Introduction
Japanese B encephalitis is a severe central nervous
system disorder caused by an arbovirus called Japanese encephalitis
virus (JEV) (1,2). JEV belongs to the
Flaviviridae family and is maintained in a zoonotic cycle
with domestic pigs and wild birds serving as reservoirs for viral
propagation. In addition, mosquitoes such as Culex
tritaeniorhynchus, Culex vishnui, Culex gelidus,
Culex fuscocephala, Culex annulirostris, Anopheles
peditaeniatus (Leicester), Anopheles barbirostris van der
Wulp, and Anopheles subpictus can serve as vectors of
JEV (3-6). This virus has a single-stranded,
positive-sense RNA genome with an open reading frame encoding for
three structural proteins, namely precursor proteins to membrane
and envelope components, capsid proteins and seven non-structural
(NS) proteins (7).
Several different vaccines are currently available
for the prevention of Japanese B encephalitis caused by JEV
(8-15), although no specific
chemotherapeutic agents exist for this disease (16,17), which disproportionately targets
young people. Therefore, the identification of a drug candidate for
development of promising antiviral therapeutics. Previous studies
have demonstrated that the novel compound CW-33 (ethyl
2-(3′,5′-dimethylanilino)-4-oxo-4,5-dihydrofuran-3-carboxylate;
compound 01 in Fig. 1) exhibits
antiviral activity with regard to the enteroviral A71 and the JEV
T1P1 strains (18-20). The present study aimed to further
examine the development of the compound CW-33 as a drug candidate
for the prevention of infection by the JEV strain T1P1.
Computer-aided drug design can increase the odds of
developing suitable lead candidates by identifying key features of
compounds required for future drug development, whereas it can
concomitantly accelerate the process of drug development. In the
present study, the compound CW-33 and its analogues were
investigated in silico using docking simulation and
quantitative structure-activity relationship (QSAR) models. Their
antiviral activities for JEV were then evaluated. The antiviral
activities of the furanonaphthoquinone derivatives with regard to
JEV are ascribed to the inhibition of the viral RNA and the viral
protein syntheses (21). The
viral NS2B-NS3 serine protease has an important role in cytoplasmic
cleavage events that occur during the viral polyprotein maturation,
and it is responsible for cleaving the viral polyprotein at the
NS2A/NS2B, NS2B/NS3, NS3/NS4A, and NS4B/NS5 junctions (22). Molecular docking simulation of JEV
protease with compound CW-33 and its analogues was performed to
investigate the ligand-protein interaction network responsible for
the antiviral activities of these compounds. A ligand-based
approach was further applied and the two dimensional (2D)-QSAR
models were developed to determine the representative molecular
descriptors related to the antiviral activities. Finally, the
following analyses were performed: Pharmacophore modeling,
comparative force field analysis (CoMFA), and comparative
similarity index analysis (CoMSIA). These processes aimed to
develop models for the investigation of the common pharmacophore
features and to identify the relationship between five
physicochemical properties of these compounds and their
corresponding antiviral activities.
Materials and methods
Data collection
The X-ray crystal structure of the JEV protease was
downloaded from the RCSB Protein Data Bank with PDB ID 4R8T
(23). The Prepare protein
protocol in the discovery studio 2.5 (DS2.5) was employed to remove
crystal waters in the crystal structure. Furthermore, this software
was used for the repair of incomplete residues and the protonation
of the JEV protease structure that was performed with the Chemistry
at Harvard Macromolecular Mechanics (CHARMM) force field (24). This force field was able to
optimize the side-chain conformation of repaired residues. The
binding site of the JEV protease was defined from the protein
cavities that were close to the active site residues of the JEV
protease and was expanded to the volume of 288.375
Å3.
The compound CW-33 and its analogues were obtained
from the Lien laboratory (School of Pharmacy, China Medical
University, Taichung, Taiwan) (18-20) (Fig.
1 and Table I). All the 15
compounds were drawn using ChemBioOffice 2010 (Perkin Elmer,
Waltham, MA, USA), and prepared by the Prepare Ligand protocol in
DS2.5 to modify their ionization state according to the
physiological ionization setting. The efficacy of the antiviral
activities of the 15 compounds was determined using the cytopathic
effect assay for JEV (18,20).
 | Table IpIC50 values of CW-33 and
its analogues with regard to the viral cytopathic effect assay
against the JEV strain T1P1. |
Table I
pIC50 values of CW-33 and
its analogues with regard to the viral cytopathic effect assay
against the JEV strain T1P1.
