Introduction
Glioblastoma (GBM) is the most common and aggressive
primary brain tumor, accounting for ~30% of primary intracranial
tumors worldwide (1). Because of
its highly invasive growth and heterogeneous nature, the average
survival time of patients with GBM is ~1 year (2). The standard GBM treatment of
surgical resection followed by radiotherapy and postoperative
chemotherapy has been improved dramatically, but the disease
prognosis remains poor (3). While
considerable progress has been made in the past decade in the
understanding of the pathology of GBM (4), the underlying pathogenic mechanism
of this tumor remains poorly understood.
Long non-coding RNAs (lncRNAs) have recently
attracted considerable attention and become an important area of
research. These RNAs are longer than 200 nucleotides and have no
protein-coding potential (5).
They can play complex and critical roles in tumor initiation and
progression (6). For instance,
the AGAP2-AS1 expression level was found to be elevated in GBM, and
it functions as an oncogenic lncRNA to modulate GBM cell
proliferation and apoptosis, suggesting that AGAP2-AS1 is a
potential therapeutic target for GBM (7). Furthermore, the lncRNA CASP5 is
upregulated in GBM tissues and promotes the malignant phenotypes of
GBM (8). These discoveries
suggested that lncRNAs could be excellent prognostic biomarkers and
potential therapeutic targets for GBM. Although recent studies have
identified some lncRNAs that exert regulatory activities during the
development of GBM, these have mainly focused on the expression
pattern of lncRNAs (9,10). At present, the regulatory
mechanism of the vast majority of lncRNAs in GBM, especially DNA
methylation, remains unclear.
DNA methylation, as an essential epigenetic
modification, is involved in a variety of biological processes and
mediates the dysregulation of gene expression (11). This modification can form a
molecular basis for the silencing of tumor suppressors and the
activation of oncogenes (12). In
general, the hypermethylation of the gene promoter can downregulate
or even silence gene expression, while the hypomethylation of the
gene promoter tends to activate gene expression. For example, a
previous study showed that the gene expression level of microRNA
(miRNA/miR)-205 decreased in GBM tissues compared to controls, and
lower expression was significantly associated with promoter
hypermethylation (13). Tabu
et al (14) found that
promoter hypomethylation was an important determinant of CD133
overexpression in GBM, and this epigenetic event may be associated
with the development of brain tumor-initiating cells expressing
CD133. Recent studies have only described the aberrant methylation
of some specific genes in GBM (15,16). However, the association between
lncRNA methylation events and transcriptional changes at a global
scale in GBM remains unknown.
With the improvement of high-throughput sequencing
technology, large-scale Illumina Infinium Human Methylation 450
BeadChips (HM450k; Illumina, Inc.) and RNA-sequencing (RNA-seq)
data have been applied for the analysis of cancer (17). The present study used a
reannotation strategy to construct the DNA methylation profile of
lncRNAs in GBM. The lncRNAs whose expression might be regulated by
aberrant promoter methylation were determined according to two
criteria, and the collective lncRNAs obtained in both scenarios
were used for further analysis, including 314 hypermethylated
lncRNAs (UhyperLncs) and 668 hypomethylated lncRNAs (UhypoLncs).
Then, a methylation-mediated lncRNA regulatory network (MLRN) and
functional analysis were used to elucidate the regulatory mechanism
of lncRNAs and predict the functions of aberrantly methylated
lncRNAs. Specially, it was found that four lncRNAs may have a good
diagnostic and prognostic function. Finally, through the
construction of drug-target association networks, the present study
provided potential therapeutic targets and small-molecule drugs for
GBM treatment. The present study enhanced the understanding of the
regulatory mechanism of lncRNAs through DNA methylation and
provided potential cancer biomarkers for the diagnosis and
treatment of GBM.
Materials and methods
Data sources
The molecular data used in the present study were
collected from various platforms. The DNA methylation data (level
three) were generated on the Infinium HM450k platform (18). The HM450k data of tumor samples of
GBM were downloaded from The Cancer Genome Atlas (TCGA; https://portal.gdc.cancer.gov) and the data of normal
samples, which contained 58 normal glial cell samples [GSE41826
(19)], were downloaded from the
Gene Expression Omnibus (GEO) (20). The normal data were used in
previous research and generated at the same organization via the
same pipeline (21). The RNA-seq
data of GBM from the Illumina RNAseqV2 platform (Illumina, Inc.)
were downloaded from TCGA, which contained 156 tumor samples and
five normal samples. To ensure the quality of the research, 50
tumor samples were specifically selected as the experimental
dataset, among which DNA methylation data and RNA-seq data were
both available. The remaining samples were used as a validation
dataset (90 DNA methylation tumor samples and 106 RNA-seq tumor
samples), where the normal samples in the experimental dataset were
also applied to the validation dataset.
