Introduction
Exposure to ionizing irradiation produces DNA damage
in cells, including DNA single strand breaks (SSBs) and double
strands breaks (DSBs). While the majority of the SSBs can be
repaired, DSBs are particularly hazardous to the cell due to the
possibility of them resulting in rearrangement of the genome, which
is lethal to the cell (1).
Radiation induces the upregulation of proteins, including nuclear
factor κ-light-chain-enhancer of activated B cells (NF-κB),
inducible nitric oxide synthases, tumor necrosis factor-α,
interleukin-1 (IL-1) and IL-6, which are associated with
inflammatory responses; additionally, this may result in DNA damage
and damage to non-targeted tissues, and result in increased risk of
carcinogenesis (2).
Heme oxygenase 1 (HO-1), an antioxidant enzyme that
exhibits low basal expression levels in the majority of cells and
tissues, is notably upregulated by a variety of oxidative stress
stimuli. The upregulation of HO-1 is generally considered to be an
adaptive cellular response against the toxicity of oxidative
stress, and has been recognized to exhibit important
anti-inflammatory functions (3-5).
Previous studies have demonstrated that the targeted overexpression
of HO-1 demonstrated beneficial effects in various experimental
animal models of inflammation (6,7).
The upregulated gene expression of HO-1 is mediated by a network of
signaling pathways, among which mitogen-activated protein kinases
(MAPKs) serve a primary role (8).
Cyclooxygenase-2 (COX-2), the inducible form of COX, is activated
by growth factors and cytokines in order to catalyze the conversion
of arachidonic acid to prostaglandins (9). The activity of COX-2 is associated
with reactive oxygen species production and inflammatory signs in
cells, which are important in mediating radiation-induced
biological effects (10). The
expression of COX-2 can be activated by ionizing radiation and its
upregulation is indicated to be associated with the decreased
sensitivity of cells to radiation (11–13). As HO-1 and COX-2 contribute to
radiation protection and are involved in the equilibrium of
oxidative stress, it is hypothesized that interconnections exist
between them in irradiated cells, which requires investigation.
The nuclear fusion of deuterium with tritium (D-T)
releases notable energy, and investigations into using fusion for
the production of electricity has been pursued for decades. A
number of experimental fusion reactors are under development
globally. As fusion reaction produces large quantities of ionizing
radiation, including high energy neutron and photon, biological
investigations associated with the health risks of fusion radiation
are important and necessary for the benefit of scientists and
workers (14). In the present
study, whether HO-1 and COX-2 are involved in the regulation of
damage caused by fusion radiation was investigated, and their
upstream regulators were identified.
Materials and methods
Chemicals
KU55933 was purchased from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA), SB203580 was purchased from Selleck
Chemicals (Houston, TX, USA), NS-398 was purchased from Abcam
(Cambridge, UK) and protoporphyrin IX zinc (II) (ZnPP) was
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
These chemicals were dissolved in 100% dimethyl sulfoxide and
stored in small aliquots at 20°C. Protease inhibitor cocktail and
protein inhibitor cocktail were purchased from Roche Diagnostics
GmbH (Mannheim, Germany) and Sigma-Aldrich (Merck KGaA),
respectively.
Cell culture and radiation
Normal human lung fibroblasts (NHLFs), an adherently
grown human primary lung fibroblast, were purchased from BeNa
Culture Collection (Beijing, China). The NHLF cells were cultured
in Dulbecco's modified Eagle's medium/F12 (GE Healthcare Life
Sciences, Little Chalfont, UK) with 10% fetal bovine serum (Clark
Bioscience, Richmond, VA, USA) and grown in a humidified 5%
CO2 incubator at 37°C. The neutron radiation appliance
was a High Intensity D-T Fusion Neutron Generator, which was
developed at the Institute of Nuclear Energy Safety Technology,
Chinese Academy of Sciences (Hefei, China) by the Fusion Design and
Study team. The production and the property of the fusion neutron
were described previously by Wu (15). Radiation doses (0, 0.01, 0.12,
0.6, and 1.2 Gy) were adjusted by setting the cells at different
linear distances from the neutron source.
Immunofluorescence staining of
γ-H2A histone family member X (γ-H2AX)
The NHLF cells were cultured on 0.17 mm-thick
glass-bottom cell culture dishes and exposed to neutron radiation.
At the desired time points (0, 0.5, 1, 3, 6, and 12 h), the cells
were washed with PBS and fixed with 4% paraformaldehyde at room
temperature for 30 min. Subsequently, the cells were washed with
PBS and permeated with 0.5% Triton X-100 at room temperature for 30
min. Following blocking with PBST (0.1% Triton X-100) containing 1%
bovine serum albumin (BSA; Sangon Biotech Co., Ltd., Shanghai,
China) and 0.1% Triton X-100 at 37 °C for 1 h, the cells were
incubated with anti-γ-H2AX antibody (1:400; cat. no.
2577; Cell Signaling Technology, Inc., Danvers, MA, USA), which was
diluted in PBST containing 1% BSA at 4°C overnight with gentle
agitation. The dishes were then washed three times with PBST for 5
min. Alexa Fluor-594-conjugated goat anti-rabbit IgG (1:800; cat.
no. 111-585-003; Jackson ImmunoResearch Laboratories, Inc., West
Grove, PA, USA) diluted in PBST containing 1% BSA was added into
the samples. Following incubation for 2 h at room temperature, the
cells were washed three times with PBST for 15 min and
counterstained with Hoechst 33342. (2 µg/ml) Images were
captured under an Olympus IX83 fluorescence microscope using a 40×
air-objective (Olympus Corporation, Tokyo, Japan) and analyzed with
Image J1.49 software (National Institutes of Health, Bethesda, MD,
USA). For γ-H2AX quantification, images captured in the Hoechst
33342 channel were used to define the nuclear region. The
fluorescence intensity of γ-H2AX in the defined nuclear region was
measured to reflect the levels of DNA DSB. At least 500 cells were
analyzed for each sample.
Immunoblot analysis
The cells were then washed twice with PBS and total
cell lysate was prepared with radioimmunoprecipitation assay buffer
containing protease inhibitors and protein phosphatase inhibitors.
