Introduction
Cardiovascular diseases remain a leading cause of
mortality worldwide due to their high morbidity and mortality
rates. It has been reported that numerous risk factors may lead to
cardiomyocyte dysfunction via the induction of apoptosis, which
eventually contributes to the development of human cardiovascular
diseases (1). Therefore,
understanding the mechanisms responsible for this apoptosis may aid
in the enhanced understanding of the pathology of these
disorders.
Hypertension, diabetes, sex differences, obesity,
smoking and high cholesterol are the most important risk factors
for cardiovascular diseases (2,3).
Recent studies have demonstrated that the abnormal accumulation of
plasma homocysteine (Hcy) is a risk factor for cardiovascular
diseases, which may lead to myocardial cell dysfunction by inducing
apoptosis (4-7). Therefore, the early identification
and timely management of risk factors is critical for reducing
morbidity and mortality in patients with cardiovascular
diseases.
Several studies have reported that connexin 43
(Cx43) can regulate cell survival and death. Cx43 can initiate
apoptotic programs that are involved in arrhythmia,
ischemia-reperfusion and heart failure (6,8).
Existing data on connexin expression confirm that Cx43 is the
principal cardiac connexin, and it forms communication channels for
electric and metabolic coupling between cardiomyocytes (9). A previous study demonstrated that
Hcy can increase the expression of Cx43 in myocardial cells
(10).
Pirfenidone [5-methyl-1-phenyl-2(1H)-pyridone; PFD]
is a pyridine compound that exerts anti-fibrotic effects in
numerous fibrotic diseases (11,12). However, the exact underlying
mechanisms of action of PFD are not yet fully understood. A
previous study identified that PFD inhibited the apoptosis of renal
tubular cells by maintaining mitochondrial membrane stability,
thereby inhibiting the mitochondrial apoptotic signaling pathway
(13). Notably, another study
demonstrated that PFD significantly reduced the electrical
stimulation-induced protein expression levels of Cx43 and α-actin 2
in bone marrow-derived mesenchymal stem cells (14). In a hepatic ischemia/reperfusion
injury model, a study identified that PFD reduced SRY-related high
mobility group-Box gene 9 overexpression-induced inflammation and
apoptosis (15). However, the
question of whether PFD is involved in the regulation of the
cardiomyocyte apoptosis signaling pathway requires further
investigation.
The aim of the present study was to investigate the
cardio-protective effects of PFD on the Hcy-induced apoptosis of
H9C2 cells. It was hypothesized that PFD protects H9C2 rat
cardiomyocytes against Hcy-induced apoptosis by modulating the Cx43
signaling pathway.
Materials and methods
Cell culture and drug administration
H9C2 rat cardio-myocytes were purchased from the
Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences. The H9C2 cells were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) and supplemented with 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) at 37°C in a humidified 5%
CO2 incubator (4).
Upon reaching 80% confluency, the cells were subjected to the
experimental procedures. H9C2 cells cultured in DMEM/FBS without
any treatment served as the control group. H9C2 cells were exposed
to Hcy (0.5, 1, 2 or 3 mmol/l; Sigma-Aldrich; Merck KGaA) for 24 h
to determine the Hcy treatment concentrations (4). The cells were pre-treated with PFD
(Sigma-Aldrich; Merck KGaA) for 30 min followed by exposure to Hcy
(3 mmol/l) for 24 h. In addition, the H9C2 cells were treated with
the Cx43 agonist, AAP10 (Chinese Peptide Co., 50 nmol/l, 1 h), or
the Cx43 inhibitor, Gap26 (cat. no. A1044, APExBIO Technology LLC,
0.5 µmol/l, 30 min), prior to treatment with Hcy. The
concentration with maximal protective effects was determined.
Cell viability assay
The viability of the H9C2 rat cardiomyocytes was
evaluated using a CCK-8 assay kit (MultiSciences Lianke), according
to the manufacturer's protocol. H9C2 cells (1×104/well)
seeded in 96-well plates were exposed to Hcy (0.5, 1, 2 or 3
mmol/l) for 24 h. For protective treatment, the cells were
pre-treated with PFD (0.1, 0.5, 1 or 1.5 mg/ml) for 30 min and
incubated with Hcy for 24 h at 37°C. Subsequently, 10 µl
CCK-8 solution were added to each well, the cells were incubated at
37°C for an additional 2 h, and cell viability was detected using a
microplate spectrophotometer (BioTek Instruments, Inc.).
Flow cytometric analysis
The Annexin V-FITC Apoptosis Detection kit
(MultiSciences Lianke) was used to detect apoptotic cells,
according to the manufacturer's protocol. Briefly, the cells were
washed with PBS and harvested by trypsinization. The cells
(1-2×106) were then incubated with 500 µl 1X
binding buffer, 5 µl Annexin V-FITC and 10 µl PI
solution. The cells were then gently shaken and incubated at 4°C
for 30 min in the dark. The quantitative analysis of apoptosis was
conducted using a flow cytometer (Bio-Rad Laboratories, Inc.).
