Introduction
At present, osteoarthritis (OA) is one of the most
common chronic and musculoskeletal diseases, and lowers patient
quality of life (1). In the
United States of America, over 26 million adults have OA and ~5.6
million cases possess lower extremity OA; ~13 million people >60
years old have radiographic OA in this region of the world
(2). Muthuri et al
(3) reported that those with
joint injury have a higher risk of developing OA than those with no
history of joint injury (4).
Additionally, genetic factors contribute to the development of OA;
the heritability of OA is >50% (5). Other factors, including obesity,
age, female gender and bone mineral density, increase the risk of
OA development (6).
OA is characterized by stiffness, joint pain and
limitation of joint movement, and occasional effusion and local
inflammation (7). Constant pain
frequently occurs in patients with late OA stage (8). Pain causes changes to the bone
structure in affected joints and disrupts the balance in mechanisms
underlying structural change, peripheral and central pain
sensitivity (9). OA patients
frequently exhibit higher levels of inflammatory cytokines, such as
interleukin (IL)-1β and tumor necrosis factor (TNF)-α (10).
MicroRNAs (miRNAs/miRs) are large family of
noncoding RNA molecules (~22 nucleotides) and are linked with
biological processes, including cellular apoptosis, proliferation
and differentiation (11,12). In addition, miRNAs bind to the
3′-untranslated region (3′-UTR) of target messenger RNA (mRNA), in
which miRNAs can block the translation of mRNA and promote the
degradation of mRNA (11). miRNAs
can regulate genes expression and signaling pathways in human
conditions, such as neuropathic pain (13). The miR-302/367 cluster is located
in the intron of the 4q25 region of chromosome 4 and is transcribed
by RNA polymerase II; the miR-302/367 cluster comprises five
members, including miR-367, -302a, -302b, -302c and -302d (14). It has been reported that the
miR-302/367 cluster serves an important role in regulating the G1-S
transition of the cell cycle (14,15). Furthermore, recent studies
supported that the miR-302/367 cluster attenuated or negatively
regulated inflammation (16,17).
To the best of our knowledge, the present study
revealed that the cartilaginous tissue of OA patients exhibited
higher miR-302d-3p expression levels compared with in normal
cartilaginous tissue. Therefore, we hypothesized that inhibition of
miR-302d-3p may prevent the progression of OA by promoting
proliferation and healing, and inhibiting inflammation in
chondrocytes in vivo.
Materials and methods
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
From March 2017 to March 2018, a total of 32
cartilaginous tissues each from patients without osteoarthritis
(OA) and with OA were obtained for analysis; in addition, 6 groups
of treated-CHON-001 cells (American Type Culture and Collection)
were acquired. The cells were treated with miR-302d-3p mimics,
miR-302d-3p mimics control, miR-302d-3p inhibitor, miR-302d-3p
inhibitor control, respectively. All patients provided written
informed consent form before the acquisition of samples. The
present study was approved by the ethics committees and health
authorities of The Second Affiliated Hospital of Henan University
of Traditional Chinese Medicine (approval. no. R201703050089). The
patients were aged between 18-60 years and were male. Patients were
included in the trial if they met clinical criteria for OA
according to The American College of Rheumatology (18). All patients with joint
deformities, rheumatoid arthritis, septic arthritis, ankylosing
spondylitis, gout, hematopoietic system, or other serious diseases
were excluded. Tissues samples were cut into pieces and RNA was
extracted using TRIzol® (Thermo Fisher Scientific, Inc.)
and a purification kit (Thermo Fisher Scientific, Inc.). Total RNA
was extracted from CHON-001 cells using TRIzol® reagent
(Thermo Fisher Scientific, Inc.). cDNAs were synthesized by cDNA
kit (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Subsequently, qPCR experiments were
performed with the SYBR Premix Ex Taq™ Real-Time PCR kit (Takara
Bio, Inc., Otsu, Japan). qPCR thermocycling conditions were:
Initial denaturation at 95°C for 1 min, followed by 50 cycle of
95°C for 30 sec, 55°C for 45 sec and 72°C for 35 sec.
