Introduction
The hallmarks of nonalcoholic steatohepatitis
(NASH), which is the progressive form of nonalcoholic fatty liver
disease (NAFLD), are inflammation and hepatocyte injury, and this
disease is currently a growing public health issue. In NASH/NAFLD,
innate immune activation serves a key role in triggering and
amplifying hepatic inflammation (1,2).
Fatty acid accumulation, in particular saturated fatty acids, in
the liver have been reported to activate a series of
pro-inflammatory signaling pathways, leading to the activation of
both the innate and adaptive types of immune response (3,4).
Previous studies revealed that toll-like receptors (TLRs), the
sensors of endogenous and microbial danger signals, are expressed
and activated in parenchymal and innate immune cells in the liver,
consequently contributing to NASH (5,6).
TLR4 is a well-known pattern recognition receptor
that serves a fundamental regulatory role in promoting the
progression of chronic liver diseases (7). Hepatic tissue injured by toxic lipid
molecules (lipotoxicity) serves an essential role in the
recruitment of innate immunity involving TLR4 (8). Additionally, the TLR4 signaling
pathway initiated through the downstream signaling ligand activates
nuclear factor-κB (NF-κB) and induces the expression of
inflammatory response-associated genes. These changes promote
signaling cascades leading to injury amplification (9). Thus, the relevance of modulating
these inflammatory signaling pathways as potential novel
therapeutic strategies for NASH urgently requires further
investigation.
As a potent immunosuppressive and anti-inflammatory
agent, celastrol (C29H38O4) has
been widely used for the treatment of various autoimmune diseases
(10,11). The latest research on celastrol
has mainly focused on ameliorating metabolic disease and relevant
organ injury. Liu et al (12) reported that celastrol is a leptin
sensitizer and a promising agent for the pharmacological treatment
of obesity. Furthermore, it has been demonstrated that celastrol
alleviated high-fat diet-mediated cardiovascular injury via
mitigating oxidative stress and improving lipid metabolism
(13). The study by Kim et
al (14) also reported that
administration of celastrol resulted in significant decreases in
adiposity in multiple organs in diabetic db/db mice. In addition,
this administration improved renal functional and structural
changes through the metabolic and NF-κB-inhibitory activity, and
the cytokine-suppressing activities in the kidney (14). Our previous study firstly
confirmed that celastrol provided a protective effect against fatty
hepatic injury in type 2 diabetic rats through suppression of the
inflammatory process (15).
However, our research only performed preliminary observational
experiments in vivo, while the definite mechanisms of
celastrol remain to be studied in vitro.
Numerous studies (5–8)
have reported that activation of the TLR4-mediated inflammatory
pathway in hepatocytes serves an important role in the early stages
of NAFLD. Therefore, it can be hypothesized that celastrol may
protect hepatocytes against lipid deposition and inflammatory
response via inhibiting TRL4 activation, and subsequently
suppressing signaling cascades and avoiding the release of
inflammatory response factors. Thus, in the present study, small
interfering RNA (siRNA) transfection was performed to evaluate the
significance of TLR4 and the contribution of the TLR4-mediated
intracellular signaling pathway to the inflammation in free fatty
acid (FFA)-induced HepG2 cells. Furthermore, the effects of
celastrol in these cells and the associated mechanisms were
investigated.
Materials and methods
Cell culture
HepG2 cells were supplied by the Department of
Immunology of Tianjin Medical University (Tianjin, China). Although
HepG2 cells originate from hepatoblastoma (16), the study by Gómez-Lechón et
al (17) demonstrated that
fat overaccumulation is induced in hepatic cells by FFA, and that
human hepatocytes and HepG2 cells behave nearly the same. HepG2
cells are, therefore, generally accepted as a promising alternative
to human hepatocytes for use as a cellular model of steatosis
(17–19). HepG2 cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin/streptomycin at 37°C and 5% CO2. Purified
celastrol was purchased from Merck KGaA (Calbiochem; Darmstadt,
Germany) and stored at −20°C. Celastrol was freshly dissolved in
10% dimethyl sulfoxide prior to use. Palmitate acid and oleic acid
were purchased from Sigma-Aldrich (Merck KGaA) and dissolved in
isopropyl alcohol. When 70–80% confluency was reached, the HepG2
cells were cultured in serum-free DMEM with 0.5% bovine serum
albumin (BSA; Sigma-Aldrich; Merck KGaA) or different
concentrations of FFA (palmitate acid: Oleic acid ratio, 1:2)
(17) or different concentrations
of celastrol, as specified below.
Cell viability assay
Cell viability was assessed using the Cell Counting
Kit-8 (CCK8) assay (Jiancheng Bioengineering Institute, Nanjing,
China). Briefly, HepG2 cells were seeded in a 96-well plate at a
density of 1×104 cells/well. Following treatment with
various concentrations of FFA (0.25, 0.5, 1.0 and 2.0 mM) or
celastrol (0.1, 0.2, 0.5, 1.0, 1.5 and 2.0 μM) in triplicate
for 20 h, 10 μl CCK8 was added to each well and incubated
for an additional 4 h at 37°C in an atmosphere containing 5%
CO2. The absorbance at 450 nm was then measured using a
microplate reader (Thermo Fisher Scientific, Inc.). The inhibition
of cytotoxicity was calculated as a fraction of the control and is
expressed as the percentage of cell viability.
Oil red O staining
Accumulation of triglycerides in the treated HepG2
cells was visualized by Oil red O staining (Sigma-Aldrich; Merck
KGaA). Briefly, cells were seeded in a 6-well plate at a density of
3×105 cells/well and then stained with freshly diluted
Oil red O solution for 30 min at room temperature. The sections
were then washed with PBS twice, and the nuclei of the cells were
counterstained with hematoxylin for 1 min. To examine lipid
accumulation, cell images were captured with a microscope (Olympus
Corporation, Tokyo, Japan).
