Introduction
Oral squamous cell carcinoma (OSCC) is one of the
most common head and neck malignancies (1), as well as one of the most common
epithelial cancers worldwide (2).
A variety of etiological factors, such as smoking and alcohol
consumption, have been associated with the development of OSCC
(3). Despite improvements and
innovations in diagnosis and treatment, the overall 5-year survival
rate of OSCC patients is <50% (4,5).
The standard treatment options for OSCC are surgery, chemotherapy
and radiotherapy, with the latter considered as an efficient
adjuvant treatment for some cases of OSCC.
Previous studies have demonstrated that tumor stem
cells are responsible for tumor metastasis (6,7),
and an increasing number of stem cell-related genes have been
proven to be involved in tumorigenesis (8,9).
MSI1 is a RNA-binding protein of the Musashi family that has been
found to be associated with certain cancers, including glioma
(10), cervical cancer (11), gastric cancer (12) and lung cancer (13,14). In addition, MSI1 appears to be a
prognostic marker for esophageal SCC (15) and glioma (16). Furthermore, MSI1 has been found to
activate the AKT (14) and Notch
(17,18) signaling pathways in certain types
of cancer; however, its role in OSCC remains unclear. The aim of
the present study was to investigate the role of MSI1 in OSCC
progression and elucidate the underlying mechanism, as well as
determine whether MSI1 acts as an oncogene in OSCC.
Materials and methods
Tissue collection
The study protocol was approved by the Ethics
Committee of the Affiliated Hospital of Nantong University
(Nantong, China). A total of 20 pairs of OSCC tissue and adjacent
healthy tissue samples were collected between March 2015 and April
2017 at the Affiliated Hospital of Nantong University. All the
patients provided written informed consent for their tissues to be
used for research purposes. None of the patients had received
radiation therapy prior to surgical resection. The collected
tissues were immediately snap-frozen in liquid nitrogen for later
use.
Cell culture, vector construction and
cell transfection
The OSCC cell lines HSC-3, Ca9-22, SAS and OSC-19
were purchased from American Type Culture Collection. The HSC-3,
SAS and OSC-19 cell lines were cultured in Dulbecco's modified
Eagle's medium (DMEM; Sigma-Aldrich; Merck KGaA) supplemented with
10% fetal bovine serum (FBS; HyClone; GE Healthcare Life Sciences),
and Ca9-22 cells were maintained in DMEM/F-12 (HyClone; GE
Healthcare Life Sciences) with 10% FBS at 37°C with 5%
CO2 in a CO2 incubator. The 293 cell line, a
vehicle for the production of adenoviral vaccines and recombinant
proteins (19), was purchased
from the Type Culture Collection of the Chinese Academy of Science
and maintained in modified Eagle's medium (MEM; HyClone; GE
Healthcare Life Sciences) supplemented with 10% FBS (HyClone; GE
Healthcare Life Sciences).
The interfering RNA for MSI1 (sh-MSI1) and negative
plasmid (sh-Ctrl) were purchased from General Biosystems Co., Ltd.
To construct the vector containing coding sequences of MSI1, cDNA
was reverse-transcribed from total RNA extracted form HSC-3 cells,
amplified with PCR with MSI1 primers and then cloned into the
pcDNA3.1 vector. Approximately 1×106 cells/per well were
seeded and grown overnight in 6-well plates. On the following day,
Lipofectamine 2000 reagent (Thermo Fisher Scientific, Inc.). was
used for transient transfection of the cells with 3.0 μg
plasmids, including sh-Ctrl, sh-MSI1, MSI1 overexpression plasmids
(MSI1) or negative empty pcDNA3.1 vector (pcDNA3.1). At 48 h
post-transfection, the cells were harvested for western blotting,
reverse transcription-quantitative pcr (RT-qPCR) analysis, flow
cytometry or other experiments at the indicated times.
For in vivo experiments, a lentiviral vector
carrying interfering RNA for MSI1, pLKD-U6-MSI1-shRNA or a negative
control vector, pLKD-U6-shRNA, and corresponding viruses
(1×108 pfu) were custom-constructed and prepared by OBiO
Technology (Shanghai) Corp., Ltd.
Luciferase reporter assay
The plasmids containing firefly luciferase reporters
and MSI1-silencing plasmids were co-transfected into 293 cells
using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.). The
cells were then lysed at 48 h after transfection and examined using
a dual-luciferase assay (Promega Corporation) according to the
manufacturer's instructions. Luciferase activity was expressed as
the ratio of firefly to Renilla luciferase activity.