Western blot analysis
The treated H9C2 cardiomyocytes were removed from
the CO2 incubator and the medium was discarded. The
cells were uniformly plated in a 6-well plate, treated, lysed and
the protein concentration was estimated by BCA protein assay. The
proteins (50 µg/lane) were separated by 10 or 12% SDS-PAGE,
then the proteins were transferred to a 0.45-µm PVDF
membrane (EMD Millipore) and immersed in blocking solution for 2 h
at room temperature. Subsequently, the membranes were incubated
overnight at 4°C with the following primary antibodies: Rabbit
anti-Cx43 (cat. no. ab11370; 1:1,000; Abcam), rabbit anti-Bax (cat.
no. ab199677; 1:1,000; Abcam), mouse anti-GAPDH (cat. no. ab8245;
1:1,000; Abcam), rabbit anti-Bcl-2 (cat. no. ab196495; 1:1,000;
Abcam) and rabbit anti-caspase-3 (cat. no. ab13847; 1:1,000;
Abcam). The blots were then washed with TBST and incubated with the
secondary antibody (cat. no. ZB-2306; cat. no. ZB-2305; 1:10,000;
Beijing Zhongshan Jinqiao Biotechnology Co.) at room temperature
for 2 h. Following 3 washes with TBST for 5 min each, the blots
were developed using enhanced chemiluminescence (ECL) reagent (GE
Healthcare Life Sciences) in a dark room. After images were
acquired, analysis was performed using Quantity One software
(Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
Each group of cells was collected and washed twice
with PBS, and total RNA was extracted using the TRIzol kit (Thermo
Fisher Scientific, Inc.). The concentration of RNA was detected
using an ultraviolet spectrophotometer (NanoDrop 2000, Thermo
Fisher Scientific, Inc.), and complementary DNA was reverse
transcribed using a capacity cDNA reverse transcription kit (Thermo
Fisher Scientific, Inc.) for qPCR. The gene primers used were as
follows: Bax forward, 5′-CCA GGA CGC ATC CAC CAA GAA G-3′ and
reverse, 5′-GCT GCC ACA CGG AAG AAG ACC-3′; Bcl-2 forward, 5′-GGT
GTG CAG ATG CCG GTT CAG-3′ and reverse, 5′-ACG GTG GTG GAG GAA CTC
TTC AG-3′; caspase-3 forward, 5′-GTA CAG AGC TGG ACT GCG GTA TTG-3′
and reverse, 5′-AGT CGG CCT CCA CTG GTA TCT TC-3′; Cx43 forward,
5′-GGA AGC ACC ATC TCC AAC TC-3′ and reverse, 5′-GTA CAG AGC TGG
ACT GCG GTA TTG-3′; and β-actin forward, 5′-CAT GTA CGT TGC TAT CCA
GGC-3′ and reverse, 5′-CTC CTT AAT GTC ACG CAC GAT-3′. The
thermocycling conditions for qPCR were as follows: UDG enzyme
activation at 50°C for 2 min, pre-denaturation at 95°C for 2 min,
denaturation at 95°C for 15 sec, annealing/extension at 60°C for 1
min, for a total of 40 cycles. The results were analyzed using the
2−ΔΔCq method (16).
Immunofluorescence assay
H9C2 rat cardiomyocytes were uniformly plated at a
density of 3×105/ml in a 6-well plate with sterile
coverslips to prepare cell slides. After 24 h, the medium was
discarded following treatment, and the cells were washed gently
with PBS and fixed with paraformaldehyde (40 g/l) for 15 min. The
H9C2 rat cardiomyocytes were rewashed with PBS and permeabilized
using Triton X-100 (2 g/l) for 3 min. Subsequently, the cells were
rewashed with PBS and incubated with BSA (Sigma-Aldrich; Merck
KGaA; 50 g/l) at room temperature for 30 min. The primary
antibodies [rabbit anti-Cx43 (cat. no. ab11370; 1:100; Abcam),
rabbit anti-Bax (cat. no. ab199677; 1:100; Abcam), rabbit
anti-Bcl-2 (cat. no. ab196495; 1:100; Abcam) and rabbit
anti-caspase-3 (cat. no. ab13847; 1:100; Abcam)] was added, and the
negative control group was incubated with PBS. The sample was
placed in a wet box overnight at 4°C. The sample was then incubated
for 30 min at 37°C, and the primary antibody was discarded. The
sample was washed with PBS on a bleaching shaker, and then
incubated with a goat anti-rabbit secondary antibody (cat. no.
12-448; 1:50; Sigma-Aldrich) for 1 h at 37°C. Subsequently, the
secondary antibody was discarded, and the sample was washed with
PBS 3 times for 5 min each time. The cells were then washed with
PBS, and the nuclei were stained with DAPI (Solarbio, Inc.) for 15
min at 37°C. Confocal microscopy (Zeiss LSM 510 META, Carl Zeiss
AG) was performed to analyze the results (AimImage Examiner; Zeiss
LSM Image Examiner version 4.2.0.121).
Statistical analysis
SPSS 20.0 (IBM Corp.) was used for all data
analyses. All values are expressed as the means ± standard error.
Analysis among multiple groups was performed by one-way analysis of
variance followed by Tukey's post hoc tests, as appropriate.
P<0.05 was considered to indicate a statistically significant
difference.