The sequence of primers employed for qPCR were
synthesized by Sangon Biotech Co., Ltd. (Table I). Data from qPCR were analyzed
with the 2−∆∆Cq method (19). miR-302d-3p or Unc-51-like kinase 1
(ULK1) mRNA expression from the tissue of one patient tissue
(without OA) was selected as the control. U6 and GAPDH were used as
internal controls.
 | Table ISequences of primers employed for
reverse transcription-quantitative polymerase chain reaction. |
Table I
Sequences of primers employed for
reverse transcription-quantitative polymerase chain reaction.
| Primer | Sequence
(5′-3′) |
|---|
| miR-302d-3p | F:
GCGTAAGTGCTTCCATGTTTGTGTGT |
| U6 | F:
CGGGTTTGTTTTGCATTTCT |
| R:
AGTCCCAGCATGAACAGCTT |
| ULK1 | F:
CCAGAGCAACATGATGG |
| R:
CCTTCCCGTCGTAGTGCT |
| GAPDH | F:
CAGCCTCAAGATCATCAGCA |
| R:
TGTGGTCATGAGTCCTTCCA |
Cell culture and transfection
CHON-001 cells are derived from human cartilage and
were obtained from the American Type Culture and Collection; the
cell line was used to investigate chondrocyte function in the
present study (20). CHON-001,
the human chondrocyte cell line, was cultured in high-glucose
Dulbecco's modified Eagle's medium (DMEM; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 0.1 mg/ml G-418 (Gibco; Thermo
Fisher Scientific, Inc.) and 10% fetal bovine serum (FBS; Gibco;
Thermo Fisher Scientific, Inc.) at 37°C at 5% CO2. in an
incubator (Thermo Fisher Scientific, Inc.). The culture medium was
denoted as complete medium.
CHON-001 cells were seeded in a 6-well plate
(Corning Inc.), and miR-302d-3p mimics or inhibitors mixed with
Lipofectamine® (Invitrogen; Thermo Fisher Scientific,
Inc.) and DMEM; cells were incubated cells for 3 h at 37°C.
Complete medium was added to cultured cells for 48 h at 37°C after
3 h incubation. The concentration of the mimics, inhibitors and
siRNAs were 50 nM. The transfection reagent was used in accordance
with the manufacturer's protocols. miR-302d-3p mimics, miR-302d-3p
mimics control, miR-302d-3p inhibitor, miR-302d-3p inhibitor
control, small interfering RNA (si)-negative control (NC) and
siULK1 were synthesized by Sigma-Aldrich (Merck KGaA). The
sequences of siRNA, mimics and inhibitors used in the present study
were as follows: siULK1: 5′-GCA CAG AGA CCG TGG G CA A-3′;
miR-302d-3p mimics: 5′-UAA GUG CUU CCA UGU UUG AGU GU-3′;
miR-302d-3p inhibitor: 5′-ACA CUC AAA CAU GGA AGC ACU UA-3′;
NC-mimics: 5′-UUC UCC GAA CGU GUC ACG UTT-3′; NC-inhibitor: 5′-CAG
UAC UUU UGU GUA GUA CAA-3′. Following transfection for 48 h, the
cells were harvested and used for the subsequent experiments.
Nontransfected cells were used as blank controls.
To further investigate the mechanism of miR-302d-3p
on the proliferation and migration of chondrocytes, the cells were
divided into 6 groups: Blank group (nontransfected cells), siNC
group (cells were transfected with siNC); siULK1 group (cells were
transfected with siULK1); inhibitors group (cells were transfected
with miR-302d-3p inhibitors); inhibitors + siNC (cells were
transfected with miR-302d-3p inhibitors and siNC) and inhibitors +
siULK1 group (cells were transfected with miR-302d-3p inhibitors
and siULK1).
Cell viability
CHON-001 cells (2×103 cells/well) were
seeded into a 96-well plate (Corning Inc.) for 24,48 and 72 h
following transfection. Cell Counting Kit-8 solution (CCK-8;
Sigma-Aldrich; Merck KGaA) was diluted with DMEM (Gibco; Thermo
Fisher Scientific, Inc.) (1:9). 100 µl CCK-8 mixture was
added to cells, which were incubated for 1 h after the medium was
discarded. The absorbance was detected at a wavelength of 450 nm
using a Multiskan spectrophotometer.
Colony formation assay
A colony formation assay was performed to evaluate
cell proliferation (21).