CCK-8 assay and BrdU incorporation
Approximately 5,000 cells were plated in 96-well
plates with 200 μl medium per well. The cells were then
transfected as indicated with sh-MSI1 (MSI1 suppression plasmid),
negative control vector (pcDNA 3.1 or sh-Ctrl) or MSI1
overexpression plasmid (MSI1) for 48 h, after which time 2
μl CCK-8 solution (Biosharp) was added to each well and the
cells were incubated for a further 4 h. The absorbance at 450 nm
was measured using a micro-plate reader (Thermo Fisher Scientific,
Inc.).
To detect BrdU incorporation, the cells transfected
with the indicated plasmids were washed thoroughly with medium and
then cultured in fresh medium containing 10 μM BrdU
(Sigma-Aldrich; Merck KGaA) for 1 h. The cells were then allowed to
grow in BrdU-free medium for another 48 h, after which time they
were harvested for detection. The harvested cells were washed with
PBS, fixed in 70% ice-cold ethanol, resuspended in 2N HCl and
incubated for 30 min at room temperature, followed by hybridization
with a mouse monoclonal anti-BrdU antibody (cat. no. ab8152,
dilution, 1:500, Abcam) overnight at 4°C. Finally, the cells were
rinsed with PBS combined with Tween-20 and incubated with
fluorescein isothiocyanate-conjugated rabbit anti-mouse
immunoglobulin antibody (Jackson ImmunoResearch), followed by
staining with 4′,6-diamidino-2-phenylindole solution for 10 min
prior to image capture.
RT-qPCR analysis
RNA was isolated from tissues and cell lines using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
First-strand cDNA was synthesized using a TIANScript RT kit
(Tiangen Biotech). Subsequently, the MSI1 expression levels were
measured using SYBR-Green™ PCR Master Mix (Invitrogen; Thermo
Fisher Scientific, Inc.), with β-actin serving as an endogenous
control. The data were analyzed using the 2−ΔΔCq method
(20). The primer sequences for
the genes were as follows: MSI1: Forward,
5′-GATCCAGGGGTTTCGGCTTC-3′ and reverse, 5′-GAAGGCCACCTTAGGGTCAA-3′;
β-actin: Forward, 5′-GATGAGATTGGCATGGCTTT-3′ and reverse,
5′-GTCACCTTCACCGTTCCAGT-3′.
Cell invasion and migration
Transwell 24-well filters (pore size, 8 μm;
BD Biosciences) were precoated with Matrigel at 37°C for 30 min.
The cells transfected with the indicated plasmids were starved in
serum-free medium overnight, and then suspended in medium
containing 2% FBS. Approximately 20,000 cells were added to the
upper chamber of the filters, and medium containing 20% FBS was
added to the lower chamber. After incubating for 24 h, the cells on
the lower surface of the membrane were fixed with cold methanol and
stained with 0.1% crystal violet solution. For the migration assay,
the cells were plated on Transwell 24-well filters in plates
without Matrigel, and the protocol was the same as that used in the
invasion assays described above. Finally, the cells in at least
five random microscopic fields were counted and photographed.
Western blotting
Cell lysates from patient tissues and transfected
cells were prepared with radioimmunoprecipitation assay lysis
buffer (Beyotime Institute of Biotechnology), and protein
concentrations were quantified using a bicinchoninic acid assay kit
(Biosharp). Using electrophoresis on 10% sodium dodecyl
sulfate-polyacrylamide gel, a total of 30 μg protein was
separated and transferred to a polyvinylidene difluoride membrane
(EMD Millipore). The membrane was then probed with antibodies
against rabbit monoclonal pS727-STAT3 (cat. no. ab32143, dilution,
1:8,000), rabbit monoclonal STAT3 (cat. no. ab32500, dilution,
1:1,000), rabbit monoclonal c-Myc (cat. no. ab32072, dilution,
1:1,000), rabbit monoclonal cyclin D1 (cat. no. ab134175, dilution,
1:25,000), rabbit monoclonal MSI1 (cat. no. ab52865, dilution,
1:1,000), rabbit monoclonal p21 (cat. no. ab109520, dilution,
1:5,000), rabbit monoclonal p27 (cat. no. ab32034, dilution,
1:5,000) or mouse monoclonal β-actin (cat. no. ab6276, dilution,
1:6,000) at 4°C overnight. All antibodies were purchased from Abcam
Biotechnology. After washing with a mixture of Tris-buffered saline
and Tween 20, the membranes were incubated with horseradish
peroxidase-conjugated anti-mouse or anti-rabbit antibody (Santa
Cruz Biotechnology, Inc.) for 2 h at room temperature. The results
were visualized using an enhanced chemiluminescence detection
system (Bio-Rad Laboratories, Inc.), and the density of each band
was analyzed using ImageJ software (National Institutes of
Health).
