Introduction
The liver is a critical organ in vertebrate animals
that is involved in multiple functions related to digestion,
detoxification, fluid and electrolyte balance, and haemostasis
(1). It plays vital roles in
controlling lipid and glucose homeostasis to meet energy needs in
response to different metabolic stresses (1). Non-alcoholic fatty liver disease
(NAFLD) is one of the most common liver diseases worldwide
(2). NAFLD is defined by hepatic
steatosis in the absence of apparent liver disease or excessive
alcohol intake. The factors leading to NAFLD remain to be fully
elucidated. Insulin resistance, excess lipid accumulation and
inflammation are known to be involved in the disease process
(3,4). Recently, several reports have
implicated hepatic mitochondrial dysfunction in the progression of
NAFLD (5-7). As the cell powerhouse, mitochondria
play an important role in lipid metabolism. Enhanced mitochondrial
fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS)
stimulation and tricarboxylic acid (TCA) cycle induction are
important in overcoming the hepatic triglyceride (TG) burden
(5). By contrast, mitochondrial
dysfunction, concomitant with molecular and structural alterations,
may result in metabolic disturbances and may contribute to NAFLD
progression (8). It has been
reported that hepatic mitochondria are structurally and molecularly
altered in NAFLD (9-13). Studies on the correlation between
mitochondrial dysfunction and NAFLD are likely to provide new
strategies for treating hepatic steatosis.
MicroRNAs (miRNAs) are small non-coding RNAs that
regulate gene expression at the post-transcriptional level
(14). It has been shown that
miRNAs participate in proliferation (15,16), apoptosis (17), inflammation (18), cancer (19) and metabolism (20,21). miR-146a-5p (miR-146a), first
described in humans by Taganov et al in 2006 (22), is a member of the miR-146 family.
A large proportion of the literature regarding miR-146a has focused
on inflammation (22,23). In addition, mounting evidence
shows that miR-146a plays important roles in cardiovascular disease
(24-26). Recently, Jin et al reported
that miR-146a was significantly decreased in non-alcoholic
steatohepatits (NASH) and that overexpression of miR-146a was
capable of improving NASH by targeting HDMCP (27). In addition, miR-146a has been
found to play an important role in liver cancer (28-30). However, there are few literature
reports regarding whether miR-146a plays a role in the development
of insulin resistance and NAFLD. Therefore, we aimed to explore the
expression level of miR-146a in fatty liver and fatty acid-treated
hepatic cells, and the relationship between miR-146a and fatty
liver and fatty acid oxidation, thus providing new insight into the
mechanism and treatment of fatty liver.
In this study, we found that miR-146a was decreased
in the liver of NAFLD mice and FFA-stimulated cells, and that
miR-146a improved glucose metabolism. Moreover, overexpression of
hepatic miR-146a attenuated lipid accumulation in the liver of high
fat diet (HFD) mice by increasing the mitochondrial density and
respiratory capacity. Mechanistic studies revealed that miR-146a
regulated mitochondrial function through its direct target gene. In
conclusion, we identified a novel function of miR-146a showing that
miR-146a could alleviate the metabolic disease in HFD mice by
targeting MED1 and enhancing mitochondrial function. These findings
indicate that miR-146a is a critical regulator of glucose and lipid
homeostasis, and may serve as a potential therapeutic target for
hepatic steatosis.
Materials and methods
Ethics statement
All animal protocols were approved by the Animal
Experimental Ethical Inspection Committee of Bengbu Medical College
(approval no. DWLL-2017-046). All in vivo experiments
described in this study were in accordance with institutional
guidelines for the care and use of animals.
Animals and treatments
The mice used for in vivo studies were all
males aged 6-10 weeks. All 43 mice were C57BL/6 and were maintained
in specific pathogen-free conditions under a consistent light-dark
cycle (lights on at 6:00 a.m. and off at 6:00 p.m.) with free
access to water and normal chow diet (SLACOM) Mice with similar
ages or from the same litters had priority of use. High-fat diet
(D12492, Research Diets) was used to feed 10 eight-week-old mice
for 3 months. During the experiments, the mice were monitored
daily. Any mice with significantly abnormal signs of rapid weight
loss, inability to eat or drink, clinical symptomatology, toxicity,
or unresponsiveness were recorded, and the data from these mice
were excluded for statistical analysis following the laboratory
animal welfare guidelines of Bengbu Medical College.
Bioinformatics analysis
The website of TargetScan (http://www.targetscan.org/vert_72/) was used to
predict the related targets of miR-146a.
Metabolic measurements
Glycogen and triglyceride levels of liver of mice
were measured with a Glycogen Assay kit (BioVision, K646-100) and
triglyceride assay kit (TriglycerideAssaykit, Sigma Aldrich Co.),
respectively. FFA concentrations and insulin levels of serum of
mice were analyzed with the NEFA C test kit (Wako Pure Chemical
Industries Ltd.) and insulin ELISA kit (Crystal Chem Inc.). ATP and
triglyceride levels of AML12 cells were determined with the Cell
Titer-Glo Luminescent kit (Promega) and triglyceride assay kit
(Triglyceride Assay kit), respectively. All experimental procedures
were performed according to the manufacturer's instructions.
Oxygen consumption and glycolysis were measured
using a XF24 instrument (Seahorse Bioscience Inc.). AML12 cells
were cultured in 20 wells of an XF24 microplate. The Mito Stress
Test kit was obtained from Seahorse Bioscience. The oxygen
consumption rate (OCR) was determined following the manufacturer's
instructions. Furthermore, the basal respiration and respiratory
capacity were calculated as described previously (31).
Prior to the fasting blood glucose tests, mice were
deprived of food for 6 h. Blood glucose levels were measured at the
indicated time points using a glucometer glucose test strip
(LifeScan). For the glucose tolerance test, the mice were deprived
of food for 16 h, and then given 2 g glucose per kilogram of body
weight intraperitoneally. Blood (~10 µl) glucose levels were
detected from tail vein after 15, 30, 60 and 120 min using a
glucometer glucose test strip (LifeScan). For the insulin tolerance
test, mice were deprived of food for 6 h, and then given 1.25 units
insulin per kilogram of body weight intraperitoneally. Blood (~10
µl) was collected from the tail and glucose levels were
measured at the indicated time points by a method similar to the
one described above for the glucose tolerance test.
Adenovirus recombination and
administration
The recombinant adenoviruses for miR-146a were
generated using the AdEasy Adenoviral Vector System (Stratagene) in
293A cells. Briefly, the stem loop sequence of miR-146a
(5′-AGCTCTGAGAACTGAATTCCATGGGTTATATCAATGTCAGACCTGTGAAATTCAGTTCTTCAGCT-3′)
and NC (5′-TCACAACCTCCTAGAAAGAGTAGATCACAACCTCCTAGAAAGAGTAGA-3′) was
cloned into pShuttle-CMV, and then pShuttle-miR-146a was recombined
into the pAd-Easy1 vector. The recombined vector was then
transfected into 293A cells to obtain the first generation virus.
The virus was further amplified in 293A cells and purified by
caesium chloride gradient centrifugation (100,000 × g, 4°C, 90 min)
to obtain a high purity adenovirus. Then 1×109 PFU of
the virus were administered to each mouse through tail vein
injection. A total of 27 mice were used for adenovirus
administration. After 14 days of virus injection, mice were
intraperitoneally injected with 0.5% pentobarbital sodium (45
mg/kg) and blood was taken from the heart. Tissues were harvested
without restricting the mice to food or water, snap-frozen in
liquid nitrogen immediately after resection and stored at
-80°C.