| Compound | R1 | R2 | R3 |
pIC50 |
|---|
| 01 (CW-33) | - | - | - | 5.09 |
| 02 | -H | -H | -F | 4.98 |
| 03 | -H | -F | -H | <3.00 |
| 04 | -F | -H | -H | 3.94 |
| 05 | -H | -H | -OMe | 4.18 |
| 06 | -H | -OMe | -H | 3.90 |
| 07 | -OMe | - H | -H | <3.00 |
| 08 | -H | -H | -Cl | 4.30 |
| 09 | -H | -Cl | -H | 3.26 |
| 10 | -Cl | -H | -H | <3.00 |
| 11 | -H | -H | -Br | 5.49 |
| 12 | -H | -Br | -H | 5.51 |
| 13 | -Br | -H | -H | 5.70 |
| 14 | -H | -Me | -H | 3.94 |
| 15 | -H | -Et | -H | 3.91 |
Molecular docking simulation
The LigandFit protocol (25) in DS 2.5 was employed to simulate
the docking poses of each compound using a shape filter and the
Monte-Carlo ligand conformation generation (24). Subsequently, the poses were
optionally minimized with the CHARMM force field (24). Similar docking poses were filtered
using the clustering algorithm. Four different scoring functions
were employed to evaluate the docking poses -PLP1 (26), -PLP2 (27), -PMF (28), and the Dock score. The scoring
functions, -PLP1, -PLP2, and -PMF, were evaluated by the sum of the
two types of the pairwise interaction, namely H-bond and steric
interactions, between the protein and the compound. The scoring
function of the Dock score was based on a force field approximation
as follows:
Dock score=−(ligand\receptor interaction energy+ligand internal energy)
The LigPlot+ program (29) was employed to generate the 2D
ligand-protein interaction diagrams.
2D-QSAR models
The 2D-QSAR models were developed by multiple linear
regression (MLR) and support vector machine (SVM) using MATLAB
(MathWorks, Natick, MA, USA) and LibSVM (National Taiwan
University) (30), respectively.
All 15 compounds displayed in Table
II were classified as the training set (10 compounds) and the
test set (5 compounds) with their pIC50 values (-log
IC50, given in terms of molar concentration). The
genetic function approximation protocol (31) in DS2.5 was employed to determine
the suitable molecular descriptors for the prediction models.
| Table IIExperimental and predicted
pIC50 values obtained by MLR and SVM models, and scoring
functions of each complex obtained by docking simulations. |
Table II
Experimental and predicted
pIC50 values obtained by MLR and SVM models, and scoring
functions of each complex obtained by docking simulations.
| Compound |
pIC50 | pIC50
(predicted)
| Scoring functions
|
|---|
| MLR | SVM | -PLP1 | -PLP2 | -PMF | Dock score |
|---|
| 01 (CW-33) | 5.09 | 4.41 | 4.16 | 76.15 | 72.63 | 46.27 | 42.021 |
| 02 | 4.98 | 4.94 | 4.92 | 74.48 | 68.18 | 37.99 | 40.49 |
| 03a | <3.00 | 4.07 | 4.25 | 74.35 | 61.59 | 46.23 | 40.434 |
| 04 | 3.94 | 3.81 | 4.00 | 74.34 | 68.52 | 36.51 | 40.272 |
| 05 | 4.18 | 4.49 | 4.21 | 64.87 | 60.85 | 51.62 | 42.795 |
| 06a | 3.90 | 3.87 | 3.89 | 71.82 | 61.05 | 56.84 | 43.857 |
| 07a | <3.00 | 3.81 | 3.86 | 79.60 | 65.39 | 55.74 | 40.432 |
| 08 | 4.30 | 4.37 | 4.24 | 67.56 | 60.28 | 52.07 | 42.201 |
| 09 | 3.26 | 3.34 | 3.78 | 72.47 | 68.86 | 47.92 | 44.907 |
| 10a | <3.00 | 3.43 | 3.79 | 77.32 | 61.04 | 44.41 | 42.746 |
| 11 | 5.49 | 5.77 | 5.55 | 77.94 | 63.42 | 56.86 | 44.851 |
| 12a | 5.51 | 5.34 | 5.48 | 68.4 | 61.67 | 50.66 | 46.429 |
| 13 | 5.70 | 5.37 | 5.50 | 67.39 | 55.68 | 46.28 | 44.888 |
| 14 | 3.94 | 3.97 | 4.00 | 68.83 | 61.38 | 51.23 | 46.782 |
| 15 | 3.91 | 4.32 | 4.10 | 79.14 | 74.3 | 46.57 | 40.94 |
Pharmacophore model
The 3D (three dimensional)-QSAR pharmacophore
generation protocol in DS2.5 was employed to generate a
pharmacophore model using the catalyst hypoGen algorithm (32). Low-energy conformations were
generated for each compound by the FAST generation protocol. The
common pharmacophore model was then constructed by a list of four
different pharmacophore features, namely H-bond donor, H-bond
acceptor, hydrophobic, and aromatic ring interactions.
CoMFA and CoMSIA models
CoMFA (33) and
CoMSIA (34) were employed with
all 15 compounds to construct 3D-QSAR models by SYBYL-X (Certara,
Princeton, NJ, USA). CoMFA was employed to evaluate the steric and
electrostatic field descriptors with the distance-dependent
dielectric method using Lennard-Jones and Coulombic potential
energies, respectively. CoMSIA was used to evaluate five
physico-chemical properties, namely steric, electrostatic,
hydrophobic, H-bond donor, and H-bond acceptor, with a Gaussian
function based on the distance. The partial least-squares (PLS)
regression was used to obtain a linear correlation between the
biological activity values and the properties of CoMFA and
CoMSIA.