Flow cytometry
For cell cycle analysis, ~2×106 cells
were harvested and fixed overnight with 70% ethanol at −20°C. After
washing twice with PBS, the cells were suspended in clean PBS with
50 μg/ml propidium iodide and 10 μg/ml RNase A for 30
min in the dark at room temperature. The cells were then analyzed
using FACStar Flow Cytometry (BD Biosciences), and the cell cycle
distribution was analyzed.
Tumor xenografts in nude mice
Five-week-old Balb/c female nude mice (Nu/Nu) were
obtained from the Laboratory Animal Center of Nantong University.
The animal experimental protocol was approved by the Institute of
Animal Care and Use Committee of Nantong University (Nantong,
China). Fresh surgical tumor tissues (F0) were collected
immediately after surgery and cut into 2-3 mm3-sized
pieces in DMEM supplemented with penicillin-streptomycin. Tumor
fragments were implanted into the right armpit of the mice. When
the tumor size reached 100-200 mm3, the samples
(referred to as F1) were cut into pieces for passaging in
vivo to create F2 xenograft tumors. When the F2 tumor size
reached 100-200 mm3, samples were collected and cut into
2-3-mm3 pieces and implanted into the right armpit of
mice to create F3. When the F3 tumor size had reached 10-20
mm3, the mice were randomly divided into three groups
(n=4 mice/group) and treated through the tail vein with different
solutions as follows: The normal saline (NS) group received 100
μl saline solution; the negative control (NC) group received
d pLKD-U6-shRNA lentivirus (1×108 pfu) in 100 μl
saline; and the last group received pLKD-U6-MSI1-shRNA lentivirus
(1×108 pfu) in 100 μl saline. Seven days later,
lentivirus administration was repeated. The tumor size and growth
rate were monitored and measured using a caliper every 5 days. The
approximate tumor volume (V) was calculated using the following
equation: V=(longest diameter x shortest
diameter2)/2.
Statistical analysis
The results are shown as the mean ± standard
deviation. The data were analyzed using the Duncan test following
an analysis of variance for multiple comparisons. Differences were
considered statistically significant when P<0.05.
Results
MSI1 is upregulated in OSCC tissues and
cell lines
To determine MSI1 expression in OSCC, RT-qPCR
analysis was conducted. The results demonstrated that the
expression of MSI1 in OSCC tissues was markedly higher compared
with that in adjacent healthy tissues (Fig. 1A). In addition, MSI1 expression in