Plasmid construction
pcDNA3.1 and pmirGLO were used to generate
pcDNA3.1-MED1 and pmirGLO-MED1-3′UTR, respectively. Then, miR-146a
primers containing EcoRI/NotI or
SacI/XbaI cloning sites were used in PCRs for the
generation of pcDNA3.1-MED1 (F: 5′-ATGAAGGCTCAGGGGGAAACCGAGG-3′ R:
5′-CTAATTGCCAATCAGGGCCA-3′) or pmirGLO-MED1-3′UTR (F:
5′-CCTAACTTTCTAAACAGACA-3′ R: 5′-ATGCTAACTCCAACAACCTG-3′). Mutation
of MED1-3′UTR was performed by a KOD-Plus mutagenesis kit (Toyobo)
following the manufacturer's instructions. The binding sites for
miR-146a were changed to 5′-TCAAGAG-3′. All the plasmids
constructed in this study were confirmed by DNA sequencing.
MiR-146a mimic (5′-UGAGAACUGAAUUCCAUGGGUU-3′) and inhibitor
(5′-AACCCAUGGAAUUCAGUUCUCA-3′) were purchased from GenePharma.
Nonsense sequences were used as mimic control
(5′-UCACAACCUCCUAGAAAGAGUAGA-3′) and antagomir control
(5′-UCUACUCUUUCUAGGAGGUUGUGA-3′).
Cell culture and reporter assay
A mouse hepatocyte cell line (AML12) (SCSP-550, Stem
Cell Bank, Chinese Academy of Sciences) was cultured in Dulbecco's
modified Eagle's medium supplemented with 10% FBS, 1 mM glutamine,
100 U/ml penicillin, and 100 mg/ml streptomycin. Cells were
maintained in a humidified incubator at 37°C and 5% CO2
atmosphere.
A dual luciferase reporter assay was performed
according to the manufacturer's instructions. miR-146a mimics (100
nM) or control (100 nM) were co-transfected with pmirGLO-MED1-3′UTR
WT or pmirGLO-MED1-3′UTR MUT in AML12 cells. Forty-eight hours
after transfection, the cells were lysed in 1.5 ml centrifuge tube
and relative luciferase activity was analyzed with the
Dual-Luciferase Reporter Assay System (Promega) on a luminometer
(Promega).
Histological analysis
For H&E staining, liver tissues were fixed with
4% paraformaldehyde. Individual lobes of liver biopsy material were
placed in processing cassettes, dehydrated through a serial alcohol
gradient, and embedded in paraffin wax blocks. Before
immunostaining, liver tissue sections were dewaxed in xylene,
rehydrated through decreasing concentrations of ethanol, and washed
in PBS prior to staining with H&E. For Oil-red O staining, the
fixed liver tissue was dehydrated by sucrose solution. Liver
sections (8 µm) were stained with Oil-red O for 8 min,
differentiated with 75% ethanol after PBS buffer washing, and
stained with hematoxylin for 90 sec. Images were obtained with a
light microscope (Olympus) and then quantified using Image Pro Plus
software (version 6.0).
RT-qPCR and mtDNA copy number
analysis
RNA from cells or tissues was extracted with TRIzol
reagent (Invitrogen). cDNA was synthesized by reverse transcription
of mRNA using the PrimeScript RT reagent kit (Takara). For miRNA
detection, total RNA was reverse-transcribed using a
miR-146a-specific stem-loop primer (Applied Biosystems) and
subsequently measured via a real-time PCR using miR-146a-specific
primer (Applied Biosystems). qPCR was performed on a 7900 HT Fast
Real-time PCR System (Applied Biosystems) using SYBR-Green (Roche).
Relative expression levels of miR-146a and mRNA were calculated
with normalization to U6 or GAPDH values,
respectively, using the 2−ΔΔCq method (32). The mtDNA copy number was
determined by comparing the ratio of mtDNA to nDNA(18S rRNA) by
RT-qPCR as described previously (33). The PCR amplification procedure
was: Step 1: 95°C for 5 min; Step 2: 95°C for 25 sec; Step 3: 60°C
for 20 sec; Step 2-3 repeats for 40 cycles. Primer sequences used
are shown in Table SI.
Western blotting
MED1 proteins were enriched by immunoprecipitation
before detection using western blot analysis. Cells or tissues were
lysed in IP lysis buffer (Beyotime, P0013) supplemented with
phosphatase inhibitor for 30 min on ice. Subsequently, the cell
lysate was centrifuged, and the supernatant was incubated with the
indicated antibodies and Protein A/G PLUS-agarose (Santa Cruz
Biotechnology sc-2003) at 4°C overnight. The immunoprecipitates
were separated by SDS-PAGE (7.5%). After electrophoresis, the
protein was transferred onto nitrocellulose membranes with 300 mA
for 3 h and blocked by 5% skim milk then probed with the various
primary antibodies (MED1, Cell Signaling Technology, no. 51613,
rabbit, polyclonal antibodies, 1:1,000; Histone H3, Ab clonal:
A2348, rabbit, polyclonal antibodies, 1:1,000) at 4°C overnight. On
the second day, the membranes were washed with TBST buffer and
probed with secondary antibodies (Jackson ImmunoResearch,
111-035-003, Peroxidase AffiniPure goat anti-rabbit IgG (H+L),
polyclonal antibodies) diluted with 5% skim milk at 1:1,500.
Detection was performed via measurement of the chemiluminescent
signal with SuperSignal Ultra. Protein quantification was analysed
by Quantity One software (Bio-Rad) and the intensity values were
normalized to β-actin. Antibodies against MED1 (Cell Signaling
Technology, no. 51613, 1:1,000) and Histone H3 (Ab clonal: A2348)
were used for western blotting.
Statistical analysis
Differences among multiple groups were assessed
using One-way analysis of variance followed by Tukey's post-hoc
comparisons. Differences between two groups were tested by the
Student's t-test. The software used were Excel and GraphPad Prism
5.0 (GraphPad). Data are presented as mean ± SD. P<0.05 was
considered statistically significant. All experiments were repeated
at least twice, and representative results are shown.
