clinical factor
(pharmaceutical therapy) identified by Cox regression analysis. The
pathway+clinic factor-based model exhibited improved performance
compared with the pathway-based model and the gene-based model in
the TCGA set, with improved P-values (2.951e-12 vs. 0.004786 vs.
6.166e-11) and AUC values (0.999 vs. 0.985 vs. 0.989). In addition,
this combined model displayed considerably better prognostic
performance compared with the prognostic three-gene signature for
patients with MGMT promoter-methylated GBM, as described in the
study of Wang et al (P=0.0033) (28).
In the present study, the pathway-based predictive
model was based on 13 prognostic pathways, such as endocytosis
pathway, insulin signaling pathway, ubiquitin-mediated proteolysis
pathway, focal adhesion and cell adhesion pathways. These pathways
might have great biological relevance to GBM prognosis. The
endocytosis pathway is an active transport form, which is
strengthened in cancer (29). The
insulin signaling pathway is critical for glucose metabolism.
Abnormal glucose metabolism has an important role in GBM growth and
chemoresistance, suggesting glucose metabolism might be a promising
target for developing GBM therapies (30). There is indeed evidence that the
total lesion glycolysis in hypoxia (hTLG) representing hypoxic
glucose metabolism is a signifi-cant prognostic factor for GBM
(31). Ubiquitin-mediated
proteolysis is a complex protein degradation process. Deregulation
of the ubiquitin system in the ubiquitin-mediated proteolysis
pathway has been demonstrated to be a causative factor of several
types of cancer (32). Focal
adhesion and cell adhesion molecules are critical determinants in
cancer cell resistance to therapy (33).
There are several potential limitations in the
present study. Firstly, the CGGA, GSE13041 and GSE74187 data-sets
do not have information concerning pharmaceutical therapy. Hence,
the pathway+clinic factor-based prognostic model was not validated
in these sets. Therefore, further analysis with more datasets is
necessary to fully test the robustness of the pathway+clinic
factor-based model. Additionally, the study only analyzed
gene-level information of 403 pathways in the GSEA repository, and
some gene information may be inevitably lost. The pathway-based
prognostic model will be applied to larger groups of GBM patients
in future studies, in order to further validate its prognostic
significance.
In conclusion, the present in silico study
presents a promising prognosis prediction model based on 13
pathways, which is constructed by a combination of PDS-based
Pathifier method and LASSO estimation-based Cox-PH model. The
pathway-based model exhibited stronger prognostic power compared
with the gene-based model. Furthermore, incorporating the clinical
information of pharmaceutical therapy to the pathway-based model
resulted in improved prognostic performance. Application of these
pathway-based prognostic models might improve stratification of GBM
patients and offer considerable potential for individualized GBM
management.
Acknowledgments
Not applicable.
Funding
No funding was received.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
RL and MW performed the data analysis and wrote the
manuscript. GZ, HZ and YZ contributed to the data analysis and
manuscript revision. ZS conceived and designed the study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kesari S: Understanding glioblastoma tumor
biology: The potential to improve current diagnosis and treatments.
Semin Oncol. 38(Suppl 4): S2–S10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Aldape K, Zadeh G, Mansouri S,
Reifenberger G and Deimling AV: Glioblastoma: Pathology, molecular
mechanisms and markers. Acta Neuropathol. 129:829–848. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McGuire S: World Cancer Report 2014.
Geneva, Switzerland: World Health Organization, International
Agency for Research on Cancer, WHO Press; 2015
Adv Nutr. 7:418–419. 2016. View Article : Google Scholar
|
|
4
|
Bleeker FE, Molenaar RJ and Leenstra S:
Recent advances in the molecular understanding of glioblastoma. J
Neurooncol. 108:11–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bao ZS, Li MY, Wang JY, Zhang CB, Wang HJ,
Yan W, Liu YW, Zhang W, Chen L and Jiang T: Prognostic value of a
nine-gene signature in glioma patients based on mRNA expression
profiling. CNS Neurosci Ther. 20:112–118. 2014. View Article : Google Scholar
|
|
6
|
Kim YW, Koul D, Kim SH, Lucioeterovic AK,
Freire PR, Yao J, Wang J, Almeida JS, Aldape K and Yung WK:
Identification of prognostic gene signatures of glioblastoma: A
study based on TCGA data analysis. Neuro Oncol. 15:829–839. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sana J, Radova L, Lakomy R, Kren L, Fadrus
P, Smrcka M, Besse A, Nekvindova J, Hermanova M, Jancalek R, et al:
Risk Score based on microRNA expression signature is independent
prognostic classifier of glioblastoma patients. Carcinogenesis.