CHON-001 cells were digested and resuspended into single cell
suspension using 0.25% trypsin-EDTA (Gibco; Thermo Fisher
Scientific, Inc.) and complete medium after the cells were treated
as aforementioned. The cells were seeded into 35 mm culture dishes
(Corning Inc.; 200 purified cells in each dish) and incubated at
37°C in a 5% CO2. incubator (Thermo Fisher Scientific,
Inc.), G418 (700 µg/ml; Abcam) was added to the medium and
mixed to detect positive cell clones for 14 days until visible cell
clones emerged. Fresh medium was replaced every 3 days. PBS was
used to wash the cells three times after the complete medium was
discarded. The cells were fixed with 4% paraformaldehyde (Beijing
Solarbio Science & Technology, Co. Ltd.) for 15 min at room
temperature and stained with Giemsa (Beijing Solarbio Science &
Technology, Co. Ltd.) for 10-30 min at room temperature. Then, the
cells were washed with PBS three times again, after which the
number of cell colonies was calculated.
Apoptosis analysis
CHON-001 cells were digested with 0.25% trypsin-EDTA
and resuspended in FBS-free DMEM after cells were treated as
aforementioned for 48 h. An apoptosis detection kit with Annexin
V-fluorescein isothiocyanate and propidium iodide (Invitrogen;
Thermo Fisher Scientific, Inc.) was employed according to the
manufacturer's protocols. The BD FACSCanto flow cytometer (BD
Biosciences) was used to analyze apoptotic cells, analysis of data
was performed using the FACSDiva software version 6.1.2 (BD
Biosciences).
Scratch-wound assay
A scratch-wound assay can be applied to evaluate
cell migration (22). CHON-001
cells were resuspended in complete medium with 1% FBS (Gibco;
Thermo Fisher Scientific, Inc.) after the cells were treated as
aforementioned for 48 h. Then, the cells were seeded in a 35-mm
plate (Corning Inc.) at a density of 5×103
cells/cm2. A sterile 200 µl pipette tips
(Sigma-Aldrich; Merck KGaA) were used to scratch the cell
monolayer, and the cells were washed with PBS. The media was
replaced with complete medium containing 1% FBS once every 12 h.
After 48 h, the scratches were observed under a light microscope
(Olympus Corporation; magnification, ×100). The distances are
different between groups at time 0 h were calculated.
Dual-luciferase reporter assay
TargetScan7.2 (http://www.targetscan.org/vert_72/) was used to
predict the target gene of miR-302d-3p, and the dual luciferase
reporter assay was used to confirm the findings. The wild-type (wt)
or mutant (mut) ULK1 3′-UTR was cloned into psi-CHECK-2 (Promega
Corporation) according to the manufacturer's instructions. Cells
were transfected with miR-302d-3p mimics or miRNA control for 48 h.
The Luciferase assay reagent II (100 µl) and 1X
Stop&Glo® reagent (100 µl; Promega
Corporation) were added to the cells, and luciferase activities
were detected using the GloMax® Discover Multimode
Microplate Reader (cat. no. GM3000; Promega Corporation) according
to the manufacturer's instructions. Luciferase activity was
normalized to Renilla luciferase activity.
Western blotting
Total protein was extracted from CHON-001 cells and
tissues with cell extraction kit (Thermo Scientific, Waltham, MA,
USA) by centrifuging samples at 4°C, 6,000 × g for 10 min. A BCA
kit was used to determine the concentration of extracted protein.
Total protein (20 µg) and a pre-stained protein ladder
(Thermo Fisher Scientific, Inc.) were separated by 10% SDS-PAGE and
transferred to polyvinylidene difluoride membranes (Sigma-Aldrich;
Merck KGaA). Membranes were stained with 1X ponceau-S (Beijing
Solarbio Science & Technology, Co. Ltd.) following transfer to
ensure consistent loading of total protein per lane. The protein
membranes were blocked with 5% bovine serum albumin (Sigma-Aldrich;
Merck KGaA) at room temperature. Primary antibodies (Table II; Cell Signaling Technologies,
Inc.) were applied and membranes were incubated at 4°C overnight.
Secondary antibodies (cat. nos. 7074 and 7076; Cell Signaling
Technologies, Inc.) were applied for 2 h at room temperature.