Results
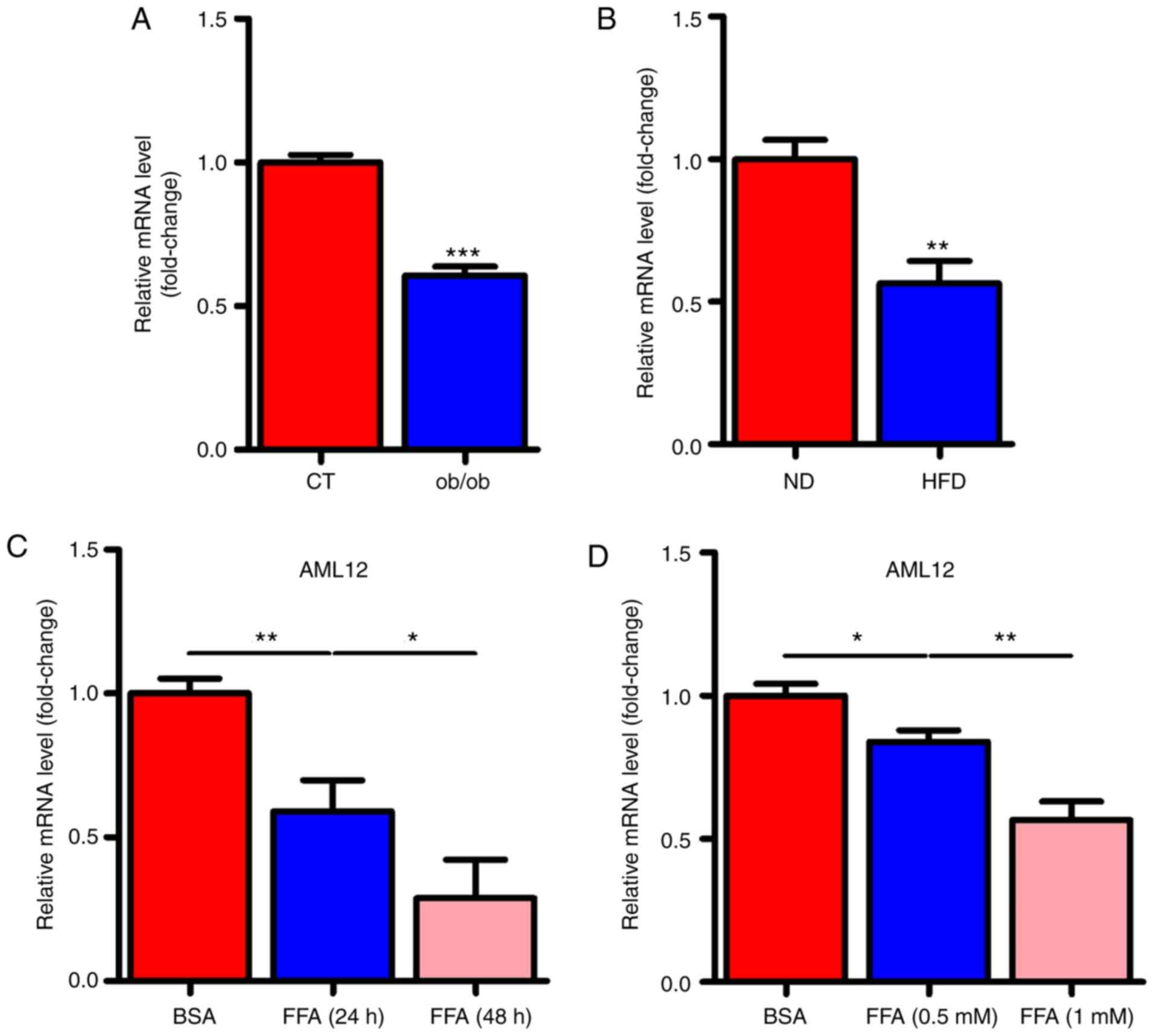
miR-146a is decreased in the liver of
NAFLD mice and FFA stimulated cells
To determine whether miR-146a played a role in the
development of metabolic syndrome, we examined the expression
levels of miR-146a in the liver of ob/ob and HFD mice. Notably, the
expression levels of miR-146a were all significantly decreased in
the liver of both ob/ob mice (Fig.
1A) and HFD mice (Fig. 1B).
Given that free fatty acid (FFA) in the liver is an important
signalling factor, we used FFA to treat AML12 cells to verify
whether FFA affected miR-146a. As expected, FFA treatment
significantly decreased the expression level of miR-146a in AML12
cells in a time- and dose-dependent manner (Fig. 1C and D). These results indicated
that miR-146a is FFA-responsive and may be involved in the
development of metabolic syndrome.
Hepatic miR-146a regulates glucose
homeostasis
To test whether miR-146a is capable of regulating
hepatic glucose metabolism, an adenoviral miR-146a (Ad-miR-146a
mimic) was used to overexpress miR-146a in the livers of HFD mice
(Fig. 2A). Notably, we found that
the fasting serum glucose level was significantly decreased on the
fifth day after the infection of Ad-miR-146a mimic. Furthermore,
overexpression of miR-146a could improved the glucose and insulin
sensitivity of HFD mice (Fig.
2C-F), further indicating that miR-146a plays an important role
in glucose metabolism. The liver mainly modulates the body's
glucose homeostasis through the regulation of hepatic glycogen
synthesis and gluconeogenesis (1). Therefore, we analyzed hepatic
glycogen content and gluconeogenesis-associated genes in the liver
of two groups using RT-qPCR. Consistent with previous results
(Fig. 2B-F), glycogen content
analysis revealed that the glycogen level was increased in the
livers of miR-146a overexpressing HFD mice (Fig. 2G) and the serum insulin level was
also significantly decreased after the overexpression of hepatic
miR-146a by Ad-miR146a mimic (Fig.
2H). Accordingly, the expression levels of gluconeogenic genes
(PEPCK, G6Pase and PGC1a) were significantly
reduced in the livers of miR-146a-overex-pressing HFD mice
(Fig. 2I). Together, these
results indicate that miR-146a regulates glucose metabolism in the
liver and that overexpression of hepatic miR-146a may alleviate
type II diabetes.
miR-146a improves lipid metabolism in the
liver of HFD mice
In addition to the improvement of glucose
metabolism, we also found that the lipid metabolism was also
improved in HFD mice after hepatic miR-146a overexpression by
infection with Ad-miR-146a mimic. We observed that lipid
accumulation was markedly reduced in the livers of HFD mice
infected with Ad-miR-146a mimic by Oil-red O staining (Fig. 3A) and H&E staining (Fig. 3B). Similarly, the liver weight and
hepatic triglycerides were all significantly decreased after the
overexpression of hepatic miR-146a in HFD mice (Fig. 3C and D). Moreover, the
overexpression of hepatic miR-146a also significantly decreased
hepatic FFA levels (Fig. 3E).
NAFLD develops as a consequence of metabolic dysregulation of de
novo lipogenesis, lipid uptake, fatty acid oxidation, and lipid
secretion (34). We further
analyxed genes related to lipid metabolism in the liver. Of note,
we found that the mRNA expression levels of genes related to fatty
acid oxidation (PPARa, Acaa2, Mcad,
Cpt1a, Hadha and Acadl) were all significantly
increased but not for genes related to de novo lipogenesis
(Fasn, Acc1, Scd1 and Srebp1c), lipid
uptake (CD36 and Fabp), and lipid secretion
(ApoA1, ApoB and Mttp) (Fig. 3F). Additionally, we found that
overexpression of miR-146a reduced lipid accumulation in AML12
cells by transfection of miR-146a mimic (Fig. 3G and H). Moreover, signicantly
increased levels of fatty acid oxidation-related genes
(PPARa, Acaa2, Mcad, Cpt1a,
Hadha and Acadl) were found in AML12 cells after
transfection of miR-146a mimic (Fig.