the OSCC cell lines was increased compared with that in normal
tongue epithelial tissues, and was highest in HSC-3 followed by
Ca9-22 cells. However, as Ca9-22 cells have been contaminated with
the MSK-922 cell line, which is of head and neck SCC origin, HSC-3
cells for further experiments. (Fig.
1B-D). These data indicated that MSI1 may contribute to OSCC
progression, but the underlying mechanism requires further
investigation.
Knockdown of MSI1 inhibits OSCC cell
proliferation, invasion and migration in vitro
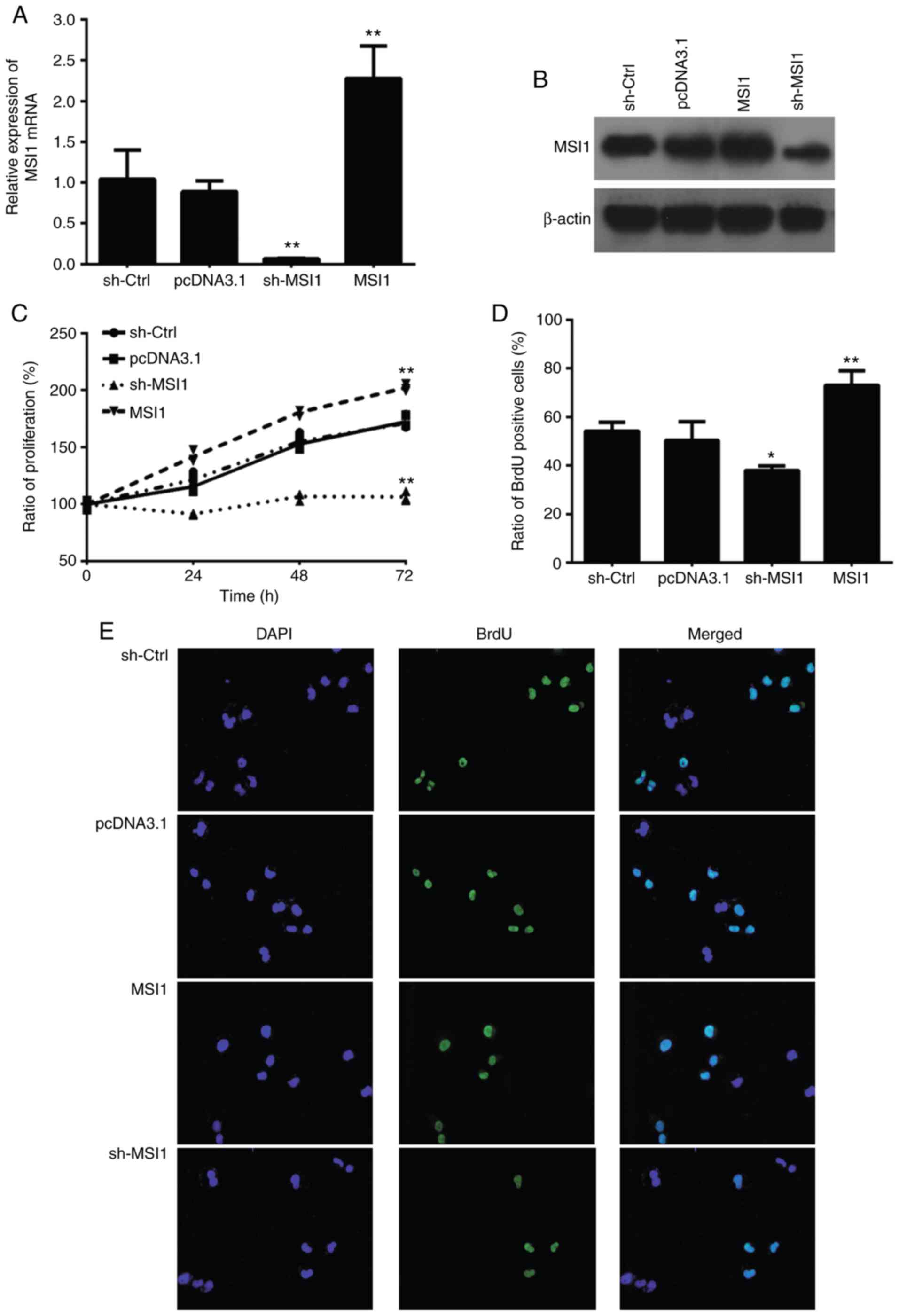
To assess the effect of MSI1 on the proliferation
and apoptosis of OSCC cells, we established HSC-3 cells with either
MSI1 suppression or MSI1 overexpression plasmids. MSI1 expression
increased in the MSI1 group, but decreased in the sh-MSI1 group, at
both the mRNA and protein levels in HSC cells (Fig. 2A and B). The CCK-8 assay then
revealed that MSI1 silencing inhibited cell proliferation compared
with control cells (Fig. 2C). In
addition, the ratio of BrdU-positive cells among HSC-3 cells with
MSI1 suppression was significantly increased compared with that in
the control cells (Fig. 2D and
E). The results also demonstrated that MSI1 suppression in OSCC
cells resulted in markedly lower invasive and migrative ability
compared with control cells (Fig.
3).
Knockdown of MSI1 inhibits the growth of
OSCC cells in vivo
To further investigate the effect of MSI1 on tumor
formation, human OSCC tissues were transplanted into nude mice. The
mice were then injected through the tail vein with saline solution
(NS), negative control (NC) or MSI1 suppression viral vector. The
increase in tumor volume was measured every 5 days. As shown in
Fig. 4, MSI1 silencing inhibited
tumor growth compared with that in the NS and NC groups. In
addition, tumor weight was lower in the sh-MSI1 group compared with
that in the NS or NC groups. Western blot analysis revealed that
the levels of MSI1 significantly decreased following injection with
the sh-MSI1 virus. These data indicate that MSI1 knockdown
inhibited the tumor-forming ability of OSCC cells in
vivo.
Knockdown of MSI1 results in OSCC cell
cycle arrest by targeting c-Myc
Cell proliferation is directly associated with
modulation of the cell cycle; therefore, the cell cycle
distribution was then analyzed using flow cytometry. As shown in
Fig. 5A and B, the proportion of
HSC-3 cells with MSI1 suppression in the S phase was markedly
higher compared with control cells.