Results
PFD prevents Hcy-induced cardiomyocyte
cytotoxicity
To investigate the effect of PFD on Hcy-induced H9C2
cell cytotoxicity, the H9C2 cells were cultured with various
concentrations (0, 0.1, 0.5, 1, 2 and 3 mmol/l) of Hcy and various
concentrations of PFD (0, 0.1, 0.5, 1 and 1.5 mg/ml) for 24 h. The
results demonstrated that exposure to 1-3 mmol/l Hcy significantly
decreased cell viability compared with the control group (Fig. 1A); treatment with PFD at 0.1-1
mg/ml demonstrated no obvious cytotoxicity towards the H9C2 cells;
however, PFD decreased cell viability at the concentration of 1.5
mg/ml (Fig. 1B). The present
study then pre-treated the cells with various concentrations of PFD
for 30 min, followed by incubation with Hcy for 24 h. Compared with
the Hcy group, the cell viability of the co-treatment group
increased in a concentration-dependent manner (Fig. 1C). Hcy at 3 mmol/l and PFD at 1
mg/ml were selected for further study. Finally, the cell morphology
in each treatment group was observed by microscopy. Cardiomyocytes
exposed to 3.0 mmol/l Hcy for 24 h exhibited a disorder of cell
alignment and extensive nuclear pyknosis; however, the cell state
of the co-treatment group was significantly improved (Fig. 1D). These results demonstrated that
PFD alleviated Hcy-induced cytotoxicity in H9C2 cells.
Effects of PFD on the Hcy-induced
apoptosis, and increased Bax/Bcl2 and cleaved caspase-3 expression
levels in H9C2 cells
To investigate the protective effects of PFD on
Hcy-induced cardiomyocyte apoptosis, H9C2 rat cardiomyocytes were
pre-treated with PFD for 30 min followed by Hcy incubation for 24
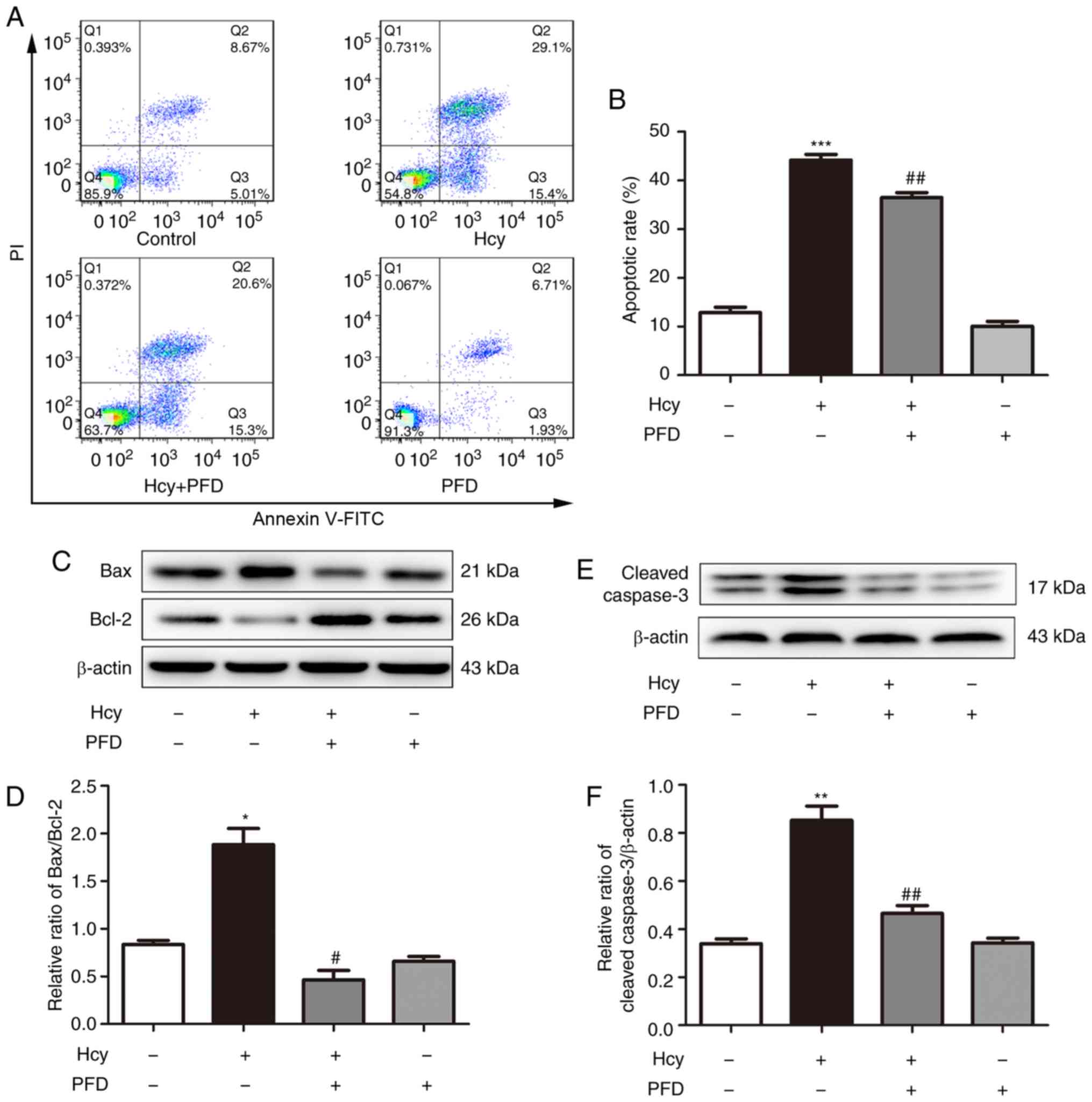
h. Annexin V/PI analysis was then performed. As presented in
Fig. 2A and B, exposure to Hcy
significantly increased the apoptotic rate of the cardiomyocytes,
while the effect was significantly inhibited following PFD
pre-treatment. Furthermore, the expression levels of related
proteins were significantly altered. Hcy upregulated the Bax/Bcl-2
ratio and increased the protein levels of cleaved caspase-3
compared with the control group (Fig.