Results and Discussion
In earlier studies, scientists have indicated that
the NS2B/NS3 protease may be a suitable target for antiviral
compounds (35). In addition, a
compound obtained from a secondary metabolite of Boesenbergia
pandurata (Schult.) has been noted for its potential antiviral
activity and possible docking pose towards the JEV NS2B/NS3
protease (36). In the present
study, molecular docking simulation of JEV protease was performed
to investigate the ligand-protein interaction network responsible
for the antiviral activities, as well as QSAR models to determine
the representative molecular descriptors and common pharmacophore
features.
Molecular docking simulation
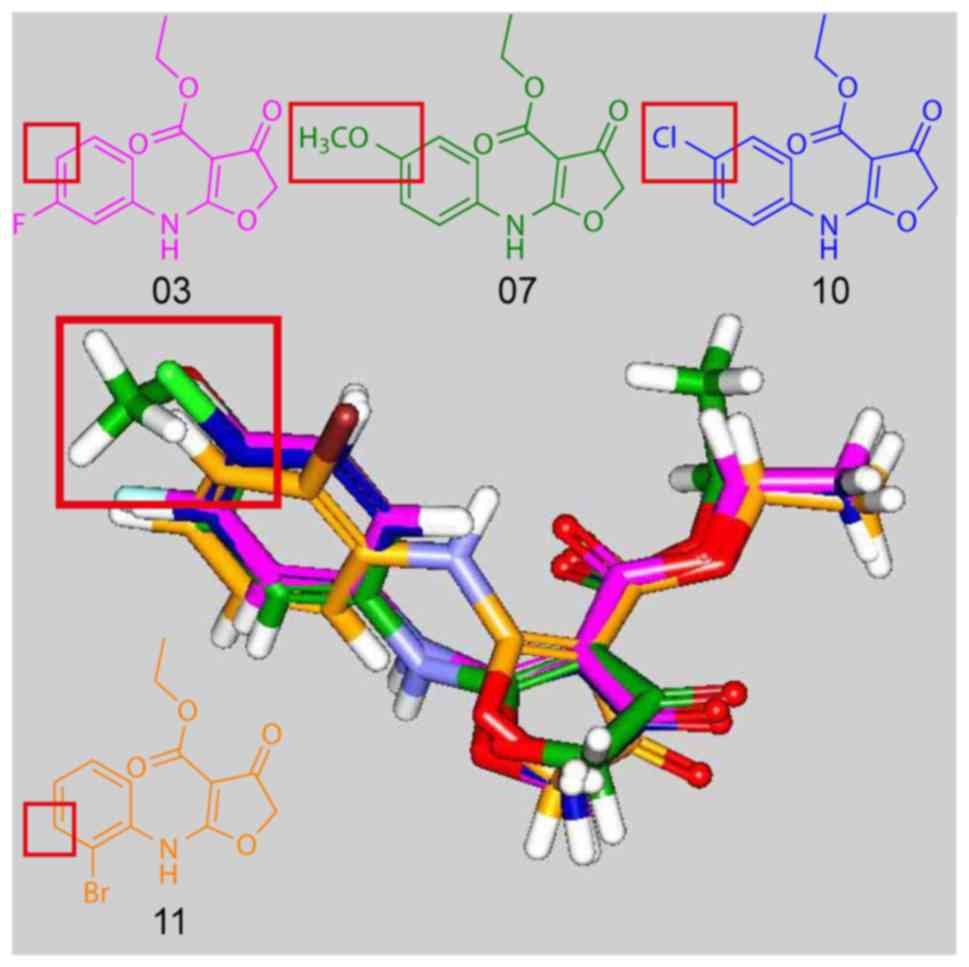
The chemical structures of compound CW-33 and its
analogues are displayed in Fig. 1
and Table I. IC50
refers to the half maximal inhibitory concentration. The results of
the docking simulations with regard to the JEV protease and the
compound CW-33 and its analogues, are reported in Table II with their corresponding
scoring values. Fig. 2 displays
the docking poses of compounds 03, 07, 10, 11. The docking poses of
compounds 03, 07, 10, which had pIC50 <3 revealed
that the amine group was rotated. This caused an alignment of the
phenyl substituents of the paramoiety of compounds 03, 07, 10 with
the meta-moiety of the phenyl substituents of compound 11. The
alignment indicated that the meta-substituent of the phenyl ring
may be more suitable for the design of active CW-33 derivatives.