3I). These results suggest that miR-146a may improve hepatic
lipid metabolism and may play an important role in fatty acid
oxidation.
 | Figure 3miR-146a attenuates lipid
accumulation in the livers of HFD mice. (A-C) Representative images
of Oil-red O staining (A) and H&E staining (B) of liver
sections and relative liver weight (n=5) (C) of normal diet fed
mice (ND) infected with Ad-NC, HFD mice infected with Ad-NC or
Ad-miR-146a mimic. Scale bar, 100 µm. (D) Relative hepatic
triglyceride (TG) levels of HFD mice infected with Ad-NC or
Ad-miR-146a mimic (n=3). (E) Relative hepatic free fatty acid (FFA)
in the HFD mice infected with Ad-NC or Ad-miR-146a mimic (n=5). (F)
The relative mRNA levels of genes related to fatty acid oxidation
(PPARa, Acaa2, Mcad, Cpt1a,
Hadha and Acadl), de novo lipogenesis
(Fasn, Acc1, Scd1 and Srebp1c), lipid
secretion (ApoA1, ApoB and Mttp) and lipid
uptake (CD36 and Fabp) in the livers of HFD mice infected with
Ad-NC or Ad-miR-146a mimic (n=5 to 6). (G) Representative images of
Oil-red O staining of AML12 cells transfected with NC or miR-146a
mimic. Scale bar, 100 µm. (H) Quantitative analysis of
cellular TAG content in AML12 cells transfected with NC or miR-146a
mimic (n=3). (I) The relative mRNA levels of genes related to fatty
acid oxidation in AML12 cells transfected with NC or miR-146a mimic
(n=3). Means ± SD are shown. *P<0.05;
**P<0.01; ***P<0.001. |
miR-146a regulates mitochondrial function
of the hepatocytes
Since we found that the expression level of fatty
acid oxidation-related genes in the liver was significantly
increased after the overexpression of miR-146a (Fig. 3F) and mitochondria play a key role
in energy metabolism by generating most of the energy via the
tricarboxylic acid cycle and oxidative phosphorylation (5), we examined whether the
overexpression of hepatic miR-146a could regulate mitochondrial
function in the liver of HFD mice. As expected, we found that the
mtDNA copy numbers were significantly increased in the livers of
HFD mice after infection of Ad-miR-146a-mimic (Fig. 4A), which indicated that miR-146a
could increase the amount of mitochondria. Consistently,
overexpression of hepatic miR-146a could increase the ATP level in
the liver of HFD mice (Fig. 4B).
Given that the nuclear respiratory factors, Nrf1 and Nrf2 are the
key transcription factors required for mitochondrial respiration,
mtDNA transcription and replication (35-38), we then measured the mRNA levels of
Nrf1 and Nrf2 and their downstream target genes (Tfam,
Plormt, Tfb1m), related to mtDNA maintenance
(39-41), and found that the expression
levels of these genes were all significantly increased in the liver
after infection with Ad-miR-146a-mimic in HFD mice (Fig. 4C). Moreover, the levels of genes
involved in the oxidative phosphorylation (OXPHOS) pathway in both
the nucleus (Ndufs3, Sdhb, Sdhc, Cox5b)
and mitochondria (ND4L, CYTB, ND1,
COII, 12SrRNA) (42-44) were significantly increased in the
liver after HFD mice were infected with Ad-miR-146a mimic (Fig. 4D). Furthermore, chemosynthetic
mimic and inhibitor of miR-146a were used to up- and downregulate,
respectively, the expression of miR-146a in AML12 cells (Fig. 4E). Consistent with the previous
results, miR-146a mimic and inhibitor increased and decreased mtDNA
copy numbers and ATP levels in AML12 cells, respectively (Fig. 4F and G). Additionally, a Seahorse
Bioscience extracellular flux analyser was used to investigate the
cellular oxygen consumption rate, an indicator of mitochondrial
oxidation. As shown in Fig. 4H and
I, bioenergetics profiles and the calculated oxygen consumption
rates indicated that overexpression of miR-146a was able to
increase the basal and maximal mitochondrial respiratory capacity,
which was consistent with the miR-146a overexpression-mediated
induction of fatty acid oxidation in the liver of HFD mice
(Fig. 3F). These results
indicated that miR-146a increased the amount of mitochondria and
promote mitochondrial oxidative phosphorylation and ATP
production.
 | Figure 4miR-146a regulates mitochondrial
function of the hepatocytes. (A and B) Relative mtDNA copy numbers
(A) and ATP levels (B) in the livers of high fat diet mice (HFD)
infected with Ad-NC or Ad-miR-146a mimic (n=5). (C) The relative
mRNA expression of nuclear respiratory factors (Nrf1 and Nrf2) and
their downstream target genes (Tfam, Plormt and
Tfb1m) in the liver of high fat diet mice (HFD) infected
with Ad-NC or Ad-miR-146a-mimic (n=5). (D) Relative mRNA expression
levels of oxidative phosphorylation pathway-related genes in
nucleus (Ndufs3, Sdhb, Sdhc and Cox5b)
and mitochondria (ND4L, CYTB, ND1, COII and 12 SrRNA) in the liver
of high-fat diet mice (HFD) a infected with Ad-NC or Ad-miR-146a
mimic (n=5). (E-G) The relative expression level of miR-146a (E)
mtDNA copy numbers (F) and ATP levels (G) in AML12 cells
transfected with NC or miR-146a-mimic, or transfected with scramble
control or miR-146a inhibitor (n=3). (H) Oxygen consumption of
AML12 cells transfected with miR-146a mimic or NC was analysed with
a Seahorse XF24 extracellular flux analyser (n=3). (I) Basal
respiration and respiratory capacity oxygen consumption rates were
measured and calculated (n=3). Means ± SD are shown.
*P<0.05; **P<0.01;
***P<0.001, vs. NC, #P<0.05;
###P<0.001, vs. scramble control. |
MED1 is a direct target gene of
miR-146a
To explore the molecular mechanism involved in the
regulation of mitochondrial function by miR-146a, we predicted the
target genes of miR-146a using TargetScan (http://www.targetscan.org/vert_72/) to identify
related putative targets. The results showed that MED1, a key
subunit of the mammalian mediator complex involved in mitochondrial
function (45), was predicted as
a target gene of miR-146a and contained a conserved binding site
(Fig. 5A). For validation, the
mouse-complete MED1 3′UTR containing the microRNA response element
(MRE) sequence was cloned into a luciferase reporter. Reporter
activity analysis revealed that overexpression of miR-146a could
decrease the luciferase activity of the reporter but had no effect
on the reporter containing the mutated MED1 3′UTR (Fig. 5B). Similarly, the mRNA and protein
levels were significantly decreased after miR-146a mimic
administration but increased with miR-146a inhibitor transfection
in AML12 cells (Fig. 5C-E). As we
expected, we found that the protein and mRNA expression levels of
MED1 were increased in the livers of HFD mice (Fig. 5F-H). Furthermore, the mRNA levels
and protein levels of MED1 were increased in FFA-treated AML12
cells in vitro (Fig.
5I-K). These results indicate that MED1 is a direct target gene
of miR-146a in murine hepatocytes.
MED1 mediates the metabolic action of
miR-146a
To determine whether MED1 mediated the effect of
miR-146a on glucose and lipid metabolism and mitochondrial
function, we employed pcDNA3-MED1, which could not be repressed by
miR-146a due to the absence of the MED1 3′UTR (Fig. 6A-E). As expected, transient
transfection and overexpression of MED1 abolished the effect of
miR-146a on lipid accumulation in AML12 cells (Fig. 6F and G). Furthermore, restoring
MED1 could also compensate the number of mitochondria and ATP
levels after miR-146a mimic administration in AML12 cells (Fig. 6H and I). Consistent with these
findings, MED1 attenuated the effect of miR-146a on the expression
levels of Nrf1, Nrf2 and their downstream target genes (Fig. 6J), and genes involved in the
oxidative phosphorylation (OXPHOS) pathway in both the nucleus
(Ndufs3, Sdhb, Sdhc and Cox5b) and
mitochondria (ND4L, CYTB, ND1, COII and
12SrRNA) (Fig. 6K) in
AML12 cells. In addition, we found that Nrf1, Nrf2 and their
downstream target genes and OXPHOS pathway-related genes were
significantly decreased with inhibiting the expression of miR-146a
(Fig. 6L and M). Notably, the
rescue of MED1 abolished the effect of miR-146a on basal and
maximal mitochondrial respiratory capacity (Fig. 6N and O). Given that the
restoration of MED1 expression could almost fully abrogate the
effect of miR-146a on mitochondrial function and lipid metabolism
in AML12 cells. We suggest that MED1 may be the primary target gene
of miR-146a that mediates the effect of miR-146a on mitochondrial
function and lipid metabolism.