2C-F); these effects were suppressed by PFD. PFD pre-treatment
reduced the level of cleaved caspase-3, suggesting that PFD can
inhibit the apoptotic signaling pathway induced by Hcy. The
expression and distribution of Bax, Bcl-2 and caspase-3 in the H9C2
cardiomyocytes was detected by immunofluorescence staining. The
results demonstrated that Bax and caspase-3 were predominantly
distributed in the cytoplasm (Fig. 3A
and D), and Bcl-2 was mainly distributed in the nuclear
membrane (Fig. 3B). The changes
in the expression of Bax, Bcl-2 and caspase-3 in each group were
consistent with the results of western blot analysis (Fig. 3C and E). At the same time, RT-qPCR
revealed an increase in the ratio of Bax/Bcl-2 and caspase-3 mRNA
following exposure to Hcy in the H9C2 cells, whereas these effects
were prevented by pre-treatment with PFD (Fig. 3F and G).
Cx43 is involved in the Hcy-induced
apoptosis of H9C2 cardiomyocytes
It has been found that Cx43 is involved in the
process of cardiomyocyte apoptosis (8). In investigating the protective
mechanisms of PFD in Hcy-induced H9C2 cell injury, it was observed
that PFD pre-treatment downregulated the Cx43 protein levels
(Fig. 4A and B). The results of
immunofluorescence staining of the H9C2 cells demonstrated that
Cx43 was predominantly distributed in the cytoplasm and cell
membrane (Fig. 4C). The Cx43
fluorescence intensity and mRNA levels of each group were
consistent with the results of western blot analysis (Fig. 4D and E). Subsequently, the H9C2
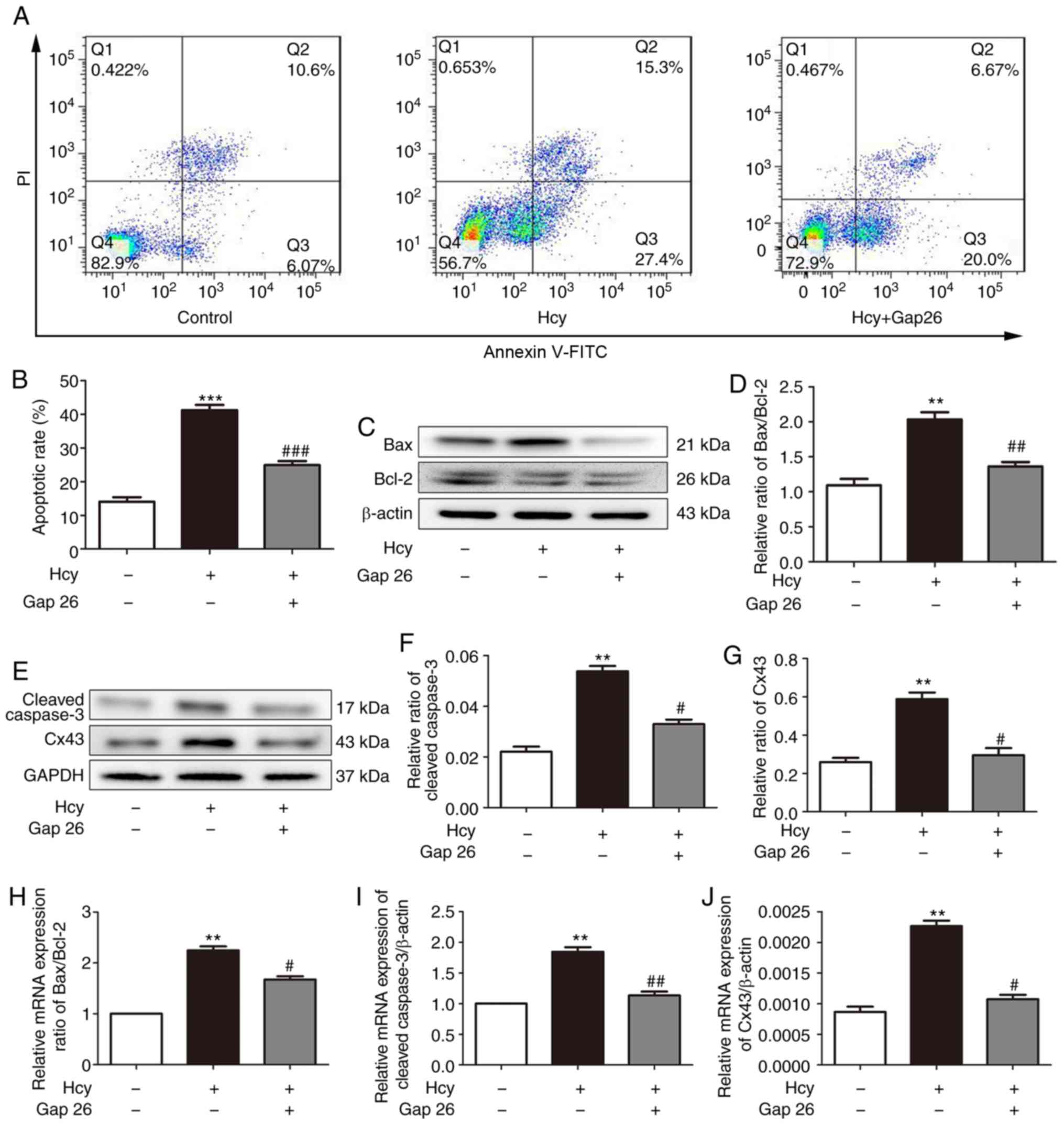
cardiomyocytes were pre-treated with the Cx43 inhibitor, Gap26 (0.5
µmol/l, 30 min), and then exposed to Hcy for 24 h for the
following experiments. As shown in Fig. 5A and B, the apoptotic rate was
significantly increased following exposure to Hcy, while it was
significantly decreased following Gap26 pre-treatment. Moreover,
Hcy increased the Bax/Bcl2 ratio and increased the expression
levels of cleaved caspase-3 and Cx43. These effects were prevented
by Gap26 (Fig. 5C-G). The changes
in the mRNA expression levels of Bax, Bcl-2, caspase-3 and Cx43 in
each group were consistent with those of western blot analysis
(Fig. 5H-J). These results
demonstrated that Cx43 is involved in Hcy-induced H9C2
apoptosis.
PFD protects the Hcy-induced apoptosis of
H9C2 cardiomyocytes via the Cx43 pathway
To investigate whether Cx43 is involved in
PFD-regulated protection against Hcy-induced H9C2 cell injury, H9C2