The human comprehensive gene annotation data were
derived from GENCODE (release 19) (22). The experimental interactions
between lncRNAs and miRNAs were collected from the starBase v2.0
database (23) and DIANA-LncBase
v2 database (24). Human miRNAs
and their targets were downloaded from starBase v2.0 and miRTarBase
(release 7.0) (25). Both
databases store manually curated collections of experimentally
supported miRNA targets. The relationships between miRNAs and
diseases were downloaded from the Human MicroRNA Disease Database
(HMDD) (26). Comprehensive
information about small molecule effects on miRNA expression was
collected from SM2miR (27).
Approved and experimentally validated drug target information were
downloaded from DrugBank (v5.1.2) (28) and PharmGKB (29). Clinical data in XML format were
downloaded from TCGA data portal for survival analysis.
Data normalization and construction of
lncRNA methylation profile
The lncRNA and mRNA expression values were
recalculated by TMM normalization and voom transformation for the
expression data in raw read count format (30,31).
To evaluate the methylation values of a given probe,
the methylation level of each probe was measured as a β-value,
which is calculated as the ratio of methylated signal to the sum of
the methylated and unmethylated signals. The range of β-values is
from 0 (unmethylated) to 1 (completely methylated). To estimate the
quality of the probe, the number of probes with missing values in
all tumor samples was calculated. In total, 89,512 probes were
removed, and the remaining missing values were filled in using the
k-nearest neighbors method (32)
with the knnImputation function in the DMwR package (https://mirrors.tuna.tsinghua.edu.cn/CRAN/src/contrib/DMwR_0.4.1.tar.gz).
Finally, 392,867 CpG sites were obtained and used for constructing
the lncRNA methylation profile.
Human genome annotation data were filtered to
extract the lncRNA promoter information. Since the regulatory
mechanism of lncRNA transcription is similar to the regulation of
coding genes, 10 kb upstream from the transcriptional start site
(TSS) was used for a relatively comprehensive range of lncRNA
promoters (17). The 392,867
probes were mapped to the lncRNA promoter regions and only the
probe closest to the TSS was used to determine the DNA methylation
level of each lncRNA promoter (17,33).
Obtaining aberrant methylation-mediated
lncRNAs
The limma package (34) was used to identify aberrantly
methylated lncRNAs and differentially expressed lncRNAs between the
tumor and normal samples, based on DNA methylation and RNA-seq
data. The P-values were corrected using the Benjamini-Hochberg
method (35), and only the
lncRNAs with a corrected P≤0.05 were considered significant. To
evaluate the correlation between methylation levels and expression
levels of aberrantly methylated lncRNAs, the Pearson correlation
coefficient (PCC) was calculated for each lncRNA between the
methylation value and the corresponding expression value.
To comprehensively analyze lncRNAs whose expression
levels were regulated by corresponding aberrant promoter
methylation, the lncRNAs were selected for further study if they
satisfied one of the following criteria: i) Hypermethylated lncRNAs
and lncRNAs with significantly downregulated expression were
overlapped as hypermethylated-underexpressed lncRNAs; similarly,
hypomethylated lncRNAs and lncRNAs with significantly upregulated
expression were overlapped as hypomethylated-highly expressed
lncRNAs; and ii) the lncRNAs with PCC <-0.2 and P<0.05 were
retained. Additionally, Student's t-tests and one-way ANOVA with
Bonferroni's correction as the post hoc test were used to compare
the validation dataset with the normal samples to obtain P-values;
P<0.05 was considered to indicate a statistically significant
difference.
Regulatory network construction and
visualization
Experimentally supported interaction pairs of
lncRNAs-miRNAs and miRNAs-targets were collected and integrated
from multiple databases. A total of 32,452 non-redundant
lncRNA-miRNA interactions and 729,567 miRNA-mRNA pairs were
retained for further analysis. A candidate methylation-mediated
lncRNA binary tuple (MlncBT) was defined as a lncRNA-mRNA
interaction pair found to interact with the same miRNA in which the
lncRNA was aberrantly methylated. In total, 3,531,816 potential
MlncBTs were identified from lncRNA-miRNA and miRNA-mRNA
interactions. To identify competing MlncBTs, the hypergeometric
distribution was calculated to evaluate the significance of the
shared miRNAs between each lncRNA and mRNA. The P-values were
adjusted by false discovery rate (FDR), and MlncBTs with FDR≤0.05
were considered significant. In total, 916,628 candidate MlncBTs
were retained for further identification.
Previous studies have indicated that increased
lncRNA expression can enhance corresponding coding gene expression
(36,37). The PCC of each MlncBT was
calculated based on the lncRNA and mRNA expression profiles.