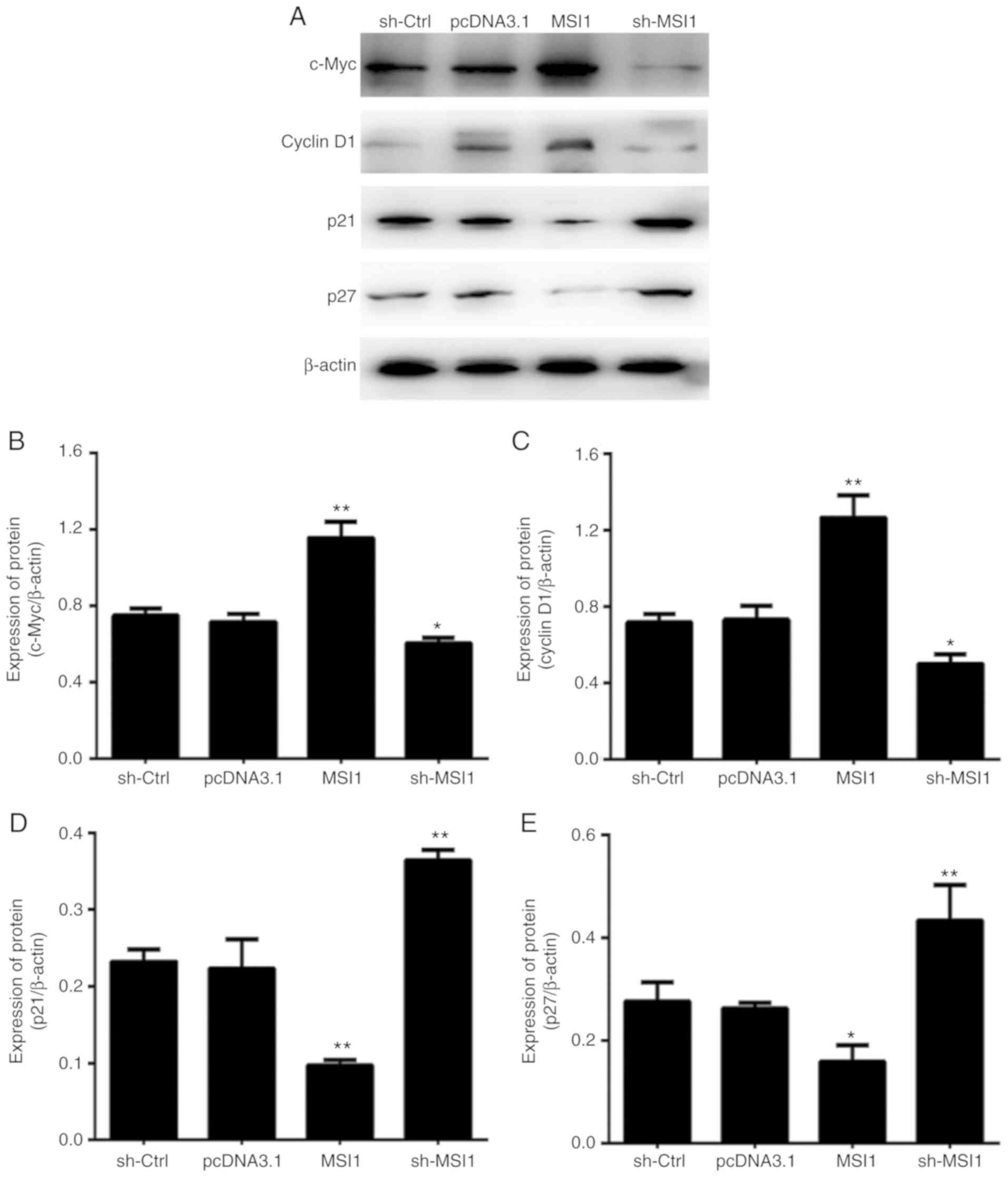
To explore the molecular mechanism by which MSI1
regulates the OSCC cell cycle, the expression levels of c-Myc,
cyclin D1, p21 and p27 were determined. Western blot analysis
revealed that the expression of both c-Myc and cyclin D1 decreased
in cells with MSI1 suppression; therefore, it was hypothesized that
MSI1 may cause cell cycle arrest in part by inhibiting the
expression of the c-Myc and cyclin D1 proteins (Fig. 6).
In addition, western blot analysis revealed that the
expression of p21 and p27 was upregulated in OSCC cells exhibiting
MSI1 suppression, which is consistent with previously reported
results (21,22). Furthermore, predictive software
(e.g., STRING, StarBase) indicated that c-Myc is also a target
downstream protein of MSI1, therefore, luciferase vectors
containing wild-type and mutant MSI1-binding sequences were next
constructed. The results revealed that the relative luciferase
activity in cells with MSI1 suppression was significantly increased
compared with that in the controls (Fig. 5C); however, no significant
difference was observed between the MSI1 suppression and control
groups in cells transfected by c-Myc mutant suppression vector.
These results indicated that MSI1 suppression promoted cell cycle
arrest, in part by binding to c-Myc.