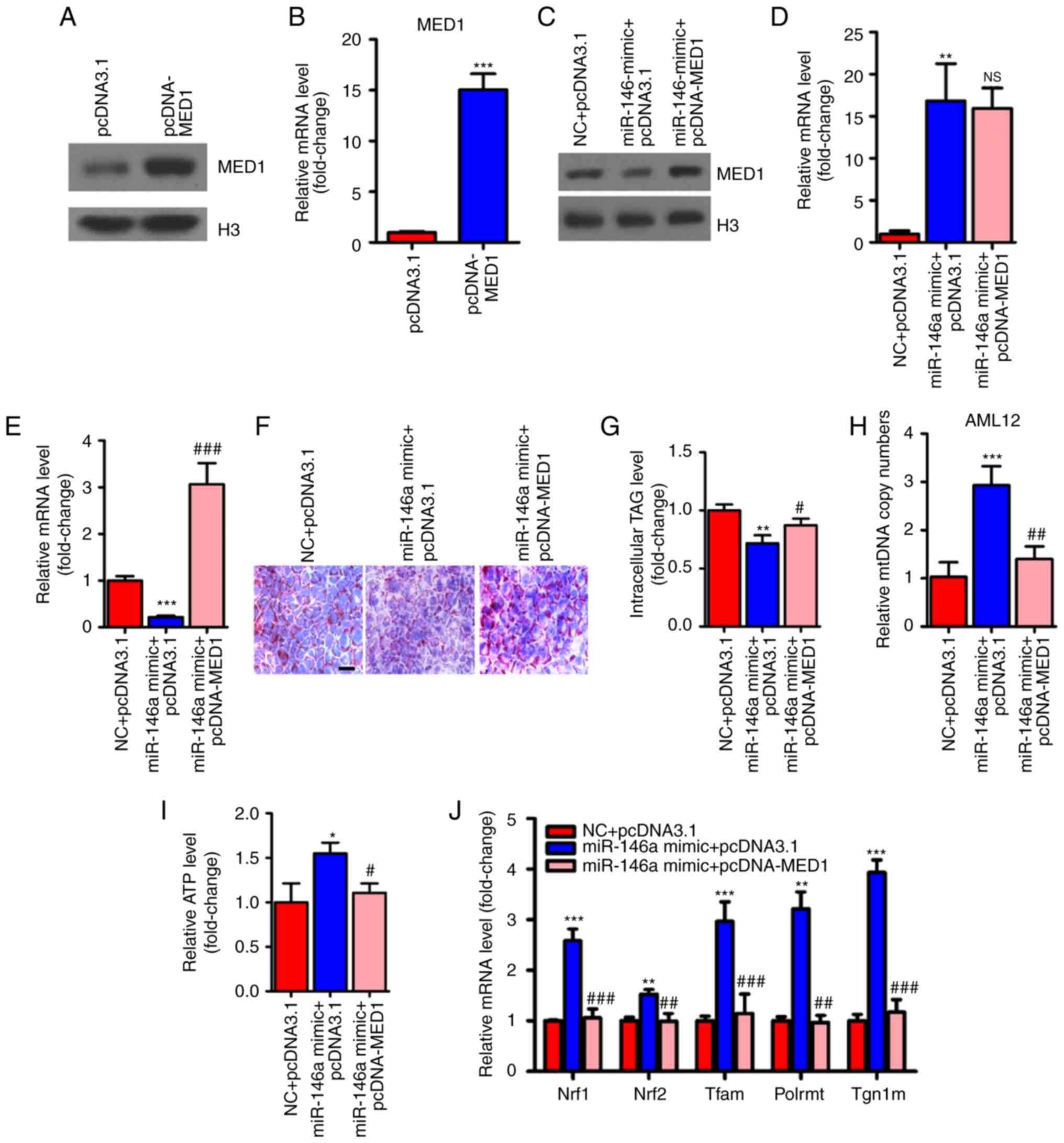 | Figure 6Restoration of MED1 inhibits the
metabolic action of miR-146a. (A and B) The protein levels (A) and
relative mRNA levels (B, n=3) of MED1 in AML12 cells transfected
with pcDNA3.1 or pcDNA-MED1. (C-E) The protein level of MED1 (C)
and relative mRNA levels of miR-146a (D, n=3) and MED1 (E, n=3) in
AML12 cells cotransfected with NC and pcDNA3.1, miR-146a mimic and
pcDNA3.1 or miR-146a mimic and pcDNA-MED1. (F and G) Representative
images of Oil-red O staining (F) and cellular TG content analysis
(G) of AML12 cells transfected with NC and pcDNA3.1, miR-146a
mimics and pcDNA3.1 or miR-146a mimic and pcDNA-MED1. Scale bar,
100 µm. (H and I) The relative mtDNA copy numbers (H) and
ATP levels (I) in AML12 cells co-transfected with NC and pcDNA3.1,
miR-146a mimic and pcDNA3.1 or miR-146a mimic and pcDNA-MED1 (n=3).
(J) The relative mRNA expression level of nuclear respiratory
factors (Nrf1 and Nrf2) and their downstream target genes
(Tfam, Plormt and Tfb1m) in AML12 cells
co-transfected with NC and pcDNA3.1, miR-146a mimic and pcDNA3.1 or
miR-146a mimic and pcDNA-MED1 (n=3). Restoration of MED1 inhibits
the metabolic action of miR-146a. (K) Relative mRNA expression of
oxidative phosphorylation pathway-related genes in the nucleus
(Ndufs3, Sdhb, Sdhc and Cox5b) and in
mitochondria (ND4L, CYTB, ND1, COII, and 12SrRNA) in AML12 cells
co-transfected with NC and pcDNA3.1, miR-146a mimic and pcDNA3.1 or
miR-146a mimic and pcDNA-MED1 (n=3). (L) The relative mRNA
expression level of nuclear respiratory factors and their
downstream target genes in AML12 cells transfected with Scramble
control or miR-146a inhibitor (n=3). (M) Relative mRNA expression
of oxidative phosphorylation pathway-related genes in the nucleus
and in mitochondria in AML12 cells transfected with Scramble
control or miR-146a inhibitor (n=3). (N) Oxygen consumption of
AML12 cells co-transfected with NC and pcDNA3.1, miR-146a mimic and
pcDNA3.1 or miR-146a mimic and pcDNA-MED1 were analyzed with a
Seahorse XF24 extracellular flux analyser (n=3). (O) Basal
respiration and respiratory capacity oxygen consumption rates were
measured and calculated (n=3). Means ± SD are shown.
*P<0.05; **P<0.01;
***P<0.001 (vs. control group transfected with NC and
pcDNA3.1), #P<0.05; ##P<0.01;
###P<0.001 (vs. the group transfected with miR-146a
mimic and pcDNA3.1). |
Discussion
NAFLD, one of the most common chronic liver diseases
worldwide, has recently raised much interest. The factors leading
to NAFLD remain to be fully elucidated. Insulin resistance, excess
lipid accumulation, inflammation and mitochondrial dysfunction are
known to be involved in the disease process (46). MicroRNAs (miRNAs) are small
non-coding RNAs that regulate gene expression at the
post-transcriptional level. Previous findings have demonstrated
that microRNAs play an important role in the progression of
metabolic disorders. An early study evaluated 4 upregulated miRNAs
(miR-103, miR-31, miR-107 and miR-126-3p) and 2 downregulated
miRNAs (miR-100 and miR-29c) in the liver tissue of ob/ob mice and
streptozotocin-induced diabetic mice (47). In addition, several studies have
shown that miR-34a is significantly elevated and miR-21 and miR-29
were significantly downregulated in the liver of ob/ob mice and
NAFLD patients (48-54). Therefore, different microRNAs play
different roles in the development of NAFLD while the decrease of
microRNA is not a general trend. Identification of essential miRNAs
that regulate metabolic processes and investigation of the specific
targets of these functional miRNAs will not only increase our
knowledge but also provide new strategies for treating metabolic
disorders.