PCC>0 and FDR≤0.05 were used as thresholds to screen out 28,721
MlncBTs comprising 262 lncRNAs and 9,441 mRNAs.
Finally, Cytoscape software (v3.7.0) (38) was used to visualize the regulatory
network and divide the network into the hypermethylated
lncRNA-mediated network and the hypomethylated lncRNA-mediated
network.
Functional prediction of lncRNAs with
different methylation patterns
Functional annotation of lncRNAs based on a
co-expression network has been verified to be effective and
accurate in previous studies (39,40). The mRNAs co-expressed with
UhyperLncs and UhypoLncs in the regulatory network were used to
perform functional enrichment analysis. Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment
analyses were performed to identify the significantly enriched
biological processes and pathways, using the R package
clusterProfiler (41). Only the
GO terms and pathways with corrected P≤0.05 were retained to assess
the potential functions of aberrantly methylated lncRNAs in
GBM.
Identification of MlncBTs associated with
GBM prognosis
To identify the clinical effect of MlncBTs, the
patients were randomly divided into a training set and a test set,
based on all the expression profile data (the sample sizes were the
same in both groups). A Cox proportional hazards regression model
was fitted to evaluate the association between the expression
profile of each gene and patient survival in GBM. The prognostic
index (PI) was adopted to classify the risk groups, as follows:
PI=∑i=1nβiXi, where n was the
number of survival correlated genes, βi was the
Cox regression coefficient for genei and
Xi was the expression level of
genei in a corresponding patient. The median PI
was used as a cut-off to divide patients in the training set into
high- and low-risk groups. This PI model and cut-off point were
also applied to the test set to divide the patients into high- and
low-risk groups. Kaplan-Meier survival analysis and the log-rank
test (P≤0.05) were performed to estimate the survival difference
between the two patient groups. Moreover, receiver operating
characteristic (ROC) analysis was performed to see whether the
lncRNAs could be used as cancer biomarkers for early diagnosis of
GBM.
Prediction of small molecule drugs for
GBM treatment
To improve the accuracy of predicting small molecule
drugs and targets, high-competing binary sub-networks were derived
from the regulatory network by applying a PCC threshold >0.5. It
was deemed that the sub-networks revealed a more stable regulatory
relationship. Since the perturbation of miRNA expression could
influence the expression level of many lncRNAs and mRNAs (42), the lncRNA-miRNA-mRNA triple
sub-networks were further constructed, with the binary sub-networks
as the background. A total of 231 GBM-related miRNAs were filtered
from HMDD and 2,583 non-redundant miRNA-small molecule associations
from SM2miR were integrated, with 1,084 associations of small
molecules that could downregulate miRNA expression and 1,499
associations of small molecules that could upregulate miRNA
expression. It was ensured that each miRNA in the sub-networks was
among the GBM-related miRNAs that were screened from HMDD. The
drug-target association network targeting miRNAs based on the
hypermethylated lncRNAs was constructed by matching 1,084
associations with the hypermethylation-mediated lncRNA sub-network.
Similarly, the drug-target association network targeting miRNAs
based on the hypomethylated lncRNAs was constructed by matching
1,499 associations with the hypomethylation-mediated lncRNA
sub-network. Furthermore, 14,256 gene-drug associations were
integrated from DrugBank and PharmGKB. These gene-drug associations
were used as seeds to match the drug-target networks targeting
miRNAs to construct drug-target networks targeting mRNAs. Finally,
drug-target association networks were constructed and illustrated
using Cytoscape software (v3.7.0).
D-lnc (43)
curates experimentally validated regulatory effects of drugs on
lncRNA expression and contains 4,960 lncRNA-drug regulatory
relationships for Homo sapiens. These lncRNA-drug regulatory
relationships were integrated and matched with the associations in
the drug-target networks that had been constructed in the present
study, in order to verify the predicted drugs and targets. All
analyses were performed using R 3.5.1 software (44).
Results
Characterization of global differences in
DNA methylation and expression
To construct the DNA methylation profile of lncRNAs
in GBM, a computational strategy was adopted to reannotate data
from Infinium 450 k arrays into human lncRNA associated promoter
regions. In the present study, 44,523 probes were located in 7,200
lncRNA promoter regions, in which each lncRNA had at least one
probe mapped to the corresponding promoter region. Although each
lncRNA had several probes mapping to the corresponding promoter
region, only the probes closest to each TSS were retained to
determine the DNA methylation status of the lncRNA promoters.
In some cancer types, previous studies have found
that genome-wide DNA hypomethylation may be observed early in
tumorigenesis, impacting genome stability and contributing to
cellular transformation (12,45). After preprocessing the profiles,
the present study focused on lncRNAs with significantly aberrant
methylation between GBM and normal samples. In total, 5,567
aberrantly methylated lncRNAs were identified, including 1,214
hypermethylated lncRNAs and 4,353 hypomethylated lncRNAs.