Intracellular triglyceride test
HepG2 cells were seeded at a density of
3×105 cells/well in a 6-well plate. Intracellular
triglyceride accumulation was determined following lipid extraction
as described previously by Schwartz and Wolins (20), followed by spectrophotometry
determination using a triglyceride assay kit (cat no. A110-2;
Nanjing Jiancheng Bioengineering Institute, Nanjing, China)
according to the manufacturer's protocol. Subsequently, the
absorbance at 510 nm was measured using a microplate reader (Thermo
Fisher Scientific, Inc.). The protein concentration was determined
using a BCA protein assay kit (Thermo Fisher Scientific, Inc.).
Values of triglyceride were normalized to the total protein
concentration. Intracellular triglyceride content (mmol/g
protein)=intracellular triglyceride concentration (mmol/l)/protein
concentration (g/l).
Knockdown of TLR4 by siRNA
HepG2 cells were transiently transfected with
fluorescein amidite (FAM)-labeled siRNA that targeted TLR4 or with
negative control siRNA obtained from GenePharma Co., Ltd.
(Shanghai, China), using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.). Three human TLR4
siRNA constructs were examined, and their sequences are shown in
Table I. Briefly, 10 μl of
20 μM siRNA and 5 μl Lipofectamine® 2000
were suspended in 250 μl of serum-free Opti-MEM medium
(Gibco; Thermo Fisher Scientific, Inc.), forming an
siRNA/Lipofectamine mixture. The mixture was then added to
2×105 HepG2 cells cultured in a 6-well plate. Following
24 h of transfection, the transfection efficiency was observed
using fluorescence microscopy (Olympus Corporation). The knockdown
effects of TLR4 were confirmed using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blotting. Following transfection, the medium was changed to
regular medium and the cells were treated with BSA, FFAs and
celastrol combinations for 24 h, as described. HepG2 cells were
divided into seven groups, including the normal control (NC), and
groups treated with 0.5 mM FFA, negative siRNA + 0.5 mM FFA, TLR4
siRNA + 0.5 mM FFA, 0.5 mM FFA + 0.5 μM celastrol, negative
siRNA + 0.5 mM FFA + 0.5 μM celastrol, and TLR4 siRNA + 0.5
mM FFA + 0.5 μM celastrol.
 | Table ISequences of TLR4 siRNA and negative
control siRNA. |
Table I
Sequences of TLR4 siRNA and negative
control siRNA.
| siRNA | Sequence |
|---|
| TLR4 siRNA1 | Forward:
5′-CCUGGUGAGUGUGACUAUUTT-3′
Reverse: 5′-AAUAGUCACACUCACCAGGTT-3′ |
| TLR4 siRNA2 | Forward:
5′-CCUGAACCUAUGAACUUUTT-3′
Reverse: 5′-AAAGUUCAUAGGGUUCAGGTT-3′ |
| TLR4 siRNA3 | Forward:
5′-GGAUUUAUCCAGGUGUGAATT-3′
Reverse: 5′-UUCACACCUGGAUAAAUCCTT-3′ |
| Negative control
siRNA | Forward:
5′-UUCUCCGAACGUGUCACGUTT-3′
Reverse: 5′-ACGUGACACGUUCGGAGAATT-3′ |
RT-qPCR analysis
Total RNA was isolated from HepG2 cells with TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). The
concentration of total RNA was determined by spectrophotometry
(TECAN Infinite F200PRO microplate reader; Tecan Group, Ltd.,
Mannedorf, Switzerland). Next, cDNA synthesis was performed using
the High-Capacity cDNA Reverse Transcription kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. qPCR was performed in a total volume of 10
μl, containing 5 μl 2X SYBR Green Premix Ex
Taq™ (Takara Bio, Inc., Otsu, Japan), 1 μl cDNA, 3
μl distilled water and 0.5 μl of each primer. qPCR
was conducted in a thermal cycler (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), with initial denaturation performed at 95°C for
3 min, followed by 45 cycles of denaturation for 10 sec at 95°C,
annealing for 30 sec at 60°C, and extension for 20 sec at 72°C. All
primers used in qPCR were synthesized by Augct DNA-Syn
Biotechnology Co., Ltd. (Beijing, China), and the human
glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene served as the
housekeeping gene. The following specific primers were used: Human
TLR4 forward, 5′-GCAATGGATCAAGGACCAGA-3′, and reverse,
5′-CTACAAGCACACTGAGGACC-3′; and human GAPDH forward,
5′-CTCCTCCACCTTTGACGCTG-3′ and reverse, 5′-TCCTCTTGTGCTCTTGCTGG-3′.
The mRNA levels were normalized against the mRNA levels of the
housekeeping gene GAPDH. Data were analyzed using the CFX Manager
software (version 1.6; Bio-Rad Laboratories, Inc.). Relative
quantification of the target gene mRNA was performed using the
2−ΔΔCq method and normalized to the internal control
(21).
