Introduction
Pulmonary fibrosis (PF) is one of the most common
chronic pulmonary diseases, and it is characterized by restrictive
functional ventilation disorder, hypoxemia and chronic progressive
diffuse lung fibrosis; it is associated with clinical features,
such as wheezing, dyspnea and dry cough. It has a high incidence
and mortality rate worldwide (1).
To date, the pathological mechanisms of PF have not been fully
elucidated (2,3); thus, no effective drug or treatment
has yet been developed.
Napsin A is an aspartic proteinase expressed not
only in type II lung cells, but also in alveolar macrophages and it
may be expressed secondary to phagocytosis (4,5).
The alveolar cavity contains active Napsin A at high levels, which
are associated with the levels of surfactant protein B (SP-B),
precursor protein proSP-B and SP-C (6). As a hallmark of the most significant
number of pulmonary adenocarcinomas, immunohistochemical analysis
for Napsin A yields negative results in the majority of squamous
cell carcinomas and adenocarcinomas of other organs (7,8).
Reportedly, its local expression not only aids in the
classification of primary pulmonary tumors as adenocarcinomas, but
also in the identification of the lung as the origin in the context
of metastatic adenocarcinoma (7,8).
In addition, elevated serum levels of Napsin A in patients with
idiopathic PF (IPF) are related to the severity of the illness
(9-11). Ueno et al (12) transfected the Napsin A gene into
293 cells and found that the cell proliferative and migratory
ability were significantly inhibited. Consistently, in a previous
study by the authors, it was also demonstrated that PF could be
suppressed by the transfection of the Napsin A gene into type II
alveolar epithelial cells, possibly through the inhibition of
integrin signal transduction (13). However, the expression of Napsin A
is significantly downregulated in the blood samples obtained from
PF patients. Investigating the mechanism by which Napsin A
expression is suppressed during PF might provide potent novel
strategies for PF treatment.
MicroRNAs (miRNAs or miRs), a family of non-coding
single-stranded small RNAs with lengths of 21-23 nucleotides, can
modulate gene expression at the post-transcriptional level by base
pairing to sequence motifs in the 3′-untranslated regions (3′-UTRs)
of target messenger RNAs (mRNAs) (14-17). Numerous studies have screened and
identified several deregulated miRNAs in lung fibrosis and other
diseases. Reportedly, miR-1343 can directly bind to the 3′-UTR of
TGF-β receptor 1 (TGFBR1) and TGFBR2 to inhibit the TGF-β signaling
pathway, thus alleviating PF (18). miR-449a has been found to target
autophagy-related Bcl2 mRNA to play an antifibrotic role in
silica-induced PF (19).
Moreover, miR-489 has been shown to bind to both MyD88 and Smad3 to
alleviate inflammation and the development of fibrosis (19,20). In summary, miRNAs may be involved
in the fibrotic development process by targeting various downstream
transcripts. Therefore, it is reasonable to hypothesize that miRNAs
may target Napsin A to exert an effect on PF development.
Herein, the online tool TargetScan was used to
identify miRNAs that reduce the expression of Napsin A by targeting
its 3′-UTR. Among 17 candidate miRNAs, the expression of miR-1290
was the most upregulated in blood samples obtained from patients
with PF (Fig. S1); moreover, to
the best of our knowledge, no study to date has reported a role for
miR-1290 in PF. To investigate the cellular functions of miR-1290
and its interaction with Napsin A, A549 cells were stimulated with
TGF-β1 and miR-1290 expression was examined. In addition, the
effects of miR-1290 on A549 proliferation and markers of fibrosis,
and the predicted binding of miR-1290 to Napsin A were examined.
The dynamic effects of miR-1290 and Napsin A on TGF-β1-induced
fibrosis were also evaluated. To determine the mechanisms through
which miR-1290 is upregulated during PF, the TransmiR v2.0 database
and ChIP-Atlas were used to identify transcription factors that are
related to PF and may bind the miR-1290 promoter to activate its
transcription. On the whole, the present study demonstrates a novel
mechanism through which Napsin A can be downregulated in PF by
regulating miRNA; miR-1290 may be a novel potential target for the
treatment of PF.
Materials and methods
Clinical patients and samples
PF was diagnosed according to the American Thoracic
Society/European Respiratory Society consensus criteria (21). The patients with PF and healthy
volunteers were enrolled at the First People's Hospital of
Changzhou from March, 2018 to March, 2019. The plasma samples were
isolated from patients (n=14; age, 58.5±10.94 years; sex: F/M
ratio, 5/9) with PF and healthy volunteers (n=20; age, 51.2±9.64;
sex: F/M, 8/12). All the experiments were approved by the Ethics
Committee of The First People's Hospital of Changzhou (ethics
number: 2018KY047). Written informed consent was also obtained from
all study participants.