Findings of the present study showed that miR-146a
play an important role in hepatic glucose and lipid metabolism. We
found that miR-146a was decreased in the liver of NAFLD mice and
FFA-stimulated cells. He et al reported that miR-146b may be
involved in target delivery to hepatocytes to improve NAFLD by
acting as a lipid cleaner and inflammation suppressor (55). MiR-146b and miR-146a belong to the
same microRNA family with two base inconsistencies in the mature
sequence, indicating that the miR-146 family plays an important
role in the progression of NAFLD by regulating different signalling
pathways. In addition, it is well known that excessive FFA
accumulation in the liver leads to an increase in oxidative stress,
which is thought to be critical for the pathogenesis of hepatic
steatosis (56). Therefore,
miR-146a may be involved in the FFA-oxidative stress-fatty liver
axis. Azizi et al reported that miR-146a was significantly
decreased under increased oxidative stress conditions (57). Therefore, FFA may inhibit the
expression level of miR-146a by increasing the level of oxidative
stress, but further investigation is needed. In addition,
overexpression of hepatic miR-146a may improve glucose tolerance
and insulin tolerance in HFD mice. Interestingly, we also found
that overexpression of miR-146a could increase mitochondrial
density and respiration capacity in AML12 cells.
Mechanistic studies revealed that MED1 is the direct
target of miR-146a, and it mediated the regulation of miR-146a on
lipid metabolism and mitochondrial function. MED1 is regarded as a
key subunit of the mammalian mediator complex. An earlier study
showed that MED1 was essential for PPAR-gamma stimulated
adipogenesis in mouse embryonic fibroblasts (58). Furthermore, Chen et al
reported that muscle-specific MED1 knockout mice showed enhanced
insulin and glucose tolerance and resistance to high-fat
diet-induced obesity through increasing mitochondrial density and
respiratory capacity (59), which
is consistent with our findings. Similarly, loss of MED1 in C2C12
cells could trigger mitochondrial biogenesis (60). In addition, Bai et al found
that MED1 was required for high-fat diet-induced and PPARγ-induced
hepatic steatosis and that loss of MED1 protects against fatty
liver under these conditions (61). On the one hand, they found that
hepatic MED1 deficiency impaired PPARγ-induced adipogenic gene
expression. On the other hand, they showed that FGF21, which
increases energy expenditure by increasing fatty acid oxidation,
was significantly increased in the liver of MED1 hepatic specific
knockout mouse after PPARγ overexpression (61). Of note, MED1 is well known to be a
coactivator of PPARα and is necessary for PPARα signaling (62,63). Additionally, the expression levels
of these factors seem to be slightly conflicting after the
overexpression of hepatic miR-146a. Therefore, the upregulation of
fatty acid oxidation-related genes by overexpression of hepatic
miR-146a may not occur mainly through PPARα but other genes, such
as FGF21 or HFN4α. Notably, whether MED1 mediates the
regulation of miR-146a on glucose metabolism still needs to be
further explored. It has been reported that there are changes in
mitochondrial morphology in diabetic patients or insulin resistance
animal models (64). Furthermore,
swollen mitochondria and decreased matrix density were observed in
rat models of insulin resistance and hypertension (65). These observations strengthen the
relationship between mitochondrial function and insulin
resistance.
Previous findings have shown that simple steatosis
is associated with an upregulation in mitochondrial oxidative
function (66-68). However, alterations in
mitochondrial energy metabolism could not completely prevent the
development of NAFLD (69).
Furthermore, reductions in mitochondrial respiratory activity and
rates of ATP synthesis were reported in both mice and human models
of long-term overnutrition that led to progressive and severe liver
disease (8-12). Therefore, it is possible to
improve NAFLD by enhancing mitochondrial respiration or the
efficiency of ATP production. It is worth noting that miR-146a can
also inhibit the development of liver cancer by targeting several
genes; therefore, overexpression of miR-146a is a better choice
than inhibition of MED1 when patients have both liver cancer and
fatty liver. Although there may be other unknown target genes of
miR-146a, such treatment may have a negative effect on the human
body (70).
In summary, our study demonstrated that
overexpression of hepatic miR-146a could improve glucose and lipid
metabolism. Additionally, MED1 mediates the effect of miR-146a on
mitochondrial function and lipid metabolism. Our findings, not only
add a new dimension to our understanding of the relationship
between mitochondrial function and NAFLD, but also provide a
potential therapeutic target for NAFLD and metabolic syndrome
disease.
Supplementary Data
Acknowledgments
Not applicable.
Funding
This study was financially supported by the National
Natural Science Foundation of China (grant no. 31700735), the
Natural Science Foundation of Anhui province (grant nos.
1808085QH264 and 1908085MB54), the Wanjiang Scholar Award of Anhui
Province (China) and the Major Natural Science Research Projects in
Anhui Universities (grant no. KJ2019ZD58).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KL and BZ designed and performed the experiments,
analysed the data and wrote the manuscript. DW and YC provided
useful advice on the manuscript and analysed the data. LQ and WW
and GL designed the experiments and wrote the manuscript. All
authors read and approved the manuscript.
Ethics approval and consent to
participate
All animal protocols were approved by the Animal
Experimental Ethical Inspection Committee of Bengbu Medical College
(approval no. DWLL-2017-046).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Trefts E, Gannon M and Wasserman DH: The
liver. Curr Biol. 27:R1147–R1151. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Targher G, Bertolini L, Padovani R,
Rodella S, Tessari R, Zenari L, Day C and Arcaro G: Prevalence of
nonalcoholic fatty liver disease and its association with
cardiovascular disease among type 2 diabetic patients. Diabetes
Care. 30:1212–1218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Byrne CD and Targher G: NAFLD: A
multisystem disease. J Hepatol. 62(1 Suppl): S47–S64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cobbina E and Akhlaghi F: Non-alcoholic
fatty liver disease (NAFLD)-pathogenesis, classification, and
effect on drug metabolizing enzymes and transporters. Drug Metab
Rev. 49:197–211. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ajith TA: Role of mitochondria and
mitochondria-targeted agents in non-alcoholic fatty liver disease.
Clin Exp Pharmacol Physiol. 45:413–421. 2018. View Article : Google Scholar
|
|
6
|
Petrosillo G, Portincasa P, Grattagliano
I, Casanova G, Matera M, Ruggiero FM, Ferri D and Paradies G:
Mitochondrial dysfunction in rat with nonalcoholic fatty liver
involvement of complex I, reactive oxygen species and cardiolipin.
Biochim Biophys Acta. 1767:1260–1267. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wei Y, Rector RS, Thyfault JP and Ibdah
JA: Nonalcoholic fatty liver disease and mitochondrial dysfunction.
World J Gastroentero. 14:193–199. 2008. View Article : Google Scholar
|
|
8
|
Satapati S, Sunny NE, Kucejova B, Fu X, He
TT, Méndez-Lucas A, Shelton JM, Perales JC, Browning JD and Burgess
SC: Elevated TCA cycle function in the pathology of diet-induced
hepatic insulin resistance and fatty liver. J Lipid Res.