The docking poses of the three compounds that exhibited optimal
biological activities, i.e. compounds 01, 09, 15, are displayed in
Fig. 3. The amine group of these
compounds contained hydrogen bonds (H-bonds) with the residues
Asn152 and Glu155, while the phenyl group established a π-cation
interaction with residue Arg76. In addition, all of the compounds
exhibited hydrophobic contacts with residues Glu74, Arg76, Trp83,
Ile147, Gly148, Asn152, Gly153, Val154, and Glu155. Finally,
compounds 01 and 15 formed additional hydrophobic contacts with
residues Phe85, Asp86, Arg87, and Leu149 due to the presence of
methyl or ethyl groups. These interactions that were either
hydrogen bonding or hydrophobic contacts, were responsible for
compound stabilization in the binding site of the enzyme.
2D-QSAR models
The genetic function approximation protocol resulted
in the identification of three representative descriptors, namely
ES_Sum_ssNH, molecular_weight, and Jurs_WNSA_2 that were used to
construct the 2D-QSAR models. The ES_Sum_ssNH descriptor is an
electrotopological state (E-state) descriptor (37,38), which denotes the E-state of the NH
group with two single bonds. It implies that the secondary amine
group is one of the most important moieties related to bioactivity.
The molecular_weight descriptor is the sum of the atomic masses of
the compounds under investigation. The Jurs_WNSA_2 descriptor
refers to the surface-weighted partial surface areas obtained by
multiplying the atomic charge-weighted positive surface area by the
total molecular solvent-accessible surface area and dividing it by
a factor of 1,000.
The MLR model was constructed with a training set of
10 compounds using the 3 representative descriptors described
above. The following linear equation was obtained:
pIC50=15.0708−6.7849*ES_Sum_ssNH+0.0671*Molecular_Weight+0.0588*Jurs_WNSA_2
The SVM model was constructed using the same
training set and representative descriptors. The test set of 5
compounds was employed to validate the accuracy of the prediction
of both 2D-QSAR models. The predicted pIC50 values
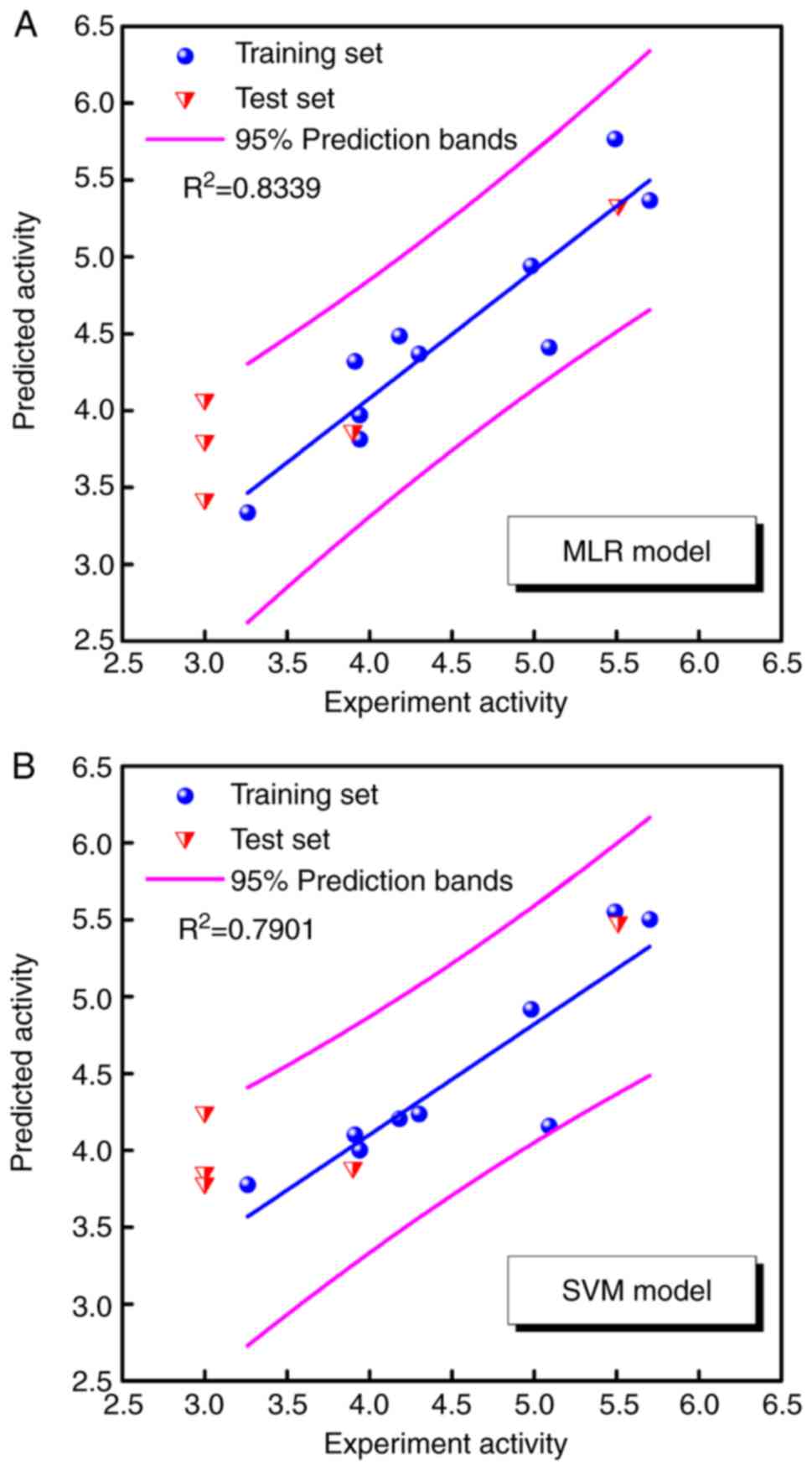
obtained by MLR and SVM models are listed in Table II. Fig. 4 illustrates the correlations
between predicted pIC50 and experimental
pIC50 in the MLR and SVM models. The square correlation
coefficients (R2) of the training set were 0.8339 and
0.7901, respectively. Both models predicted favorable bioactivities
for CW-33 and its analogues with the exception of compounds 03, 07,
and 10, which exhibited pIC50 values <3. The data
implied that the poor bioactivities of compounds 03, 07, and 10
were not associated with these three representative descriptors.