Primary and secondary antibodies were diluted with TBST with
Tween-20, as specified by the supplier. An ECL kit (Sigma-Aldrich;
Merck KGaA) was employed to visualize proteins. Stains were
developed with X-ray film (Fuji, Tokyo, Japan). The densitometry
was performed using the Bio-Rad ChemiDoc system with Image Lab
software version 6.0 (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
| Table IIPrimary antibodies employed for
western blotting. |
Table II
Primary antibodies employed for
western blotting.
| Antibody | Cat. no. | Target protein
weight (kDa) |
|---|
| ULK1 | 8,054 | 150 |
| IκBα | 4,814 | 39 |
| p-IκBα | 9,246 | 40 |
| p65 | 8,242 | 65 |
| p-p65 | 3,039 | 65 |
| GAPDH | 4,292 | 37 |
Statistical analysis
The aforementioned experiments were independently
repeated at least three times. The results were presented as the
mean ± standard deviation. Analysis was conducted with one way
ANOVA using SPSS 21.0 (IBM Corp.) followed by Tukey's Honest
Significant Difference post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression of miR-302d-3p and ULK1 in
patient samples, and the effects of miR-302d-3p inhibitor or mimics
on CHON-001 cell proliferation
Cartilaginous tissue of patients with OA had
significantly higher miR-302d-3p expression levels compared with in
normal cartilaginous tissue (Fig.
1A). Additionally, significantly lower expression levels of
ULK1 were detected in the cartilaginous tissue of patients with OA
(Fig. 1B-D). Transfection of
cells with miR-302d-3p mimics resulted in a significant increase in
miR-302d-3p expression (Fig. 1E)
and significantly decreased CHON-001 cell proliferation compared
with in the blank control (Fig.
1F). On the contrary, miR-302d-3p inhibitor decreased
miR-302d-3p expression and promoted CHON-001 cell proliferation
compared with cells transfected with miR-302d-3p mimics (Fig. 1F).
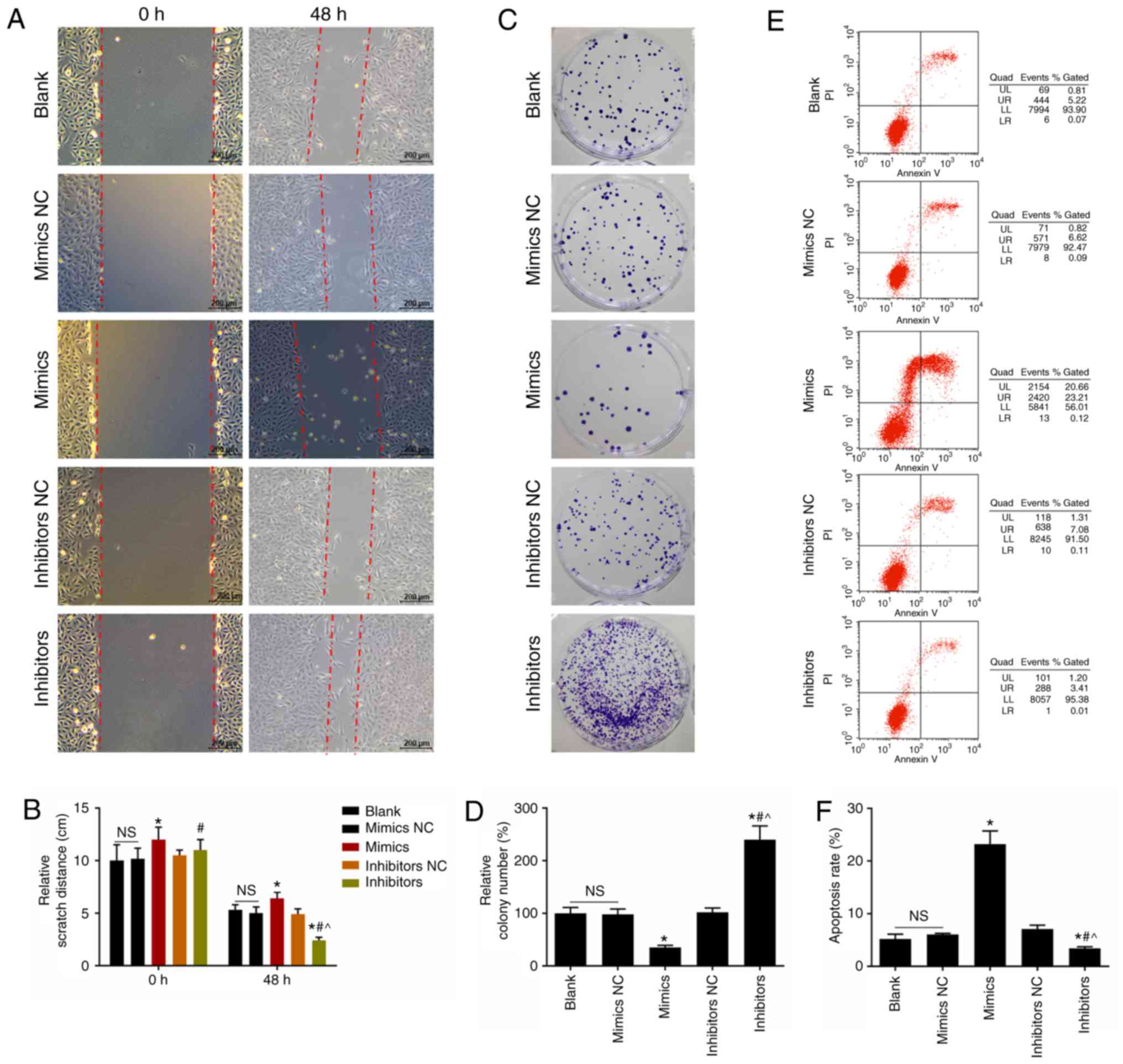
Effects of miR-302d-3p inhibitor and
mimics on CHON-001 cell migration, colony number and apoptosis
miR-302d-3p inhibitor significantly reduced the
scratch distance and promoted CHON-001 cell migration compared with
the nontransfected control and miR-302d-3p mimics groups (Fig. 2A-B). The miR-302d-3p mimics group
had long scratch distance and lower ability of migration in
CHON-001 cells (Fig. 2A and B).
Cells transfected with miR-302d-3p inhibitor exhibited a