Meanwhile, hierarchical clustering analysis of these aberrantly
methylated lncRNAs was performed according to the DNA methylation
level (Fig. 1A). It was noted
that these lncRNAs markedly differed between the tumor and normal
tissues, and the number of hypomethyl-ated lncRNAs was much greater
than that of hypermethylated lncRNAs. These results indicated that
lncRNAs exhibit a more hypomethylated pattern during the occurrence
and development of GBM. This global lncRNA hypomethylation may
cause oncogene activation and genomic instability, and initiate
tumorigenesis (46,47). Moreover, 2,567 differentially
expressed lncRNAs were obtained in GBM compared to corresponding
normal samples, in which 713 lncRNAs were upregulated, and 1,854
lncRNAs were downregulated. The expression levels of the
differentially expressed lncRNAs are shown in a heatmap (Fig. 1B).
Exploring aberrant methylation regulation
patterns of lncRNAs in GBM
Since aberrant promoter methylation silences tumor
suppressor genes or activates oncogenes (48), it was considered that aberrant
promoter methylation of lncRNAs might be an important epigenetic
regulator of lncRNA expression in GBM. To comprehensively analyze
the regulatory effects of lncRNA aberrant promoter methylation on
their expression in GBM, two criteria with unique biological and
statistical significance were considered (Fig. 2A). For the first criterion, 213
hypermethylated-underexpressed lncRNAs were obtained by overlapping
the hypermethylated lncRNAs and significantly underexpressed
lncRNAs. Similarly, 288 hypomethylated-highly expressed lncRNAs
were identified by overlapping the hypomethylated lncRNAs and
significantly highly expressed lncRNAs. For the second criterion,
564 significantly negatively correlated lncRNAs were screened out
by calculating the PCC of each aberrantly methylated lncRNA between
methylation and expression level. The collective lncRNAs obtained
in both scenarios was used for further analysis (Table SI), including 314 hypermethylated
lncRNAs and 668 hypomethylated lncRNAs. Notably, 83 lncRNAs
satisfied both criteria, among which 45 were hypermethylated and 38
were hypomethylated (Fig. S1).
It was considered that the aberrant promoter methylation of these
83 lncRNAs was more likely to be an epigenetic regulator of their
expression.
A previous report suggested that different biotypes
of lncRNAs perform distinct functions (49). Therefore, the two categories of
lncRNAs were subdivided based on their location with respect to
protein-coding genes to determine their types (Fig. 2B). The results showed that the
majority of these lncRNAs were from antisense to protein-coding
loci (antisense, 49.39%) and intergenic regions (lincRNAs,
40.53%).
To further dissect the promoter methylation patterns
of lncRNAs associated with lncRNA expression, DNA methylation
levels and expression levels between tumor and normal control
samples in the validation dataset were used to evaluate statistical
differences based on the aforementioned lncRNAs. As expected, it
was found that UhyperLncs had higher methylation levels and lower
expression levels in the tumor samples (Fig. 2C). On the contrary, the overall
methylation levels of UhypoLncs in tumors tended to decrease, but
the overall expression levels rose compared with normal control
samples (Fig. 2D). These results
indicated that aberrant promoter methylation of lncRNAs could
affect their expression, and these lncRNAs may be cancer biomarkers
for diagnosis and treatment in GBM. Furthermore, the DNA
methylation levels and expression levels between UhyperLncs,
UhypoLncs and non-significantly aberrantly methylated lncRNAs
(nsamLncs, Fig. 2E and F) were
observed; the overall methylation level of UhyperLncs was higher,
but the overall expression level of UhyperLncs was lower compared
with UhypoLncs. Notably, it was found that the nsamLncs, among the
three groups, had the highest methylation level but the lowest
expression level.
Construction of the MLRN and functional
annotation
Previous studies have demonstrated that aberrant
methylation of lncRNA promoters in tumors can lead to silencing or
activation of lncRNA expression, and that dysregulation of lncRNA
expression can regulate the expression of mRNAs by sharing common
miRNA-binding sites with mRNAs (37). To further analyze how lncRNA
dysregulation contributes to GBM progression through
methylation-mediated epigenetic regulation, a comprehensive MLRN
was constructed. The MLRN contained 262 lncRNAs, 9,441 mRNAs, and
28,721 MlncBTs. The MLRN was divided into a highly
methylation-mediated lncRNA regulatory network (high MLRN) and a
low methylation-mediated lncRNA regulatory network (low MLRN) based
on the hypermethylated lncRNAs and hypomethylated lncRNAs that were
singled out (Fig. 3A and B).