Cell lines and cell transfection
A human non-small cell lung cancer cell line (A549;
ATCC® CCL-185) and a normal human lung epithelial cell
line (BEAS-2B; ATCC® CRL-9609) were purchased from ATCC
and cultured in F-12K medium (for A549 cells, cat. no. 30-2004,
ATCC) or bronchial epithelial basal medium BEBM (for BEAS-2B cells,
Lonza/Clonetics Corp.) supplemented with 10% FBS. The cells were
cultured at 37°C with 5% v/v CO2. For TGF-β1 treatment,
cells were stimulated with 10 ng/ml TGF-β1 for 48 h.
miR-1290 expression in target cells was assessed by
transfection with 50 nM miR-1290 mimics or 100 nM miR-1290
inhibitor (RiboBio). Napsin A was knocked down by transfection with
20 nM si-Napsin A (RiboBio). CREB1 overexpression was achieved by
transfection with 1 µg/ml CREB1 overexpression vector (CREB1
OE, RiboBio). All transfections were performed using Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.). After 48 h, the
cells were harvested for further experiments. The sequences are
listed in Table SI.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA extraction, reverse transcription, and PCR
were performed following previously described methods (22,23) using the miScript Reverse
Transcription kit (Qiagen, Inc.) and SYBR-Green PCR Master Mix
(Qiagen, Inc.). RNU6B expression or GAPDH expression was used as an
endogenous control for miRNA or mRNA determination. The
2−ΔΔCq method (24)
was used to analyze relative fold changes. The primer sequences are
listed in Table SI.
Determination of cell viability by MTT
assay
Cell viability was examined by MTT assay following
previously described methods (25). The OD values were measured at 490
nm using microplate reader (Bio-Rad Laboratories, Inc.). The cell
viability of the untreated cells (control) was defined as 100%.
Western blot analysis
The protein levels of α-smooth muscle actin (α-SMA),
Collagen I and Napsin A were examined by western blot analysis
following previously described methods (25). A total of 50 µg protein
samples were then separated by 10% SDS-PAGE and transferred onto
nitrocellulose membrane (Bio-Rad Laboratories, Inc.). The membranes
were blocked with 5% non-fat milk in TBST (0.1% Tween-20) for 2 h
at room temperature and incubated with the following antibodies:
Anti-α-SMA (1:1,000, ab5694, Abcam), anti-Collagen I (1:1,000,
ab34710), anti-Napsin A (1:1,000, ab73021, Abcam), anti-GAPDH
(1:1,000, ab8245, Abcam), anti-AKT (1:1,000, 10176-2-AP,
Proteintech), ani-p-AKT (1:1,000, 66444-1-Ig, Proteintech) and
goat-anti-rabbit or mouse HRP-conjugated secondary antibodies
(SA00001-1 and SA00001-2, Proteintech). Signals were visualized
using enhanced chemiluminescence (ECL) substrates (Merck KGaA)
using GAPDH as an endogenous reference protein. ImageJ software
version 1.8.0 (National Institute of Health) was used for
densitometric analysis.
Luciferase reporter assay
The Napsin A 3′-UTR was amplified by PCR and was
then cloned downstream of the Renilla psiCHECK2 vector
(Promega Corp.); the product was named wt-Napsin A 3′-UTR. To
generate a Napsin A 3′-UTR mutant reporter, the predicted miR-1290
binding site was mutated to remove the complementarity, and this
type of reporter vector was named mut-Napsin A 3′-UTR. 293 cells
(ATCC) were co-transfected with miR-1290 mimics or miR-1290
inhibitor and wt-Napsin A 3′UTR or mut-Napsin A 3′UTR, and the
Dual-Luciferase Reporter Assay System (Promega Corp.) was employed
to determine the luciferase activity. Renilla lucif-erase
activity was normalized to Firefly luciferase activity for each
transfected well of cells.
Transcription factors predicted to regulate miR-1290
were identified using the TransmiR v2.0 database (http://www.cuilab.cn/transmir) and ChIP-Atlas
(http://chip-atlas.org/enrichment_analysis). miR-1290
with the mutant or wild-type seed region was synthesized and cloned
into the Renilla psiCHECK2 vector (Promega Corp.). A total
of 7 nucleotides in the seed region were mutated to obtain the
mutant sequence. The CREB1 protein-coding sequence was cloned into
the pcDNA3.1 vector (Applied Biosystems). 293 cells were
co-transfected with pcDNA3.1/CREB1 and wild- or mutant-miR-1290
vectors. After 48 h, the cells were harvested and luciferase
activity assays were performed in the following treatment with
TGF-β1 or following no treatment.
Immunofluorescence (IF) staining
α-SMA protein expression was examined by IF staining
using anti-α-SMA antibody (ab5694, Abcam) according to previously
described methods (26). The
cells were incubated with FITC-conjugated secondary antibody
(Beyotime Institute of Biotechnology) for 1 h in the dark at room
temperature. Nuclei were stained with DAPI (Beyotime Institute of
Biotechnology) for 5 min at room temperature. Images were observed
and acquired by a fluorescence microscope (Nikon Corp.). Green
fluorescence indicates α-SMA expression and blue fluorescence
indicates nuclei.
Statistical analysis
Data from at least 3 independent experiments were
processed using SPSS 17.0 (IBM, Inc.) and are presented as the
means ± SD. Student's t-tests were used for statistical comparisons
between 2 groups. One-way ANOVA followed by Tukey's multiple
comparison test was used to estimate the differences among >2
groups. Pearson's correlation analysis was also used to determine
the correlation between the expression of miR-1290 and Napsin A in
the plasma samples. Values of P<0.05 and P<0.01 were
considered to indicate statistically significant and highly
statistically significant differences, respectively.