Protein concentrations were determined using a BCA kit (Sangon
Biotech Co., Ltd.). The cell lysate (50 µg) was resolved
using 10% or 12% SDS-PAGE and transferred onto a polyvinylidene
fluoride (PVDF) membrane. Following blocking in tris-buffered
saline with 0.1% Tween 20 with 1% skim milk, the PVDF membrane was
incubated with primary antibodies at 4°C overnight. The primary
antibodies used were as follows: α-tubulin (1:20,000; cat. no.
ab108629; Abcam); phosphorylated (p)-ataxia telangiectasia mutated
(p-ATM; 1:1,000; cat. no. DR1002; Merck KGaA); Cox-2 (1:1,000; cat.
no. 160112; Cayman Chemical Company, Ann Arbor, MI, USA); β-actin
(1:500; cat. no. sc-8432; Santa Cruz Biotechnology, Inc.); p-p38
(1:1,000; cat. no. 612280; BD Biosciences, Franklin Lakes, NJ,
USA); and HO-1 (1:1,000; cat. no. 10701; ProteinTech Group, Inc.,
Chicago, IL, USA). Subsequently, the membrane was washed three
times with PBS with 0.1% Tween 20 and incubated with corresponding
horseradish peroxidase-conjugated secondary antibodies (goat
anti-rabbit, cat. no. 111-035-003 or goat anti-mouse, cat. no.
115-035-003; both 1:200,000; Jackson ImmunoResearch Laboratories,
Inc.) for 2 h at room temperature. The protein bands were
visualized using enhanced chemiluminescent substrate (Wuhan Boster
Biological Technology, Ltd., Wuhan, China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA (100 ng/µl) was extracted using
RNAiso reagent (Takara Bio, Inc, Otsu, Japan). The total RNA was
added into the reaction mixture from One-Step SYBR®
PrimeScript™ PLUS RT-PCR kit. The reaction mixture was as follows:
Reaction volume, 20 µl; 2× One Step SYBR RT-PCR Buffer, 10
µl; Takara Ex Taq HS Mix, 1.2 µl; PrimeScript PLUS
RTase Mix, 0.4 µl; Forward Primer (10 µM), 0.8
µl; Reverse Primer (10 µM), 0.8 µl; Rox
Reference Dye, 0.4 µl; Total RNA, 2 µl; RNase Free
ddH2O, 4.4 µl. RT-qPCR analysis was performed on
a StepOne™· Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) according to 2−ΔΔCq
method (16). Reverse
transcription was performed at 42°C for 10 min. The PCR conditions
were as follows: 95°C for 1 min, 50°C for 30 sec, 72°C for 30 sec,
35 cycles. The primers for HO-1 were: Forward 5′-CTG TGT AAC CTC
TGC TGT TCC-3′ and reverse 5′-CCA CAC TAC CTG AGT CTA CC-3′. The
primers for human β-actin were: Forward 5′-CCT GGC ACC CAG CAC
AAT-3′ and reverse 5′-GGG CCG GAC TCG TCA TAC-3′.
Statistical analysis
All data are presented as the mean ± standard
deviation of at least three independent experiments performed in
triplicate. Statistical significance between two groups was
evaluated using Student's t-test with GraphPad Prism 5 (GraphPad
Software, Inc., San Diego, CA, USA). Statistical significance
between multiple groups was evaluated using one-way analysis of
variance with SPSS 12.0 software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Induction of DNA DSBs by neutron
radiation
The induced levels of DNA DSBs in the irradiated
NHLF cells were evaluated to assess the toxicity of D-T neutron
radiation. The cells were exposed to 0.01, 0.6 and 1.2 Gy neutrons
and, 1 h later, an immunofluorescence staining assay was performed
to analyze the DSB levels. As shown in Fig. 1A, there was a dose-dependent
tendency for the induction of DSB in the dose range used in the
experiment. Subsequently, the levels of DSBs at different time
intervals following 0.6 Gy neutron radiation were detected
(Fig. 1B). A significant increase
in the number of DSBs was observed 30 min following radiation
exposure, and enhanced DSB formation was sustained to the 6 h time
point. At 12 h post-radiation, the DSB levels decreased, reflecting
the repair of neutron-induced DNA damage.
Upregulation of HO-1-alleviates
neutron-induced DSBs
HO-1 serves important biological roles in
maintaining cellular oxidative equilibrium and has been reported to
protect cells against stress induced damage; therefore, whether
neutron radiation stimulated the expression of HO-1was determined.
Following 6 or 12 h of exposure to 0.6 Gy neutrons, the NHLF cells
were collected and the expression of HO-1 in total cell lysates was
detected by western blot analysis. As shown in Fig. 2A, upregulation of HO-1 was
observed 6 h post-radiation. At the 12-h time point, the expression
level of HO-1 was enhanced by 75%, compared with that in the
control. Subsequently, the effect of HO-1 on radiation-induced DSB
formation was determined. Pretreatment with Znpp increased DSB
formation by ~30% in the 0.6 Gy neutron-irradiated cells (Fig. 2B). These results demonstrated that
induction of the expression of HO-1 by neutron radiation alleviated
DNA damage.
COX-2 mediates the upregulation of HO-1
in irradiated cells
COX-2 has been reported to be upregulated in
ionizing irradiated cells and the inhibition of COX-2 results in
enhanced toxicity; therefore, whether COX-2 affected the expression
of HO-1 in neutron irradiated cells was investigated. The
expression level of COX-2 was detected at 6 and 12 h following
radiation exposure. As shown in Fig.
3A, COX-2 was upregulated by 30% at 6 h compared to 0 h in the
irradiated cells. Subsequently, the protein and mRNA levels of HO-1
were detected in irradiated cells treated with COX-2 inhibitor
NS-398. The cells were on radiation-induced DSB formation was then
investigated. Treatment of the NHLF cells with COX-2 inhibitor
NS-398 increased neutron-induced DSB formation (Fig. 3D). The DSB formation rate
increased by ~15% in the cells pretreated with NS-398 and then
exposed to 0.6 Gy neutron irradiation. These results demonstrated
that the induction of the expression of COX-2 by neutron radiation
alleviated DNA damage.