There were 76 UhyperLncs, 4,110 mRNAs and 8,879 MlncBTs in the high
MLRN and 186 UhypoLncs, 8,178 mRNAs, 19,842 MlncBTs in the low
MLRN.
Although tumorigenesis is a complicated dynamic
process, a recent study demonstrated that the dysregulation of
lncRNA methylation plays critical and complex roles during the
development and progression of tumors (21). These aberrantly methylated lncRNAs
mediate diverse biological functions, such as metabolism, cell
apoptosis, angiogenesis, or many other cancer-related functions
(50,51).
As the functional prediction of lncRNAs is hampered
by the shortage of annotated information, functional annotation
analysis of lncRNAs has frequently been conducted based on the
guilt by association principle (52). To dissect whether different
methylation patterns of lncRNAs in GBM correspond to distinct
biological functions, the present study annotated lncRNAs with
significantly enriched functional terms among the protein-coding
genes that were co-expressed with the lncRNAs (53). A sum of 555 GO terms and 101 KEGG
pathways were identified to be associated with the UhyperLncs, and
1,006 enriched GO terms and 66 pathways were obtained for the
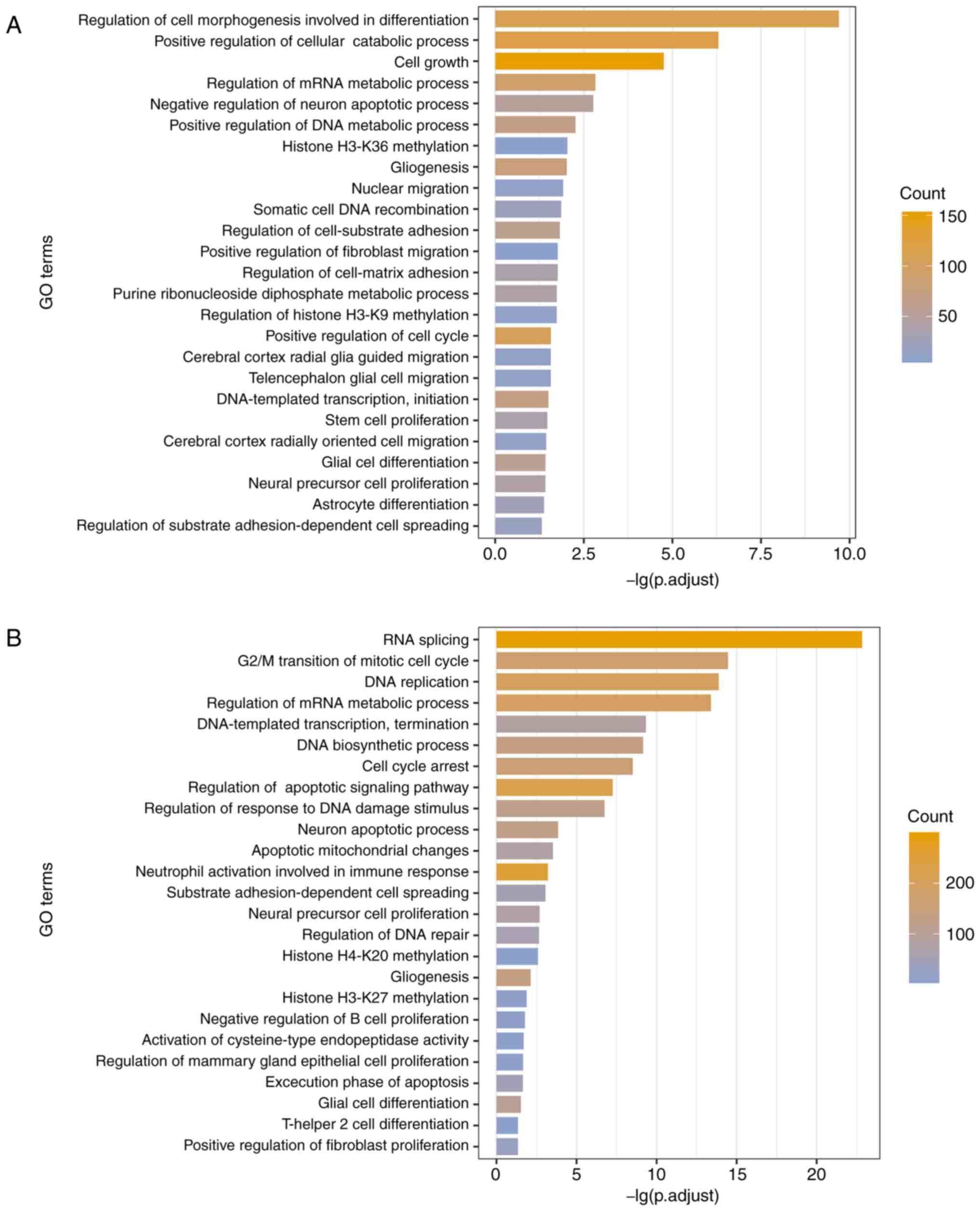
UhypoLncs. The results indicated that GO terms enriched by the
UhyperLncs and the UhypoLncs involved cell proliferation, cell
adhesion, cell apoptosis, cellular biological processes and
methylation of related proteins, which are closely associated with
the development of tumors (54)
(Fig. 4A and B). Also, cell
migration-related processes (such as 'nuclear migration' and
'positive regulation of fibroblast migration') were enriched in the
UhyperLncs, while more biological transcription-related processes
(such as 'RNA splicing' and 'regulation of DNA repair') were
enriched in the UhypoLncs. For the KEGG pathway analysis, it was
found that UhyperLncs and UhypoLncs were enriched in many
cancer-related KEGG pathways (Fig. 4C
and D). The UhyperLncs were mainly enriched in 'ErbB signaling
pathway', 'FoxO signaling pathway', 'Wnt signaling pathway' and
'MAPK signaling pathway'. The UhypoLncs were mainly enriched in
'cell cycle', 'Alzheimer disease', 'colorectal cancer' and 'renal
cell carcinoma'.
