Introduction
Cardiovascular diseases (CVDs) are a common threat
to human health and were the leading cause of human mortality
between 2009 and 2011 worldwide (1,2).
Ischemic heart disease, which accounts for a large subset of CVDs,
leads to hypoxia and, ultimately, cardiomyocyte death (3). Timely reperfusion is the main
therapeutic strategy to resuscitate the ischemic or hypoxic
myocardium (4). Unfortunately,
reperfusion also elicits a number of adverse reactions, including
ischemia/reperfusion (I/R) injury (5,6).
An increasing body of evidence suggests that apoptosis is initiated
shortly after the onset of ischemia and becomes markedly enhanced
during reperfusion (7,8). Therefore, strategies aimed at
preventing or delaying cardiomyocyte apoptosis may be advisable for
the treatment of ischemic heart disease, especially myocardial I/R
injury.
Multiple biological processes and cell signaling
pathways are involved in myocardial I/R injury. The increased
generation of oxygen free radicals is a major contributor to the
initiation and development of I/R injury (9). Excessive reactive oxygen species
(ROS) production causes the opening of the mitochondrial
permeability transition pore, DNA damage and lipid peroxidation,
leading to apoptosis (10).
Peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α
(PGC-1α) has been demonstrated to suppress oxidative stress and
delay the progression of heart failure (11). Nuclear factor erythroid 2-related
factor 2 (Nrf2) promotes the transcription of key antioxidant
genes, such as heme oxygenase-1 (HO-1) and NADPH quinone oxido
reductase 1 (NQO1), to prevent cardiomyocyte damage from oxidative
stress (12). PGC-1α/Nrf2
signaling is as a potential target for cardioprotection in
myocardial I/R injury (13).
Several pro-inflammatory cytokines in the serum have also been
demonstrated to be upregulated during myocardial I/R injury,
including tumor necrosis factor-α (TNF-α) (14). TNF-α contributes to post-ischemic
myocardial dysfunction by directly inhibiting contractility and
inducing cardiomyocyte apoptosis (15). The PPARγ/PGC-1α/TNF-α pathway has
been implicated in the attenuation of oxygen-glucose
deprivation/reperfusion (OGD/R)-induced myocardial injury (16). Therefore, the use of antioxidant
and anti-inflammatory compounds is considered to be a promising
therapeutic strategy for ischemic heart disease.
Melatonin (Mel; N-acetyl-5-methoxytryptamine), which
is mainly produced by the pineal gland, is a powerful endogenous
antioxidant due to its direct free-radical scavenging and indirect
antioxidant activities (17,18). Due to its amphiphilic property,
Mel can easily penetrate all morphophysiological barriers and enter
all subcellular compartments to influence the physiological
functions of the majority of organs, including the heart (19). Previous studies have indicated
that Mel serves a pivotal role in the treatment of myocardial I/R
injury (20–24). Mel exerts its cardioprotective
effects through its antioxidant and antiapoptotic properties
(22–24) and the preservation of
mitochondrial function (25).
However, the underlying mechanisms through which Mel prevents
OGD/R-induced cardiomyocyte damage remain to be further
elucidated.
The present study aimed to investigate the potential
protective effects of Mel on OGD/R-induced H9c2 cardiomyocyte
injury and the underlying molecular mechanisms.
Materials and methods
Cell culture
A H9c2 rat cardiomyocyte cell line was purchased
from American Type Culture Collection (cat. no. CRL-1446) and
cultured in Dulbecco’s modified Eagle’s medium (DMEM; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (Thermo
Fisher Scientific, Inc.), 2 mM glutamine, 100 U/ml penicillin and
100 mg/ml streptomycin (all from Merck KGaA) at 37°C in a
humidified incubator containing 5% CO2.
Cell transfection
The small interfering RNA (siRNA) duplexes
corresponding to rat PGC-1α (siPGC-1α), Nrf2 (siNrf2) and the
negative control siRNA (siNC) were purchased from Santa Cruz
Biotechnology, Inc. siRNAs (100 nM) were transfected into H9c2
cells using Lipofectamine® 2000 reagent (Thermo Fisher
Scientific, Inc.), according to the manufacturer’s instructions. In
brief, H9c2 cells (5×105) were plated in a 6-well plate
and cultured for 24 h. When the cells reached 80% confluence,
Lipofectamine 2000 was added to the medium without serum and
incubated for 5 min at room temperature. The diluted siRNA was
gently mixed with the medium containing Lipofectamine 2000 and
incubated for 20 min at room temperature prior to adding the
mixture to each well containing cells and medium. The transfected
cells were cultured at 37°C in a CO2 incubator for 24 h
prior to subsequent experiments. Untransfected cells were used as a
blank control, and cells transfected with siNC were used as a
negative control.
OGD/R cell model and Mel treatment
H9c2 cells were transfected with siPGC-1α, siNrf2 or
siNC mimic 24 h prior to OGD stimulation. For the OGD/R
experiments, when the transfected H9c2 cells reached 70–80%
confluence, the culture medium was replaced by glucose-free DMEM
(Thermo Fisher Scientific, Inc.) and the cells were cultured in an
anaerobic chamber (95% N2 and 5% CO2) at 37°C
for 4 h. The cells were then incubated with normal culture medium
under normoxic conditions (95% air and 5% CO2) at 37°C
for the indicated duration (6, 12, 24 and 48 h) as reperfusion.
Different concentrations of Mel (0.01, 0.1, 1 and 10 mM; Merck
KGaA) or 1 μg/ml TNF-α antibody (cat. no. 11948; Cell
Signaling Technology, Inc.) was added into the culture medium at
the initiation of reperfusion. Untreated cells were used as the
control.
Cell viability assay
Cell viability was measured by Cell Counting Kit-8
(CCK-8; Beyotime Institute of Biotechnology) according to the
manufacturer’s instructions. In brief, H9c2 cells were seeded into
96-well plates at a density of 1×104 cells/well.
Following treatment, 10 μl CCK-8 solution was added to each
well and incubated for 2 h at 37°C. The absorbance was measured at
450 nm using a microplate reader (Bio-Rad Laboratories, Inc.) to
calculate the number of viable cells.
Measurement of lactate dehydrogenase
(LDH) and creatine kinase myocardial band (CK-MB) activity
The levels of LDH and CK-MB in the supernatant of
H9c2 cells were assessed using commercial kits (Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer’s
instructions. Briefly, H9c2 cells were subjected to 4 h of OGD
followed by reperfusion for 24 h. Different concentrations of Mel
(0.01, 0.1, 1 and 10 mM) were added to the culture medium at the
initiation of reperfusion and incubated for the indicated durations
(6, 12, 24 and 48 h). The cells were harvested and centrifuged at
250 × g at room temperature for 2 min. A total of 100 μl
working solution was added to each well and reacted for 30 min at
room temperature. The reaction was stopped by adding 50 μl
stop solution to each well. Absorbance was measured at 450 and 340
nm using a microplate reader (Bio-Rad Laboratories, Inc.).