cardiomyocytes were pre-treated with the Cx43 agonist, AAP10 (50
nmol/l, 1 h) and Hcy for the following experiments. Compared with
the Hcy group, the rate of apoptosis decreased following
pre-treatment with PFD, and this effect was prevented by AAP10
(Fig. 6A and B). Furthermore, Hcy
increased the ratio of Bax/Bcl2 and upregulated the expression
levels of cleaved caspase-3 and Cx43. These effects were prevented
by PFD. Furthermore, the effects of PFD were prevented by AAP10
(Figs. 6C-F, and 7A and B). Compared with PFD
pre-treatment, the Cx43 agonist, AAP10, also increased the mRNA
ratio of Bax/Bcl-2, and increased the mRNA expression levels of
cleaved caspase-3 and Cx43 (Fig.
7C-E). The results demonstrated that PFD protects H9C2
cardiomyocytes against Hcy-induced apoptosis via the Cx43
pathway.
Discussion
The present study observed that PFD attenuates
Hcy-induced cardiomyocyte apoptosis by modulating the Cx43
signaling pathway. A recent study reported that PFD can reduce
tubular cell apoptosis by inhibiting mitochondrial apoptotic
signaling pathways (13).
However, the mechanism of the protective effect of PFD on
myocardial apoptosis has not yet been determined.
In recent years, apoptosis has been found to be
widely involved in the pathological processes of various
cardiovascular diseases, such as atherosclerosis, heart failure and
arrhythmias. Elevated levels of Hcy in plasma are a risk factor for
cardiovascular disease and can cause a range of pathological
processes. Several studies have demonstrated that Hcy can activate
the apoptotic signaling pathway in cardiomyocytes (4-6).
In the present study, first, cytotoxicity was measured to obtain
understanding into the Hcy-induced toxic effects on cardiomyocytes.
The results demonstrated that exposure to 3 mmol/l Hcy can
significantly reduce cell viability. Cardiomyocyte apoptosis can be
regulated by Bcl-2 family proteins (17,18). Numerous studies have reported that
Hcy can enhance apoptosis in a variety of cell types, including
endothelial cells, mesangial cells and human umbilical vein
endothelial cells (19-21). The present study found that Hcy
increased the ratio of Bax/Bcl-2, promoted caspase-3 activation and
induced cardiomyocyte apoptosis.
PFD has been shown to reduce the fibrosis of
different organs, including the lungs, kidney and heart (22,23). A recent study demonstrated that
PFD exerts cardiovascular protective effects in a variety of
noxious stimulation models (24).