However, the three representative descriptors were associated with
the antiviral activities with regard to the JEV protease.
Pharmacophore model
The present study aimed to investigate the common
pharmacophore features based on the 3D structures of all 15
compounds. The results of the best pharmacophore hypothesis are
illustrated in Fig. 5A with the
pharmacophore features. The pharmacophore model included two H-bond
acceptor features, one aromatic ring feature, and one hydrophobic
feature. The mapping of the pharmacophore features onto compounds
01 and 15 suggested that the oxygen atoms of the carboxylate and
the 4-oxo-4,5-di-hydrofuran moieties matched the H-bond acceptors
features. The ethyl moiety matched the hydrophobic feature and the
phenyl moiety matched the aromatic ring feature (Fig. 5B and C). This was in line with the
results of the docking simulation, suggesting that the phenyl
moiety of each compound exhibited a π-cation interaction with
residue Arg76. In addition, the oxygen atoms of the carboxylate
moiety formed H-bonds with residue Asn152, and the ethyl moiety
exhibited hydrophobic contacts with residue Glu74.
CoMFA and CoMSIA models
To determine the correlation between the efficacy of
antiviral activity and the compound functional groups, the CoMFA
and CoMSIA models were constructed using all 15 compounds that were
aligned to the pharmacophore model described above. Following PLS
analysis, the CoMFA model with three components exhibited
significant steric fields (100% contribution), whereas the CoMSIA
model with four components indicated significant steric,
hydrophobic, and H-bond donor fields (15.9, 63.2 and 20.9%
contribution, respectively). The predicted pIC50 values
for the two significant CoMFA and CoMSIA models are listed in
Table III. The correlations
between predicted pIC50 vs. experimental
pIC50 values for the two models are displayed in
Fig. 6. The square correlation
coefficients (R2) were 0.7607 and 0.8234, respectively.
The pIC50 values of compounds 03, 07 and 10 were >3,
indicating that the CoMFA and CoMSIA models could partially predict
the estimation of this endpoint. Compound 03 exhibited predicted
pIC50 values of 2.72 and 2.58 in the CoMFA and CoMSIA
models, respectively. Compound 07 exhibited a predicted
pIC50 value of 2.80 in the CoMSIA model, and compound 10
demonstrated a predicted pIC50 value of 2.78 in the
CoMFA model. The data derived by these two models may explain the
poor antiviral efficacy of compounds 03, 07, and 10 and may predict
their corresponding pIC50.
| Table IIIExperimental and predicted
pIC50 values obtained by CoMFA and CoMSIA models. |
Table III
Experimental and predicted
pIC50 values obtained by CoMFA and CoMSIA models.
| Compound |
pIC50 | pIC50
(predicted)
|
|---|
| CoMFA C | oMSIA |
|---|
| 01 (CW-33) | 5.09 | 4.73 | 4.57 |
| 02 | 4.98 | 5.01 | 4.68 |
| 03 | <3.00 | 2.72 | 2.58 |
| 04 | 3.94 | 4.71 | 4.41 |
| 05 | 4.18 | 4.47 | 4.40 |
| 06 | 3.90 | 3.75 | 3.66 |
| 07 | <3.00 | 3.67 | 2.80 |
| 08 | 4.30 | 4.78 | 4.65 |
| 09 | 3.26 | 3.36 | 3.31 |
| 10 | <3.00 | 2.78 | 3.74 |
| 11 | 5.49 | 4.93 | 5.07 |
| 12 | 5.51 | 5.37 | 5.60 |
| 13 | 5.70 | 4.81 | 5.24 |
| 14 | 3.94 | 4.39 | 4.13 |
| 15 | 3.91 | 3.74 | 4.37 |
Fig. 7 illustrates
the CoMFA and CoMSIA contour maps for compound CW33, with the
favorable and unfavorable regions (80 and 20%, respectively) using
SD*coefficients for each field.