Results
miR-1290 expression is upregulated in
plasma samples from patients with PF and TGF-β1-stimulated A549
cells
Before investigating the cellular functions of
miR-1290, the first step of the present study was to evaluate
Napsin A and miR-1290 expression within blood samples obtained from
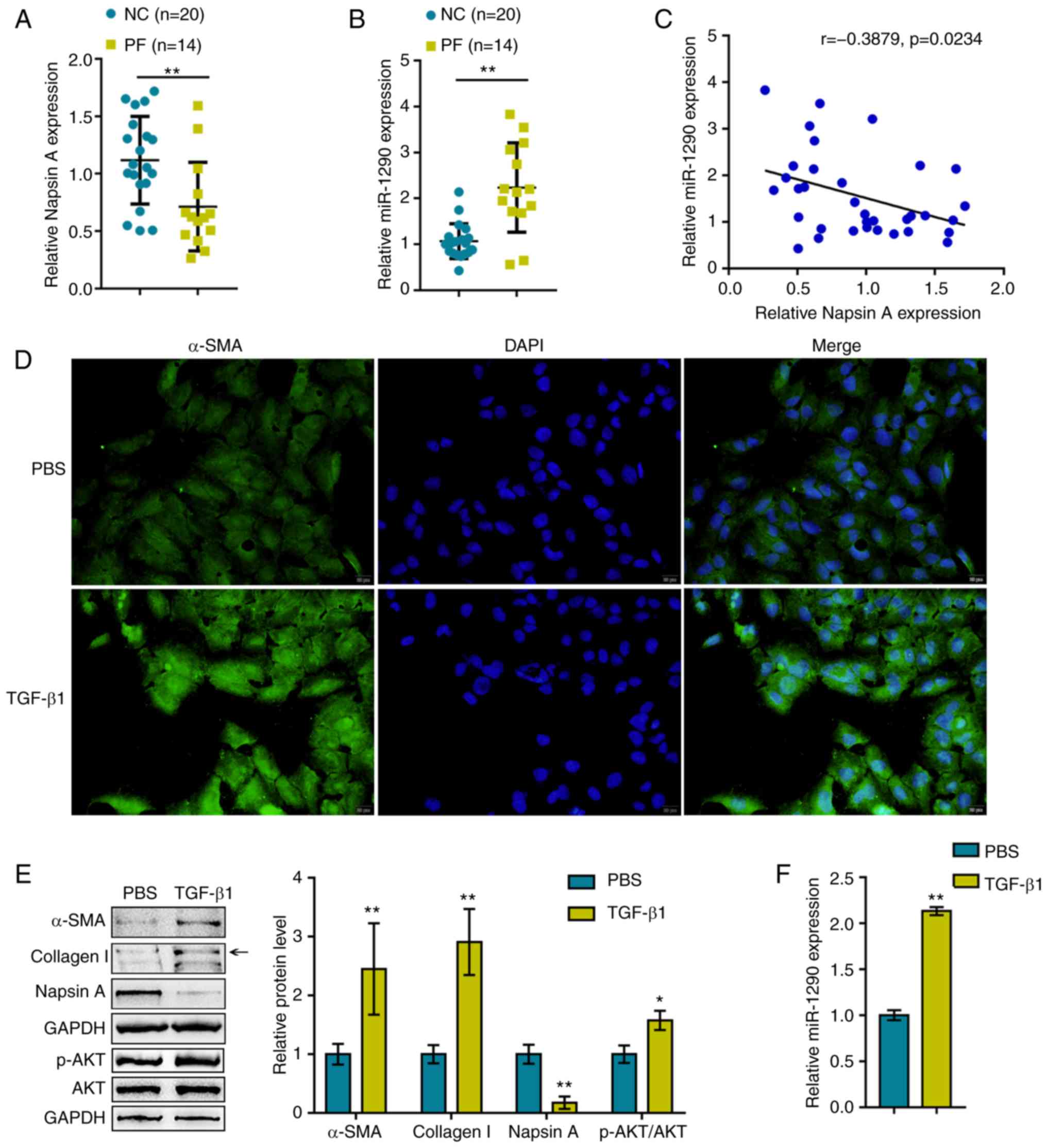
healthy donors and patients with PF. As shown in Fig. 1A and B, compared to the healthy
donors, the expression of Napsin A was significantly decreased,
whereas the expression of miR-1290 was increased within the plasma
samples obtained from patients with PF. Moreover, the expression
levels of miR-1290 negatively correlated with Napsin A expression
levels in the samples (Fig.
1C).
Previous studies have demonstrated that TGF-β1 is
related to the AKT pathway in PF (27), lung cancer (28), live fibrosis (29). Based on this, the present study
examined the association between TGF-β1 and the AKT pathway in A549
cells under TGF-β1 stimulation. The A549 cells were treated with
TGF-β1 to generate a cell model of TGF-β1-induced fibrosis, which
was validated by IF staining and western blot analysis for α-SMA,
Collagen I, Napsin A and AKT (total AKT and phosphorylated AKT)
protein expression levels. As shown in Fig. 1D and E, the α-SMA, Collagen I and
p-AKT protein levels were significantly upregulated by TGF-β1
stimulation, indicating fibrotic changes in the A549 cells. The
protein levels of Napsin A were decreased upon TGF-β1 stimulation.