35:2756–2762. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ma S, Kosorok MR, Huang J and Dai Y:
Incorporating higher-order representative features improves
prediction in network-based cancer prognosis analysis. BMC Med
Genomics. 4:52011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bild AH, Yao G, Chang JT, Wang Q, Potti A,
Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al:
Oncogenic pathway signatures in human cancers as a guide to
targeted therapies. Nature. 439:353–357. 2006. View Article : Google Scholar
|
|
10
|
Chin L, Hahn WC, Getz G and Meyerson M:
Making sense of cancer genomic data. Genes Dev. 25:534–555. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Drier Y, Sheffer M and Domany E:
Pathway-based personalized analysis of cancer. Proc Natl Acad Sci
USA. 110:6388–6393. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang S, Yee C, Ching T, Yu H and Garmire
LX: A novel model to combine clinical and pathway-based
transcriptomic information for the prognosis prediction of breast
cancer. PLoS Comput Biol. 10:e10038512014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan W, Zhang W, You G, Zhang J, Han L, Bao
Z, Wang Y, Liu Y, Jiang C, Kang C, et al: Molecular classification
of gliomas based on whole genome gene expression: A systematic
report of 225 samples from the Chinese Glioma Cooperative Group.
Neuro Oncol. 14:1432–1440. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun Y, Zhang W, Chen D, Lv Y, Zheng J,
Lilljebjörn H, Ran L, Bao Z, Soneson C, Sjögren HO, et al: A glioma
classification scheme based on coexpression modules of EGFR and
PDGFRA. Proc Natl Acad Sci USA. 111:3538–3543. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parrish RS and Spencer HJ III: Effect of
normalization on significance testing for oligonucleotide
microarrays. J Biopharm Stat. 14:575–589. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang P, Wang Y, Hang B, Zou X and Mao JH:
A novel gene expression-based prognostic scoring system to predict
survival in gastric cancer. Oncotarget. 7:55343–55351.
2016.PubMed/NCBI
|
|
18
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng
Z, Zhu G, Qi J, Ma H, Nian H and Wang Y: RNA-seq analyses of
multiple meri-stems of soybean: Novel and alternative transcripts,
evolutionary and functional implications. BMC Plant Biol.
14:1692014. View Article : Google Scholar
|
|
20
|
Tilford CA and Siemers NO: Gene set
enrichment analysis. Methods Mol Biol. 563:99–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Goel MK, Khanna P and Kishore J:
Understanding survival analysis: Kaplan-Meier estimate. Int J
Ayurveda Res. 1:274–278. 2010. View Article : Google Scholar
|
|
22
|
Ohgaki H: Epidemiology of brain tumors.
Methods Mol Biol. 472:323–342. 2009. View Article : Google Scholar
|
|
23
|
Abraham G, Kowalczyk A, Loi S, Haviv I and
Zobel J: Prediction of breast cancer prognosis using gene set
statistics provides signature stability and biological context. BMC
Bioinformatics. 11:2772010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
van den Akker EB, Passtoors WM, Jansen R,
van Zwet EW, Goeman JJ, Hulsman M, Emilsson V, Perola M, Willemsen
G, Penninx BW, et al: Meta-analysis on blood transcriptomic studies
identifies consistently coexpressed protein-protein interaction
modules as robust markers of human aging. Aging Cell. 13:216–225.
2014. View Article : Google Scholar
|
|
25
|
Lee E, Chuang HY, Kim JW, Ideker T and Lee
D: Inferring pathway activity toward precise disease
classification. PLoS Comput Biol. 4:e10002172008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pittman J, Huang E, Dressman H, Horng CF,
Cheng SH, Tsou MH, Chen CM, Bild A, Iversen ES, Huang AT, et al:
Integrated modeling of clinical and gene expression information for
personalized prediction of disease outcomes. Proc Natl Acad Sci
USA. 101:8431–8436. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cheng F, Prat A, Parker JS, Liu Y, Carey
LA, Troester MA and Perou CM: Building prognostic models for breast
cancer patients using clinical variables and hundreds of gene
expression signatures. BMC Med Genomics. 4:32011. View Article : Google Scholar
|
|
28
|
Wang W, Zhang L, Wang Z, Yang F, Wang H,
Liang T, Wu F, Lan Q, Wang J and Zhao J: A three-gene signature for
prognosis in patients with MGMT promoter-methylated glioblastoma.
Oncotarget. 7:69991–69999. 2016.PubMed/NCBI
|
|
29
|
Mellman I and Yarden Y: Endocytosis and
cancer. Cold Spring Harb Perspect Biol. 5:a0169492013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shen H, Decollogne S, Dilda PJ, Hau E,
Chung SA, Luk PP, Hogg PJ and McDonald KL: Dual-targeting of
aberrant glucose metabolism in glioblastoma. J Exp Clin Cancer Res.
34:142015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Toyonaga T, Yamaguchi S, Hirata K,
Kobayashi K, Manabe O, Watanabe S, Terasaka S, Kobayashi H, Hattori
N, Shiga T, et al: Hypoxic glucose metabolism in glioblastoma as a
potential prognostic factor. Eur J Nucl Med Mol Imaging.
44:611–619. 2017. View Article : Google Scholar
|
|
32
|
Hoeller D and Dikic I: Targeting the
ubiquitin system in cancer therapy. Nature. 458:438–444. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eke I and Cordes N: Focal adhesion
signaling and therapy resistance in cancer. Semin Cancer Biol.
31:65–75. 2015. View Article : Google Scholar
|