Flow cytometry to determine
apoptosis
Apoptosis was measured using an Annexin
V-fluorescein isothiocyanate (FITC) and propidium iodide (PI)
detection kit (BD Biosciences). Briefly, following the indicated
treatments, H9c2 cells were harvested, washed thrice with cold PBS
and resuspended in binding buffer (BD Biosciences), followed by
staining with 5 μl Annexin V-FITC in the dark for 10 min at
37°C. The cells were then incubated with 10 μl PI solution
in the dark for 30 min. The apoptotic cells were determined by flow
cytometry (BD FACScalibur; BD Biosciences) and analyzed using
CellQuest software (BD Biosciences).
Measurement of mitochondrial membrane
potential (MMP)
5,5′,
6,6′-Tetracholoro-1,1′3,3′-tetraethybenzimidazole-carbocyanide
iodine (JC-1; Beyotime Institute of Biotechnology) was used to
measure the MMP of H9c2 cells according to the manufacturer’s
instructions. Briefly, following the indicated treatments, H9c2
cells were harvested and resuspended in 1 ml culture medium (DMEM +
FBS) with JC-1 fluorescent dye. Following incubation at 37°C for 20
min, the cells were centrifuged at 600 × g at 4°C for 3 min, and
the supernatant was removed. The cells were resuspended in 1X JC-1
buffer for observation under a fluorescent microscope at ×100
magnification (Carl Zeiss AG). In each sample, five random fields
were selected for observation and analysis.
Measurement of caspase-3 and caspase-9
activity
The activity of caspase-3 and caspase-9 in H9c2
cells subjected to different treatments was measured by Caspase
Activity kits (Beyotime Institute of Biotechnology) using the
substrate peptides acetyl-Asp-Glu-Val-Asp p-nitroanilide
(Ac-DEVD-pNA) and acetyl-Leu-Glu-His-Asp p-nitroanilide
(Ac-LEHD-pNA), respectively. In brief, following treatment, the
cells were lysed using RIPA lysis buffer (Beyotime Institute of
Biotechnology), and the supernatants were mixed with buffer
(Beyotime Institute of Biotechnology) containing the substrate
peptides and incubated at 37°C for 2 h. The release of pNA was
quantified by determining the absorbance at 405 nm using a
microplate reader (Bio-Rad Laboratories, Inc.). The caspase
activity was expressed as a relative percentage of the control
value.
Determination of ROS production
Intracellular ROS levels were monitored using
2′,7′-dichlorofluorescein diacetate (DCFH-DA; Thermo Fisher
Scientific, Inc.) according to the manufacturer’s instructions. In
brief, following the indicated treatments, H9c2 cells were loaded
with 10 μM DCFH-DA at 37°C for 30 min and washed with PBS
thrice. Absorbance was measured at 485 and 525 nm using a
fluorescent microplate reader (Carl Zeiss AG) to determine the
intensity of DCF fluorescence.
Measurement of malondialdehyde (MDA)
production and superoxide dismutase (SOD), catalase (CAT) and
glutathione peroxidase (GSH-Px) activity levels
The MDA content and SOD, CAT and GSH-Px activity
levels were measured using commercial kits (Nanjing Jiancheng
Bioengineering Institute) according to the manufacturers’
instructions. In brief, following the indicated treatments, H9c2
cells were lysed using RIPA lysis buffer (Beyotime Institute of
Biotechnology) and the supernatants were collected to determine MDA
content and SOD, CAT and GSH-Px activity levels. The absorbance was
measured at 530 (MDA), 450 (SOD), 405 (CAT) and 412 (GSH-Px) nm
using a microplate reader (Bio-Rad Laboratories, Inc.).
Western blot analysis
Following treatment, H9c2 cells were washed twice
with PBS, lysed in RIPA buffer (Beyotime Institute of
Biotechnology) and centrifuged at 1,000 × g at 4°C for 3 min. The
supernatants were collected and quantified for protein
concentration using a BCA kit (Beyotime Institute of
Biotechnology). Equal amounts (30 μg/lane) of protein from
each sample were separated by sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred onto polyvinylidene difluoride
membranes (EMD Millipore). The membranes were blocked with 5%
non-fat milk in TBST buffer (50 mM Tris, pH 7.4, 250 mM NaCl, 0.1%
Tween-20) for 1 h at room temperature and probed with antibodies
against PGC-1α (1:1,000; cat. no. ab54481), Nrf2 (1:2,000; cat. no.
ab137550), HO-1 (1:1,000; cat. no. ab189491) and NQO1 (1:1,000;
cat. no. ab97385; all from Abcam), caspase-3 (1:1,000; cat. no.
14220), cleaved (cl)-caspase-3 (1:1,000; cat. no. 9664), cytochrome
c (1:1,000; cat. no. 11940), Bax (1:1,000; cat. no. 5023), Bcl-2
(1:1,000; cat. no. 3498), TNF-α (1:1,000; cat. no. 6945) and
β-actin (1:1,000; cat. no. 4967; all from Cell Signaling
Technology, Inc.) overnight at 4°C. Following washing with TBST
thrice, the membranes were incubated with horseradish
peroxidase-conjugated anti-rabbit IgG (1:2,000; cat. no. 7074; Cell
Signaling Technology, Inc.) for 1 h at room temperature and
visualized by enhanced chemiluminescence reaction reagents (EMD
Millipore). Protein band densities were quantified using Quantity
One software (Bio-Rad Laboratories, Inc.). The results were
normalized to the internal control β-actin and calculated as
fold-changes compared with the control group in each
experiment.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from H9c2 cells following
indicated treatments using TRIzol® reagent (Thermo
Fisher Scientific Inc.). Complementary DNA was synthesized by RT
reaction with PrimeScript RT Master Mix (Takara Biotechnology Co.,
Ltd.). The reverse transcription protocol used was 37°C for 15 min
and 85°C for 30 sec. qPCR assay was performed using SYBR Premix Ex
Taq kit (Takara Biotechnology Co., Ltd.) according to the
manufacturer’s instructions. The amplification conditions were as
follows: 95°C for 10 min, followed by 40 cycles of 95°C for 10 sec
and 60°C for 60 sec. Reactions were conducted on the ABI Prism 7500
Real-Time PCR System (Thermo Fisher Scientific, Inc.). The mRNA
levels were calculated using the 2−ΔΔCq method (26). GAPDH was used for normalization.
The primers used were as follows: TNF-α forward,
5′-CCTCTTCTCATTCCTGCTCG-3′ and reverse, 5′-GGTATGAAATGGCAAATCGG-3′;
interleukin (IL)-6 forward, 5′-CTGCGCAGCTTTAAGGAGTTC-3′ and
reverse, 5′-TCTGAGGTGCCCATGCTACA-3′; IL-1β forward,
5′-CAACCAACAAGTGATATTCTCCATG-3′and reverse,
5′-GATCCACACTCTCCAGCTGCA-3′; IL-8 forward,
5′-GGCAGCCTTCCTGATTTCTG-3′ and reverse,
5′-CTTGGCAAAACTGCACCTTCA-3′; monocyte chemotactic protein-1 (MCP-1)
forward, 5′-CTCTCGCCTCCAGCATGAA-3′ and reverse,
5′-GGGAATGAAGGTGGCTGCTA-3′; GAPDH forward,
5′-CCATCACCATCTTCCAGGAG-3′ and reverse,
5′-CCTGCTTCACCACCTTCTTG-3′.