Western blotting
Proteins of HepG2 cell samples were extracted using
a radioimmunoprecipitation assay buffer (Thermo Fisher Scientific,
Inc.) on ice, according to the manufacturer's protocol, and the
protein concentrations were then measured using a BCA protein assay
kit (Thermo Fisher Scientific, Inc.). An equal amount of protein
from each sample was subjected to 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and then transferred to
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Membranes were blocked with 5% milk for 2 h at room
temperature and subsequently incubated overnight at 4°C with
primary antibodies against TLR4 (1:1,000; cat no. ab13867; Abcam,
Cambridge, UK), myeloid differentiation primary response 88 (MyD88;
1:500; cat no. ab2064; Abcam), phospho-NF-κBp65 (1:2,000; cat no.
ab86299; Abcam), NF-κBp65 (1:2,000; cat no. ab16502; Abcam),
interleukin (IL)-1β (1:1,000; cat no. ab2105; Abcam) and tumor
necrosis factor α (TNFα; 1:1,000; cat no. ab8348; Abcam), as well
as the internal control β-actin (1:10,000; cat no. KM9001T; Tianjin
Sungene Biotech Co., Ltd., Tianjin, China). Next, membranes were
washed twice for 10 min in 1X TBST and then incubated with
horseradish peroxidase-conjugated goat anti-rabbit or goat
anti-mouse secondary antibodies (all 1:5,000; Tianjin Sungene
Biotech Co., Ltd.) at room temperature for 2 h. Membranes were then
washed twice for 10 min in 1X TBST. Proteins were visualized by
enhanced chemiluminescence (EMD Millipore), and blots were scanned
in a dark room. Densitometry analysis of bands was performed with
the ImageJ software (National Institutes of Health, Bethesda, MD,
USA).
Statistical analysis
Statistical analysis was performed using SPSS
version 17.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism
version 5.0 software (GraphPad Software, Inc., La Jolla, CA, USA).
Quantitative data are expressed as the mean ± standard error of the
mean. The statistical significance of the data obtained was
evaluated using the one-way analysis of variance, followed by the
least significant difference-test. A value of P<0.05 was
considered to denote a statistically significant difference.
Results
Effects of celastrol on triglycerides
accumulation in FFAs-induced HepG2 cells
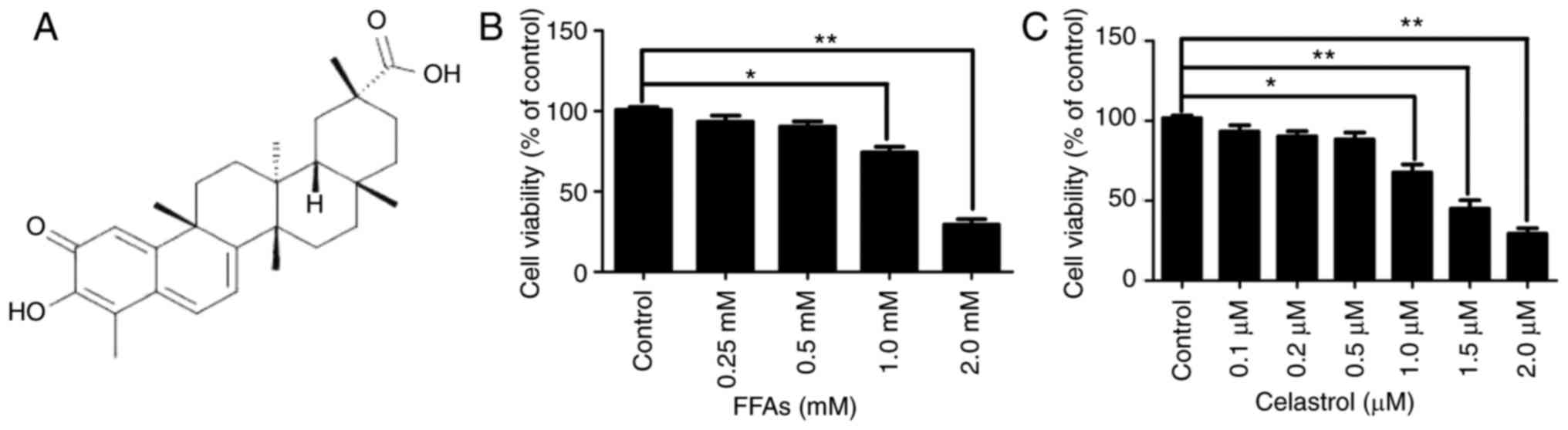
The chemical structure of celastrol is presented in
Fig. 1A. The cytotoxic effects of
various concentrations FFA (0.25, 0.5, 1.0 and 2.0 mM) and
celastrol (0.1, 0.2, 0.5, 1.0, 1.5 and 2.0 μM) in HepG2
cells were initially investigated. A CCK8 assay was used to
quantify the effects on HepG2 cell growth. As illustrated in
Fig. 1B, there were no
significant difference in cell viability between the NC group and
cells treated with 0.25 or 0.5 mM FFA. However, HepG2 cells treated
with 1.0 and 2.0 mM FFA exhibited a significant decrease in cell
viability by ~25.7 and ~69.3%, respectively (both P<0.05).
Therefore, 0.5 mM FFA was used as the optimal concentration for the
cell model of hepatic steatosis. Furthermore, as illustrated in
Fig. 1C, there were no
significant difference in cell viability between the NC, and the
0.1, 0.2 and 0.5 μM celastrol-treated groups. However, HepG2
cells treated with 1.0, 1.5 and 2.0 μM celastrol exhibited a
significant decrease in cell viability by approximately 22.3, 55
and 70.7%, respectively (all P<0.05).
Oil red O staining was then used to evaluate the
effect of celastrol on lipid deposition. As shown in Fig. 2A–E, HepG2 cells in the 0.5 mM
FFA-treated group presented widespread deposition of red lipid
droplets as compared with the NC group. By contrast, the lipid
droplets were significantly decreased in the FFA + celastrol (0.1,
0.2 and 0.5 μM) treatment groups, particularly in the 0.5
μM celastrol-treated group. As shown in Fig. 2F, the levels of intracellular
triglycerides were significantly higher in 0.5 mM FFA-induced HepG2
cells compared with those of the NC group. In addition,
intracellular triglyceride levels were decreased in the
celastrol-treated group in a dose-dependent manner, with 0.5
μM celastrol exhibiting the highest efficacy. Therefore,
based on the data of the present study, 0.5 μM celastrol was
selected as the optimal concentration for subsequent
experiments.