In addition, the expression of miR-1290 was significantly
upregulated by TGF-β1 stimulation (Fig. 1F), suggesting that miR-1290
participates in TGF-β1-induced fibrosis.
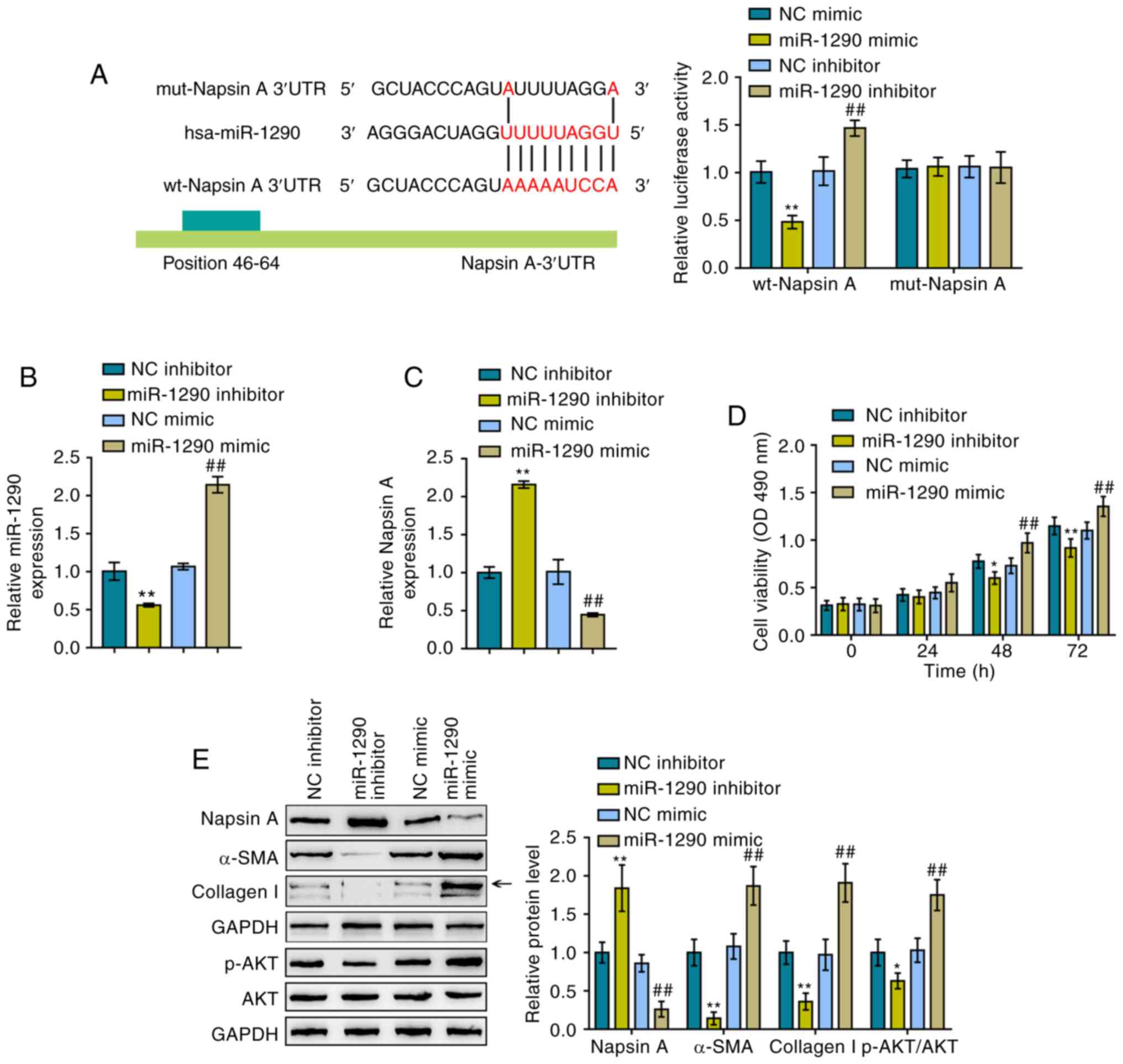
miR-1290 directly targets Napsin A to
modulate A549 cell proliferation and TGF-β1-induced fibrosis
To further determine the interaction between
miR-1290 and Naspin A, luciferase reporter assays were performed.
As described in the Materials and methods section, two different
types of Napsin A 3′-UTR luciferase reporter vectors were
constructed, the wild-type and mutant-type, and these were named
wt-Napsin A 3′-UTR and mut-Napsin A 3′-UTR, respectively (Fig. 2A). These vectors were
co-transfected into 293 cells with miR-1290 mimics or a miR-1290
inhibitor and luciferase activity examined. miR-1290 overexpression
induced a significant decrease in the luciferase activity of
wild-type Napsin A 3′-UTR, which was increased by the inhibition of
miR-1290; mutating the putative miR-1290 binding site abolished the
changes in luciferase activity (Fig.
2A). Based on these data, miR-1290 directly targeted the Napsin
A 3′-UTR. Subsequently, miR-1290 mimics/inhibitor were transfected
into the cells to achieve miR-1290 overexpression/inhibition in
A549 cells and RT-qPCR was performed to verify the transfection
efficiency (Fig. 2B).