significant increase in colony number than the miR-302d-3p mimics
group, which suggested that miR-302d-3p downregulation promoted the
proliferation of CHON-001 cells (Fig.
2C and D). miR-302d-3p mimics significantly promoted apoptosis
compared with nontransfected cells, but was suppressed in response
to miR-302d-3p inhibitor transfection (Fig. 2E and F).
siULK1 suppresses CHON-001 cell
proliferation
TargetScan7.2 was used to predict target genes of
ULK1, which revealed miR-302d-3p as a potential target (Fig. 3A). This segment of the ULK1-3′UTR
sequence (AGCACUU) is complementary to the miR-302d-3p wt sequence
(UCGUGAA). The results of the dual luciferase report assay showed
that miR-302d-3p mimics significantly decreased the luciferase
activity of psi-CHECK-2 compared with the blank group, which
supported that miR-302d-3p could bind to the 3′-UTR of ULK1
(Fig. 3B). Furthermore, siULK1
significantly inhibited ULK1 expression, while miR-302d-3p
inhibitor increased ULK1 expression (Fig. 3C). siULK1 significantly inhibited
the proliferation of CHON-001 cells compared with the inhibitor and
siNC group, and miR-302d-3p inhibitor induced CHON-001 cell
proliferation (Fig. 3D).
siULK1 suppresses CHON-001 cell migration
and colony number
The siULK1 group and miR-302d-3p inhibitor + siULK1
group had longer scratch distances compared that other groups,
which suggested that siULK1 inhibited CHON-001 cell migration
(Fig. 4A and B). Compared with
the blank group, siULK1 significantly decreased CHON-001 cell
colony number, whereas this was promoted by miR-302d-3p inhibitor,
indicating that siULK1 reduced cell proliferation (Fig. 4C and D). In addition, siULK1
significantly promoted cell apoptosis compared with the
nontransfected cell group (Fig. 4E
and F).
Effects of miR-302d-3p inhibitor and
siULK1 on the expression of phosphorylated (p)-IκBα, IκBα, p-p65
and p65 in CHON-001 cells
siULK1 significantly promoted the protein expression
of p-IκBα and p-p65 compared with nontransfected cells, which
supported that ULK1 knockdown may be associated with the activation
of IκBα and p65 in CHON-001 cells (Fig. 5A and B). miR-302d-3p inhibitor
significantly suppressed the expression levels of p-IκBα and p-p65
compared with the nontransfected cell group, which indicated that
miR-302d-3p inhibitor suppressed the activation of IκBα and p65 in
CHON-001 cells (Fig. 5A and
B).