Effects of celastrol on the expression
levels of TLR4 and downstream key inflammatory mediators in
FFA-induced HepG2 cells
To examine the anti-inflammatory effect of
celastrol, HepG2 cells were treated with 0.5 μM celastrol,
followed by stimulation with 0.5 mM FFA for 24 h. RT-qPCR and
western blotting were subsequently used to quantify the effects of
celastrol on TLR4 signaling in the FFA-induced HepG2 cells. As
illustrated in Fig. 3A and B,
FFAs signifi-cantly increased the expression levels of TLR4 mRNA
and protein compared with those of the NC group. However,
co-treatment with 0.5 μM celastrol greatly inhibited TLR4
transcripts and protein expression levels. Similar to TLR4, the
protein expression levels of downstream inflammatory mediators
MyD88 (Fig. 3C), p-NF-κBp65
(Fig. 3D), IL-1β (Fig. 3E) and TNFα (Fig. 3F) were significantly increased in
the FFA-induced HepG2 cells compared with those in the NC group,
and were markedly decreased following celastrol
co-administration.
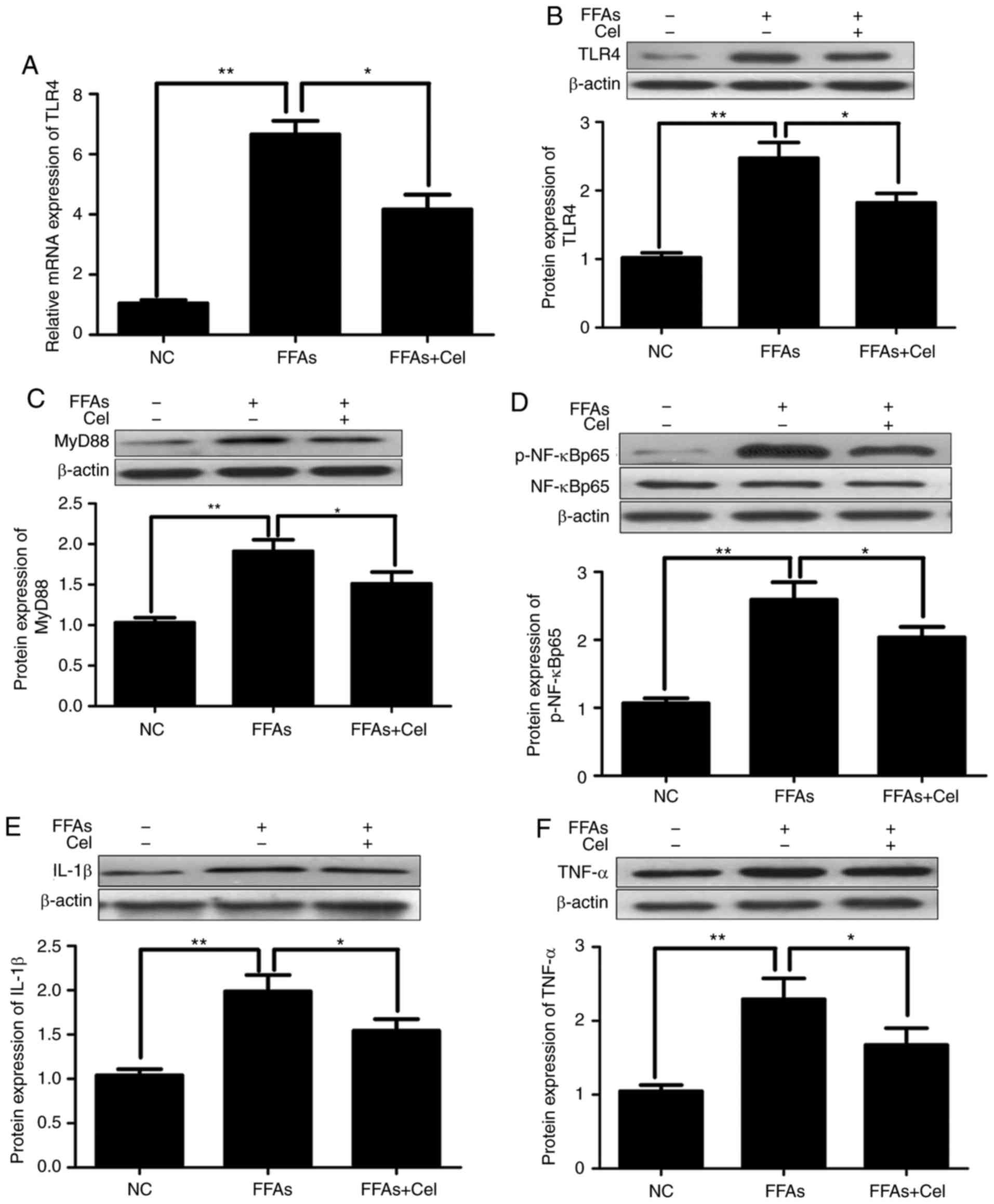 | Figure 3Celastrol administration
downregulated TLR4 and downstream signaling factors in the
FFA-induced HepG2 cells. HepG2 cells were cultured with 0.5
μM celastrol and 0.5 mM FFA for 24 h, and the transcripts
and protein expression levels of inflammatory mediators were
analyzed by reverse transcription-quantitative polymerase chain
reaction and western blotting. (A) Relative mRNA and (B) protein
expression levels of TLR4. Relative protein expression levels of
(C) MyD88, (D) phospho-NF-κBp65 and total NF-κBp65 (p-NF-κBp65