Knockdown of MSI1 inhibits the activation
of the STAT3 signaling pathway
A large body of evidence has shown that STAT3
expression plays a key role in cancer cell survival, growth and
invasion. As shown in Fig. 7,
activation of STAT3 at Ser-727 was increased in OSCC tissues
compared with that in adjacent healthy tissues, whereas STAT3 at
Ser-727 was inhibited in HSC-3 cells following transfection with
MSI1-silencing plasmids. These results indicated that MSI1
suppressed OSCC growth in part by inhibiting the activation of
STAT3 signaling.
Discussion
Previous studies have proven that RNA-binding
proteins are crucial for cell proliferation and apoptosis during
the process of tumorigenesis (23-25). As a member of the MSI family of
RNA-binding proteins, MSI1 was found to be overexpressed in several
types of cancer, including non-small-cell lung cancer (14), osteosarcoma (21), and esophageal SCC (17).
In the present study, that the expression of MSI1
was found to be significantly increased in OSCC tissues and cell
lines, which suggested that MSI1 is likely implicated in OSCC. To
further determine the mechanism through which MSI1 regulates OSCC
cell proliferation, invasion or cell cycle arrest, MSI1 was either
silenced or overexpressed in OSCC cells (HSC-3). The results
demonstrated that MSI1 suppression significantly inhibited the
proliferation and invasive capacity of cells in vitro, and
significantly suppressed tumor growth in vivo. These data
indicated that MSI1 acts as an oncogene, promoting cell
proliferation and tumor growth. In addition, the results of the
cell cycle analysis demonstrated that MSI1 suppression induced cell
cycle arrest, which is considered to be an important factor in
regulating cancer progression. Our findings were in agreement with
those of previous studies on other types of tumors (26,27).
Based on StarBase, STRING, and previous studies,
proteins associated with cell cycle arrest (e.g., p21 and p27l) and
cell apoptosis (c-Myc) were the target RNAs of MSI1, and it has
been reported that both p21 and p27 were the downstream regulators
of MSI1 in osteosarcoma (21);
however, no studies have focused on the regulating mechanisms
connecting MSI1 and c-Myc. The present study demonstrated that the
expression of c-Myc and cyclin D1 was downregulated in HSC-3 cells
with MSI1 suppression compared with that in control cells. In
addition, luciferase assay demonstrated that MSI1 was able to
directly bind to the consensus sequence of c-Myc 3′-UTR in OSCC
cells. As a type of oncogene, c-Myc phosphoprotein can interact
with the pre-replicative complex at the S phase of the cell cycle,
and c-Myc silencing can cause cell-cycle arrest at the S phase and
promote apoptosis in cancer cells, as previously shown in gastric
cancer and esophageal SCC (28,29). Thus, MSI1 appears to cause cell
cycle arrest in part by inhibiting the expression of c-Myc.
STAT3 has been proven to be a master regulator of
several cancer hallmarks and enablers (30), and its activity is increased in
~50% of all cancers (31). In the
present study, we found that STAT3 activation at Ser-727 was
inhibited in OSCC tissues compared with adjacent healthy tissues,
which is in accordance with the results reported by Deepak et
al (31), Gkouveris et
al (32), and others. In
addition, STAT3 at Ser-727 was inhibited in HSC-3 cells following
transfection with MSI1-silencing plasmids, and MSI1 suppression
significantly decreased the invasiveness of HSC-3 cells.
Accordingly, it may be hypothesized that MSI1 inhibits the invasion
of OSCC cells by downregulating p-STAT3. As previously reported,
aberrant regulation of STAT3 in oral cancer tumorigenesis promotes
malignant behavior by regulating cell cycle progression, invasion
and resistance to standard therapies (33); however, whether MSI1 silencing can
regulate the progression of OSCC cell resistance by inhibiting
STAT3 activation signaling remains unclear. The role of the
MSI1/STAT3 axis in OSCC chemo-resistance requires elucidation in
future studies. In addition, although patient-derived xenograft
(PDX) models can retain the histological and genetic
characteristics of their donor tumors, and have been shown to be
the preferred preclinical tool in translational cancer research
compared with other conventional models, there was a limitation in
the number of mice used in the present study. We hope to improve
the accuracy of the results of in vivo experiments in future
studies.
In conclusion, the results of the present study
revealed that MSI1 is highly expressed in OSCC tissues, and that
MSI1 silencing inhibits cell proliferation and tumor formation by
cell cycle arrest, involving activation of c-Myc. In addition, MSI1
suppression inhibited the activation of STAT3 signaling, which
plays an important role in OSCC, including OSCC chemo- and
radioresistance. These findings uncovered a potential target in the
clinical treatment of OSCC, but the potential role of MSI1 in OSCC
chemoresistance requires further investigation. Furthermore, PDX
models generated from human tumor samples may retain the
histological and genetic characteristics of their tumors, and are
the preferred preclinical tool compared with conventional models
(34); however, there was still a
limitation regarding the number of mice in this study, and the
results of in vivo experiments must be verified in future
studies.