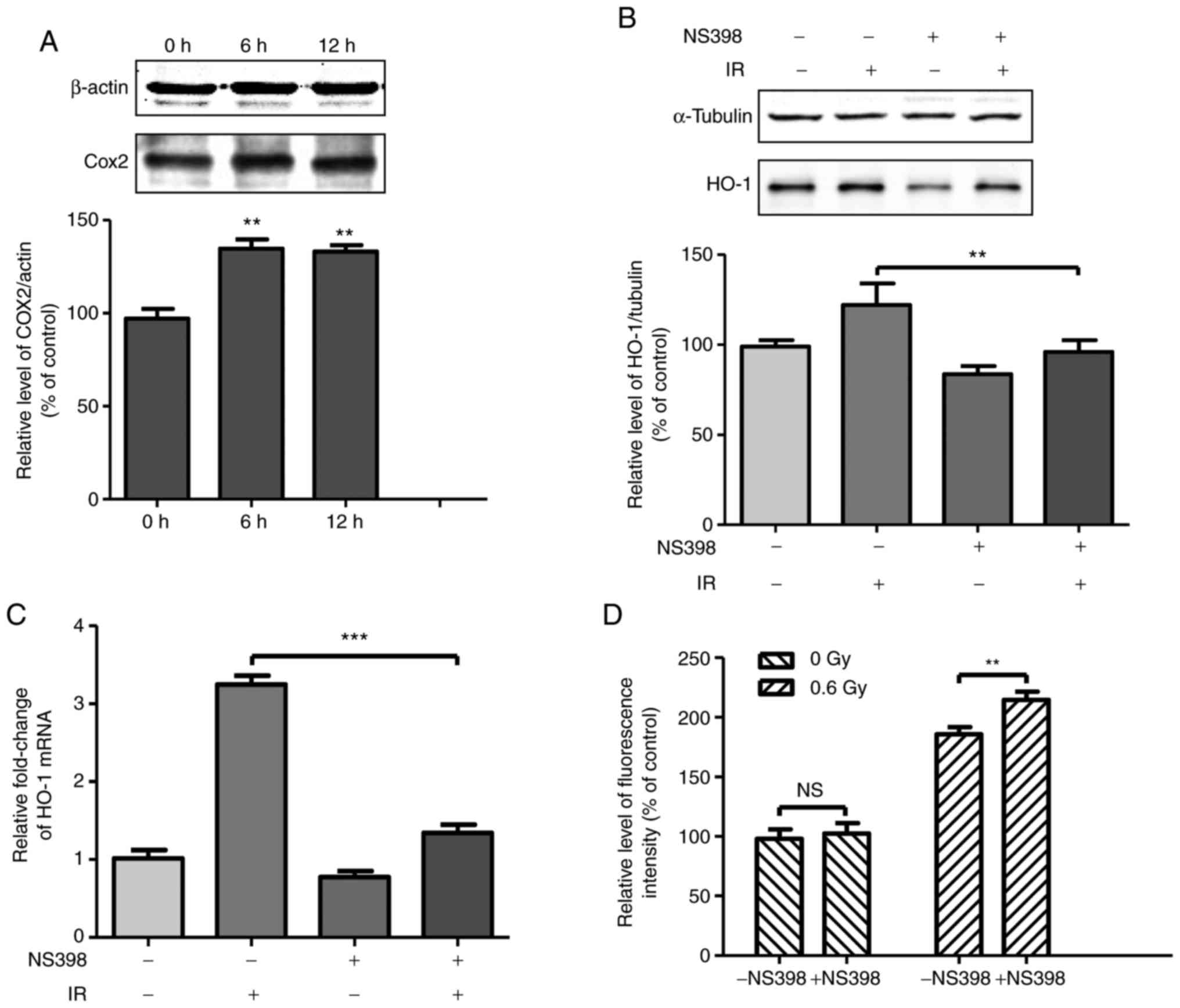 | Figure 3Upregulation of COX-2 by fusion
radiation promotes the expression of HO-1. (A) Cells were exposed
to 0.6 Gy fusion neutron. At the indicated time points, the
irradiated cells were collected and the expression levels of COX-2
were determined by western blot analysis. Cells were treated with
COX-2 inhibitor NS-398 (final concentration, 40 µM) for 1 h
prior to fusion radiation exposure. At 6 h following exposure to
radiation, (B) protein and (C) mRNA levels of HO-1 were detected by
western blot and reverse transcription-quantitative polymerase
chain reaction analyses, respectively. (D) Cells were treated with
COX-2 inhibitor NS-398 (final concentration, 40 µM) for 1 h
prior to fusion radiation exposure. At 1 h following exposure to
radiation, the cells were fixed for H2AX (pSer139)
immunostaining. **P<0.01, ***P<0.001.
COX-2, cyclooxygenase 2; HO-1, heme oxygenase 1; IR, irradiation;
NS, not significant. |
COX-2/HO-1 is regulated by p38 in
irradiated cells
p38, a member of the MAPKs, is reported to be
involved in ionization radiation-induced biological responses. To
determine the upstream regulator of COX-2, phosphorylation was
first determined. As shown in Fig.
4A, the phosphorylation of p38 showed a tendency to increase
following radiation exposure, to a level of 1.5-fold at the 2 h
time point and 2.2-fold at the 8 h time point, compared with the
control group. To determine the association between the activation
of p38 and the expression of COX-2, the cells were treated with p38
inhibitor SB203580 for 1 h prior to radiation exposure. At 6 h
post-radiation exposure, the expression levels of COX-2 and HO-1 in
the total cell lysate were detected and are shown in Fig. 4B and C. The results demonstrated
that the induction of COX-2 and HO-1 was significantly suppressed
by SB203580, indicating that the activation of p38 by radiation
exposure contributed to the induced expression of COX-2 and HO-1.
Subsequently, the expressions levels of DSB formation in the
irradiated cells treated with p38 inhibitor SB203580 were detected.