Enzyme-linked immunosorbent assay
(ELISA)
Following the indicated treatments, the culture
media were collected and centrifuged at 600 × g at room temperature
for 5 min. The levels of TNF-α, IL-6, IL-1β, IL-8 and MCP-1 in the
supernatants were determined using commercially available ELISA
kits (R&D systems, Inc.) according to the manufacturer’s
instructions and expressed as pg/ml.
Statistical analysis
The data are presented as the mean ± SD from three
independent experiments. The results were compared by one-way
analysis of variance, followed by least significant difference
post-hoc test using SPSS 16.0 software (SPSS, Inc.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Mel inhibits OGD/R-induced H9c2 cell
injury
First, the potential effects of Mel on the viability
of OGD/R-treated H9c2 cells were explored. CCK-8 assay results
demonstrated that OGD/R significantly reduced the viability of H9c2
cells compared with that of the control group. Mel (0.01, 0.1, 1
and 10 mM) significantly attenuated the decrease in
OGD/R-stimulated H9c2 cell viability (Fig. 1A). Next, the activity levels of
LDH and CK-MB, two indicators of cardiomyocyte injury, were
measured using commercial kits. A notable OGD/R-induced increase in
the activity levels of LDH and CK-MB was observed in H9c2 cells,
whereas Mel (0.01, 0.1, 1 and 10 mM) significantly prevented injury
(Fig. 1B and C). In addition, Mel
markedly enhanced cell viability (Fig. 1D) and decreased LDH and CK-MB
activity (Fig. 1E and F) in
OGD/R-exposed H9c2 cells. These results indicated that Mel
protected H9c2 cells from OGD/R-induced damage. Mel (1 mM)
treatment for 24 h was chosen for subsequent experiments due to the
observed effects against OGD/R-induced H9c2 cell injury.
 | Figure 1Mel protects H9c2 cells from
OGD/R-induced injury. (A–C) H9c2 cells were subjected to 4 h of OGD
followed by reperfusion for 24 h. Mel (0.01, 0.1, 1 and 10 mM) was
added to the culture medium at the initiation of reperfusion. (A)
The viability of H9c2 cells was measured by CCK-8 assay. H9c2 cell
injury was evaluated by measuring (B) LDH and (C) CK-MB activity.
(D–F) H9c2 cells were subjected to 4 h of OGD, followed by
reperfusion for the indicated durations (6, 12, 24 and 48 h). Mel
(1 mM) was added to the culture medium at the initiation of
reperfusion. (D) CCK-8 assay was conducted to determine cell
viability. The activity of (E) LDH and (F) CK-MB was measured using
commercial kits. Data are expressed as the mean ± SD from three
independent experiments. *P<0.05 vs. control;
#P<0.05 vs. OGD/R. Mel, melatonin; OGD/R,
oxygen-glucose deprivation/reperfusion; CCK-8, Cell Counting Kit-8;
LDH, lactate dehydrogenase; CK-MB, creatine kinase myocardial band;
OD, optical density. |
Mel suppresses OGD/R-induced apoptosis of
H9c2 cells
OGD/R can result in cardiomyocyte apoptosis
(27). Flow cytometry results
demonstrated that OGD/R increased the percentage of apoptotic cells
compared with that in the control group, whereas Mel reduced the
OGD/R-induced apoptosis of H9c2 cells (Fig. 2A and B). The MMP in H9c2 cells
subjected to OGD/R was decreased; however, this effect was markedly
attenuated in the presence of Mel (Fig. 2C). To assess the involvement of
caspase activation in the inhibition of OGD/R-induced apoptosis by
Mel, the activity levels of caspase-3 and caspase-9 in the
mitochondria-mediated apoptosis pathway were examined. Caspase-3
and caspase-9 activity levels were enhanced by OGD/R exposure
compared with the control group (Fig.
2D and E). However, Mel significantly reduced the OGD/R-induced
caspase-3 and caspase-9 activity. Consistent with the change in
caspase-3 activity, the protein expression levels of cl-caspase-3
and cytochrome c were increased in OGD/R-insulted H9c2 cells. Mel
reversed the upregulation of cl-caspase-3 and cytochrome c
expression induced by OGD/R (Fig.
2F). Western blot analysis also revealed significant
upregulation of Bax and downregulation of Bcl-2 following OGD/R
insult in H9c2 cells, which was reversed by Mel treatment (Fig. 2F). These results suggested that
Mel inhibited OGD/R-induced H9c2 cell apoptosis.
 | Figure 2Mel prevents OGD/R-induced apoptosis
in H9c2 cells. H9c2 cells were subjected to 4 h of OGD, followed by
reperfusion for 24 h. Mel (1 mM) was added to the culture medium at
the initiation of reperfusion. (A) Flow cytometry was performed to
analyze apoptosis. (B) Percentage of apoptotic cells in (A). (C)
Measurement of MMP with JC-1 fluorescent staining. (D) Caspase-3
and (E) caspase-9 activities were determined using commercial kits.
(F) Representative western blot results of cl-caspase-3, caspase-3,
cytochrome c, Bax and Bcl-2. β-actin was used as the loading
control. Data are expressed as the mean ± SD from three independent
experiments. *P<0.05 vs. control;
#P<0.05 vs. OGD/R. Mel, melatonin; OGD/R,
oxygen-glucose deprivation/reperfusion; MMP, mitochondrial membrane
potential; JC-1,
5,5′,6,6′-tetracholoro-1,1′3,3′-tetraethybenzimidazole-carbocyanide
iodine; cl, cleaved. |
Mel reduces OGD/R-induced oxidative
stress in H9c2 cells
OGD/R induces oxidative stress (28). As demonstrated in Fig. 3A, OGD/R promoted intracellular ROS
production in H9c2 cells compared with that in the control group.
Of note, Mel significantly inhibited OGD/R-induced ROS generation.
In addition, the content of MDA, which is an indicator of lipid
peroxidation, was significantly reduced by Mel in OGD/R-insulted
H9c2 cells (Fig. 3B). Compared
with the OGD/R group, Mel also significantly increased the activity
of endogenous antioxidant enzymes, including SOD (Fig. 3C), CAT (Fig. 3D) and GSH-Px (Fig. 3E) in H9c2 cells. These results
demonstrated that Mel exerted an antioxidant effect in
OGD/R-insulted H9c2 cells.
Mel elicits an antioxidant effect via the
activation of PGC-1α/Nrf2 signaling in OGD/R-insulted H9c2
cells
PGC-1α knockdown reduced the expression of Nrf2,
HO-1 and NQO-1, whereas Nrf2 knockdown suppressed HO-1 and NQO-1
expression in OGD/R-stimulated H9c2 cells (Fig. 4A). OGD/R led to an upregulation of
PGC-1α, Nrf2, HO-1 and NOQ1 expression in H9c2 cells. Mel further
enhanced the levels of PGC-1α, Nrf2, HO-1and NOQ1 in OGD/R-insulted
H9c2 cells (Fig. 4B). In
addition, the Mel-induced decrease in ROS generation in
OGD/R-exposed H9c2 cells was attenuated by PGC-1α or Nrf2 silencing
(Fig. 4C). PGC-1α or Nrf2
knockdown counteracted the Mel-induced increase in the viability of
OGD/R-exposed H9c2 cells (Fig.