Consistently, miR-1290 overexpression inhibited the expression of
Napsin A, while miR-1290 inhibition promoted it (Fig. 2C).
The cellular functions of miR-1290 were then
evaluated under TGF-β1 stimulation. As shown in Fig. 2D, miR-1290 overexpression
significantly promoted the proliferation of the A549 cells, whereas
miR-1290 inhibition suppressed it; in other words, miR-1290
overexpression enhanced TGF-β1-stimulated A549 cell growth, while
miR-1290 inhibition decreased the growth. To evaluate the
cytotoxicity of miR-1290 against normal cells, the viability of the
human normal lung epithelial cell line, BEAS-2B, was examined by
MTT assay (Fig. S2). The
transfection efficiency of miR-1290 mimics and miR-1290 inhibitor
in BEAS-2B cells is illustrated in Fig. S2A. According to the results of
MTT assay (Fig. S2B), miR-1290
overexpression markedly promoted the proliferation of BEAS-2B
cells, whereas miR-1290 inhibition restrained it. In addition,
miR-1290 overexpression decreased the Napsin A protein levels and
increased the protein levels of α-SMA, Collagen I and p-AKT in A549
cells. However, miR-1290 inhibition exerted opposing effects on the
protein levels of Napsin A, α-SMA, Collagen I and p-AKT under
TGF-β1 stimulation in A549 cells (Fig. 2E). In summary, miR-1290 may
directly bind to the 3′-UTR of Napsin A, and miR-1290 inhibition
improves TGF-β1-induced fibrosis.
Dynamic effects of miR-1290 and Napsin A
on A549 cell proliferation and TGF-β1-induced fibrotic changes
After confirming the effects of miR-1290 on
TGF-β1-induced fibrotic changes, the dynamic effects of miR-1290
and its downstream target, Napsin A, on A549 proliferation and
TGF-β1-induced fibrotic changes were evaluated. The A549 cells were
trans-fected with si-Napsin A to knock down its expression and
RT-qPCR was then performed to verify the transfection efficiency
(Fig. 3A). The A549 cells were
co-transfected with miR-1290 inhibitor/si-Napsin A, and Napsin A
expression, cell viability, and Napsin A, α-SMA and Collagen I
protein levels were then evaluated. The TGF-β1-induced suppression
of Napsin A expression was significantly reversed by transfection
with miR-1290 inhibitor, whereas it was further suppressed by
Napsin A knockdown; Napsin A knockdown significantly attenuated the
effects of the miR-1290 inhibitor (Fig. 3B).
As regards cellular functions, miR-1290 inhibition
reduced, whereas Napsin A knockdown increased TGF-β1-induced A549
cell proliferation; the effect of miR-1290 inhibitor was also
reversed by Napsin A knockdown (Fig.
3C). Consistently, TGF-β1 decreased the Napsin A levels,
whereas it increased the α-SMA, Collagen I and p-AKT levels, and
this effect was attenuated by treatment with the miR-1290
inhibitor, but enhanced by Napsin A knockdown; Napsin A knockdown
significantly attenuated the effect of the miR-1290 inhibitor
(Fig. 3D). In summary, these
findings demonstrate that miR-1290 regulates TGF-β1-induced
fibrotic changes via Napsin A.
TGF-β1-induced CREB1 expression promotes
the transcription of miR-1290
To further investigate the mechanisms through which
TGF-β1 induces the expression of miR-1290, the online tools,
TransmiR v2.0 database and ChIP-Atlas, were used to identify
transcription factors that may regulate miR-1290 expression and can
be induced by TGF-β1 stimulation; CREB1 was identified (30,31). The miR-1290 luciferase reporter
vector was constructed as described in the Materials and methods
section and was co-transfected into 293 cells with pcDNA3.1/CREB1.
Luciferase activity was determined in the presence or absence of
TGF-β1 treatment. As shown in Fig.
4A, CREB1 overexpression significantly enhanced the luciferase
activity of the miR-1290 reporter vector and TGF-β1 treatment
further enhanced the CREB1 overexpression-induced increase in
luciferase activity.
To validate the effects of CREB1 on miR-1290, the
CREB1-overexpressing vector was transfected into the A549 cells to
induce CREB1 overexpression and RT-qPCR was then performed to
verify the transfection efficiency (Fig. 4B). As predicted, CREB1
overexpression significantly promoted the expression of miR-1290
(Fig. 4C). Therefore, a novel
mechanism is demonstrated through which TGF-β1-induced CREB1
upregulation promotes the transcription of miR-1290, thus
inhibiting Napsin A expression and promoting fibrotic changes in
A549 cells (Fig. 4D).
Discussion
Herein, miR-1290 was regarded as an upstream
regulatory miRNA, that reduces the expression of Napsin A by
binding to its 3′-UTR. miR-1290 was upregulated in PF blood samples
and in TGF-β1-stimulated A549 cells. miR-1290 can directly target
Napsin A, significantly promote A549 proliferation and increase the
protein levels of markers of fibrosis. Napsin A knockdown exerted
effects on A549 proliferation and TGF-β1-induced fibrosis that were
similar to those observed following miR-1290 overexpression; more
importantly, Napsin A knockdown significantly reversed the effects
of miR-1290 inhibition, indicating that miR-1290 promotes
TGF-β1-induced fibrosis by targeting Napsin A. Moreover,
TGF-β1-induced CREB1 overexpression promoted the transcription of
miR-1290 in A549 cells.