levels were normalized to total NF-κBp65 levels), (E) IL-1β, and
(F) TNFα were assayed by western blotting. Data are expressed as
the mean ± standard error of the mean (n=6). *P<0.05
and **P<0.01. FFA, free fatty acid; TLR4, toll-like
receptor 4; MyD88, myeloid differentiation primary response 88;
NF-κB, nuclear factor-κB; IL-1β, interleukin 1β; TNFα, tumor
necrosis factor α. |
Effects of transfection with TLR4 siRNA
in HepG2 cells
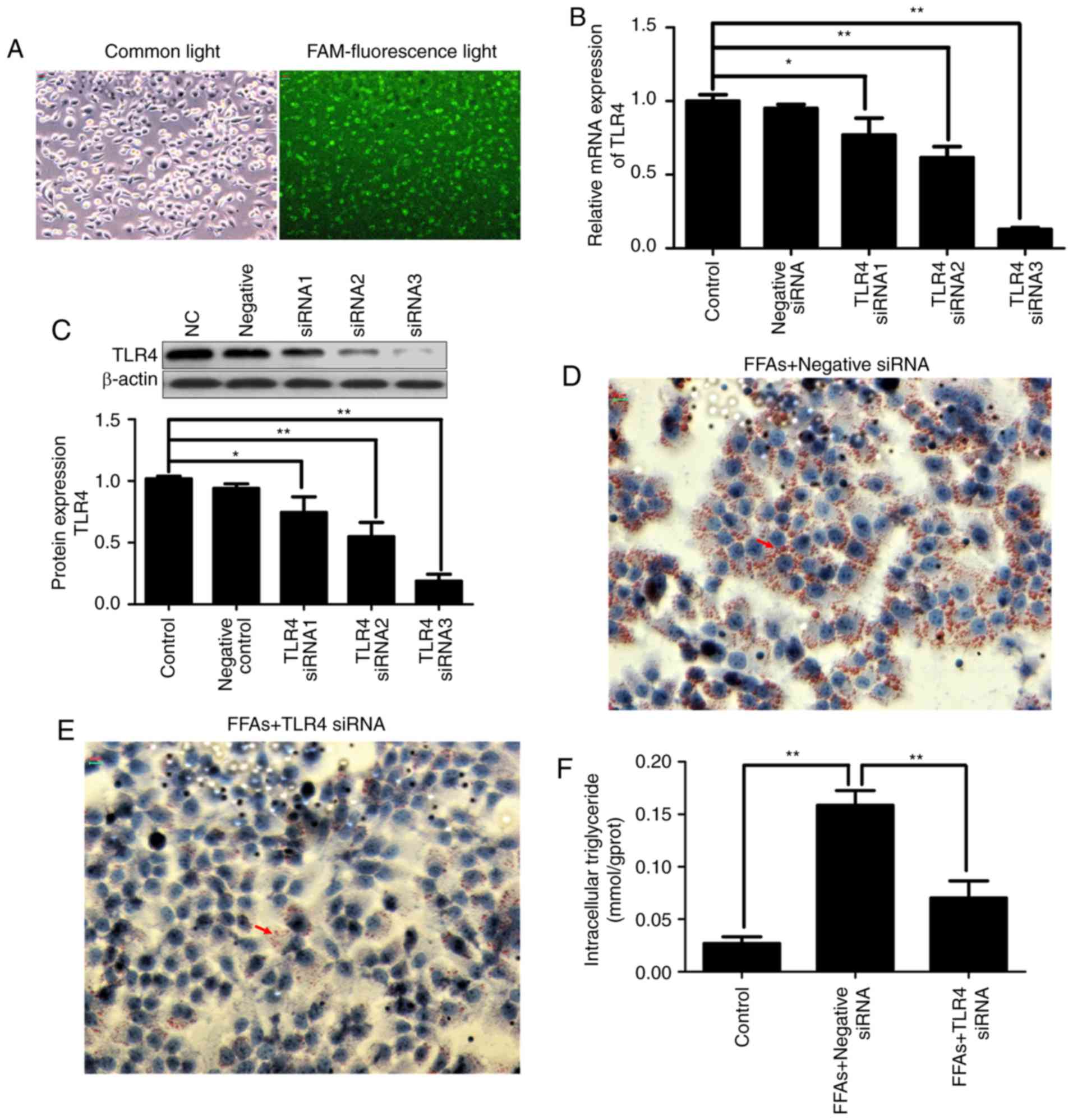
FAM-labeled siRNA was transfected into HepG2 cells
with Lipofectamine® 2000. After 24 h of transfection,
the transfection efficiency was observed using fluorescence
microscopy. As shown in Fig. 4A,
fluorescent particles within the cells indicated that siRNA was
transfected successfully into the HepG2 cells.
In order to evaluate the contribution of TLR4 to the
inflammatory activation in the FFA-induced HepG2 cells and silence
the expression of disease gene, three TLR4 siRNA constructs were
transiently transfected into HepG2 cells using
Lipofectamine® 2000. As shown in Fig. 4B and C, transfection with three
different TLR4 siRNA constructs reduced the transcription and
protein expression of TLR4 in the HepG2 cells by varying degrees.
More specifically, TLR4 siRNA3 exhibited the strongest knockdown
effect, inhibiting TLR4 mRNA and protein expression by ~80%.
Therefore, TLR4 siRNA3 was selected for use in subsequent
experiments. Notably, no effect on TLR4 expression was observed in
HepG2 cells transfected with negative control siRNA.