Preincubation with SB203580 increased the DSB formation rate by
~13% in the 0.6 Gy neutron-irradiated cells (Fig. 4D). These results demonstrated that
the induction of the expression of p38 by neutron radiation
alleviated DNA damage.
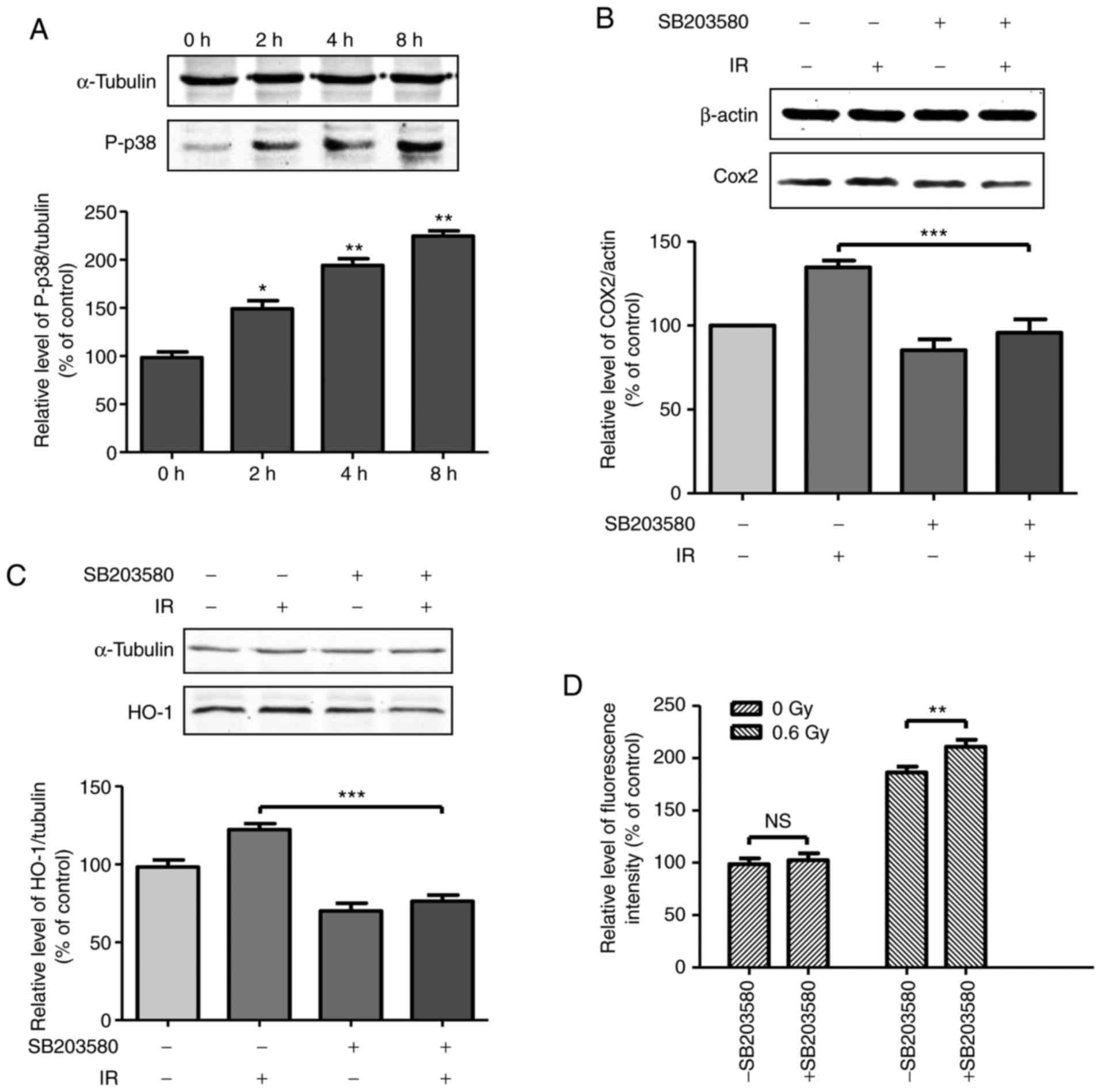 | Figure 4Activation of p38 MAPK contributes to
the upregulation of COX-2 and HO-1 in cells exposed to fusion
radiation. (A) Cells were exposed to 0.6 Gy fusion neutron. At the
indicated time points, irradiated cells were collected and the
expression levels of phosphorylated p38 were determined by western
blot analysis. Cells were pre-incubated with p38 inhibitor SB203580
(final concentration, 20µM) for 1 h prior to fusion
radiation exposure. At 6 h following exposure to radiation, cells
were collected. The expression levels of (B) COX-2 and (C) HO-1
were detected by western blot analysis. (D) Cells were
pre-incubated with p38 inhibitor SB203580 (final concentration,
20µM) for 1 h prior to fusion radiation exposure. At 1 h
following exposure to radiation, the cells were fixed for
H2AX (pSer139) immunostaining. *P<0.05,
**P<0.01, ***P<0.001. COX-2,
cyclooxygenase 2; HO-1, heme oxygenase 1; p-p38, phosphorylated
p38; IR, irradiation; NS, not significant. |
ATM DNA damage response stimulates
p-p38/COX-2/HO-1
ATM is a DNA damage response kinase and it is
rapidly activated through autophosphorylation in irradiated cells.
Following neutron radiation exposure, the phosphorylation of ATM
was observed at 30 min and was enhanced up to 3 h post-radiation
exposure (Fig. 5A). Subsequently,
the ATM inhibitor, KU55933, was used to investigate whether the
enhanced expression of COX-2 and HO-1 was due to the activation of
ATM. The cells were treated with KU55933 for 0.5 h prior to
irradiation, and at 6 h post-radiation, the levels of COX-2 and
HO-1 were detected. As shown in Fig.
5B and C, the radiation-induced expression of COX-2 and HO-1
was suppressed, indicating the ATM-regulated DNA damage response
contributed to COX-2- and HO-1-associated protection against
neutron-induced toxicity. Furthermore, the phosphorylation of p38
following treatment with KU55933 in irradiated cells was
identified, which is shown in Fig.