Since CW-33 and its analogues have similar chemical
structures, we focused on the contour maps of the phenyl moiety of
CW-33. In the CoMFA model, the results indicated that the favored
steric fields were in close proximity to the meta-moieties of the
phenyl substituents, whereas the disfavored steric fields were in
close proximity to the para-moieties of the phenyl substituents
(Fig. 7A). Specific favored
steric fields were identified close to the meta- and ortho-moieties
of the phenyl substituents in the CoMSIA model (Fig. 7B). The data further indicated that
the hydrophobic substituents that were beneficial to the
bioactivity were found in the meta- and para-positions of the
phenyl ring (Fig. 7C). In
contrast to the hydrophobic sites, the current model did not
provide information regarding the regions responsible for H-bonding
residing in close proximity to the phenyl moiety of CW-33 (Fig. 7D). In line with the results of
docking simulation, the data implied that the hydrophobic
substituents in the meta- and ortho-positions of the phenyl rings
were beneficial to the potency of the antiviral compounds.
In an earlier study, there was no suitable 3D
structure of NS2B protease, so the authors built the 3D model of
NS2B protein using homology modelling (36). In the present study, the X-ray
crystal structure of the JEV protease was employed from the RCSB
Protein Data Bank to increase the precision of docking simulation.
The current results indicated that the residues Glu155, Arg76, and
Glu74 have important roles in the docking of compounds. In case
that these key residues are mutant, homology modelling can be used
to generate a reliable mutant type JEV protease for docking
simulation to indicate that some compounds may change their docking
poses to interact with other residues, while others may fail in
docking. However, the effects of mutation in their bioactivities
should still be confirmed by in vitro and in vivo
experimental studies.
As previous studies have indicated that the compound
CW-33 and its analogues exhibit antiviral activity with regard to
the JEV T1P1 strain, this study aimed to indicate the key features
of compounds which may be related to their antiviral activities.
The results provided some beneficial suggestions for further
studies of synthesis of CW-33 analogues on JEV therapeutics.
Because the compounds employed to perform the QSAR models in the
present study were obtained from previous studies and the compound
CW-33 and its analogues have similar main scaffolds, these QSAR
models would be not suitable for compounds which have very
different chemical scaffold than compound CW-33. Additionally, the
docking poses of compounds in the domain of JEV protease indicated
the suitable docking poses and interactions with JEV protease and
provided some beneficial suggestions to increase the binding
interactions between compounds and JEV protease. However, the
docking simulation cannot be used to evaluate the antiviral
activities of compounds, so it will be necessary to perform in
vivo or in vitro experiments to evaluate the antiviral
activities of compounds which are synthesised in further
studies.
As suggested by docking simulation and 2D-QSAR
models, the secondary amine group is a significant moiety required
for antiviral bioactivity that can form H-bonds with residue
Glu155. The sum of the atomic masses and the charged
surface-weighted partial surface areas were also related to the
antiviral activities of the compounds towards JEV. The
pharmacophore model further suggested that the aromatic ring
features in the phenyl moieties of each compound formed π-cation
interactions with residue Arg76. In addition, the H-bond acceptor
features were notably the oxygen atoms of the carboxylate moiety
that could form a H-bond with residue Asn152. The hydrophobic
feature of the ethyl moiety exhibited hydrophobic contacts with
residues Glu74. The CoMFA and CoMSIA models indicated that the
hydrophobic substituents in the meta-moieties of the phenyl rings
were beneficial for the antiviral activity of the compounds
investigated. The results offer important insight that can be used
for further studies on JEV therapeutics.
Abbreviations:
|
JEV
|
Japanese encephalitis virus
|
|
NS
|
non-structural
|
|
QSAR
|
quantitative structure-activity
relationship
|
|
CoMFA
|
comparative force field analysis
|
|
CoMSIA
|
comparative similarity indices
analysis
|
|
H-bond
|
hydrogen bond
|
|
E-state
|
electrotopological state
|
|
MLR
|
multiple linear regression
|
|
SVM
|
support vector machine
|
|
CHARMM
|
Chemistry at Harvard Macromolecular
Mechanics
|
|
PLS
|
partial least-squares
|
Funding
This study was supported by the China Medical
University (grant no. CMU107-S-33), the Ministry of Science and
Technology of Taiwan (grant nos. MOST 105-2320-B-039-032, MOST
106-2320-B-039-011 and MOST 107-2320-B-039-058) and by the China
Medical University Hospital (grant nos. DMR-107-135, DMR-107-136,
DMR-108-108 and DMR-108-142).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
KCC, ACH, JYG, CWL and JCL conceived and designed
the experiments, performed the experiments, analyzed the data and
wrote the paper. YFL and CWL performed the cytopathic effect assay,
determined the efficacy of antiviral activity and calculated the
IC50 values. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
CWL, ACH and JCL have produced patents on CW-33.