Identification of four small nucleolar
RNA host gene (SNHG) family lncRNAs associated with good
prognosis
Previous research discovered that hub genes play
essential roles in networks, and the top 10–20% of the nodes in
networks are usually defined as hubs (36). Since hub nodes in the low MLRN had
more degrees compared to those in the high MLRN, it was decided to
analyze the hub nodes in the low MLRN. The present study found that
SNHG16 was the hub lncRNA with the highest degree (degree, 1,012)
in the low MLRN and another lncRNA, SNHG7, was also a hub node
(degree, 272) in the low MLRN. Some lncRNAs in the SNHG family have
been reported to be involved in glioma as potential oncogenes, and
the dysregulation of their expression may promote the growth of
glioma. For instance, the expression of SNHG7 was found to be
upregulated in GBM tissues, and SNHG7 knockdown markedly suppressed
cell proliferation and migration, while inducing cell apoptosis in
GBM (54). Another lncRNA,
SNHG18, was found to strengthen glioma cell radioresistance, and
the expression was observably upregulated in clinical glioma
tissues (55).
Notably, the present study found that SNHG16, SNHG7
and two other lncRNAs (SNHG9 and SNHG18) that were not in the MLRN
all belonged to the UhypoLncs group, which meant that they were all
hypomethylated in GBM and their increased expression in GBM might
be affected by the corresponding promoter hypomethylation. It has
been reported that SNHG7 expression is significantly increased in
hypopharyngeal cancer, and that metformin decreases SNHG7
expression by mediating hypermethylation of the SNHG7 promoter
(56), but the methylation status
of the other three lncRNAs has not yet been reported, to the best
of our knowledge. The present study also validated the methylation
states of all probes annotated to these four lncRNA promoters. The
results indicated that the methylation level of all CpGs on the
SNHG7 promoter (Fig. 5A) and
SNHG18 promoter (Fig. 5B) was
significantly lower than that in paired normal samples. The
methylation level of CpGs on the SNHG9 promoter and SNHG16 promoter
was mostly decreased in GBM samples (Fig. S2A and B). Moreover, CpGs
annotated to the four lncRNA promoters were more significant when
they were closer to the TSS, which revealed that the CpGs closer to
TSS were key in determining the methylation status of the lncRNA
promoter. The diagnostic value of the four lncRNAs was further
appraised to see whether they could be used as cancer biomarkers
for early diagnosis of GBM. ROC analysis was performed according to
the methylation and expression of the lncRNAs. The overall area
under the ROC curve values for the diagnostic potential of the
methylation and expression of these lncRNAs in GBM were >0.8
(Fig. 5C and D), which suggested
that they could distinguish GBM samples from normal samples and
might become diagnostic cancer biomarkers for GBM, especially
SNHG16.