5D. In irradiated cells pretreated with KU55933, the
phosphorylation of p38 was significantly inhibited. These results
indicated that the activation of p38 was controlled by ATM in
neutron-irradiated cells, and also confirmed the role of p38 in
upregulating the expression of COX-2 and HO-1.
 | Figure 5ATM DNA damage response is
responsible for the upregulation of p-p38/COX-2/HO-1. (A) Cells
were exposed to 0.6 Gy fusion neutron. At the indicated time
points, irradiated cells were collected, and the expression levels
of phosphorylated ATM were determined by western blot analysis.
Cells were treated with ATM inhibitor KU55933 (final concentration,
25 µM) for 0.5 h prior to fusion radiation exposure. At 6 h
following exposure to radiation, the cells were collected and the
expression levels of (B) COX-2 and (C) HO-1 were detected by
western blot analysis. (D) At 2 h following pretreatment with 25
µM KU55933 and fusion radiation, cells were collected and
the levels of p-p38 were determined by western blot analysis.
**P<0.01, ***P<0.001. ATM, ataxia
telangiectasia mutated; COX-2, cyclooxygenase 2; HO-1, heme
oxygenase 1; IR, irradiation. |
Discussion
The notable energy release from the process of D-T
nuclear fusion has attracted the attention of nuclear physicists
due to its possible significant influence on the methods of
supplying energy in the future. The 14.1 MeV neutron produced
during the reaction of D-T fusion is a potential health risk due to
ionization and damaging tissues; therefore, an understanding of the
biological effects induced by the 14.1 MeV neutron is important for
protection and medicinal treatment against neutron radiation risks.
However, current knowledge on the biological effects and the
underlying mechanisms remains limited. In the present study, the
level of DNA DSBs, which is a typical DNA damage marker for
ionizing radiation, was assessed to detect the level of damage
induced by fusion radiation in NHLFs. The results demonstrated that
the DSB levels were radiation dose-dependent in the range of 0–1.2
Gy. Compared with gamma-rays, the linear energy transfer of
neutrons is notably higher, which means neutron deposits increase
energy when traversing cells and causes increased ionization and
DSBs (17). Broustas et al
(18) compared gene expression
profiles in a human peripheral blood model to neutron and X-ray
radiation. They identified 125 genes that responded significantly
to the two types of radiation as a function of dose, with the
magnitude of response to neutrons generally being increased,
compared with that observed following X-ray exposure (18). The relative biological effects
induced by neutrons varied with the neutron energy. Tanaka et
al (19) compared the
efficacies of neutron with energy from 0.18–2.30 MeV in the
induction of comet DNA and chromosome aberrations. They concluded
that 0.37 MeV neutron caused of the most DNA damage, whereas the
2.30 MeV neutron caused the least damage (19); however, the biological effects
induced by neutrons with energies >10 MeV, for example, the 14.1
MeV fusion neutron, remain to be fully elucidated. The results of
the present study demonstrated that the neutron emitted from D-T
fusion was effective in causing DNA damage; therefore, its
biological effects warrant further examination.
HO-1, an essential enzyme in heme catabolism,
cleaves heme to form biliverdin, carbon monoxide (CO) and ferrous
iron. The transcription of HO-1 can be mediated by redox-dependent
transcription factors, including NF-E2-related factor 2 (NRF2),
NF-κB, and activating protein-1 (8). It has been shown to protect cells
from various stresses-induced toxicity. HO-1-derived CO is an
anti-inflammatory molecule. A previous study indicated that CO
exposure facilitated the homologous recombination repair of DNA
damage through an ATM/ataxia telangiectasia and Rad3-related
protein (ATR)-dependent pathway (20). Chen et al (21) reported that, in low dose
α-particle irradiated A549 cells, HO-1 was upregulated in a
NRF2-dependent manner, and its increased expression conferred A549
resistance following high dose α-particle radiation. Furthermore,
scavenging CO reduced the resistance to radiation (21). Additionally, in a study on
radiation-induced bystander effects, Han et al (22) determined that CO (released from CO
release molecule 2) treatment resulted in the decreased formation
of DSBs in bystander cells. A report by Singh et al
(23) demonstrated that treatment
with the mixture of podophyllotoxin and rutin reduced the levels of
oxidative stress and apoptotic cell death in gamma-ray irradiated
mouse bone marrow and spleen. Additionally, the NRF2-mediated
upregulation of HO-1 partially contributed to the protective role
of podophyllotoxin and rutin (23). The results of the present study
indicated that the inhibition of HO-1 activity exacerbated fusion
radiation-induced DNA damage, confirmed the role of HO-1 in
radiation protection, and indicated that the targeted upregulation
of HO-1 may be a potential method to decrease the risk of fusion
radiation to health.
The expression of COX-2 is upregulated in numerous
types of cancer. Furthermore, its product prostaglandin H2 is
converted by prostaglandin E2 (PGE2) synthase into PGE2, which in
turn can stimulate cancer progression. Consequently, COX-2 is a
target for the development of drugs that prevent and treat cancer.
Previous studies have reported that the targeted inhibition of
COX-2 improved the efficacy of cancer radiotherapy, indicating the
protective role of COX-2 in irradiated tumor cells (9–13).
Hofer et al (24)
determined in animal experiments that, following gamma-ray
radiation, male COX-2-knockout mice exhibited attenuated
hematopoiesis, compared with wild-type mice. Additionally, it was
concluded that the genetic disruption of COX-2 has a positive
effect in hematopoiesis under basal conditions, but is detrimental
following radiation exposure (24). Ozbilgin et al (25) indicated that the increased
expression of COX-1 and COX-2 in the urothelium of mice may prevent
bladder damage from acute gamma-ray radiation, and benefits the
differentiation and restoration of the urothelium. Zuo et al
(26) reported that, treatment of
mouse epidermal cells with a low concentration of arsenite
increased the expression of COX-2, and this induction of COX-2
resulted in decreased levels of apoptosis following ultraviolet B
treatment. These data indicated that, although COX-2 is considered
to mediate inflammation and promote carcinogenesis, its
upregulation is beneficial under a number of stress conditions. To
the best of our knowledge, there has been no previous report on the
functions of COX-2 in neutron-irradiated cells. The present study
indicated that COX-2 may exert a protective effect through
stimulating the expression of HO-1, in order to decrease the DSB
levels in cells exposed to fusion radiation, which is consistent
with the aforementioned data.