 | Figure 5Effects of siULK1 and miR-302d-3p
inhibitor on the expression of p-IκBα, IκBα, p-p65 and p65 in
CHON-001 cells. (A and B) Western blotting for the detection of
p-IκBα, IκBα, p-p65 and p65 protein expression after transfection
with siULK1, miR-302d-3p inhibitor alone or in combination for 48
h. The values were presented as the mean ± standard deviation; data
were analyzed by one way ANOVA. *P<0.05 vs. Blank
group; ^P<0.05 vs. inhibitor + siNC group and
#P<0.05 vs. siULK1 group. miR, microRNA; NC, negative
control; p, phosphorylated; si, small interfering RNA; ULK1,
Unc-51-like kinase 1. |
Discussion
ULK1 is serine/threonine protein kinase (STK) and
has five ULK1 homologs, including ULK1, ULK2, ULK3, ULK4 and STK36;
ULK1 and ULK2 have been reported to be involved in the mechanism of
autophagy (23,24). A recent report showed that
activation of ULK4 inhibited autophagy and inflammatory responses
(25). In our study, the
cartilaginous tissue of OA patients had lower ULK1 expression
levels, but miR-302d-3p was upregulated. Articular cartilage
supports joint lubrication, and chondrocytes maintain the balance
between the degradation and synthesis of the extracellular matrix
(26). Musumeci et al
supported that chondrocyte apoptosis was positively associated with
cartilage destruction in patients with OA (27). Therefore, we explored the effects
of miR-302d-3p inhibitor on chondrocyte proliferation and apoptosis
in vitro. The results indicated that miR-302d-3p inhibitor
inhibited the apoptosis and promoted the proliferation of
chondrocytes. On the contrary, miR-302d-3p mimics promoted
apoptosis and inhibited proliferation.
Cell migration serves a critical role in biological
processes, including immune responses, tissue regeneration, wound
healing and cancer metastasis (28). A scratch-wound assay is one simple
method to measure cell migration by means of cellular movement
(29). Chondrocyte migration is
required for the repair of cartilage (30). Cartilage injury leads to the
development of degenerative joint diseases such as OA (31). miR-302d-3p inhibitor promoted
chondrocyte migration, but was inhibited in response to miR-302d-3p
mimics.
We used TargetScan7.2 to predict the target gene of
miR-302d-3p, and a dual luciferase reporter assay was conducted.
Our results revealed that ULK1 was one target gene of miR-302d-3p;
RT-qPCR and western blotting verified that miR-302d-3p inhibitor
increased ULK1 expression and siULK1 decreased ULK1 expression.
Furthermore, siULK1 inhibited the proliferation and migration of
chondrocytes, and miR-302d-3p inhibitor promoted chondrocyte
proliferation.
p65/RelA is a subunit of nuclear factor (NF)-κB and
is regarded as an important member of the NF-κB pathway (32). IL-1 and TNF-α are well regarded as
pro-inflammatory factors and are promptly released in injured
tissues (33). A study has
suggested that inflammatory factors, such as TNF-α and chemokines,
can be induced by NF-κB activation in chondrocytes and synovial
cells (34). NF-κB is activated
by multifarious stimulation, including Toll-like receptors, growth
factor receptors, oxidation and genotoxic stress (35). IκB binds NF-κB dimers in the
cytoplasm, preventing the NF-κB proteins from translocating to the
nucleus to regulate gene expression; the IκB proteins include:
IκBα, IκBβ and IκBε (36).
Therefore, the induction of NF-κB may depend on the phosphorylation
of IκBs (37). siULK1 promoted
the phosphorylation of IκBα and p65, while miR-302d-3p inhibitor
suppressed phosphorylation of IκBα and p65 in chondrocytes, which
indicated that miR-302d-3p inhibitor decreased chondrocyte
inflammation.
In conclusion, we reported upregulated miR-302d-3p
and decreased ULK1 mRNA expression levels in the cartilaginous
tissue of OA patients. Additionally, inhibition of miR-302d-3p
promoted the proliferation and migration, and inhibited the
apoptosis of chondrocytes, suppressing inflammation. This may be
due to the upregulated expression of ULK1. Inhibition of ULK1 had
adverse effects compared with inhibition of miR-302d-3p in
chondrocytes. Thus, downregulation of miR-302d-3p or upregulation
of ULK1 may be considered as potential therapeutic strategies for
preventing and treating OA; however, further investigation using
gene knock-out models and miR-302d-3p inhibitor treatment in
vivo is required.
Funding
No funding was received.
Availability of data and materials
The analyzed datasets generated during the study are
available from the corresponding author on reasonable request.
Authors' contributions
Substantial contributions to conception and design:
SW, YZhe and ZH; data acquisition, data analysis and
interpretation: ZW, YZha and LW; drafting the article or critically
revising it for important intellectual content: LW, SW, YZhe.