Furthermore, several studies have reported that
SNHG16 is highly expressed in glioma tissues, and SNHG16 can
upregulate the expression of some coding genes by interacting with
miRNAs (57–59). In the present study, the lncRNA
SNHG16 interacted with many mRNAs by combining with the target
miRNAs in the low MLRN. To evaluate whether these MlncBTs were
prognostic factors for GBM, the expression profiles were combined
with clinical annotations and a subset of MlncBTs that
significantly correlated with the overall survival of GBM was
identified. It was found that two MlncBTs related to SNHG16 had a
significant effect on survival [SNHG16-heat repeat containing 1
(HEATR1) and SNHG16-mitochondrial E3 ubiquitin protein ligase 1
(MUL1)]. HEATR1 is an mRNA related to GBM, and its expression in
GBM tissues is significantly higher than that in normal samples
(60), but the expression of the
mRNA MUL1 in GBM has not been reported, to the best of our
knowledge. Kaplan-Meier survival analysis of the training set
indicated that the two MlncBTs could divide the GBM patients into
two different risk groups with high and low PIs (Fig. 5E and F). The GBM patients in the
test set could be separated into two groups based on the two
MlncBTs (Fig. 5G and H). Taken
together, it was concluded that the hypomethylation of the SNHG16
promoter might contribute to the activation of its expression, and
then the high expression of SNHG16 may upregulate HEATR1 and MUL1
expression via the miRNA sponge mechanism. At the same time, these
findings suggested their potential as novel prognostic signatures
in GBM. All of the prognostic MlncBTs are summarized in Table SII.
Prediction of small molecule drugs for
GBM treatment
As precision medicine becomes increasingly relevant
in healthcare, the field of pharmacogenomics also continues to gain
prominence in the clinical setting (29). It was inferred that some potential
small molecules and targets that could be used for the treatment of
patients with GBM may be identified by constructing drug-target
association networks targeting miRNAs (Fig. 6A and B). In the hypermethylated
sub-network, these potential small molecules may indirectly promote
lncRNA expression by inhibiting the corresponding expression of
miRNAs to achieve the purpose of treating GBM (Fig. 6A); for example, lenalidomide
(downregulates hsa-miR-449a), emodin (downregulates hsa-miR-137)
and marine fungal metabolite 1386A (down-regulates hsa-miR-320a).
Previous studies have shown that lenalidomide and emodin may be
used for the treatment of GBM (61,62). Besides, experiments have
demonstrated that marine fungal metabolite 1386A has strong
cytotoxicity towards cancer cells and can alter the miRNA profiles
of MCF-7 breast cancer cells (63). It was deduced that 1836A might be
a novel potential small-molecule treatment for GBM. In the
hypomethylated sub-network, these potential small molecules might
indirectly inhibit lncRNA expression by promoting the corresponding
expression of miRNAs to treat GBM (Fig. 6B). Temozolomide (TMZ) is widely
used to treat GBM, and a previous study demonstrated that some
miRNAs, including hsa-miR-137, were significantly upregulated in
GBM after treatment with both TMZ and Olea europaea leaf
extract (64). Studies have
revealed that the lncRNAs SNHG7, SNHG16 and ZFAS1 are upregulated
in GBM or glioma tissues compared with non-tumor brain tissues
(54,58,65), and cisplatin has shown good
performance in patients with recurrent GBM (66). It was inferred that cisplatin
might upregulate the expression of these lncRNAs by downregulating
the expression of hsa-miR-449a. Notably, the present results
indicated that TMZ may upregulate hsa-miR-137 and further
down-regulate SNHG16. Furthermore, the present study sought to
identify a combined treatment with different targets for use in
patients with GBM by constructing drug-target networks targeting
mRNAs (Fig. 6C and D). A total of
70 drugs (39 mRNA targets) were acquired in the hypermethylated
sub-network (Fig. 6C) and 209
drugs (112 mRNA targets) in the hypomethylated sub-network
(Fig. 6D). The partial validation
results of the D-lncRNA database are discussed in the
Discussion.
Discussion
Increasing evidence suggests that lncRNAs play
crucial roles in carcinogenesis (6). In recent years, with the development
of high-throughput sequencing technology, epigenetic regulation has
become a hotspot in biomedical research. DNA methylation is an
important pattern of epigenetic regulation, which can be involved
in different biological processes by regulating the transcriptional
activity of genes, including the occurrence and development of
tumors (11).
The present study comprehensively investigated the
changes in DNA methylation of lncRNA promoters in GBM. DNA
methylation and expression profiles from TCGA and GEO were
integrated to study the methylation regulation pattern of lncRNAs.
The present study found that many lncRNAs in GBM exhibited
epigenetic dysregulation in promoter regions, most of which were
hypomethylated. There is evidence that reduced expression of
lncRNAs is due to hypermethylation inhibition and increased
expression of lncRNAs is due to hypomethylation activation
(51). Sang et al
(67) preliminarily explored the
functions and pathways of coding genes in hepatocellular carcinoma
by overlapping differentially methylated mRNAs and differentially
expressed mRNAs. Therefore, to establish a strategy for predicting
lncRNAs whose expression level might be associated with aberrant
promoter methylation, the present study not only obtained
hypermethylated-underexpressed lncRNAs and hypomethylated-highly
expressed lncRNAs, but also retained lncRNAs with a significant
negative correlation between methylation and expression level in
GBM samples. In total, 314 hypermethylated lncRNAs and 668
hypomethylated lncRNAs were used for further analysis. A recent
study predicted six lncRNAs that might improve the prognosis of GBM
by using weighted gene co-expression network analysis, Cox
regression and L1-LASSO penalization (68). Notably, one of the six lncRNAs
(PRRT3-AS1) was among the hypermethylated lncRNAs identified in the
present study. The results suggested that the aberrantly methylated
lncRNAs screened in the present study may be biomarkers of GBM
prognosis. Moreover, it was worth noting that 83 lncRNAs satisfied
both criteria, and it was thought that these lncRNAs would have
greater value in the future to experimentally verify their
epigenetic regulation.