ATM is a well-known DNA damage-response protein. In
cultured cells, elevated DSBs activate ATM through the
autophosphorylation of serine 1981. Additionally, it regulates the
functions of proteins involved in cell cycle checkpoints, apoptotic
cell death and DNA damage repair, including p53, BRCA1 and nibrin
(27). In the present study, the
phosphorylation of ATM was enhanced by fusion radiation, which is
consistent with previous results observed in cells exposed to other
types of ionizing radiation (28). As COX-2 was upregulated, whether
its expression was under the control of the activation of ATM
following fusion radiation was investigated. Park et al
(29) reported that gefitinib, a
small-molecular epidermal growth factor receptor tyrosine kinase
inhibitor, increased the radiation sensitivity of non-small cell
lung cancer cell lines NCI-H460 and VMRC-LCD through inhibiting the
activation of ATM. Additionally, the overexpression of COX-2 in
NCI-H460 cells reduced the radiation sensitivity induced by
gefitinib; however, no direct association was determined between
the activation of ATM and expression of COX-2 (29). Chacko et al (30) determined that the hydro-alcoholic
extract of Clerodendron infortunatum (CIE) reduces the total
body gamma-ray radiation exposure in mice. Additionally, in the
intestinal tissue of irradiated animals, following CIE treatment,
the expression levels of ATM, but not its phosphorylated form, were
elevated; however, the expression of COX-2 was reduced (30). In a previous study by Kim and Pyo
(31), it was reported that
treatment with the mixture of 17-AAG, an inhibitor of heat shock
protein 90, and celecoxib, an inhibitor of COX-2, increased the
sensitivity of various human cancer cells to radiation through
downregulating ATM and ATR (31).
These previous studies indicated that the associations between the
activation of ATM and expression of COX-2 are variable in
irradiated cells. In the present study, the ATM inhibitor
suppressed the upregulation of COX-2 and HO-1, indicating a
regulatory role of the ATM DNA response on the expression on COX-2
in cells exposed to fusion radiation. Additionally, it was
clarified that p38 was a mediator of ATM and COX-2. p38 is a member
of the MAPKs and responds to a variety of stress stimuli, including
cytokines, radiation and heat shock. Its activation in irradiated
cells is associated with radiation-induced cell cycle arrest and
apoptosis (32,33). Acheva et al (34) used a 3D organotypic skin model to
investigate the mechanisms of radiation-induced inflammatory
responses following localized irradiation, and determined that the
rapid activation of NF-κB, phosphorylated p38 and COX-2 were
upregulated in the irradiated and bystander areas of the 3D
cultures (34). However, in a
study by Hung et al (35),
it was demonstrated that the induction of COX-2 under endoplasmic
reticulum stress was controlled by NF-κB, which was regulated by
phosphorylated p38 (35).
Furthermore, Tessner et al (36) determined that p38 is critical for
the enhanced transcription and expression of COX-2 in
gamma-ray-irradiated human epithelial cells (36), which was consistent with the
observation by Hung et al (35). Additionally, Hu et al
(37) observed in an in
vitro epithelial wounding model that the production of PGE2 was
increased in a time-dependent manner via the activation of COX-2,
which was stimulated by the phosphorylation of extracellular
signal-regulated protein kinase 1/2, another member of the MAPK
family (37). The results of the
present study demonstrated that COX-2 and its mediated upregulation
of HO-1 were regulated by activated p38, and that p38 was also
associated with the ATM DNA damage response, which upregulated the
expression of COX-2/HO-1 in cells exposed to fusion radiation.
These reports indicate that multiple regulatory mechanisms of MAPK
members on the expression of COX-2 may be correlated with specific
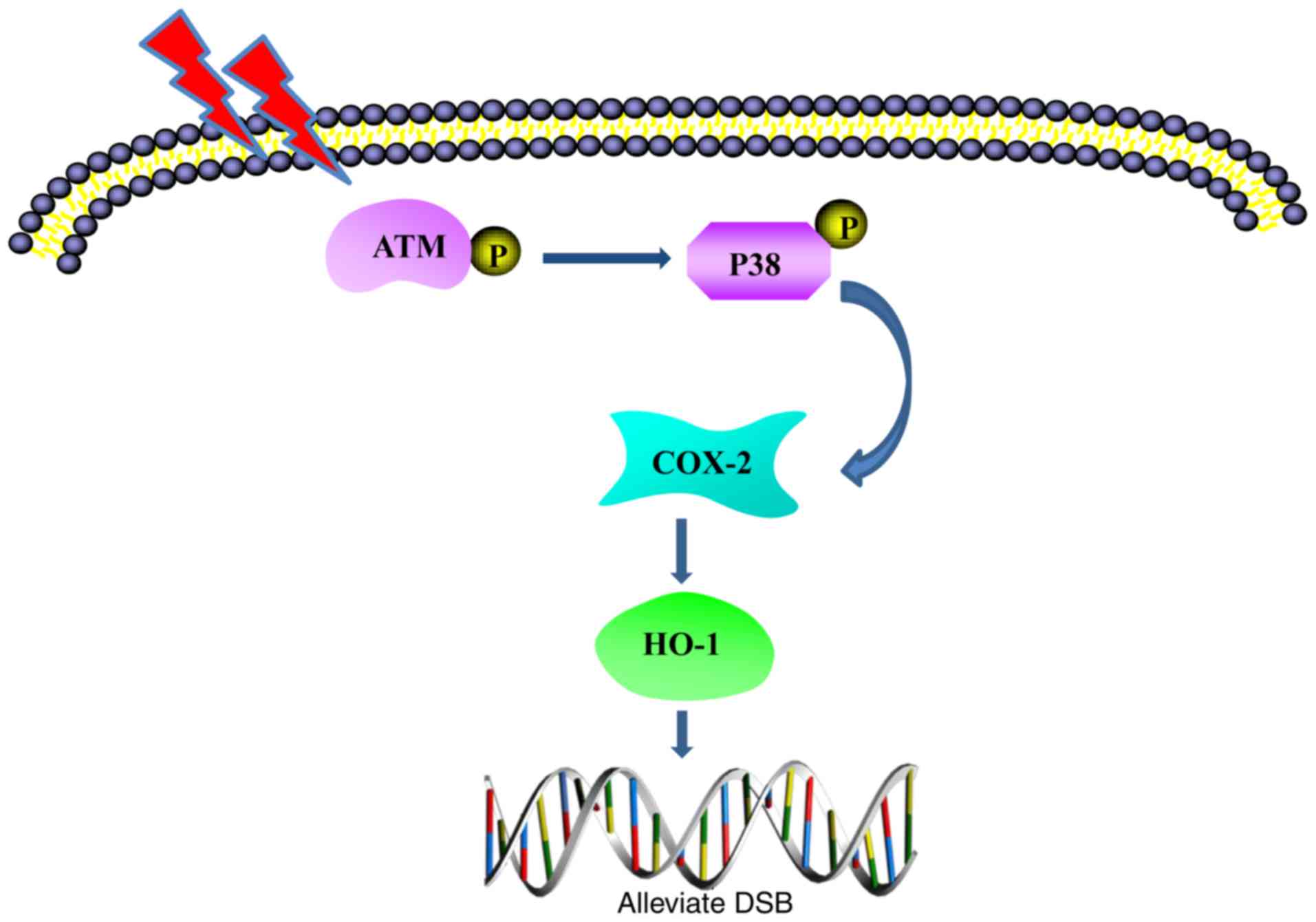
stress conditions. A hypothetic model is depicted in Fig. 6.