Agreement to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of the work are
appropriately investigated and resolved: YZhe, YZha and ZH. All
authors approved the final version to be published.
Ethics approval and consent to
participate
The present study was approved by the ethics
committees and health authorities of The Second Affiliated Hospital
of Henan University of Traditional Chinese Medicine (approval. no.
R201703050089). All procedures performed in studies involving human
participants were conducted in accordance with the ethical
standards of the institutional and/or national research committee,
and with the 1964 Helsinki declaration and its later amendments or
comparable ethical standards. Written informed consent was obtained
from all patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no conflicts of interest.
Abbreviations:
|
ULK1
|
Unc-51-like kinase 1
|
|
OA
|
osteoarthritis
|
|
RT-qPCR
|
reverse transcription-quantitative
polymerase chain reaction
|
|
miRNAs
|
microRNAs
|
|
STK
|
serine/threonine protein kinase
|
Acknowledgments
Not applicable.
References
|
1
|
Salaffi F, Carotti M, Stancati A and
Grassi W: Health-related quality of life in older adults with
symptomatic hip and knee osteoarthritis: A comparison with matched
healthy controls. Aging Clin Exp Res. 17:255–263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brown TD, Johnston RC, Saltzman CL, Marsh
JL and Buckwalter JA: Posttraumatic osteoarthritis: A first
estimate of incidence, prevalence, and burden of disease. J Orthop
Trauma. 20:739–744. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Muthuri SG, McWilliams DF, Doherty M and
Zhang W: History of knee injuries and knee osteoarthritis: A
meta-analysis of observational studies. Osteoarthritis Cartilage.
19:1286–1293. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thomas AC, Hubbard-Turner T, Wikstrom EA
and Palmieri-Smith RM: Epidemiology of posttraumatic
osteoarthritis. J Athl Train. 52:491–496. 2017. View Article : Google Scholar :
|
|
5
|
Spector TD and MacGregor AJ: Risk factors
for osteoarthritis: Genetics. Osteoarthritis Cartilage. 12(Suppl
A): pp. S39–S44. 2004, View Article : Google Scholar
|
|
6
|
Litwic A, Edwards MH, Dennison EM and
Cooper C: Epidemiology and burden of osteoarthritis. Br Med Bull.
105:185–199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Woolf AD: Driving musculoskeletal health
for Europe: EUMUSC. NET Reumatismo. 63:1–4. 2011.
|
|
8
|
Felson DT: Developments in the clinical
understanding of osteoarthritis. Arthritis Res Ther. 11:2032009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pereira D, Ramos E and Branco J:
Osteoarthritis. Acta Med Port. 28:99–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shen J, Abu-Amer Y, O'Keefe RJ and
McAlinden A: Inflammation and epigenetic regulation in
osteoarthritis. Connect Tissue Res. 58:49–63. 2017. View Article : Google Scholar :
|
|
11
|
Su LC, Huang AF, Jia H, Liu Y and Xu WD:
Role of microRNA-155 in rheumatoid arthritis. Int J Rheum Dis.
20:1631–1637. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Acunzo M and Croce CM: MicroRNA in cancer
and Cachexia-A mini-review. J Infect Dis. 212(Suppl 1): pp.
S74–S77. 2015, View Article : Google Scholar :
|
|
13
|
Jiangpan P, Qingsheng M, Zhiwen Y and Tao
Z: Emerging role of microRNA in neuropathic pain. Curr Drug Metab.
17:336–344. 2016. View Article : Google Scholar
|
|
14
|
Gao Z, Zhu X and Dou Y: The miR-302/367
cluster: A comprehensive update on its evolution and functions.
Open Biol. 5:1501382015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Baskerville S, Shenoy A, Babiarz
JE, Baehner L and Blelloch R: Embryonic stem cell-specific
microRNAs regulate the G1-S transition and promote rapid
proliferation. Nat Genet. 40:1478–1483. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiao L, Jiang L, Hu Q and Li Y: MiR-302e
attenuates allergic inflammation in vitro model by targeting RelA.
Biosci Rep. 38:BSR201800252018. View Article : Google Scholar
|
|
17
|
Ma T, Liu X, Cen Z, Xin C, Guo M, Zou C,
Song W, Xie R, Wang K, Zhou H, et al: MicroRNA-302b negatively
regulates IL-1beta production in response to MSU crystals by
targeting IRAK4 and EphA2. Arthritis Res Ther. 20:342018.