53:1080–1092. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cortez-Pinto H, Chatham J, Chacko VP,
Arnold C, Rashid A and Diehl AM: Alterations in liver ATP
homeostasis in human nonalcoholic steatohepatitis-A pilot study.
JAMA. 282:1659–1664. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schmid AI, Szendroedi J, Chmelik M, Krssak
M, Moser E and Roden M: Liver ATP synthesis is lower and relates to
insulin sensitivity in patients with type 2 diabetes. Diabetes
Care. 34:448–453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abdelmalek MF, Lazo M, Horska A, Bonekamp
S, Lipkin EW, Balasubramanyam A, Bantle JP, Johnson RJ, Diehl AM,
Clark JM, et al: Higher dietary fructose is associated with
impaired hepatic adenosine triphosphate homeostasis in obese
individuals with type 2 diabetes. Hepatology. 56:952–960. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Szendroedi J, Chmelik M, Schmid AI,
Nowotny P, Brehm A, Krssak M, Moser E and Roden M: Abnormal hepatic
energy homeostasis in type 2 diabetes. Hepatology. 50:1079–1086.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fritsch M, Koliaki C, Livingstone R,
Phielix E, Bierwagen A, Meisinger M, Jelenik T, Strassburger K,
Zimmermann S, Brockmann K, et al: Time course of postprandial
hepatic phosphorus metabolites in lean, obese, and type 2 diabetes
patients. Am J Clin Nutr. 102:1051–1058. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hackl H, Burkard TR, Sturn A, Rubio R,
Schleiffer A, Tian S, Quackenbush J, Eisenhaber F and Trajanoski Z:
Molecular processes during fat cell development revealed by gene
expression profiling and functional annotation. Genome Biol.
6:R1082005. View Article : Google Scholar
|
|
16
|
Yuan Y, Zeng ZY, Liu XH, Gong DJ, Tao J,
Cheng HZ and Huang SD: MicroRNA-203 inhibits cell proliferation by
repressing ΔNp63 expression in human esophageal squamous cell
carcinoma. BMC Cancer. 11:572011. View Article : Google Scholar
|
|
17
|
Lima RT, Busacca S, Almeida GM, Gaudino G,
Fennell DA and Vasconcelos MH: MicroRNA regulation of core
apoptosis pathways in cancer. Eur J Cancer. 47:163–174. 2011.
View Article : Google Scholar
|
|
18
|
Liston A, Linterman M and Lu LF: MicroRNA
in the adaptive immune system, in sickness and in health. J Clin
Immunol. 30:339–346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liang T, Liu C and Ye Z: Deep sequencing
of small RNA repertoires in mice reveals metabolic
disorders-associated hepatic miRNAs. PLoS One. 8:e807742013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rottiers V and Naar AM: MicroRNAs in
metabolism and metabolic disorders. Nat Rev Mol Cell Biol.
13:239–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taganov KD, Boldin MP, Chang KJ and
Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an
inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA. 103:12481–12486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cobb BS, Hertweck A, Smith J, O'Connor E,
Graf D, Cook T, Smale ST, Sakaguchi S, Livesey FJ, Fisher AG and
Merkenschlager M: A role for Dicer in immune regulation. J Exp Med.
203:2519–2527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Dong Y, Chen S, Zhang G, Zhang M,
Gong Y and Li X: Circulating MicroRNA-146a and MicroRNA-21 predict
left ventricular remodeling after ST-elevation myocardial
infarction. Cardiology. 132:233–241. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiong XD, Cho M, Cai XP, Cheng J, Jing X,
Cen JM, Liu X, Yang XL and Suh Y: A common variant in pre-miR-146
is associated with coronary artery disease risk and its mature
miRNA expression. Mutat Res. 761:15–20. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jazdzewski K, Murray EL, Franssila K,
Jarzab B, Schoenberg DR and de la Chapelle A: Common SNP in
pre-miR-146a decreases mature miR expression and predisposes to
papillary thyroid carcinoma. Proc Natl Acad Sci USA. 105:7269–7274.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jin X, Liu J, Chen YP, Xiang Z, Ding JX
and Li YM: Effect of miR-146 targeted HDMCP up regulation in the
pathogenesis of nonalcoholic steatohepatitis. PLoS One.
12:e01742182017. View Article : Google Scholar
|
|
28
|
Bleau AM, Redrado M, Nistal-Villan E,
Villalba M, Exposito F, Redin E, de Aberasturi AL, Larzabal L,
Freire J, Gomez-Roman J and Calvo A: miR-146a targets c-met and
abolishes colorectal cancer liver metastasis. Cancer Lett.
414:257–267. 2018. View Article : Google Scholar
|
|
29
|
Sun X, Zhang J, Hou Z, Han Q, Zhang C and
Tian Z: miR-146a is directly regulated by STAT3 in human
hepatocellular carcinoma cells and involved in anti-tumor immune
suppression. Cell Cycle. 14:243–252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang X, Ye ZH, Liang HW, Ren FH, Li P,
Dang YW and Chen G: Down-regulation of miR-146a-5p and its
potential targets in hepatocellular carcinoma validated by a TCGA-
and GEO-based study. FEBS Open Bio. 7:504–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Canto C, Jiang LQ, Deshmukh AS, Mataki C,
Coste A, Lagouge M, Zierath JR and Auwerx J: Interdependence of
AMPK and SIRT1 for metabolic adaptation to fasting and exercise in
skeletal muscle. Cell Metab. 11:213–219. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
33
|
Schneider L, Giordano S, Zelickson BR, S
Johnson M, A Benavides G, Ouyang X, Fineberg N, Darley-Usmar VM and
Zhang J: Differentiation of SH-SY5Y cells to a neuronal phenotype
changes cellular bioenergetics and the response to oxidative
stress. Free Radic Biol Med. 51:2007–2017. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu W, Cao H, Yan J, Huang R and Ying H:
'Micro-managers' of hepatic lipid metabolism and NAFLD. Wiley
Interdiscip Rev RNA. 6:581–593. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bonawitz ND, Clayton DA and Shadel GS:
Initiation and beyond: Multiple functions of the human
mitochondrial transcription machinery. Mol Cell. 24:813–825. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Piantadosi CA, Carraway MS, Babiker A and
Suliman HB: Heme oxygenase-1 regulates cardiac mitochondrial
biogenesis via Nrf2-mediated transcriptional control of nuclear
respiratory factor-1. Circ Res. 103:1232–1240. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Scarpulla RC, Vega RB and Kelly DP:
Transcriptional integration of mitochondrial biogenesis. Trends
Endocrinol Metab. 23:459–466. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Suliman HB, Sweeney TE, Withers CM and
Piantadosi CA: Co-regulation of nuclear respiratory factor-1 by
NFkappaB and CREB links LPS-induced inflammation to mitochondrial
biogenesis. J Cell Sci. 123:2565–2575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Campbell CT, Kolesar JE and Kaufman BA:
Mitochondrial transcription factor A regulates mitochondrial
transcription initiation, DNA packaging, and genome copy number.