In conclusion, the present study demonstrated the
involvement of HO-1 in alleviating fusion radiation-induced DNA
damage in NHLFs. Induction of the pro-inflammatory protein COX-2 by
fusion radiation contributed to the upregulation of HO-1.
Furthermore, the ATM DNA damage response was investigated, which
was activated by fusion radiation, and was demonstrated to be
important in the enhanced expression of HO-1 and COX-2 through
stimulating the activation of p38 MAPK. The results of the present
study provide novel information on fusion radiation-induced
biological effects and potential targets for decreasing the health
risks.
Acknowledgments
The authors would like to thank the other members of
the Fusion Design and Study team for their assistance in the
present study.
Funding
The present study was supported by the National
Magnetic Confinement Fusion Science Program of China (grant no.
2014GB112006), the Natural Science Foundation of Anhui Province of
China (grant no. 1508085SME220), the National Natural Science
Foundation of China (grant nos. 11575232 and 31370842), the
International Partnership Program of Chinese Academy of Sciences
(grant no. 116134KYSB20160084) and the Innovative Program of
Development Foundation of Hefei Center for Physical Science and
Technology (grant no. 2016FXCX005).
Availability of data and materials
The analyzed data sets during the study are
available from the corresponding author upon reasonable
request.
Authors' contributions
JW, XY and HL designed the experiments. XY, HL and
XJ performed the biological experiments. CJ, ZX, TL and ZW
performed the deuterium-tritium fusion radiation of biological
samples. XY, HL and JW analyzed the results and wrote the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lomax ME, Folkes LK and O'Neill P:
Biological consequences of radiation-induced DNA damage: Relevance
to radiotherapy. Clin Oncol (R Coll Radiol). 25:578–585. 2013.
View Article : Google Scholar
|
|
2
|
Najafi M, Motevaseli E, Shirazi A, Geraily
G, Rezaeyan A, Norouzi F, Rezapoor S and Abdollahi H: Mechanisms of
inflammatory responses to radiation and normal tissues toxicity:
Clinical implications. Int J Radiat Biol. 94:335–356. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gozzelino R, Jeney V and Soares MP:
Mechanisms of cell protection by heme oxygenase-1. Annu Rev
Pharmacol Toxicol. 50:323–354. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vile GF, Basu-Modak S, Waltner C and
Tyrrell RM: Heme oxygenase 1mediates an adaptive response to
oxidative stress in human skin fibroblasts. Proc Natl Acad Sci USA.
91:2607–2610. 1994. View Article : Google Scholar
|
|
5
|
Otterbein LE and Choi AM: Heme oxygenase:
Colors of defense against cellular stress. Am J Physiol Lung Cell
Mol Physiol. 279:L1029–L1037. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ryter SW, Alam J and Choi AM: Heme
oxygenase-1/carbon monoxide: From basic science to therapeutic
applications. Physiol Rev. 86:583–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Soares MP, Marguti I, Cunha A and Larsen
R: Immunoregulatory effects of HO-1: How does it work? Curr Opin
Pharmacol. 9:482–489. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paine A, Eiz-Vesper B, Blasczyk R and
Immenschuh S: Signaling to heme oxygenase-1 and its
anti-inflammatory therapeutic potential. Biochem Pharmacol.
80:1895–1903. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pang LY, Hurst EA and Argyle DJ:
Cyclooxygenase-2: A role in cancer stem cell survival and
repopulation of cancer cells during therapy. Stem Cells Int.
2016:20487312016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tang FR and Loke WK: Molecular mechanisms
of low dose ionizing radiation-induced hormesis, adaptive
responses, radio-resistance, bystander effects, and genomic
instability. Int J Radiat Biol. 91:13–27. 2015. View Article : Google Scholar
|
|
11
|
Salehifar E and Hosseinimehr SJ: The use
of cyclooxygenase-2 inhibitors for improvement of efficacy of
radiotherapy in cancers. Drug Discov Today. 21:654–662. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rosser CJ, Gaar M and Porvasnik S:
Molecular fingerprinting of radiation resistant tumors: Can we
apprehend and rehabilitate the suspects? BMC Cancer. 9:2252009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komaki R, Liao Z and Milas L: Improvement
strategies for molecular targeting: Cyclooxygenase-2 inhibitors as
radiosensitizers for non-small cell lung cancer. Semin Oncol.