In a cardiac hypertrophic mouse model induced by Ang II infusion,
echocardiography exhibited a significant increase in left
ventricular hypertrophy in the vehicle group compared with the
control group. Notably, PFD significantly inhibited this effect
(24). These results demonstrated
that PFD exerts a certain protective effect on cardiovascular
disease; however, the specific mechanisms have not yet been
determined. In this study, it was found that treatment with PFD at
0.1-1 mg/ml had no significant effect on H9c2 cell viability. At
the same time, compared with Hcy treatment alone, the cell
viability of the PFD pre-treatment group increased in a
concentration-dependent manner. Chen et al found that PFD
significantly inhibited HK2 cell apoptosis in a dose-dependent
manner (13). The results of the
present study are consistent with those of this previous study. Of
note, it was found that the PFD (1.5 mg/ml) and Hcy co-treatment
group exhibited an enhanced viability even they exhibited
cytotoxicity. This study found similar findings to those of
previous studies. Tsuchiya et al found that PFD increased
the survival rate of LPS-treated rats at 24 h dose dependently;
however, PFD did not lead to a further increase in the survival
rate at 650 mg/kg (25).
Gefitinib and cisplatin are commonly used anticancer drugs that
inhibit the growth of tumor cells. Tsai et al found that the
simultaneous administration of gefitinib and cisplatin in a panel
of previously untreated non-small cell lung cancer cell lines
caused overall antagonism, which was directly associated with
gefitinib sensitivity (26).
Thus, it was hypothesized that co-treatment with PFD and Hcy may
counteract the toxic effects; however, the specific mechanisms
warrant further investigation. Therefore, PFD can alleviate
Hcy-induced cytotoxicity in H9C2 cells.
In a nephrotoxicity animal model, researchers
observed that PFD was capable of partly reversing the pro-apoptotic
properties of chronic cyclosporine A (CsA) by decreasing the
expression of p53 and Fas-L and increasing that of the survival
gene Bcl-xL (27). Another study
found that PFD treatment inhibited caspase-9 and caspase-3 cleavage
in renal proximal tubular cells (13). The results of the present study
revealed that PFD pre-treatment reduced the ratio of Bax/Bcl-2 in
the Hcy-induced cardiomyocyte apoptosis model. At the same time,
the activation of caspase-3 was suppressed by PFD. These results
demonstrate that PFD can protect against Hcy-induced H9C2
myocardial cell apoptosis by reducing the expression of
pro-apoptotic factors; however, the specific regulatory mechanisms
have not yet been elucidated.
Cell communication refers to the transmission of
information from one cell to another through a medium, which
produces a corresponding response. The maintenance of homeostasis
is controlled by utilizing the interaction between extracellular,
intracellular and intercellular signaling pathways (28). Gap junctions are a special
membrane structure that connects adjacent cells. These junctions
have a wide distribution range and exist in the majority of animal
cells. Connexin is the basic building block of the gap junction
channel (17). Cx43 is widely
involved in the development of diseases, such as the regulation of
cell growth, proliferation, apoptosis and homeo-stasis (17,29). Connexin mimetic peptides, such as
Gap26, are known to be inhibitors of gap junction channels
(30). It has been indicated that
the Cx43 agonist resists-arrhythmias peptide, AAP10, improves
gap-junctional intercellular coupling in human cardiomyocytes and
prevents separation caused by acidification (31). A recent study demonstrated that
Cx43 is a key regulator of cardiomyocyte apoptosis under
pathological and physiological conditions (8). To investigate the protective
mechanisms of PFD in Hcy-induced H9C2 cell injury, the present
study found a significant decrease in the Cx43 protein level
following PFD pre-treatment. Subsequently, the H9C2 cardiomyocytes
were pre-treated with the Cx43 inhibitor, Gap26, and incubated with
Hcy. Hcy increased the ratio of Bax/Bcl2 and upregulated the
protein expression levels of cleaved caspase-3 and Cx43. These
effects were prevented by Gap26. In a model of intestinal damage, a
previous study found that intestinal damage can be significantly
relieved following administration of Cx43 inhibitors (32). Another study demonstrated that
stretch-induced apoptosis in human trabecular meshwork cells is
accompanied by upregulation of Cx43 (33). The present findings indicated that
Cx43 is involved in the apoptosis of Hcy-induced H9C2
cardiomyocytes. The beneficial effect of PFD was then abolished
using the connexin agonist AAP10. The present study found that Hcy
induces apoptosis of H9C2 cardiomyocytes through Cx43, and this
phenomenon can be prevented by PFD, which can aggravate
cardiomyocyte apoptosis following administration of the connexin
agonist AAP10.
Cx43 phosphorylation/dephosphorylation plays an
important role in regulating cell survival and death processes.
Evidence has also demonstrated that high levels of phosphorylated
Cx43 mainly regulates cell survival. Cx43 dephosphorylation is
involved in numerous pathological processes. Yang et al
found that Cx43 phosphorylation at S282 mediates cardiomyocyte
survival and S282 dephosphorylation induces cardiomyocyte apoptosis
under normal conditions (8). The
present study had some limitations. It is important to perform an
assay for Cx43 phosphorylation in future studies.
In conclusion, the present study elucidated the
protective role of PFD in cardiomyocyte apoptosis. The findings
demonstrated that PFD modulates the expression of apoptosis-related
proteins through the Cx43 signaling pathway to attenuate
Hcy-induced apoptosis in H9C2 cells. These results provide new
insight into the clinical application of PFD in the treatment of
cardiovascular diseases.