Over the past decade, emerging evidence has revealed
potential biomarkers for the prediction of PF. According to
microarray profiles, multiple miRNAs, including miR-29 (32,33), miR-326 (34), miR-98 (35) and miR-let-7d (36), may participate in the pathogenesis
of PF. Herein, the expression of miR-1290 was significantly
upregulated in blood samples obtained from patients with PF and in
A549 cells upon TGF-β1 stimulation, and the overexpression of
miR-1290 promoted A549 cell proliferation and fibrosis marker
proteins levels under TGF-β1 stimulation conditions, indicating the
potential of miR-1290 as a novel biomarker for PF. Previously,
miR-1290 expression has been reported to be significantly
upregulated by Matrigel and may thus be cancer-related. Matrigel is
considered to be a medium that is rich in extracellular matrix
(ECM) components and capable of altering cellular cell phenotypes
and gene expression (37).
miR-1290 expression is abnormally upregulated in non-small cell
lung cancer (NSCLC); thus, serum levels of miR-1290 may serve as an
underlying prognostic biomarker for NSCLC (38). However, to the best of our
knowledge, this is the first study to report the upregulation of
miR-1290 in blood samples from patients with PF. miR-1290 may thus
play a critical role in PF progression.
As has already been mentioned, miR-1290 expression
is significantly upregulated by TGF-β1 stimulation. Upon TGF-β1
treatment, miR-1290 overexpression further enhanced the promotive
effects of TGF-β1 on A549 cell growth and α-SMA and Collagen I
protein levels. In other words, miR-1290 may antagonize
TGF-β1-induced fibrotic changes in A549 cells. More importantly, as
predicted by an online tool, miR-1290 overexpression significantly
inhibited the protein levels of Napsin A. Previously, it was
demonstrated that Napsin A overexpression significantly reversed or
attenuated TGF-β1-induced fibrotic changes in A549 cells (39) Herein, it was demonstrated that
miR-1290 targets the 3′-UTR of Napsin A to exert a negative
regulatory effect on the expression of Napsin A upon TGF-β1
stimulation. The effects of miR-1290 inhibition on TGF-β1-induced
fibrotic changes in A549 cells were reversed by Napsin A silencing,
indicating that miR-1290 promotes TGF-β1-induced fibrosis by
targeting Napsin A.
Since the expression of miR-1290 was significantly
increased in the serum obtained from patients with PF, the
mechanisms of the abnormal miR-1290 upregulation in serum from
patients with PF were further investigated. According to
bioinformatics analyses, CREB1 may activate the transcription of
miR-1290 via targeting its promoter region. Inflammation, cell
growth, differentiation, adaptation and survival, as well as other
cell functions, have been shown to be modulated by CREB (40). It has been revealed that an
increase in CREB activity can be related to the pathologic
mechanism of asthma, changes in cognitive memory, and the process
of chronic obstructive pulmonary disease (COPD) (41). Herein, the predicted binding of
CREB1 to miR-1290 was validated by revealing that TGF-β1-induced
CREB1 overexpression promoted the expression of miR-1290 by
targeting the promoter region.
In conclusion, the present study demonstrates that
TGF-β1-induced CREB1 overexpression significantly upregulates
miR-1290 expression, therefore antagonizing TGF-β1-induced fibrotic
changes in A549 cells through the miR-1290 downstream target,
Napsin A. The CREB1/miR- 1290/Napsin A axis may thus be a potent
target in the treatment of TGF-β1-induced PF. However, further
in vivo studies and clinical investigations are warranted to
confirm these findings.
Supplementary Data
Funding
The present study was supported by the 2018 Applied
Basic Research Program of Changzhou Science and Technology Bureau
(CJ20189021).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
SG and JZ made substantial contributions to the
conception and design of the study. SG and YW contributed to the
experimental design and study implementation. QZ analyzed and
interpreted the data. SG and QZ drafted the manuscript. JZ revised
the study critically for important intellectual content. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures performed in experiments involving
human participants were in accordance with the ethical standards of
the Ethics Committee of The First People's Hospital of Changzhou.
All participants signed informed consent forms.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Lepparanta O, Sens C, Salmenkivi K,
Kinnula VL, Keski-Oja J, Myllärniemi M and Koli K: Regulation of
TGF-β storage and activation in the human idiopathic pulmonary
fibrosis lung. Cell Tissue Res. 348:491–503. 2012. View Article : Google Scholar
|
|
2
|
Ni S, Wang D, Qiu X, Pang L, Song Z and
Guo K: Bone marrow mesenchymal stem cells protect against
bleomycin-induced pulmonary fibrosis in rat by activating Nrf2
signaling. Int J Clin Exp Pathol. 8:7752–7761. 2015.PubMed/NCBI
|
|
3
|
Antoniou KM, Margaritopoulos GA and
Siafakas NM: Pharmacological treatment of idiopathic pulmonary
fibrosis: From the past to the future. Eur Respir Rev. 22:281–291.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Uchida A, Samukawa T, Kumamoto T, Ohshige
M, Hatanaka K, Nakamura Y, Mizuno K, Higashimoto I, Sato M and
Inoue H: Napsin A levels in epithelial lining fluid as a diagnostic
biomarker of primary lung adenocarcinoma. BMC Pulm Med. 17:1952017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu J, Zhang Y, Ding T, Cheng R, Gong W,
Guo Y, Luo Y, Pan Y, Zhai Q, Sun W, et al: Napsin A expression in
subtypes of thyroid tumors: Comparison with lung adenocarcinomas.