View Article : Google Scholar
|
|
18
|
Altman R, Asch E, Bloch D, Bole G,
Borenstein D, Brandt K, Christy W, Cooke TD, Greenwald R and
Hochberg M: Development of criteria for the classification and
reporting of osteoarthritis. Classification of osteoarthritis of
the knee. Diagnostic and Therapeutic Criteria Committee of the
American Rheumatism Association. Arthritis Rheum. 29:1039–1049.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Lim HD, Kim YS, Ko SH, Yoon IJ, Cho SG,
Chun YH, Choi BJ and Kim EC: Cytoprotective and anti-inflammatory
effects of melatonin in hydrogen peroxide-stimulated CHON-001 human
chondrocyte cell line and rabbit model of osteoarthritis via the
SIRT1 pathway. J Pineal Res. 53:225–237. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hu W, Zhang W, Li F, Guo F and Chen A:
miR-139 is up-regulated in osteoarthritis and inhibits chondrocyte
proliferation and migration possibly via suppressing EIF4G2 and
IGF1R. Biochem Biophys Res Commun. 474:296–302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Justus CR, Leffler N, Ruiz-Echevarria M
and Yang LV: In vitro cell migration and invasion assays. J Vis
Exp. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zachari M and Ganley IG: The mammalian
ULK1 complex and autophagy initiation. Essays Biochem. 61:585–596.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin MG and Hurley JH: Structure and
function of the ULK1 complex in autophagy. Curr Opin Cell Biol.
39:61–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He Y, She H, Zhang T, Xu H, Cheng L, Yepes
M, Zhao Y and Mao Z: p38 MAPK inhibits autophagy and promotes
microglial inflammatory responses by phosphorylating ULK1. J Cell
Biol. 217:315–328. 2018. View Article : Google Scholar :
|
|
26
|
Musumeci G, Castrogiovanni P, Trovato FM,
Weinberg AM, Al-Wasiyah MK, Alqahtani MH and Mobasheri A:
Biomarkers of chondrocyte apoptosis and autophagy in
osteoarthritis. Int J Mol Sci. 16:20560–20575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Musumeci G, Aiello FC, Szychlinska MA, Di
Rosa M, Castrogiovanni P and Mobasheri A: Osteoarthritis in the
XXIst century: Risk factors and behaviours that influence disease
onset and progression. Int J Mol Sci. 16:6093–6112. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Park JS, Rhau B, Hermann A, McNally KA,
Zhou C, Gong D, Weiner OD, Conklin BR, Onuffer J and Lim WA:
Synthetic control of mammalian-cell motility by engineering
chemotaxis to an orthogonal bioinert chemical signal. Proc Natl
Acad Sci USA. 111:5896–5901. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cory G: Scratch-wound assay. Methods Mol
Biol. 769:25–30. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Onuora S: Regenerative medicine. PBMCs
stimulate chondrocyte migration and cartilage repair. Nat Rev
Rheumatol. 11:5632015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jiang Y and Tuan RS: Origin and function
of cartilage stem/progenitor cells in osteoarthritis. Nat Rev
Rheumatol. 11:206–212. 2015. View Article : Google Scholar
|
|
32
|
Chen S, Jiang S, Zheng W, Tu B, Liu S,
Ruan H and Fan C: RelA/p65 inhibition prevents tendon adhesion by
modulating inflammation, cell proliferation, and apoptosis. Cell
Death Dis. 8:pp. e27102017, View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol. 1:pp.
a0016512009, View Article : Google Scholar
|
|
34
|
Malfait AM: Osteoarthritis year in review
2015: Biology. Osteoarthritis Cartilage. 24:21–26. 2016. View Article : Google Scholar
|
|
35
|
Christian F, Smith EL and Carmody RJ: The
regulation of NF-kappaB subunits by phosphorylation. Cells.
5:E122016. View Article : Google Scholar
|
|
36
|
Fernandez G, Zaikos TD, Khan SZ, Jacobi
AM, Behlke MA and Zeichner SL: Targeting IkappaB proteins for HIV
latency activation: The role of individual IkappaB and NF-kappaB
proteins. J Virol. 87:3966–3978. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hinz M and Scheidereit C: The IkappaB
kinase complex in NF-kappaB regulation and beyond. EMBO Rep.
15:46–61. 2014. View Article : Google Scholar : PubMed/NCBI
|