Funding
The present study was supported by the Priority
Academic Program Development of Jiangsu Higher Education
Institutions (grant no. PAPD2014-34) and the Jiang Su Province
Medical Innovation Team Project (grant no. CXTDA2017036).
Availability of data and materials
All the datasets generated and analyzed during the
present study are included in the published manuscript.
Authors' contributions
CFW and HCZ performed the experimental study and
data collection. CFW, XMF and XMS analyzed and interpreted the
data. CHW and HCZ wrote and reviewed the manuscript. XMF and YNW
revised the manuscript and provided material support. YNW conceived
and supervised the whole project. All the authors have read and
approved the final version of this manuscript.
Ethics approval and consent to
participate
The study protocol was approved by the Ethics
Committee of the Affiliated Hospital of Nantong University
(Nantong, China) and all the patients provided written informed
consent for their tissues to be used for the purposes of this
study. All animal experiments were approved by the Institute of
Animal Care and Use Committee of Nantong University (Nantong,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests to disclose.
Acknowledgments
Not applicable.
References
|
1
|
Brocklehurst PR, Baker SR and Speight PM:
Oral cancer screening: What have we learnt and what is there still
to achieve? Future Oncol. 6:299–304. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kessler P, Grabenbauer G, Leher A,
Bloch-Birkholz A, Vairaktaris E and Neukam FW: Neoadjuvant and
adjuvant therapy in patients with oral squamous cell carcinoma
long-term survival in a prospective, non-randomized study. Br J
Oral Maxillofac Surg. 46:1–5. 2008. View Article : Google Scholar
|
|
4
|
Gupta S, Kong W, Peng Y, Miao Q and
Mackillop WJ: Temporal trends in the incidence and survival of
cancers of the upper aerodigestive tract in Ontario and the United
States. Int J Cancer. 125:2159–2165. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kapoor A and Kumar S: Cancer stem cell: A
rogue responsible for tumor development and metastasis. Indian J
Cancer. 51:282–289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Jiang CQ and Fan LF: Correlation
of Musashi-1, Lgr5, and pEGFR expressions in human small intestinal
adenocarcinomas. Tumour Biol. 36:6075–6082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu XF, Yang WT, Xu R, Liu JT and Zheng
PS: Cervical cancer cells with positive Sox2 expression exhibit the
properties of cancer stem cells. PLoS One. 9:e870922014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu XS, Xi HQ and Chen L: Lgr5 is a
potential marker of colorectal carcinoma stem cells that correlates
with patient survival. World J Surg Oncol. 10:2442012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen HY, Lin LT, Wang ML, Tsai KL, Huang
PI, Yang YP, Lee YY, Chen YW, Lo WL, Lan YT, et al: Musashi-1
promotes chemoresistant granule formation by PKR/eIF2α signalling
cascade in refractory glioblastoma. Biochim Biophys Acta Mol Basis
Di. 1864:1850–1861. 2018. View Article : Google Scholar
|
|
10
|
Gong P, Wang Y, Gao Y, Gao M, Liu L, Qu P,
Jin X and Gao Q: Msi1 promotes tumor progression by
epithelial-to-mesenchymal transition in cervical cancer. Hum
Pathol. 65:53–61. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guan A, Wang H, Li X, Xie H, Wang R, Zhu Y
and Li R: MiR-330-3p inhibits gastric cancer progression through
targeting MSI1. Am J Transl Res. 8:4802–4811. 2016.PubMed/NCBI
|
|
12
|
Liu L, Qiu F, Chen J, Wu D, Nong Q, Zhou Y
and Lu J: Functional polymorphism in the MSI1 gene promoter confers
a decreased risk of lung cancer in chinese by reducing MSI1
expression. Curr Genomics. 19:375–383. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lang Y, Kong X, He C, Wang F, Liu B, Zhang
S, Ning J, Zhu K and Xu S: Musashi1 promotes non-small cell lung
carcinoma malignancy and chemoresistance via activating the Akt
signaling pathway. Cell Physiol Biochem. 44:455–466. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Qin G, Lian J, Yue D, Chen X, Nan S, Qi Y,
Li B, Cui G, Li X, Zhao S and Zhang Y: Musashi1, a potential
prognostic marker in esophageal squamous cell carcinoma. Oncol Rep.
38:1724–1732. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dahlrot RH: The prognostic value of
clinical factors and cancer stem cell-related markers in gliomas.