Endocr Pathol. 31:39–45. 2020. View Article : Google Scholar
|
|
6
|
Beck J, Miller MA, Frank C, DuSold D and
Ramos-Vara JA: Surfactant Protein A and Napsin A in the
immunohistochemical characterization of canine pulmonary
carcinomas: Comparison with thyroid transcription factor-1. Vet
Pathol. 54:767–774. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hirano T, Gong Y, Yoshida K, Kato Y,
Yashima K, Maeda M, Nakagawa A, Fujioka K, Ohira T, Ikeda N, et al:
Usefulness of TA02 (napsin A) to distinguish primary lung
adenocarcinoma from metastatic lung adenocarcinoma. Lung Cancer.
41:155–162. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bishop JA, Sharma R and Illei PB: Napsin A
and thyroid transcription factor-1 expression in carcinomas of the
lung, breast, pancreas, colon, kidney, thyroid, and malignant
mesothelioma. Hum Pathol. 41:20–25. 2010. View Article : Google Scholar
|
|
9
|
Samukawa T, Hamada T, Uto H, Yanagi M,
Tsukuya G, Nosaki T, Maeda M, Hirano T, Tsubouchi H and Inoue H:
The elevation of serum napsin A in idiopathic pulmonary fibrosis,
compared with KL-6, surfactant protein-A and surfactant protein-D.
BMC Pulm Med. 12:552012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
du Bois RM, Weycker D, Albera C, Bradford
WZ, Costabel U, Kartashov A, Lancaster L, Noble PW, Raghu G, Sahn
SA, et al: Ascertainment of individual risk of mortality for
patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care
Med. 184:459–466. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
du Bois RM, Weycker D, Albera C, Bradford
WZ, Costabel U, Kartashov A, King TE Jr, Lancaster L, Noble PW,
Sahn SA, et al: Forced vital capacity in patients with idiopathic
pulmonary fibrosis: Test properties and minimal clinically
important difference. Am J Respir Crit Care Med. 184:1382–1389.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ueno T, Linder S and Elmberger G: Aspartic
proteinase napsin is a useful marker for diagnosis of primary lung
adenocarcinoma. Br J Cancer. 88:1229–1233. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zheng JX, Guan SH, Xu Q, Tang Y, Liu JZ
and Lu XT: Effect of Napsin A transfection into type II alveolar
epithelial cells on pulmonary fibrosis. Zhonghua Yi Xue Za Zhi.
90:3294–3299. 2010.In Chinese.
|
|
14
|
Ambros V: microRNAs: Tiny regulators with
great potential. Cell. 107:823–826. 2001. View Article : Google Scholar
|
|
15
|
Chapman EJ and Carrington JC:
Specialization and evolution of endogenous small RNA pathways. Nat
Rev Genet. 8:884–896. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen K and Rajewsky N: The evolution of
gene regulation by transcription factors and microRNAs. Nat Rev
Genet. 8:93–103. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo H, Ingolia NT, Weissman JS and Bartel
DP: Mammalian microRNAs predominantly act to decrease target mRNA
levels. Nature. 466:835–840. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stolzenburg LR, Wachtel S, Dang H and
Harris A: miR-1343 attenuates pathways of fibrosis by targeting the
TGF-β receptors. Biochem J. 473:245–256. 2016. View Article : Google Scholar
|
|
19
|
Han R, Ji X, Rong R, Li Y, Yao W, Yuan J,
Wu Q, Yang J, Yan W, Han L, et al: miR-449a regulates autophagy to
inhibit silica-induced pulmonary fibrosis through targeting Bcl2. J
Mol Med (Berl). 94:1267–1279. 2016. View Article : Google Scholar
|
|
20
|
Wu Q, Han L, Yan W, Ji X, Han R, Yang J,
Yuan J and Ni C: miR-489 inhibits silica-induced pulmonary fibrosis
by targeting MyD88 and Smad3 and is negatively regulated by lncRNA
CHRF. Sci Rep. 6:309212016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Raghu G, Rochwerg B, Zhang Y, Garcia CA,
Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, et
al: An official ATS/ERS/JRS/ALAT clinical practice guideline:
Treatment of idiopathic pulmonary fibrosis. An update of the 2011
clinical practice guideline. Am J Respir Crit Care Med. 192:e3–19.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aslani S, Mahmoudi M, Garshasbi M,
Jamshidi AR, Karami J and Nicknam MH: Evaluation of DNMT1 gene
expression profile and methylation of its promoter region in
patients with ankylosing spondylitis. Clin Rheumatol. 35:2723–2731.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Asadi M, Shanehbandi D, Mohammadpour H,
Hashemzadeh S and Sepehri B: Expression level of miR-34a in tumor
tissue from patients with esophageal squamous cell carcinoma. J
Gastrointest Cancer. 50:304–307. 2019. View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
25
|
Liu H, Deng H, Zhao Y, Li C and Liang Y:
LncRNA XIST/miR-34a axis modulates the cell proliferation and tumor
growth of thyroid cancer through MET-PI3K-AKT signaling. J Exp Clin
Cancer Res. 37:2792018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang TH, Liang LZ, Liu XL, Wu JN, Su K,
Chen JY and Zheng QY: LncRNA UCA1/miR-124 axis modulates
TGFβ1-induced epithelial-mesenchymal transition and invasion of
tongue cancer cells through JAG1/Notch signaling. J Cell Biochem.