Continuous FFA stimulation caused lipid deposition;
however, when TLR4 was knocked down, FFA stimulation was also
blocked. As shown in Fig. 4D and
E, the increased deposition of lipid droplets in the
FFA-induced HepG2 cells was significantly decreased in the TLR4
siRNA-transfected group by Oil red O staining. As shown in Fig. 4F, the increased intracellular
triglyceride content in the FFA-induced HepG2 cells was also
significantly decreased in the TLR4 siRNA-transfected group.
Celastrol protects HepG2 cells partly via
the TLR4 signaling pathway
Subsequently, experiments attempting to clarify the
mechanisms underlying the modifications of FFA-induced cytokine
production by celastrol were conducted. TLR4 siRNA was used to
study the role of TLR4 in FFA-induced inflammation. In order to
eliminate the interference of transfection reagents, negative siRNA
was established as a control. Thus, the expression levels of
downstream inflammatory mediators were investigated subsequent to
exposure to BSA, 0.5 mM FFA, or a mixture of 0.5 mM FFA and 0.5
μM celastrol for 24 h in HepG2 cells transfected with
negative control siRNA or TLR4 siRNA. As shown in Fig. 5, significantly increased
activation of MyD88 (Fig. 5A),
p-NF-κBp65 (Fig. 5B), IL-1β
(Fig. 5C) and TNFα (Fig. 5D) was observed in HepG2 cells
following treatment with 0.5 mM FFA. This effect was partly
dependent on TLR4, since knockdown of TLR4 alleviated the action of
FFA in activating these downstream inflammatory mediators.
Celastrol treatment also resulted in the inhibition of FFA-induced
inflammatory response. Furthermore, celastrol and TLR4 siRNA
co-treatment demonstrated enhanced suppressive effects as compared
with each treatment alone.
 | Figure 5Effects of celastrol and TLR4 siRNA
on the TLR4 signaling cascade pathway in the FFA-induced HepG2
cells. In order to understand the relative mechanisms of celastrol
on ameliorating inflammatory response in the steatotic HepG2 cells,
the expression levels of downstream inflammatory mediators
following exposure to BSA, 0.5 mM FFA, or a mixture of FFA and 0.5
μM celastrol for 24 h in HepG2 cells transfected with
negative control siRNA or TLR4 siRNA were investigated. Relative
protein expression levels of (A) MyD88, (B) phospho-NF-κBp65 and
total NF-κBp65 (p-NF-κBp65 was normalized to total NF-κBp65 level),
(C) IL-1β and (D) TNFα were analyzed by western blotting. Data are
expressed as the mean ± standard error of the mean (n=6).
*P<0.05 and **P<0.01. FFA, free fatty
acid; TLR4, toll-like receptor 4; siRNA, small interfering RNA;
MyD88, myeloid differentiation primary response 88; NF-κB, nuclear
factor-κB; IL-1β, interleukin 1β; TNFα, tumor necrosis factor
α. |
Discussion
In a previous study by our group, it was
demonstrated that administration of celastrol exhibited
significantly suppressive effects on the levels of triglycerides,
total cholesterol of hepatic tissue, and reduced steatohepatitis
and macrophage infiltration in type 2 diabetic rats (15). Similar results were also observed
in the present study in vitro. The lipid deposition and
intracellular triglyceride contents were significantly higher in
the FFA-induced HepG2 cells compared with those of the NC group and
markedly decreased in the celastrol-treated groups, with a higher
dose of celastrol exhibiting increased efficacy.
The activation of immune and inflammatory signaling
pathways is considered to be of central importance in the
pathogenesis of NAFLD/NASH (22,23). Accumulating evidence in rodents
and in hepatocyte cultures suggested that altered TLR4 signaling is
a key pathway factor, and has an important regulatory role in
stimulating the immune and inflammatory response-associated gene
expression in the pathogenesis of NAFLD, and has been implicated in
the progression to NASH (24,25). Xu et al (26) previously reported increased
hepatic expression of TLR4 mRNA in rats fed with a high-fat diet
compared with their control counterparts. The increase in hepatic
TLR4 mRNA was associated with the appearance of NASH in this rat
model (26). Furthermore, when
fed with a methionine/choline-deficient diet, the most widely
accepted experimental model of NASH, TLR4-deficient mice exhibited
less severe hepatic injury and reduced accumulation of
intra-hepatic lipids compared with wild-type mice (27). The study by Sharifnia et al
(28) also provided evidence that
TLR4 was upregulated in a large cohort of NASH patients compared
with those suffering from NAFLD. All these findings indicated that
activation of the TLR4 signaling pathway was critically involved in
the pathogenesis of NASH. The accumulation of FFA causes the
activation of TLR4 signaling (29). Consistent with previous findings,
the current study observed that the expression levels of TLR4 mRNA
and protein were significantly increased in the FFA-induced HepG2
cells compared with those in the NC cells.
TLR4 signaling is initiated through two different
pathways, the MyD88-dependent and the MyD88-independent pathways,
and the latter is mediated by toll/IL-1 receptor domain-containing
adaptor inducing interferon-β (6). MyD88 is a critical downstream
signaling ligand of the TLR4 receptor complex, while it is also an
important adapter protein of the NF-κB signaling pathway, which
contributes to the expression of inflammatory genes. The triggering
of MyD88 pathway ultimately leads to nuclear translocation of
NF-κBp65 and the activation of inflammatory cascades that produce
various proinflammatory cytokines, including TNFα and IL-1β
(30). The study of Sharifnia
et al (28) in a human
hepatocyte culture model suggested that NF-κB activation was at
least partially driven by hepatocyte-mediated TLR4 signaling in
response to LPS and palmitate acid, with similar results observed
in high-fat diet mice (31). In
the present study, along with the upregulation of TLR4, the
expression levels of its downstream mediators MyD88 and p-NF-κBp65,
as well as the cytokines IL-1β and TNFα, were consistently
upregulated by FFA induction.