Funding
The study was supported by the National Natural
Science Foundation of China (grant nos. 81860286 and 81660271 to
KM), Shihezi University International Science and Technology
Cooperation Promotion Project (grant no. GJHZ201603 to KM), Corps
Young and Middle-aged Science and Technology Innovation Leadership
Program of China (grant no. 2016BC006 to KM) and Hospital Level Key
Area Innovation Team Project (The First Affiliated Hospital of
Shihezi University Medical College, grant no. TJ2016-001 to
LW).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
KM and LW conceived and designed the experiments.
YO, JS and LL were involved in the study design. KC, LC, YO, LZ and
XL performed the experiments. KC and LC analyzed the data. KC, YO
and LC wrote and revised the manuscript. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank the Key Laboratory
of Xinjiang Endemic and Ethnic Diseases, the 3rd Department of
Cardiology, the First Affiliated Hospital of the Medical College,
Shihezi University and the Departments of Physiology and
Pathophysiology of Shihezi University School of Medicine for their
assistance.
References
|
1
|
Shekhar A, Heeger P, Reutelingsperger C,
Arbustini E, Narula N, Hofstra L, Bax JJ and Narula J: Targeted
imaging for cell death in cardiovascular disorders. JACC Cardiovasc
Imaging. 11:476–493. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Madonna R, Balistreri CR, De Rosa S,
Muscoli S, Selvaggio S, Selvaggio G, Ferdinandy P and De Caterina
R: Impact of sex differences and diabetes on coronary
atherosclerosis and ischemic heart disease. J Clin Med. 8:pii: E98.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Luo F, Das A, Chen J, Wu P, Li X and Fang
Z: Metformin in patients with and without diabetes: A paradigm
shift in cardiovascular disease management. Cardiovasc Diabetol.
18:542019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aminzadeh A and Mehrzadi S:
Cardioprotective effect of levosimendan against
homocysteine-induced mitochondrial stress and apoptotic cell death
in H9C2. Biochem Biophys Res Commun. 507:395–399. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan CD, Sun JY, Fu XT, Hou YJ, Li Y, Yang
MF, Fu XY and Sun BL: Astaxanthin attenuates homocysteine-induced
cardiotoxicity in vitro and in vivo by inhibiting mitochondrial
dysfunction and oxidative damage. Front Physiol. 8:10412017.
View Article : Google Scholar
|
|
6
|
Zhang Z, Zhao L, Zhou Y, Lu X, Wang Z,
Wang J and Li W: Taurine ameliorated homocysteine-induced H9C2
cardiomyocyte apoptosis by modulating endoplasmic reticulum stress.
Apoptosis. 22:647–661. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ostrakhovitch EA and Tabibzadeh S:
Homocysteine and age-associated disorders. Ageing Res Rev.
49:144–164. 2019. View Article : Google Scholar
|
|
8
|
Yang Y, Yan X, Xue J, Zheng Y, Chen M, Sun
Z, Liu T, Wang C, You H and Luo D: Connexin43 dephosphorylation at
serine 282 is associated with connexin43-mediated cardiomyocyte
apop-tosis. Cell Death Differ. 26:1332–1345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Vozzi C, Dupont E, Coppen SR, Yeh HI and
Severs NJ: Chamber-related differences in connexin expression in
the human heart. J Mol Cell Cardiol. 31:991–1003. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li H, Brodsky S, Kumari S, Valiunas V,
Brink P, Kaide J, Nasjletti A and Goligorsky MS: Paradoxical
overexpression and translocation of connexin43 in
homocysteine-treated endothelial cells. Am J Physiol Heart Circ
Physiol. 282:H2124–H2133. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Komiya C, Tanaka M, Tsuchiya K, Shimazu N,
Mori K, Furuke S, Miyachi Y, Shiba K, Yamaguchi S, Ikeda K, et al:
Antifibrotic effect of pirfenidone in a mouse model of human
nonalcoholic steatohepatitis. Sci Rep. 7:447542017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Stahnke T, Kowtharapu BS, Stachs O,
Schmitz KP, Wurm J, Wree A, Guthoff RF and Hovakimyan M:
Suppression of TGF-β pathway by pirfenidone decreases extracellular
matrix deposition in ocular fibroblasts in vitro. PLoS One.
12:e01725922017. View Article : Google Scholar
|
|
13
|
Chen JF, Liu H, Ni HF, Lv LL, Zhang MH,
Zhang AH, Tang RN, Chen PS and Liu BC: Improved mitochondrial
function underlies the protective effect of pirfenidone against
tubulointerstitial fibrosis in 5/6 nephrectomized rats. PLoS One.
8:e835932013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
He X, Li L, Tang M, Zeng Y, Li H and Yu X:
Biomimetic electrical stimulation induces rat bone marrow
mesenchymal stem cells to differentiate into cardiomyocyte-like
cells via TGF-beta 1 in vitro. Prog Biophys Mol Biol. 148:47–53.