Acknowledgments
Not applicable.
References
|
1
|
Unni SK, Růžek D, Chhatbar C, Mishra R,
Johri MK and Singh SK: Japanese encephalitis virus: From genome to
infectome. Microbes Infect. 13:312–321. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Simon LV and Kruse B: Encephalitis,
Japanese. StatPearls. StatPearls Publishing; Treasure Island, FL:
2017
|
|
3
|
van den Hurk AF, Ritchie SA and Mackenzie
JS: Ecology and geographical expansion of Japanese encephalitis
virus. Annu Rev Entomol. 54:17–35. 2009. View Article : Google Scholar
|
|
4
|
Karunaratne SH and Hemingway J:
Insecticide resistance spectra and resistance mechanisms in
populations of Japanese encephalitis vector mosquitoes, Culex
tritaeniorhynchus and Cx. gelidus, in Sri Lanka. Med Vet Entomol.
14:430–436. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thenmozhi V, Rajendran R, Ayanar K,
Manavalan R and Tyagi BK: Long-term study of Japanese encephalitis
virus infection in Anopheles subpictus in Cuddalore district, Tamil
Nadu, South India. Trop Med Int Health. 11:288–293. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar K, Arshad SS, Selvarajah GT, Abu J,
Toung OP, Abba Y, Bande F, Yasmin AR, Sharma R, Ong BL, et al:
Prevalence and risk factors of Japanese encephalitis virus (JEV) in
livestock and companion animal in high-risk areas in Malaysia. Trop
Anim Health Prod. 50:741–752. 2018. View Article : Google Scholar :
|
|
7
|
Solomon T, Ni H, Beasley DW, Ekkelenkamp
M, Cardosa MJ and Barrett AD: Origin and evolution of Japanese
encephalitis virus in southeast Asia. J Virol. 77:3091–3098. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schiøler KL, Samuel M and Wai KL: Vaccines
for preventing Japanese encephalitis. Cochrane Database Syst Rev.
CD0042632007.PubMed/NCBI
|
|
9
|
Turtle L and Driver C: Risk assessment for
Japanese encephalitis vaccination. Hum Vaccin Immunother.
14:213–217. 2018. View Article : Google Scholar :
|
|
10
|
Jelinek T, Burchard GD, Dieckmann S,
Bühler S, Paulke-Korinek M, Nothdurft HD, Reisinger E, Ahmed K,
Bosse D, Meyer S, et al: Short-term immunogenicity and safety of an
accelerated pre-exposure prophylaxis regimen with Japanese
encephalitis vaccine in combination with a rabies vaccine: A phase
III, multicenter, observer-blind study. J Travel Med. 22:225–231.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Singh A, Mitra M, Sampath G, Venugopal P,
Rao JV, Krishnamurthy B, Gupta MK, Sri Krishna S, Sudhakar B, Rao
NB, et al: A Japanese encephalitis vaccine from India induces
durable and cross-protective immunity against temporally and
spatially wide-ranging global field strains. J Infect Dis.
212:715–725. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vu TD, Nguyen QD, Tran HTA, Bosch-Castells
V, Zocchetti C and Houillon G: Immunogenicity and safety of a
single dose of a live attenuated Japanese encephalitis chimeric
virus vaccine in Vietnam: A single-arm, single-center study. Int J
Infect Dis. 66:137–142. 2018. View Article : Google Scholar
|
|
13
|
Fan YC, Lin JW, Liao SY, Chen JM, Chen YY,
Chiu HC, Shih CC, Chen CM, Chang RY, King CC, et al: Virulence of
Japanese encephalitis virus genotypes I and III, Taiwan. Emerg
Infect Dis. 23:1883–1886. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Connor B and Bunn WB: The changing
epidemiology of Japanese encephalitis and New data: The
implications for new recommendations for Japanese encephalitis
vaccine. Trop Dis Travel Med Vaccines. 3:142017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ginsburg AS, Meghani A, Halstead SB and
Yaich M: Use of the live attenuated Japanese Encephalitis vaccine
SA 14-14-2 in children: A review of safety and tolerability
studies. Hum Vaccin Immunother. 13:2222–2231. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Solomon T, Dung NM, Kneen R, Gainsborough
M, Vaughn DW and Khanh VT: Japanese encephalitis. J Neurol
Neurosurg Psychiatry. 68:405–415. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hegde NR and Gore MM: Japanese
encephalitis vaccines: Immunogenicity, protective efficacy,
effectiveness, and impact on the burden of disease. Hum Vaccin
Immunother. 13:1–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang SH, Lien JC, Chen CJ, Liu YC, Wang
CY, Ping CF, Lin YF, Huang AC and Lin CW: Antiviral activity of a
novel compound CW-33 against Japanese encephalitis virus through
inhibiting intracellular calcium overload. Int J Mol Sci.