4D). PGC-1α or Nrf2 knockdown also reversed the Mel-mediated
reduction in apoptosis in OGD/R-insulted H9c2 cells (Fig. 4E). These results demonstrated that
the antioxidant effects of Mel were dependent on the activation of
PGC-1α/Nrf2 signaling in OGD/R-insulted H9c2 cells.
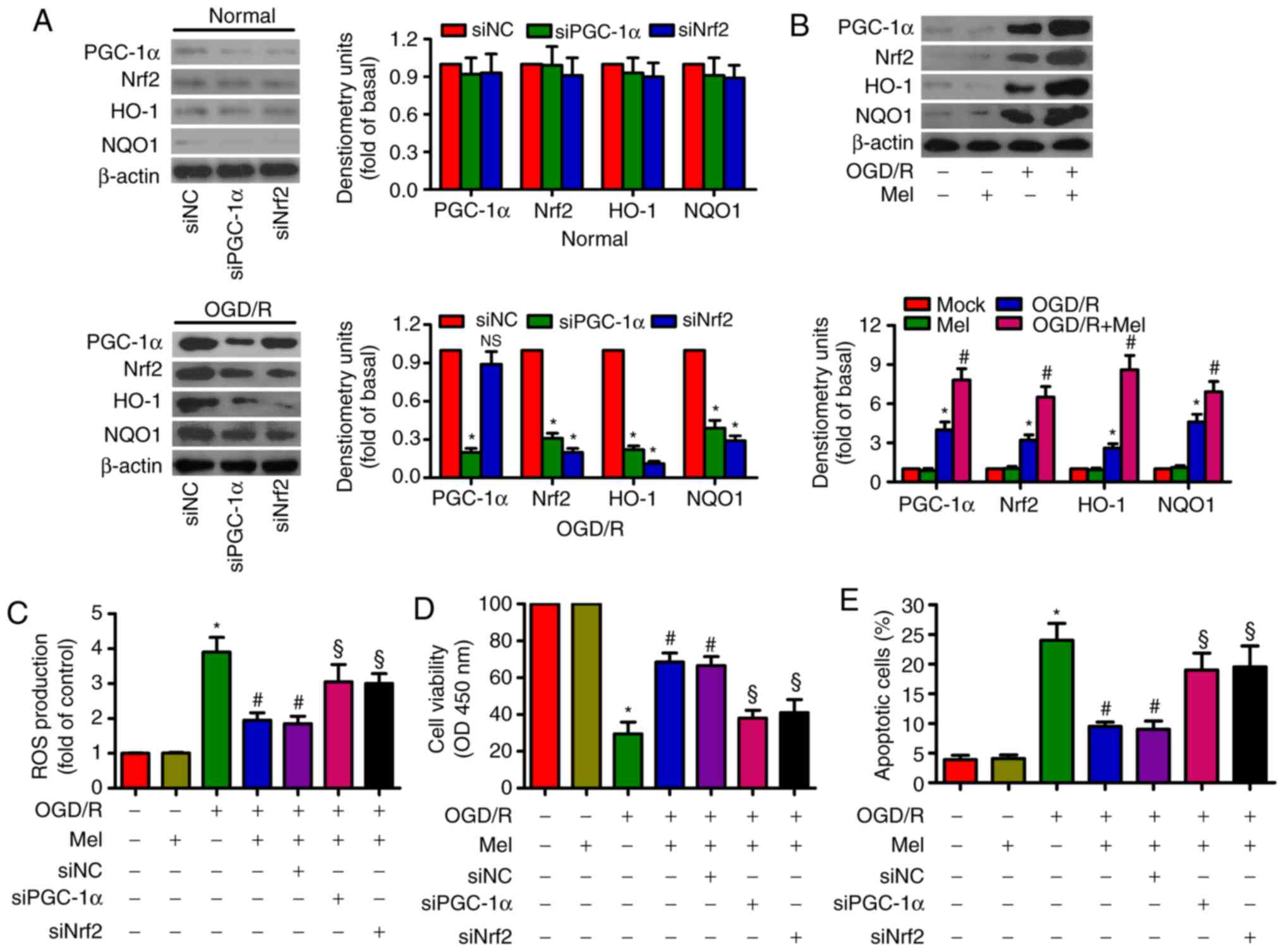 | Figure 4Mel activates PGC-1α/Nrf2 signaling
to exert antioxidant effects on OGD/R-insulted H9c2 cells. (A) H9c2
cells were transfected with 100 nM siNC, siPGC-1αor siNrf2 for 24 h
and subjected to 4 h of OGD, followed by reperfusion for 24 h. The
expression of PGC-1α, Nrf2, HO-1 and NOQ1 was analyzed by western
blotting. β-actin was used as an endogenous control. (B) H9c2 cells
were subjected to 4 h of OGD followed by reperfusion for 24 h. Mel
(1 mM) was added to the culture medium at the initiation of
reperfusion. Western blot analysis was performed to detect the
expression of PGC-1α, Nrf2, HO-1and NOQ1. β-actin was used as the
endogenous control. (C–E) H9c2 cells were transfected with 100 nM
siPGC-1α or siNrf2 for 24 h and subjected to 4 h of OGD, followed
by reperfusion for 24 h. Mel (1 mM) was added to the culture medium
at the initiation of reperfusion. (C) ROS production was assessed
using a 2′,7′-dichlorofluorescein diacetate fluorescent probe. (D)
Cell viability and (E) apoptosis were measured by Cell Counting
Kit-8 and flow cytometry, respectively. Data are expressed as the
mean ± SD from three independent experiments. *P<0.05
vs. control; #P<0.05 vs. OGD/R; §P<0.05
vs. OGD/R + Mel or OGD/R + Mel + siNC. Mel, melatonin; PGC-1α,
peroxisome proliferator-activated receptor gamma coactivator-1α;
Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme
oxygenase-1; OGD/R, oxygen-glucose deprivation/reperfusion; ROS,
reactive oxygen species; si, small interfering RNA; NC, negative
control. |
Mel represses the OGD/R-induced
expression and release of pro-inflammatory cytokines in H9c2
cells
To examine whether Mel serves an inhibitory role in
OGD/R-induced inflammation, the expression and release of several
pro-inflammatory cytokines in H9c2 cells was measured by RT-qPCR
and ELISA. The RT-qPCR results revealed that the expression of
TNF-α (Fig. 5A), IL-6 (Fig. 5B), IL-1β (Fig. 5C), IL-8 (Fig. 5D) and MCP-1 (Fig. 5E) were considerably higher in the
OGD/R-exposed H9c2 cells compared with those in the control cells,
and that Mel partially reversed the OGD/R-induced increase in the
TNF-α, IL-6, IL-1β, IL-8 and MCP-1 levels. Consistently, the
production of TNF-α (Fig. 5F),
IL-6 (Fig. 5G), IL-1β (Fig. 5H), IL-8 (Fig. 5I) and MCP-1 (Fig. 5J) was significantly suppressed by
Mel in OGD/R-treated H9c2 cells. These results demonstrated that
Mel reduced the levels of pro-inflammatory cytokines in
OGD/R-exposed H9c2 cells.