Dan Med J. 61:B49442014.PubMed/NCBI
|
|
16
|
Moghbeli M, Forghanifard MM, Sadrizadeh A,
Mozaffari HM, Golmakani E and Abbaszadegan MR: Role of Msi1 and
MAML1 in regulation of notch signaling pathway in patients with
esophageal squamous cell carcinoma. J Gastrointest Cancer.
46:365–369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pastò A, Serafin V, Pilotto G, Lago C,
Bellio C, Trusolino L, Bertotti A, Hoey T, Plateroti M, Esposito G,
et al: NOTCH3 signaling regulates MUSASHI-1 expression in
metastatic colorectal cancer cells. Cancer Res. 74:2106–2118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
19
|
Niu J, Zhao X, Liu Q and Yang J: Knockdown
of MSI1 inhibited the cell proliferation of human osteosarcoma
cells by targeting p21 and p27. Oncol Lett. 14:5271–5278.
2017.PubMed/NCBI
|
|
20
|
Jadhav S, Ajay AK, Trivedi P, Seematti J,
Pellegrini K, Craciun F and Vaidya VS: RNA-binding protein Musashi
Homologue 1 regulates kidney fibrosis by translational inhibition
of p21 and Numb mRNA. J Biol Chem. 291:14085–14094. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Abdelmohsen K, Srikantan S, Kuwano Y and
Gorospe M: miR-519 reduces cell proliferation by lowering
RNA-binding protein HuR levels. Proc Natl Acad Sci USA.
105:20297–20302. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Busà R, Paronetto MP, Farini D,
Pierantozzi E, Botti F, Angelini DF, Attisani F, Vespasiani G and
Sette C: The RNA-binding protein Sam68 contributes to proliferation
and survival of human prostate cancer cells. Oncogene.
26:4372–4382. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sutherland LC, Rintala-Maki ND, White RD
and Morin CD: RNA binding motif (RBM) proteins: A novel family of
apoptosis modulators? J Cell Biochem. 94:5–24. 2005. View Article : Google Scholar
|
|
24
|
Shi C and Zhang Z: miR-761 inhibits tumor
progression by targeting MSI1 in ovarian carcinoma. Tumour Biol.
37:5437–5443. 2016. View Article : Google Scholar
|
|
25
|
Akasaka Y, Saikawa Y, Fujita K, Kubota T,
Ishikawa Y, Fujimoto A, Ishii T, Okano H and Kitajima M: Expression
of a candidate marker for progenitor cells, Musashi-1, in the
prolif-erative regions of human antrum and its decreased expression
in intestinal metaplasia. Histopathology. 47:348–356. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dominguez-Sola D, Ying CY, Grandori C,
Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J and
Dalla-Favera R: Non-transcriptional control of DNA replication by
c-Myc. Nature. 448:445–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Cheng J, Xie D, Ding X, Hou H,
Chen X, Er P, Zhang F, Zhao L, Yuan Z, et al: NS1-binding protein
radiosensitizes esophageal squamous cell carcinoma by
transcriptionally suppressing c-Myc. Cancer Commun (Lond).
38:332018. View Article : Google Scholar
|
|
28
|
Mesquita FP, Pinto LC, Soares BM, de Sousa
Portilho AJ, da Silva EL, de Farias Ramos IN, Khayat AS,
Moreira-Nunes CA, Bezerra MM, de Lucas Chazin E, et al: Small
benzothiazole molecule induces apoptosis and prevents metastasis
through DNA interaction and c-MYC gene supression in diffuse-type
gastric adenocarcinoma cell line. Chem Biol Interact. 294:118–127.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Redell MS and Tweardy DJ: Targeting
transcription factors for cancer therapy. Curr Pharm Des.
11:2873–2887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Deepak Roshan VG, Sinto MS, Thomas S and
Kannan S: Cyclin D1 overexpression associated with activation of
STAT3 in oral carcinoma patients from South India. J Cancer Res
Ther. 14:403–408. 2018.PubMed/NCBI
|
|
32
|
Gkouveris I, Nikitakis N, Avgoustidis D,
Karanikou M, Rassidakis G and Sklavounou A: ERK1/2, JNK and STAT3
activation and correlation with tumor differentiation in oral SCC.
Histol Histopathol. 32:1065–1076. 2017.PubMed/NCBI
|
|
33
|
Li R, You S, Hu Z, Chen ZG, Sica GL, Khuri
FR, Curran WJ, Shin DM and Deng X: Inhibition of STAT3 by
niclosamide synergizes with erlotinib against head and neck cancer.
PLoS One. 8:e746702013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun S and Zhang Z: Patient-derived
xenograft platform of OSCC: A renewable human bio-bank for
preclinical cancer research and a new co-clinical model for
treatment optimization. Front Med. 10:104–110. 2016. View Article : Google Scholar : PubMed/NCBI
|