120:10495–10504. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gimenez A, Duch P, Puig M, Gabasa M,
Xaubet A and Alcaraz J: Dysregulated collagen homeostasis by matrix
stiffening and TGF-β1 in fibroblasts from idiopathic pulmonary
fibrosis patients: Role of FAK/Akt. Int J Mol Sci. 18:E24312017.
View Article : Google Scholar
|
|
28
|
Jiang W, Xu Z, Yu L, Che J, Zhang J and
Yang J: MicroRNA-144-3p suppressed TGF-β1-induced lung cancer cell
invasion and adhesion by regulating the Src-Akt-Erk pathway. Cell
Biol Int. Apr 30–2019.Epub ahead of print.
|
|
29
|
Mi XJ, Hou JG, Jiang S, Liu Z, Tang S, Liu
XX, Wang YP, Chen C, Wang Z and Li W: Maltol mitigates
thioacetamide-induced liver fibrosis through TGF-β1-mediated
activation of PI3K/Akt signaling pathway. J Agric Food Chem.
67:1392–1401. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jang YS, Kim JH, Seo GY and Kim PH: TGF-β1
stimulates mouse macrophages to express APRIL through Smad and
p38MAPK/CREB pathways. Mol Cells. 32:251–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Singh R, Shankar BS and Sainis KB:
TGF-β1-ROS-ATM-CREB signaling axis in macrophage mediated migration
of human breast cancer MCF7 cells. Cell Signal. 26:1604–1615. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Herrera J, Beisang DJ, Peterson M, Forster
C, Gilbertsen A, Benyumov A, Smith K, Korenczuk CE, Barocas VH,
Guenther K, et al: Dicer1 deficiency in the idiopathic pulmonary
fibrosis fibroblastic focus promotes fibrosis by suppressing
MicroRNA biogenesis. Am J Respir Crit Care Med. 198:486–496. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X, Xu K, Yang XY, Liu J, Zeng Q and
Wang FS: Upregulated miR-29c suppresses silica-induced lung
fibrosis through the Wnt/beta-catenin pathway in mice. Hum Exp
Toxicol. 37:944–952. 2018. View Article : Google Scholar
|
|
34
|
Xu T, Yan W, Wu Q, Xu Q, Yuan J, Li Y, Li
P, Pan H and Ni C: miR-326 inhibits inflammation and promotes
autophagy in silica-induced pulmonary fibrosis through targeting
TNFSF14 and PTBP1. Chem Res Toxicol. 32:2192–2203. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gao SY, Zhou X, Li YJ, Liu WL, Wang PY,
Pang M, Xie SY and Lv CJ: Arsenic trioxide prevents rat pulmonary
fibrosis via miR-98 overexpression. Life Sci. 114:20–28. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huleihel L, Ben-Yehudah A, Milosevic J, Yu
G, Pandit K, Sakamoto K, Yousef H, LeJeune M, Coon TA, Redinger CJ,
et al: Let-7d microRNA affects mesenchymal phenotypic properties of
lung fibroblasts. Am J Physiol Lung Cell Mol Physiol.
306:L534–L542. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Price KJ, Tsykin A, Giles KM, Sladic RT,
Epis MR, Ganss R, Goodall GJ and Leedman PJ: Matrigel basement
membrane matrix influences expression of microRNAs in cancer cell
lines. Biochem Biophys Res Commun. 427:343–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jin JJ, Liu YH, Si JM, Ni R and Wang J:
Overexpression of miR-1290 contributes to cell proliferation and
invasion of non small cell lung cancer by targeting interferon
regulatory factor 2. Int J Biochem Cell Biol. 95:113–120. 2018.
View Article : Google Scholar
|
|
39
|
Zheng JX, Guan SH, Xu Q, Liu JZ and Song
P: Inhibition of epithelial-mesenchymal transition in A549 cell by
transfected Napsin A. Chin Med J (Engl). 125:2734–2740. 2012.
|
|
40
|
Sirotkin AV, Benco A, Mlyncek M, Harrath
AH, Alwasel S and Kotwica J: The involvement of the
phosphorylatable and nonphosphorylatable transcription factor
CREB-1 in the control of human ovarian cell functions. C R Biol.
342:90–96. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mroz RM, Holownia A, Chyczewska E, Drost
EM, Braszko JJ, Noparlik J, Donaldson K and Macnee W:
Cytoplasm-nuclear trafficking of CREB and CREB phosphorylation at
Ser133 during therapy of chronic obstructive pulmonary disease. J
Physiol Pharmacol. 58(Suppl 5): S437–S444. 2007.
|