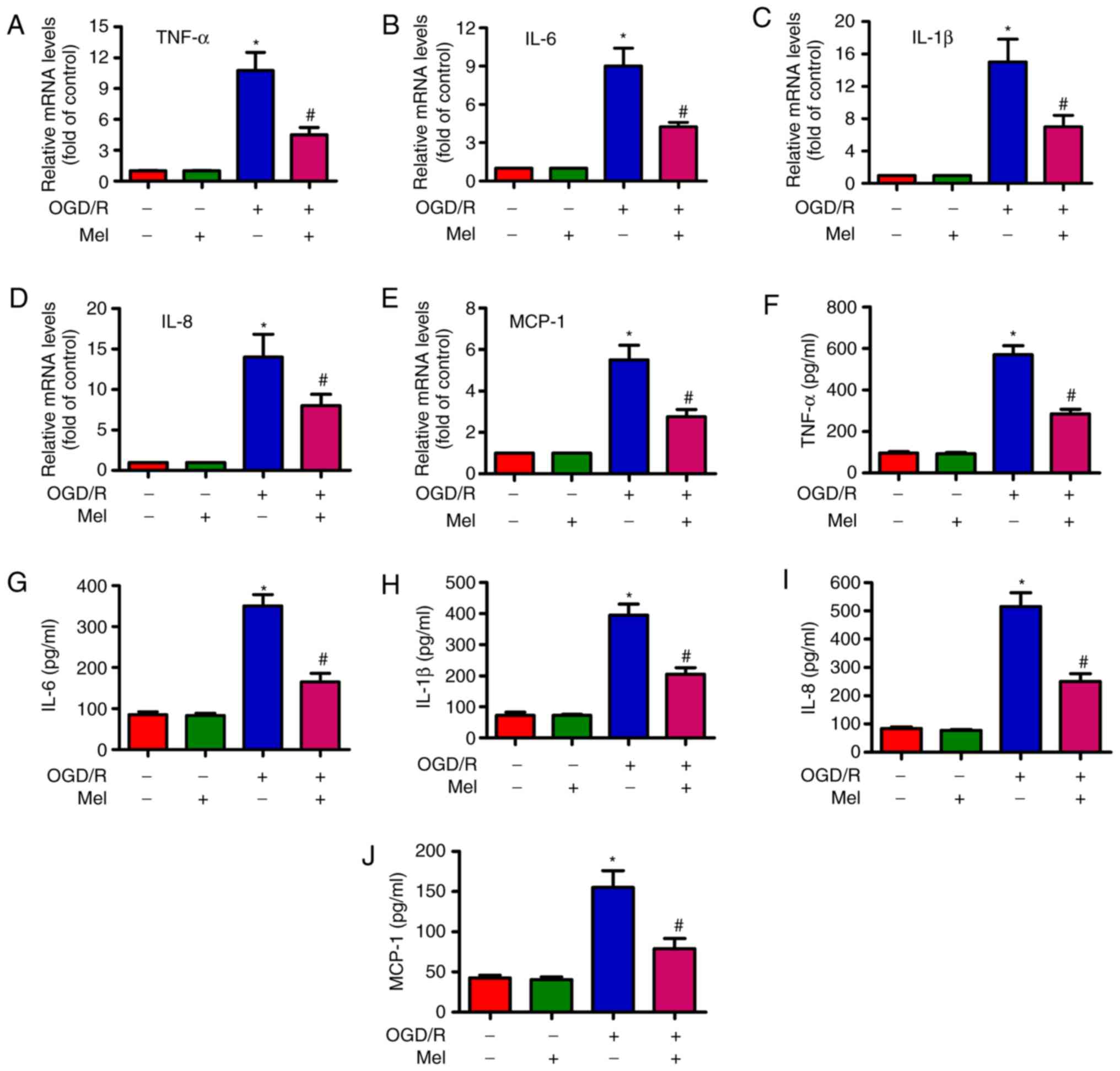 | Figure 5Mel reduces the level of
pro-inflammatory cytokines in OGD/R-insulted H9c2 cells. H9c2 cells
were subjected to 4 h of OGD followed by reperfusion for 24 h. Mel
(1 mM) was added to the culture medium at the initiation of
reperfusion. (A–E) The mRNA expression levels of (A) TNF-α, (B)
IL-6, (C) IL-1β, (D) IL-8 and (E) MCP-1 were analyzed by reverse
transcription-quantitative PCR. (F–J) The release of (F) TNF-α, (G)
IL-6, (H) IL-1β, (I) IL-8 and (J) MCP-1 in the supernatants was
measured by ELISA. Data are expressed as the mean ± SD from three
independent experiments. *P<0.05 vs. control;
#P<0.05 vs. OGD/R. Mel, melatonin; OGD/R,
oxygen-glucose deprivation/reperfusion; TNF-α, tumor necrosis
factor-α; IL, interleukin; MCP, monocyte chemotactic protein. |
Mel prevents the OGD/R-induced apoptosis
of H9c2 cells by regulating the PGC-1α/TNF-α signaling pathway
PGC-1α can decrease TNF-α production in myotubes via
TNF-α stimulation (29). TNF-α is
implicated in OGD/R-induced H9c2 cell apoptosis (27). As demonstrated in Fig. 6A, siPGC-1α transfection led to an
upregulation of TNF-α expression in OGD/R-exposed H9c2 cells. OGD/R
induced a significant increase in PGC-1α and TNF-α expression in
H9c2 cells, whereas Mel further increased PGC-1α expression but
reduced the level of TNF-α (Fig.
6B). In OGD/R-insulted H9c2 cells, the Mel-mediated decrease in
the production of TNF-α was reversed by PGC-1α knockdown. However,
the addition of the TNF-α antibody reduced TNF-α production
(Fig. 6C). In addition, PGC-1α
silencing resulted in a decrease in the Mel-induced enhancement in
the viability of OGD/R-exposed H9c2 cells, which was rescued by the
addition of the TNF-α antibody (Fig.
6D). Incubation with the TNF-α antibody reversed the PGC-1α
depletion-induced increase in OGD/R-stimulated H9c2 cell apoptosis
(Fig. 6E). These results revealed
that Mel exerted an anti-apoptotic effect in OGD/R-treated H9c2
cells via the PGC-1α/TNF-α signaling pathway.
 | Figure 6Mel inhibits apoptosis in
OGD/R-treated H9c2 cells by regulating PGC-1α/Nrf2 signaling. (A)
H9c2 cells were transfected with 100 nM siNC or siPGC-1α for 24 h
and subjected to 4 h of OGD, followed by reperfusion for 24 h. The
expression of TNF-α was analyzed by western blot analysis. β-Actin
was used as the endogenous control. (B) H9c2 cells were subjected
to 4 h of OGD followed by reperfusion for 24 h. Mel (1 mM) was
added to the culture medium at the initiation of reperfusion.