Biochim Biophys Acta. 1819:921–929. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Falkenberg M, Gaspari M, Rantanen A,
Trifunovic A, Larsson NG and Gustafsson CM: Mitochondrial
transcription factors B1 and B2 activate transcription of human
mtDNA. Nat Genet. 31:289–294. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
41
|
Larsson NG, Wang J, Wilhelmsson H, Oldfors
A, Rustin P, Lewandoski M, Barsh GS and Clayton DA: Mitochondrial
transcription factor A is necessary for mtDNA maintenance and
embryogenesis in mice. Nat Genet. 18:231–236. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dhar SS, Ongwijitwat S and Wong-Riley MT:
Nuclear respiratory factor 1 regulates all ten nuclear-encoded
Subunits of cytochrome c oxidase in neurons. J Biol Chem.
283:3120–3129. 2008. View Article : Google Scholar
|
|
43
|
Kelly DP and Scarpulla RC: Transcriptional
regulatory circuits controlling mitochondrial biogenesis and
function. Genes Dev. 18:357–368. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Scarpulla RC: Transcriptional paradigms in
mammalian mitochondrial biogenesis and function. Physiol Rev.
88:611–638. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jia Y, Viswakarma N and Reddy JK: Med1
subunit of the mediator complex in nuclear receptor-regulated
energy metabolism, liver regeneration, and hepatocarcinogenesis.
Gene Expr. 16:63–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leite NC, Salles GF, Araujo AL,
Villela-Nogueira CA and Cardoso CR: Prevalence and associated
factors of non-alcoholic fatty liver disease in patients with
type-2 diabetes mellitus. Liver Int. 29:113–119. 2009. View Article : Google Scholar
|
|
47
|
Li S, Chen X, Zhang H, Liang X, Xiang Y,
Yu C, Zen K, Li Y and Zhang CY: Differential expression of
microRNAs in mouse liver under aberrant energy metabolic status. J
Lipid Res. 50:1756–1765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ding J, Li M, Wan X, Jin X, Chen S, Yu C
and Li Y: Effect of miR-34a in regulating steatosis by targeting
PPARα expression in nonalcoholic fatty liver disease. Sci Rep.
5:137292015. View Article : Google Scholar
|
|
49
|
Xu Y, Zalzala M, Xu J, Li Y, Yin L and
Zhang Y: A metabolic stress-inducible miR-34a-HNF4 α pathway
regulates lipid and lipoprotein metabolism. Nat Commun. 6:74662015.
View Article : Google Scholar
|
|
50
|
Derdak Z, Villegas KA, Harb R, Wu AM,
Sousa A and Wands JR: Inhibition of p53 attenuates steatosis and
liver injury in a mouse model of non-alcoholic fatty liver disease.
J Hepatol. 58:785–791. 2013. View Article : Google Scholar :
|
|
51
|
Ahn J, Lee H, Jung CH and Ha T: Lycopene
inhibits hepatic steatosis via microRNA-21-induced downregulation
of fatty acid-binding protein 7 in mice fed a high-fat diet. Mol
Nutr Food Res. 56:1665–1674. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sun C, Huang F, Liu X, Xiao X, Yang M, Hu
G, Liu H and Liao L: miR-21 regulates triglyceride and cholesterol
metabolism in non-alcoholic fatty liver disease by targeting HMGCR.
Int J Mol Med. 35:847–853. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
He Y, Huang C, Lin X and Li J: MicroRNA-29
family, a crucial therapeutic target for fibrosis diseases.
Biochimie. 95:1355–1359. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Pogribny IP, Starlard-Davenport A,
Tryndyak VP, Han T, Ross SA, Rusyn I and Beland FA: Difference in
expression of hepatic microRNAs miR-29c, miR-34a, miR-155, and
miR-200b is associated with strain-specific susceptibility to
dietary nonalcoholic steatohepatitis in mice. Lab Invest.
90:1437–1446. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
He S, Guo W, Deng F, Chen K, Jiang Y, Dong
M, Peng L and Chen X: Targeted delivery of microRNA 146b mimic to
hepatocytes by lactosylated PDMAEMA nanoparticles for the treatment
of NAFLD. Artif Cells Nanomed Biotechnol. 46(Suppl 2): S217–S228.
2018. View Article : Google Scholar
|
|
56
|
Paradies G, Paradies V, Ruggiero FM and
Petrosillo G: Oxidative stress, cardiolipin and mitochondrial
dysfunction in nonalcoholic fatty liver disease. World J
Gastroentero. 20:14205–14218. 2014. View Article : Google Scholar
|
|
57
|
Azizi R, Soltani-Zangbar MS, Sheikhansari
G, Pourmoghadam Z, Mehdizadeh A, Mahdipour M, Sandoghchian S,
Danaii S, Koushaein L, Samadi Kafil H and Yousefi M: Metabolic
syndrome mediates inflammatory and oxidative stress responses in
patients with recurrent pregnancy loss. J Reprod Immunol.
133:18–26. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ge K, Guermah M, Yuan CX, Ito M, Wallberg
AE, Spiegelman BM and Roeder RG: Transcription coactivator TRAP220
is required for PPAR gamma 2-stimulated adipogenesis. Nature.
417:563–567. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Chen W, Zhang X, Birsoy K and Roeder RG: A
muscle-specific knockout implicates nuclear receptor coactivator
MED1 in the regulation of glucose and energy metabolism. Proc Natl
Acad Sci USA. 107:10196–10201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yu J, Xiao Y, Liu J, Ji Y, Liu H, Xu J,
Jin X, Liu L, Guan MX and Jiang P: Loss of MED1 triggers
mitochondrial biogenesis in C2C12 cells. Mitochondrion. 14:18–25.
2014. View Article : Google Scholar
|
|
61
|
Bai L, Jia Y, Viswakarma N, Huang J,
Vluggens A, Wolins NE, Jafari N, Rao MS, Borensztajn J, Yang G and
Reddy JK: Transcription coactivator mediator subunit MED1 is
required for the development of fatty liver in the mouse.
Hepatology. 53:1164–1174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kornberg RD: The molecular basis of
eukaryotic transcription. Proc Natl Acad Sci USA. 104:12955–12961.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Malik S and Roeder RG: Dynamic regulation
of pol II transcription by the mammalian Mediator complex. Trends
Biochem Sci. 30:256–263. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q,
Ren C, Ponomarenko A and DeCarli LM: Model of nonalcoholic
steatohepatitis. Am J Clin Nutr. 79:502–509. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim JA, Wei Y and Sowers JR: Role of
mitochondrial dysfunction in insulin resistance. Circ Res.
102:401–414. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Miele L, Grieco A, Armuzzi A, Candelli M,
Forgione A, Gasbarrini A and Gasbarrini G: Hepatic mitochondrial
beta-oxidation in patients with nonalcoholic steatohepatitis
assessed by 13C-octanoate breath test. Am J Gastroenterol.
98:2335–2336. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Iozzo P, Bucci M, Roivainen A, Någren K,
Järvisalo MJ, Kiss J, Guiducci L, Fielding B, Naum AG, Borra R, et
al: Fatty acid metabolism in the liver, measured by positron
emission tomography, is increased in obese individuals.
Gastroenterology. 139:846–856. e1–6. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sunny NE, Parks EJ, Browning JD and
Burgess SC: Excessive hepatic mitochondrial TCA cycle and
gluconeogenesis in humans with nonalcoholic fatty liver disease.
Cell Metab. 14:804–810. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sunny NE, Bril F and Cusi K: Mitochondrial
adaptation in nonalcoholic fatty liver disease: Novel mechanisms
and treatment strategies. Trends Endocrinol Metab. 28:250–260.
2017. View Article : Google Scholar
|
|
70
|
Rong M, He R, Dang Y and Chen G:
Expression and clinicopathological significance of miR-146a in
hepatocellular carcinoma tissues. Ups J Med Sci. 119:19–24. 2014.
View Article : Google Scholar :
|