2019. View Article : Google Scholar
|
|
15
|
Fan XD, Zheng HB, Fan XS and Lu S:
Increase of SOX9 promotes hepatic ischemia/reperfusion (IR) injury
by activating TGF-β1. Biochem Biophys Res Commun. 503:215–221.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCq method. Methods. 25:402–408. 2001. View Article : Google Scholar
|
|
17
|
Decrock E, Vinken M, De Vuyst E, Krysko
DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V and Leybaert
L: Connexin-related signaling in cell death: To live or let die?
Cell Death Differ. 16:524–536. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pollack M, Phaneuf S, Dirks A and
Leeuwenburgh C: The role of apoptosis in the normal aging brain,
skeletal muscle, and heart. Ann N Y Acad Sci. 959:93–107. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li J, Luo M, Xie N, Wang H and Wang J:
Bioinformatics-based analysis of the involvement of AC005550.3,
RP11-415D17.3, and RP1-140K8.5 in homocysteine-induced vascular
endothelial injury. Am J Transl Res. 10:2126–2136. 2018.PubMed/NCBI
|
|
20
|
Majumder S, Ren L, Pushpakumar S and Sen
U: Hydrogen sulphide mitigates homocysteine-induced apoptosis and
matrix remodelling in mesangial cells through Akt/FOXO1 signalling
cascade. Cell Signal. 61:66–77. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Hong Y, Zhang C, Shen Y, Pan YS,
Chen RZ, Zhang Q and Chen YH: Picroside II attenuates
hyperhomocysteinemia-induced endothelial injury by reducing
inflammation, oxidative stress and cell apoptosis. J Cell Mol Med.
23:464–475. 2019. View Article : Google Scholar
|
|
22
|
Neri T, Lombardi S, Faìta F, Petrini S,
Balìa C, Scalise V, Pedrinelli R, Paggiaro P and Celi A:
Pirfenidone inhibits p38-mediated generation of procoagulant
microparticles by human alveolar epithelial cells. Pulm Pharmacol
Ther. 39:1–6. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Z, Liu X, Wang B, Nie Y, Wen J, Wang Q
and Gu C: Pirfenidone suppresses MAPK signalling pathway to reverse
epithelial-mesenchymal transition and renal fibrosis. Nephrology
(Carlton). 22:589–597. 2017. View Article : Google Scholar
|
|
24
|
Yamazaki T, Yamashita N, Izumi Y, Nakamura
Y, Shiota M, Hanatani A, Shimada K, Muro T, Iwao H and Yoshiyama M:
The antifibrotic agent pirfenidone inhibits angiotensin II-induced
cardiac hypertrophy in mice. Hypertens Res. 35:34–40. 2012.
View Article : Google Scholar
|
|
25
|
Tsuchiya H, Kaibori M, Yanagida H,
Yokoigawa N, Kwon AH, Okumura T and Kamiyama Y: Pirfenidone
prevents endo-toxin-induced liver injury after partial hepatectomy
in rats. J Hepatology. 40:94–101. 2004. View Article : Google Scholar
|
|
26
|
Tsai CM, Chen JT, Stewart DJ, Chiu CH, Lai
CL, Hsiao SY, Chen YM and Chang KT: Antagonism between gefitinib
and cisplatin in non-small cell lung cancer cells why randomized
trials failed? J Thorac Oncol. 6:559–568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shihab FS, Bennett WM, Yi H and Andoh TF:
Effect of pirfeni-done on apoptosis-regulatory genes in chronic
cyclosporine nephrotoxicity. Transplantation. 79:419–426. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Scemes E, Spray DC and Meda P: Connexins,
pannexins, innexins: Novel roles of 'hemi-channels'. Pflugers Arch.
457:1207–1226. 2009. View Article : Google Scholar
|
|
29
|
Cooreman A, Van Campenhout R, Ballet S,
Annaert P, Van Den Bossche B, Colle I, Cogliati B and Vinken M:
Connexin and pannexin (hemi)channels: Emerging targets in the
treatment of liver disease. Hepatology. 69:1317–1323. 2019.
View Article : Google Scholar
|
|
30
|
Wang N, De Bock M, Antoons G, Gadicherla
AK, Bol M, Decrock E, Evans WH, Sipido KR, Bukauskas FF and
Leybaert L: Connexin mimetic peptides inhibit Cx43 hemichannel
opening triggered by voltage and intracellular Ca2+ elevation.
Basic Res Cardiol. 107:3042012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hagen A, Dietze A and Dhein S: Human
cardiac gap-junction coupling: Effects of antiarrhythmic peptide
AAP10. Cardiovasc Res. 83:405–415. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zou Z, Liu B, Zeng L, Yang X, Huang R, Wu
C, Zhu H, Gao Y, Yuan D and Yu J: Cx43 inhibition attenuates
sepsis-induced intestinal injury via downregulating ROS transfer
and the activation of the JNK1/Sirt1/FoxO3a signaling pathway.
Mediators Inflamm. 2019:78543892019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tellios N, Feng M, Chen N, Liu H, Tellios
V, Wang M, Li X, Chang CA and Hutnik C: Mechanical stretch
upregulates connexin43 in human trabecular meshwork cells. Clin Exp
Ophthalmol. 47:787–794. 2019.PubMed/NCBI
|