Western blot analysis was conducted to detect the expression of
PGC-1α and TNF-α. β-Actin was used as the endogenous control. (C–E)
H9c2 cells were transfected with 100 nM siPGC-1α or treated with a
TNF-α antibody (1 μg/ml) for 24 h and subjected to 4 h of
OGD, followed by reperfusion for 24 h. Mel (1 mM) was added to the
culture medium at the initiation of reperfusion. (C) TNF-α
production was measured by ELISA. (D) Cell viability and (E)
apoptosis were evaluated by Cell Counting Kit-8 and flow cytometry
assays, respectively. Data are expressed as the mean ± SD from
three independent experiments. *P<0.05 vs. control;
#P<0.05 vs. OGD/R; §P<0.05 vs. OGD/R +
Mel or OGD/R + Mel + siNC; ◆P<0.05 vs. OGD/R + Mel or
OGD/R + Mel + siNC; ■P<0.05 vs. OGD/R + Mel or OGD/R
+ Mel + siNC. Mel, melatonin; OGD/R, oxygen-glucose
deprivation/reperfusion; PGC-1α, peroxisome proliferator-activated
receptor gamma coactivator-1α; Nrf2, nuclear factor erythroid
2-related factor 2; TNF-α, tumor necrosis factor-α; TNF-α Ab, TNF-α
antibody; si, small interfering RNA; NC, negative control. |
Discussion
The main results of the present study were as
follows: i) Mel exhibited cardioprotective effects in
OGD/R-stimulated H9c2 cardiomyocytes, as confirmed by the increased
cell viability and decreased LDH and CK-MB activity levels; ii) Mel
reduced OGD/R-induced H9c2 cell apoptosis via the intrinsic
apoptotic pathway; iii) Mel alleviated OGD/R-induced oxidative
stress in H9c2 cells; iv) the antioxidant effects of Mel were
mediated by activating PGC-1α/Nrf2 signaling in OGD/R-exposed H9c2
cells; v) Mel inhibited OGD/R-induced inflammatory response in H9c2
cells; vi) Mel elicited its antiapoptotic effects in OGD/R-insulted
H9c2 cells via the regulation of the PGC-1α/TNF-α signaling
pathway. Overall, Mel was demonstrated to inhibit OGD/R-induced
H9c2 cardiomyocyte injury by inhibiting oxidative stress and
inflammation.
Myocardial I/R injury is mainly caused by
cardiomyocyte damage or death. LDH and CK-MB are constitutively
expressed in myocardial cells and cannot shuttle across cytoplasmic
membranes in the normal physiological state; however, they are
released when cells are damaged or dead (30). Therefore, the activity of LDH and
CK-MB in the culture media represents the extent of OGD/R-caused
H9c2 cell injury. In the present study, decreased viability and
increased LDH and CK-MB activity was observed in OGD/R-treated H9c2
cells. Mel protected the H9c2 cells from OGD/R-induced damage.
Cardiomyocyte apoptosis serves a primary role in the pathogenesis
of myocardial I/R injury (31).
Myocardial apoptosis is a complicated process mediated by a series
of enzymes and molecules, including the opening of the
mitochondrial permeability transition pore, release of cytochrome c
and activation of caspases (32).
The Bcl-2 family of proteins have emerged as the key regulatory
components of the apoptotic process; the Bcl-2 family comprises
antiapoptotic proteins (such as Bcl-2 and Bcl-xL) and proapoptotic
molecules (such as Bax and Bak), which function primarily to
protect or disrupt the integrity of the mitochondrial membrane and
control the release of proapoptotic proteins (33). Mitochondrial dysfunction and MMP
loss are early events in the apoptotic process (34). I/R stimulation leads to the
opening of the mitochondrial permeability transition pore and
release of cytochrome c from the intermembrane space of
mitochondria (35). Once
released, cytochrome c binds to the cytosolic protein Apaf1
facilitating the formation of the apoptosome complex, which results
in the activation of caspase-9 and subsequent activation of
caspase-3; the activation of caspase-3 triggers apoptosis (36). In the present study, OGD/R led to
a significant increase in the activity levels of caspase-3 and
caspase-9, loss of MMP and apoptosis in H9c2 cells. However, Mel
reduced the apoptotic rate and the activity levels of caspase-3 and
caspase-9 and attenuated the loss of MMP. Mel decreased the levels
of proapoptotic cl-caspase-3, cytochrome c and Bax and enhanced the
antiapoptotic Bcl-2 expression in OGD/R-exposed H9c2 cells. These
results suggested that Mel exerted antiapoptotic effects in
OGD/R-injured cardiomyocytes.
Myocardial I/R injury induces oxidative stress,
which causes apoptosis in cardiovascular cells (37). Excessive amounts of ROS during the
reperfusion period may damage cell structure and facilitate
myocardial apoptosis (38). MDA,
which is a secondary product of lipid peroxidation, is a biomarker
of oxidative stress and indicates free radical production and
consequent tissue damage (39).
Antioxidant enzymes, such as SOD, CAT and GSH-Px, act as a
compensatory mechanism for hyperoxidation that protects against
oxidative injury (40). However,
myocardial I/R leads to the collapse of the antioxidant system and
increases the vulnerability of the myocardium to oxygen free
radicals (41). The results of
the present study demonstrated that the myocardial protective
effect of Mel was mediated by the inhibition of oxidative stress,
as indicated by the reduction of ROS production and MDA content,
and the enhancement of cellular antioxidant enzymes, including CAT,
SOD and GSH-Px. Mitochondria are the principal sites of ROS
production (42). PGC-1α and Nrf2
are the major regulators of mitochondrial biogenesis and activity
(43,44). PGC-1α and Nrf2 regulate the
expression of certain antioxidant-related genes, which remove ROS
through sequential enzymatic reactions (11,12). A previous study has demonstrated
that Mel ameliorated myocardial I/R injury by activating the 5′
AMP-activated protein kinase/PGC-1α/SIRT3 signaling pathway
(23). In addition, Mel protected
H9c2 cells against OGD/R-induced oxidative damage through the
Nrf2/NQO1 pathway (24). A
compound hydroxysafflor yellow A that interferes with the
PGC-1α/Nrf2 pathway serves an antioxidant role in
isoproterenol-damaged H9c2 cells (13). Consistent with these findings, the
results of the present study demonstrated that the repressive
effects of Mel on ROS generation and apoptosis in OGD/R-insulted
H9c2 cells were attenuated by the knockdown of PGC-1α or Nrf2,
which suggested that Mel inhibited oxidative stress and apoptosis
in H9c2 cells by activating thePGC-1α/Nrf2 signaling pathway.
I/R injury induces the production and release of
several pro-inflammatory cytokines and chemokines (27,45,46). TNF-α upregulation in an ischemic
region induced TNF-α expression in the neighboring normal
myocardium, which resulted in amplified cytokine effects (47). Of note, TNF-α induces the
activation of nuclear factor-κB and subsequent upregulation of
other inflammatory mediators (48). TNF-α is a pro-inflammatory
cytokine that has a potent pro-apoptotic function through binding
to its cell surface receptor TNFR1 (49). A neutralizing antibody against
TNF-α has been demonstrated to exert antiapoptotic effects in
cardiomyocytes (50). OGD/R
resulted in a significant increase in the levels of TNF-α, IL-6 and
MCP-1 in H9c2 cells (46). Yang
et al (16) reported that
several pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6
and IL-8, are upregulated by OGD/R in H9c2 cells. Consistently with
these results, high levels of TNF-α, IL-6, IL-1β, IL-8 and MCP-1
were observed in OGD/R-treated H9c2 cells. Mel reversed the
increase in these pro-inflammatory mediators. Recently, the
PPARγ/PGC-1α/TNF-α signaling pathway has been implicated in the
inflammatory response of OGD/R-insulted H9c2 cells (16). In the present study, the
Mel-mediated reduction in TNF-α release and apoptosis was
counteracted by PGC-1α knockdown and rescued by TNF-α antibody
treatment in OGD/R-exposed H9c2 cells. These data indicated that
Mel suppressed the apoptosis of OGD/R-damaged H9c2 cells through
the PGC-1α/TNF-α signaling pathway.