In order to directly determine whether TLR4 mediated
the increased activities of downstream inflammatory factors upon
FFA exposure, the effect of lossing the function of TLR4 was
examined in HepG2 cells. TLR4 siRNA was transfected into HepG2
cells, and RT-qPCR and western blotting revealed ~80% effective
TLR4 knockdown. Negative control siRNA did not affect the
expression of TLR4, which indicated that the transfection effect of
TLR4 siRNA was achieved through TLR4 gene inhibition, but not the
transfection reagent. Next, the present study sought to determine
whether TLR4 transcription was responsible for the activation of
downstream inflammatory factor, as suggested by the silencing
inhibition study.
Continuous FFA stimulation in the present study
activated the TLR4 signaling pathways and caused lipid deposition;
however, when TLR4 was knocked down, FFA stimulation was also
blocked. The increased deposition of lipid droplets and
intracellular triglyceride content in the FFA-induced HepG2 cells
were significantly decreased in the TLR4 siRNA-transfected groups.
As a consequence of limited TLR4 involvement, downstream mediators
MyD88, p-NF-κBp65, IL-1β and TNFα also exhibited partial
attenuation. Taken together, these results confirmed our earlier
findings that the activation of inflammatory factors in a lipotoxic
environment was partially mediated through TLR4 signaling. Thus,
targeting TLR4 may provide a promising intervention strategy for
the prevention or treatment of NAFLD.
Similar to the siRNA effect, the data of the present
study demonstrated that celastrol treatment markedly abolished the
FFA-induced TLR4-mediated signaling cascade pathways. In addition,
the results found that co-treatment with TLR4 siRNA and celastrol
further attenuated the expression levels of downstream mediators
compared with each treatment alone. These results suggested that
celastrol may ameliorate the hepatic inflammation through other
pathways in addition to TLR4. Previous studies have demonstrated
that upregulation and activation of several other TLR family
members were also involved in the innate immune response and the
formation of inflammation (4,5).
In total, 12 of the 13 members of the TLR family, with the
exception of TLR3, associate with the common adaptor molecule MyD88
through interaction of their intracellular toll/IL-1 receptor
domains to trigger inflammatory responses. Studies in animal
models, as well as in human NAFLD patients, have indicated the
likelihood for TLR2, TLR5, TLR6 and TLR9 to participate in the
onset or progression of NAFLD (4,32,33).
A limitation of the present study is that only a
preliminary experimental approach was used. Whether other TLR
family members directly inhibit hepatic inflammation requires
further investigation. Furthermore, the liver is an important
natural immune organ composed of a diverse array of cell types,
while it is also enriched in various immune cells, the majority of
which have the potential to be involved in inflammation. It is
clear that there are complex immune and inflammatory pathways that
result in the progression of NASH, involving signaling in various
cell types that are stimulated by pathogen- or damage-associated
molecular patterns, as well as interaction between different cells
(4,6). Thus, further research is required in
Kupffer cells, and our future studies will address the effect and
mechanisms of celastrol on the inflammatory response, which should
provide valuable insights into the development in NAFLD.
In conclusion, the TLR4-mediated signaling cascade
pathway may be a useful novel therapeutic target in the progression
of NAFLD. The present study provided evidence that celastrol
exerted its protective effects through improved triglyceride
accumulation, as well as inhibition of pro-inflammatory processes.
Elucidating the underlying mechanisms by which celastrol modulates
hepatic steatosis may provide the molecular basis for developing
therapeutic agents against NAFLD.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81470187).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
LC conceived and designed the experiments. CL and YX
performed the data analysis. LH conducted all experiments and wrote
the manuscript. BS revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Arrese M, Cabrera D, Kalergis AM and
Feldstein AE: Innate immunity and inflammation in NAFLD/NASH. Dig
Dis Sci. 61:1294–1303. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tiniakos DG, Vos MB and Brunt EM:
Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu
Rev Pathol. 5:145–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang S, Rutkowsky JM, Snodgrass RG,
Ono-Moore KD, Schneider DA, Newman JW, Adams SH and Hwang DH:
Saturated fatty acids activate TLR-mediated proinflmmatory
signaling pathways. J Lipid Res. 53:2002–2013. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ganz M and Szabo G: Immune and
inflammatory pathways in NASH. Hepatol Int. 7(Suppl 2): S771–S781.
2013. View Article : Google Scholar
|
|
5
|
Petrasek J, Csak T and Szabo G: Toll-like
receptors in liver disease. Adv Clin Chem. 59:155–201. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bieghs V and Trautwein C: Innate immune
signaling and gut-liver interactions in non-alcoholic fatty liver
disease. Hepatobiliary Surg Nutr. 3:377–385. 2014.
|
|
7
|
Kiziltas S: Toll-like receptors in
pathophysiology of liver diseases. World J Hepatol. 8:1354–1369.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Farrell GC, van Rooyen D, Gan L and
Chitturi S: NASH is an inflammatory disorder: Pathogenic,
prognostic and therapeutic implications. Gut Liver. 6:149–171.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Tu Q, Yan W, Xiao D, Zeng Z,
Ouyang Y, Huang L, Cai J, Zeng X, Chen YJ and Liu A: CXC195
suppresses proliferation and inflammatory response in LPS-induced
human hepatocellular carcinoma cells via regulating
TLR4-MyD88-TAK1-mediated NF-κB and MAPK pathway. Biochem Biophys
Res Commun. 456:373–379. 2015. View Article : Google Scholar
|
|
10
|
Venkatesha SH, Astry B, Nanjundaiah SM, Yu
H and Moudgil KD: Suppression of autoimmune arthritis by
Celastrus-derived Celastrol through mdoulation of pro-inflammatory
chemokines. Bioorg Med Chem. 20:5229–5234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y, Cao L, Xu LM, Cao FF, Peng B,
Zhang X, Shen YF, Uzan G and Zhang DH: Celastrol ameliorates EAE
induction by suppressing pathogenic T cell responses in the
peripheral and central nervous systems. J Neuroimmune Pharmacol.