17:E13862016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang CY, Huang AC, Hour MJ, Huang SH, Kung
SH, Chen CH, Chen IC, Chang YS, Lien JC and Lin CW: Antiviral
potential of a novel compound CW-33 against enterovirus A71 via
inhibition of viral 2A protease. Viruses. 7:3155–3171. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lien JC, Wang CY, Lai HC, Lu CY, Lin YF,
Gao GY, Chen KC, Huang AC, Huang SH and Lin CW: Structure analysis
and antiviral activity of CW-33 analogues against Japanese
encephalitis virus. Sci Rep. 8:165952018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takegami T, Simamura E, Hirai K and Koyama
J: Inhibitory effect of furanonaphthoquinone derivatives on the
replication of Japanese encephalitis virus. Antiviral Res.
37:37–45. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bera AK, Kuhn RJ and Smith JL: Functional
characterization of cis and trans activity of the Flavivirus
NS2B-NS3 protease. J Biol Chem. 282:12883–12892. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weinert T, Olieric V, Waltersperger S,
Panepucci E, Chen L, Zhang H, Zhou D, Rose J, Ebihara A, Kuramitsu
S, et al: Fast native-SAD phasing for routine macromolecular
structure determination. Nat Methods. 12:131–133. 2015. View Article : Google Scholar
|
|
24
|
Brooks BR, Bruccoleri RE, Olafson BD,
States DJ, Swaminathan S and Karplus M: CHARMM: A program for
macromolecular energy minimization and dynamics calculations. J
Comput Chem. 4:187–217. 1983. View Article : Google Scholar
|
|
25
|
Venkatachalam CM, Jiang X, Oldfield T and
Waldman M: LigandFit: A novel method for the shape-directed rapid
docking of ligands to protein active sites. J Mol Graph Model.
21:289–307. 2003. View Article : Google Scholar
|
|
26
|
Gehlhaar DK, Verkhivker GM, Rejto PA,
Sherman CJ, Fogel DB, Fogel LJ and Freer ST: Molecular recognition
of the inhibitor Ag- 1343 by Hiv-1 protease: Conformationally
flexible docking by evolutionary programming. Chem Biol. 2:317–324.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gehlhaar DK, Bouzida D and Rejto Paul A:
Reduced dimensionality in ligand-protein structure prediction:
Covalent inhibitors of serine proteases and design of site-directed
combinatorial libraries. Rational Drug Design American Chemical
Society. 292–311. 1999. View Article : Google Scholar
|
|
28
|
Muegge I and Martin YC: A general and fast
scoring function for protein- ligand interactions: A simplified
potential approach. J Med Chem. 42:791–804. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laskowski RA and Swindells MB: LigPlot+:
Multiple ligand-protein interaction diagrams for drug discovery. J
Chem Inf Model. 51:2778–2786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan RE, Chen PH and Lin CJ: Working set
selection using second order information for training support
vector machines. J Mach Learn Res. 6:1889–1918. 2005.
|
|
31
|
Rogers D and Hopfinger AJ: Application of
genetic function approximation to quantitative structure-activity
relationships and quantitative structure-property relationships. J
Chem Inf Comput Sci. 34:8541994. View Article : Google Scholar
|
|
32
|
Li H, Sutter J and Hoffman R: HypoGen: An
automated system for generating 3D predictive pharmacophore models.
Pharmacophore Perception, Development and Use in Drug Design. Guner
O: International University Line; La Jolla: 2000
|
|
33
|
Cramer RD, Patterson DE and Bunce JD:
Comparative molecular field analysis (CoMFA). 1. Effect of shape on
binding of steroids to carrier proteins. J Am Chem Soc.
110:5959–5967. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Klebe G, Abraham U and Mietzner T:
Molecular similarity indices in a comparative analysis (CoMSIA) of
drug molecules to correlate and predict their biological activity.
J Med Chem. 37:4130–4146. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang CC, Hsieh YC, Lee SJ, Wu SH, Liao CL,
Tsao CH, Chao YS, Chern JH, Wu CP and Yueh A: Novel dengue virus-
specific NS2B/NS3 protease inhibitor, BP2109, discovered by a
high-throughput screening assay. Antimicrob Agents Chemother.
55:229–238. 2011. View Article : Google Scholar
|
|
36
|
Seniya C, Mishra H, Yadav A, Sagar N,
Chaturvedi B, Uchadia K and Wadhwa G: Antiviral potential of
4-hydroxypanduratin A, secondary metabolite of Fingerroot,
Boesenbergia pandurata (Schult.), towards Japanese Encephalitis
virus NS2B/NS3 protease. Bioinformation. 9:54–60. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hall LH, Mohney B and Kier LB: The
electrotopological state: Structure information at the atomic level
for molecular graphs. J Chem Inf Comput Sci. 31:76–82. 1991.
View Article : Google Scholar
|
|
38
|
Hall LH and Kier LB: The E-state as the
basis for molecular structure space definition and structure
similarity. J Chem Inf Comput Sci. 40:784–791. 2000. View Article : Google Scholar : PubMed/NCBI
|