In conclusion, the results of the present study
demonstrated that Mel inhibited OGD/R-induced oxidative stress,
inflammation and apoptosis in H9c2 cells. Mechanistically, the
Mel-induced protection of H9c2 cells was dependent on the
PGC-1α/Nrf2 and PGC-1α/TNF-α signaling pathways. Overall, this
study provided a deep insight into the protective effects of Mel
against myocardial I/R injury.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
YG and ML contributed to the conception and design
of the present study. WZ, KL and HW conducted all the experiments.
WZ and KL drafted the manuscript and revised it critically for
important intellectual content. WZ, HW and ML interpreted and
analyzed the data. YG and ML provided final approval of the version
to be published. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Steptoe A and Kivimäki M: Stress and
cardiovascular disease. Nat Rev Cardiol. 9:360–370. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ford ES and Caspersen CJ: Sedentary
behaviour and cardiovascular disease: A review of prospective
studies. Int J Epidemiol. 41:1338–1353. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu NB, Wu M, Chen C, Fujino M, Huang JS,
Zhu P and Li XK: Novel molecular targets participating in
myocardial ischemia-reperfusion injury and cardioprotection.
Cardiol Res Pract. 2019:6935147. 2019. View Article : Google Scholar
|
|
4
|
Heusch G: Molecular basis of
cardioprotection: Signal transduction in ischemic pre-, post-, and
remote conditioning. Circ Res. 116:674–699. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hausenloy DJ and Yellon DM: Targeting
myocardial reperfusion injury-the search continues. N Engl J Med.
373:1073–1075. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ibanez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia/reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Whelan RS, Kaplinskiy V and Kitsis RN:
Cell death in the pathogenesis of heart disease: mechanisms and
significance. Annu Rev Physiol. 72:19–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cannistra M, Ruggiero M, Zullo A, Gallelli
G, Serafini S, Maria M, Naso A, Grande R, Serra R and Nardo B:
Hepatic ischemia reperfusion injury: A systematic review of
literature and the role of current drugs and biomarkers. Int J
Surg. 33(Suppl 1): S57–S70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsutsui H, Kinugawa S and Matsushima S:
Oxidative stress and heart failure. Am J Physiol Heart Circ
Physiol. 301:H2181–H2190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kärkkäinen O, Tuomainen T, Mutikainen M,
Lehtonen M, Ruas JL, Hanhineva K and Tavi P: Heart specific PGC-1α
deletion identifies metabolome of cardiac restricted metabolic
heart failure. Cardiovasc Res. 115:107–118. 2019. View Article : Google Scholar
|
|
12
|
Zhou S, Sun W, Zhang Z and Zheng Y: The
role of Nrf2-mediated pathway in cardiac remodeling and heart
failure. Oxid Med Cell Longev. 2014:260429. 2014. View Article : Google Scholar
|
|
13
|
Chen M, Wang M, Yang Q, Wang M, Wang Z,
Zhu Y, Zhang Y, Wang C, Jia Y, Li Y and Wen A: Antioxidant effects
of hydroxysafflor yellow A and acetyl-11-keto-β-boswellic acid in
combination on isoproterenol-induced myocardial injury in rats. Int
J Mol Med. 37:1501–1510. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang J, Xia F, Zhao H, Peng K, Liu H,
Meng X, Chen C and Ji F: Dexmedetomidine-induced cardioprotection
is mediated by inhibition of high mobility group box-1 and the
cholinergic anti-inflammatory pathway in myocardial
ischemia-reperfusion injury. PLoS One. 14:e02187262019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen ZW, Qian JY, Ma JY, Chang SF, Yun H,
Jin H, Sun AJ, Zou YZ and Ge JB: TNF-α-induced cardiomyocyte
apoptosis contributes to cardiac dysfunction after coronary
microembolization in mini-pigs. J Cell Mol Med. 18:1953–1963. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y, Li J, Hou Z, Yu Y and Yu B: KLF5
overexpression attenuates cardiomyocyte inflammation induced by
oxygen-glucose deprivation/ reperfusion through the
PPARγ/PGC-1α/TNF-α signaling pathway. Biomed Pharmacother.
84:940–946. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Y, Sun Y, Yi W, Li Y, Fan C, Xin Z,
Jiang S, Di S, Qu Y, Reiter RJ and Yi D: A review of melatonin as a
suitable antioxidant against myocardial ischemia-reperfusion injury
and clinical heart diseases. J Pineal Res. 57:357–366. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reiter RJ, Mayo JC, Tan DX, Sainz RM,
Alatorre-Jimenez M and Qin L: Melatonin as an antioxidant: under
promises but over delivers. J Pineal Res. 61:253–278. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tordjman S, Chokron S, Delorme R, Charrier
A, Bellissant E, Jaafari N and Fougerou C: Melatonin: Pharmacology,
functions and therapeutic benefits. Curr Neuropharmaco. 15:434–443.
2017. View Article : Google Scholar
|
|
20
|
Yu L, Sun Y, Cheng L, Jin Z, Yang Y, Zhai
M, Pei H, Wang X, Zhang H, Meng Q, et al: Melatonin
receptor-mediated protection against myocardial
ischemia/reperfusion injury: Role of SIRT1. J Pineal Res.
57:228–238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dwaich KH, Al-Amran FG, Al-Sheibani BI and
Al-Aubaidy HA: Melatonin effects on myocardial ischemia-reperfusion
injury: Impact on the outcome in patients undergoing coronary
artery bypass grafting surgery. Int J Cardiol. 221:977–986. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu L, Fan C, Li Z, Zhang J, Xue X, Xu Y,
Zhou G, Yang Y and Wang H: Melatonin rescues cardiac thioredoxin
system during ischemia-reperfusion injury in acute hyperglycemic
state by restoring Notch1/Hes1/Akt signaling in a membrane
receptor-dependent manner. J Pineal Res. 62:2017. View Article : Google Scholar
|
|
23
|
Yu L, Gong B, Duan W, Fan C, Zhang J, Li
Z, Xue X, Xu Y, Meng D, Li B, et al: Melatonin ameliorates
myocardial ischemia/reperfusion injury in type 1 diabetic rats by
preserving mitochondrial function: Role of AMPK-PGC-1α-SIRT3
signaling. Sci Report. 7:413372017. View Article : Google Scholar
|
|
24
|
Zhai M, Li B, Duan W, Jing L, Zhang B,
Zhang M, Yu L, Liu Z, Yu B, Ren K, et al: Melatonin ameliorates
myocardial ischemia reperfusion injury through SIRT3-dependent
regulation of oxidative stress and apoptosis. J Pineal Res.