10:506–516. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu J, Lee J, Salazar Hernandez MA,
Mazitschek R and Ozcan U: Treatment of obesity with celastrol.
Cell. 161:999–1011. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhu F, Li C, Jin XP, Weng SX, Fan LL,
Zheng Z, Li WL, Wang F, Wang WF, Hu XF, et al: Celastrol may have
an anti-atherosclerosis effect in rabbit experimental carotid
atherosclerosis model. Int J Clin Exp Med. 7:1684–1691. 2014.
|
|
14
|
Kim JE, Lee MH, Nam DH, Song HK, Kang YS,
Lee JE, Kim HW, Cha JJ, Hyun YY, Han SY, et al: Celastrol, an NF-κB
inhibitor, improves insulin resistance and attenuates renal injury
in db/db mice. PLoS One. 8:e620682013. View Article : Google Scholar
|
|
15
|
Han LP, Li CJ, Sun B, Xie Y, Guan Y, Ma ZJ
and Chen LM: Protective effects of celastrol on diabetic liver
injury via TLR4/MyD88/NF-κB signaling pathway in type 2 diabetic
rats. J Diabetes Res. 2016:26412482016. View Article : Google Scholar
|
|
16
|
López-Terrada D, Cheung SW, Finegold MJ
and Knowles BB: HepG2 is a hepatoblastoma-derived cell line. Hum
Pathol. 40:1512–1515. 2009. View Article : Google Scholar
|
|
17
|
Gómez-Lechón MJ, Donato MT,
Martínez-Romero A, Jiménez N, Castell JV and O'Connor JE: A human
hepatocellular in vitro model to investigate steatosis. Chem Biol
Interact. 165:106–116. 2007. View Article : Google Scholar
|
|
18
|
Xu F, Li Z, Zheng X, Liu H, Liang H, Xu H,
Chen Z, Zeng K and Weng J: SIRT1 mediates the effect of GLP-1
receptor agonist exenatide on ameliorating hepatic steatosis.
Diabetes. 63:3637–3646. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheng S, Liang S, Liu Q, Deng Z, Zhang Y,
Du J, Zhang Y, Li S, Cheng B and Ling C: Diosgenin prevents
high-fat diet-induced rat non-alcoholic fatty liver disease through
the AMPK and LXR signaling pathways. Int J Mol Med. 41:1089–1095.
2018.
|
|
20
|
Schwartz DM and Wolins NE: A simple and
rapid method to assay triacylglycerol in cells and tissues. J Lipid
Res. 48:2514–2520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Takaki A, Kawai D and Yamamoto K:
Molecular mechanisms and new treatment strategies for non-alcoholic
steatohepatitis (NASH). Int J Mol Sci. 15:7352–7379. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Szabo G and Petrasek J: Inflammasome
activation and function in liver disease. Nat Rev Gastroenterol
Hepatol. 12:387–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kesar V and Odin JA: Toll-like receptors
and liver disease. Liver Int. 34:184–196. 2014. View Article : Google Scholar
|
|
25
|
Bieghs V and Trautwein C: The innate
immune response during liver inflammation and metabolic disease.
Trends Immunol. 34:446–452. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu ZJ, Fan JG, Wang XP and Wang GL:
Upregulating expressions of hepatic lipopolysaccharide receptors in
nonalcoholic steato-hepatitic rats. Zhonghua Gan Zang Bing Za Zhi.
14:49–52. 2006.In Chinese. PubMed/NCBI
|
|
27
|
Rivera CA, Adegboyega P, van Rooijen N,
Tagalicud A, Allman M and Wallace M: Toll-like receptor-4 signaling
and Kupffer cells play pivotal roles in the pathogenesis of
non-alcoholic steato-hepatitis. J Hepatol. 47:571–579. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sharifnia T, Antoun J, Verriere TG, Suarez
G, Wattacheril J, Wilson KT, Peek RM Jr, Abumrad NN and Flynn CR:
Hepatic TLR4 signaling in obese NAFLD. Am J Physiol Gastrointest
Liver Physiol. 309:G270–G278. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shen YF, Zhang X, Wang Y, Cao FF, Uzan G,
Peng B and Zhang DH: Celastrol targets IRAKs to block Toll-like
receptor 4-mediated nuclear factor-κB activation. J Integr Med.
14:203–208. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mehal WZ: The inflammasome in liver injury
and non-alcoholic fatty liver disease. Dig Dis. 32:507–515. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang N, Wang H, Yao H, Wei Q, Mao XM,
Jiang T, Xiang J and Dila N: Expression and activity of the
TLR4/NF-κB signaling pathway in mouse intestine following
administration of a short-term high-fat diet. Exp Ther Med.
6:635–640. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Arias-Loste MT, Iruzubieta P, Puente Á,
Ramos D, Santa Cruz C, Estébanez Á, Llerena S, Alonso-Martín C, San
Segundo D, Álvarez L, et al: Increased expression profile and
functionality of TLR6 in peripheral blood mononuclear cells and
hepatocytes of morbidly obese patients with non-alcoholic fatty
liver disease. Int J Mol Sci. 17:E18782016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Miura K and Ohnishi H: Role of gut
microbiota and Toll-like receptors in nonalcoholic fatty liver
disease. World J Gastroenterol. 20:7381–7391. 2014. View Article : Google Scholar : PubMed/NCBI
|