31(Suppl 1): S47–S53. 2004. View Article : Google Scholar
|
|
14
|
Nie B, Ni M and Wei S: Individual dose due
to radioactivity accidental release from fusion reactor. J Hazard
Mater. 327:135–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu Y: Development of high intensity D-T
fusion neutrongenerator HINEG. Int J Energy Res. 42:68–72. 2016.
View Article : Google Scholar
|
|
16
|
Bustin SA, Benes V, Garson JA, Hellemans
J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL,
et al: The MIQE guidelines: Minimum information for publication of
quantitative real-time PCR experiments. Clin Chem. 54:611–622.
2009. View Article : Google Scholar
|
|
17
|
Tran V and Little MP: Dose and dose rate
extrapolation factors for malignant and non-malignant health
endpoints after exposure to gamma and neutron radiation. Radiat
Environ Biophys. 56:299–328. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Broustas CG, Xu Y, Harken AD, Chowdhury M,
Garty G and Amundson SA: Impact of neutron exposure on global gene
expression in a human peripheral blood model. Radiat Res.
187:433–440. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tanaka K, Gajendiran N, Endo S, Komatsu K,
Hoshi M and Kamada N: Neutron energy-dependent initial DNA damage
and chromosomal exchange. J Radiat Res. 40(Suppl): S36–S44. 1999.
View Article : Google Scholar
|
|
20
|
Otterbein LE, Hedblom A, Harris C,
Csizmadia E, Gallo D and Wegiel B: Heme oxygenase-1 and carbon
monoxide modulate DNA repair through ataxia-telangiectasia mutated
(ATM) protein. Proc Natl Acad Sci USA. 108:14491–14496. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen N, Wu L, Yuan H and Wang J:
ROS/Autophagy/Nrf2 pathway mediated low-dose radiation induced
radio-resistance in human lung adenocarcinoma A549 cell. Int J Biol
Sci. 11:833–844. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han W, Yu KN, Wu LJ, Wu YC and Wang HZ:
Mechanism of protection of bystander cells by exogenous carbon
monoxide: Impaired response to damage signal of radiation-induced
bystander effect. Mutat Res. 709–710:1–6. 2011. View Article : Google Scholar
|
|
23
|
Singh A, Yashavarddhan MH, Kalita B,
Ranjan R, Bajaj S, Prakash H and Gupta ML: Podophyllotoxin and
Rutin modulates ionizing radiation-induced oxidative stress and
apoptotic cell death in mice bone marrow and spleen. Front Immunol.
8:1832017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hofer M, Hoferová Z, Dušek L, Souček K and
Gruzdev A: Hematological profile of untreated or ionizing
radiation-exposed cyclooxygenase-2-deficient mice. Physiol Res.
66:673–676. 2017.PubMed/NCBI
|
|
25
|
Ozbilgin MK, Onal T, Ozcan C, Temel M,
Aktas C, Gareveran MS, Uluer ET, Inan S and Kurtman C: Effects of
cyclooxygenase on the urothelium of the urinary bladder of mice
exposed to pelvic radiation. Anal Quant Cytopathol Histpathol.
38:103–110. 2016.PubMed/NCBI
|
|
26
|
Zuo Z, Ouyang W, Li J, Costa M and Huang
C: Cyclooxygenase-2 (COX-2) mediates arsenite inhibition of
UVB-induced cellular apoptosis in mouse epidermal Cl41 cells. Curr
Cancer Drug Targets. 12:607–616. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Clouaire T, Marnef A and Legube G: Taming
tricky DSBs: ATM on duty. DNA Repair (Amst). 56:84–91. 2017.
View Article : Google Scholar
|
|
28
|
Thompson LH: Recognition, signaling, and
repair of DNA double-strand breaks produced by ionizing radiation
in mammalian cells: The molecular choreography. Mutat Res.
751:158–246. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park SY, Kim YM and Pyo H: Gefitinib
radiosensitizes non-small cell lung cancer cells through inhibition
of ataxia telangiectasia mutated. Mol Cancer. 9:2222010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chacko T, Menon A, Majeed T, Nair SV, John
NS and Nair CKK: Mitigation of whole-body gamma radiation-induced
damages by Clerodendron infortunatum in mammalian organisms. J
Radiat Res. 58:281–291. 2017.
|
|
31
|
Kim YM and Pyo H: Cooperative enhancement
of radiosensitivity after combined treatment of
17-(allylamino)-17-demethoxygel-danamycin and celecoxib in human
lung and colon cancer cell lines. DNA Cell Biol. 31:15–29. 2012.
View Article : Google Scholar :
|
|
32
|
Dent P, Yacoub A, Fisher PB, Hagan MP and
Grant S: MAPK pathways in radiation responses. Oncogene.
22:5885–5896. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Munshi A and Ramesh R: Mitogen-activated
protein kinases and their role in radiation response. Genes Cancer.
4:401–408. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Acheva A, Schettino G and Prise KM:
Pro-inflammatory signaling in a 3D organotypic skin model after low
LET irradiation-NF-κB, COX-2 activation, and impact on cell
differentiation. Front Immunol. 8:822017. View Article : Google Scholar
|
|
35
|
Hung JH, Su IJ, Lei HY, Wang HC, Lin WC,
Chang WT, Huang W, Chang WC, Chang YS, Chen CC and Lai MD:
Endoplasmic reticulum stress stimulates the expression of
cyclooxygenase-2 through activation of NF-kappaB and pp38
mitogen-activated protein kinase. J Biol Chem. 279:46384–46392.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tessner TG, Muhale F, Schloemann S, Cohn
SM, Morrison AR and Stenson WF: Ionizing radiation up-regulates
cyclooxygenase-2 in I407 cells through p38 mitogen-activated
protein kinase. Carcinogenesis. 25:37–45. 2004. View Article : Google Scholar
|
|
37
|
Hu YP, Peng YB, Zhang YF, Wang Y, Yu WR,
Yao M and Fu XJ: Reactive oxygen species mediated prostaglandin E2
contributes to acute response of epithelial injury. Oxid Med Cell
Longev. 2017:41238542017. View Article : Google Scholar :
|