63:2017. View Article : Google Scholar
|
|
25
|
Gao L, Zhao YC, Liang Y, Lin XH, Tan YJ,
Wu DD, Li XZ, Ye BZ, Kong FQ, Sheng JZ and Huang HF: The impaired
myocardial ischemic tolerance in adult offspring of diabetic
pregnancy is restored by maternal melatonin treatment. J Pineal
Res. 61:340–352. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Wu WY, Wang WY, Ma YL, Yan H, Wang XB, Qin
YL, Su M, Chen T and Wang YP: Sodium tanshinone IIA silate inhibits
oxygen-glucose deprivation/recovery-induced cardiomyocyte apoptosis
via suppression of the NF-κB/TNF-α pathway. Br J Pharmacol.
169:1058–1071. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu H, Guan Q, Guo L, Zhang H, Pang X,
Cheng Y, Zhang X and Sun Y: Gypenosides alleviate myocardial
ischemia-reperfusion injury via attenuation of oxidative stress and
preservation of mitochondrial function in rat heart. Cell Stress
Chaperones. 21:429–437. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eisele PS, Salatino S, Sobek J, Hottiger
MO and Handschin C: The peroxisome proliferator-activated receptor
γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional
activity of NF-κB in skeletal muscle cells. J Biol Chem.
288:2246–2260. 2013. View Article : Google Scholar
|
|
30
|
Gürgün C, Ildizli M, Yavuzgil O, Sin A,
Apaydin A, Cinar C and Kültürsay H: The effects of short term
statin treatment on left ventricular function and inflammatory
markers in patients with chronic heart failure. Int J Cardiol.
123:102–107. 2008. View Article : Google Scholar
|
|
31
|
Xia P, Liu Y and Cheng Z: Signaling
pathways in cardiac myocyte apoptosis. Biomed Res Int.
2016:9583268. 2016. View Article : Google Scholar
|
|
32
|
He X, Li S, Liu B, Susperreguy S, Formoso
K, Yao J, Kang J, Shi A, Birnbaumer L and Liao Y: Major
contribution of the 3/6/7 class of TRPC channels to myocardial
ischemia/reperfusion and cellular hypoxia/reoxygenation injuries.
Proc Natl Acad Sci USA. 114:E4582–E4591. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kinnally KW, Peixoto PM, Ryu SY and Dejean
LM: Is mPTP the gatekeeper for necrosis, apoptosis, or both?
Biochim Biophys Acta. 1813.616–622. 2011.
|
|
36
|
Broughton BR, Reutens DC and Sobey CG:
Apoptotic mechanisms after cerebral ischemia. Stroke. 40:e331–e339.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lorgis L, Zeller M, Dentan G, Sicard P,
Richard C, Buffet P, L’Huillier I, Beer JC, Cottin Y, Rochette L
and Vergely C: The free oxygen radicals test (FORT) to assess
circulating oxidative stress in patients with acute myocardial
infarction. Atherosclerosis. 213:616–621. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bartz RR, Suliman HB and Piantadosi CA:
Redox mechanisms of cardiomyocyte mitochondrial protection. Front
Physiol. 6:2912015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ozer MK, Parlakpinar H, Cigremis Y, Ucar
M, Vardi N and Acet A: Ischemia-reperfusion leads to depletion of
glutathione content and augmentation of malondialdehyde production
in the rat heart from overproduction of oxidants: Can caffeic acid
phenethyl ester (CAPE) protect the heart? Mol Cell Biochem.
273:169–175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jain AK, Mehra NK and Swarnakar NK: Role
of antioxidants for the treatment of cardiovascular diseases:
Challenges and opportunities. Curr Pharm Des. 21:4441–4455. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Vijayasarathy K, Shanthi Naidu K and
Sastry BK: Melatonin metabolite 6-Sulfatoxymelatonin, Cu/Zn
superoxide dismutase, oxidized LDL and malondialdehyde in unstable
angina. Int J Cardiol. 144:315–317. 2010. View Article : Google Scholar
|
|
42
|
Kausar S, Wang F and Cui H: The role of
mitochondria in reactive oxygen species generation and its
implications for neurodegenerative diseases. Cells. 7:E2742018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gureev AP, Shaforostova EA and Popov VN:
Regulation of mitochondrial biogenesis as a way for active
longevity: Interaction between the Nrf2 and PGC-1α signaling
pathways. Front Genet. 10:4352019. View Article : Google Scholar
|
|
44
|
Lai L, Wang M, Martin OJ, Leone TC, Vega
RB, Han X and Kelly DP: A role for peroxisome
proliferator-activated receptor γ coactivator 1 (PGC-1) in the
regulation of cardiac mitochondrial phospholipid biosynthesis. J
Biol Chem. 289:2250–2259. 2014. View Article : Google Scholar
|
|
45
|
Zhang R, Xu L, Zhang D, Hu B, Luo Q, Han
D, Li J and Shen C: Cardioprotection of ginkgolide B on myocardial
ischemia/reperfusion-induced inflammatory injury via regulation of
A20-NF-κB pathway. Front Immunol. 9:28442018. View Article : Google Scholar
|
|
46
|
Ma L, Liu H, Xie Z, Yang S, Xu W, Hou J
and Yu B: Ginsenoside Rb3 protects cardiomyocytes against
ischemia-reperfusion injury via the inhibition of JNK-mediated
NF-κB pathway: A mouse cardiomyocyte model. PLoS One.
9:e1036282014. View Article : Google Scholar
|
|
47
|
Saito Y, Watanabe K, Fujioka D, Nakamura
T, Obata JE, Kawabata K, Watanabe Y, Mishina H, Tamaru S, Kita Y,
et al: Disruption of group IVA cytosolic phospholipase A(2)
attenuates myocardial ischemia-reperfusion injury partly through
inhibition of TNF-α-mediated pathway. Am J Physiol Heart Circ
Physiol. 302:H2018–H2030. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gray CB, Suetomi T, Xiang S, Mishra S,
Blackwood EA, Glembotski CC, Miyamoto S, Westenbrink BD and Brown
JH: CaMKIIδ subtypes differentially regulate infarct formation
following ex vivo myocardial ischemia/reperfusion through NF-κB and
TNF-α. J Mol Cell Cardiol. 103:48–55. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Qian Q, Cao X, Wang B, Qu Y, Qian Q, Sun Z
and Feng F: TNF-α-TNFR signal pathway inhibits autophagy and
promotes apoptosis of alveolar macrophages in coal worker’s
pneumoconiosis. J Cell Physiol. 234:5953–5963. 2019. View Article : Google Scholar
|
|
50
|
Li S, Jiao X, Tao L, Liu H, Cao Y, Lopez
BL, Christopher TA and Ma XL: Tumor necrosis factor-alpha in
mechanic trauma plasma mediates cardiomyocyte apoptosis. Am J
Physiol Heart Circ Physiol. 293:H1847–H1852. 2007. View Article : Google Scholar : PubMed/NCBI
|