Functional analysis of lncRNAs has revealed that
they can regulate gene expression at the transcriptional,
post-transcriptional and epigenetic levels (1), and a competing endogenous
relationship is one way in which lncRNAs influence the expression
of mRNAs, by binding to the target miRNAs of mRNAs (36). The construction of the MLRN
provided a global perspective to study the interactions between
mRNAs and differentially methylated lncRNAs. Although it was
confirmed that epigenetic modification of lncRNAs may be a
modulator of their expression, it was unclear what important roles
these lncRNAs may play in GBM pathogenesis. In the present study,
both hypermethylated and hypomethylated lncRNAs were involved in
many biological processes and pathways related to tumorigenesis and
progression in GBM. Notably, it was found that aberrant methylation
of either UhyperLncs or UhypoLncs may perturb specific common
pathways, such as 'ErbB signaling pathway', 'FoxO signaling
pathway', 'mTOR signaling pathway', 'glioma' and 'apoptosis'. As
the most enriched pathway, ErbB [epidermal growth factor receptor
(EGFR)] amplification and mutations are the most common oncogenic
events in GBM, and oncogenic EGFR induces DNA methylation-mediated
transcriptional silencing of tumor suppressors (69).
Furthermore, it was found that four SNHG family
lncRNAs had an excellent diagnostic effect and clinical prognostic
value. Three of the four lncRNAs (SNHG7, SNHG16, SNHG18) have been
reported as oncogenic lncRNAs in glioma or GBM, and were highly
expressed in tumors (54,55,58). In the present study, the
expression of SNHG9 was upregu-lated in GBM, thus it was assumed
that SNHG9 may also be a potentially carcinogenic lncRNA. Moreover,
it was found that the four lncRNAs were hypomethylated, and one
lncRNA (SNHG7) has been reported to improve the survival of
patients with hypopharyngeal cancer by inhibiting the
hypomethylation of its promoter (56). Furthermore, two MlncBTs
(SNHG16-HEATR1 and SNHG16-MUL1) were associated with a good
prognosis. Finally, the drug-target association networks were
constructed to provide potential small molecule drugs and targets
for the precise treatment of GBM. Additionally, it was found that
doxorubicin can inhibit GAS5 expression by upregulating
hsa-miR-449a in the present results. In the D-lnc database, it was
also found that doxorubicin can downregulate GAS5 expression (data
not shown).
There is no denying that the present research has
some shortcomings which should be addressed. Since GBM is different
from general tumors, sample acquisition is a problem, and
aberrantly methylated lncRNA research should be replicated in a
larger cohort. Moreover, due to technical and time constraints, it
was not possible to verify the association between these lncRNA
expression changes and promoter methylation events. Meanwhile, the
lack of analysis of the high MLRN was a limitation to the present
study. Further studies in animal models of GBM and brain tissues
from patients with GBM may help to solve these issues.
In conclusion, the present study investigated the
regulatory mechanism and functions of DNA methylation of lncRNAs in
GBM by integrating multi-omics data. The identified GBM-related or
clinically relevant lncRNAs (MlncBTs) might help to improve
understanding of the mechanisms of tumorigenesis and progression in
GBM, and could be further evaluated for use as cancer biomarkers.
Meanwhile, the present study provides an insight into the discovery
of potential drug targets for GBM treatment.
Supplementary Data
Funding
This study was supported by The Cooperation Project
of Basic Clinical Scientific Research in Capital Medical University
[grant nos. 17JL61 and 17JL(TT2X)] and The Scientific Research
Common Program of Beijing Municipal Commission of Education (grant
no. KM201710025010).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JJ, DL and LZ conceived and designed the study. JJ
and XZ wrote the manuscript. JJ, XZ and YA made substantial
contributions to the acquisition of data. JJ, LL and QL analyzed
and interpreted the data. LL, DL and LZ reviewed and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Li Q, Jia H, Li H, Dong C, Wang Y and Zou
Z: lncRNA and mRNA expression profiles of glioblastoma multiforme
(GBM) reveal the potential roles of lncRNAs in GBM pathogenesis.
Tumour Biol. 37:14537–14552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Szulzewsky F, Arora S, de Witte L, Ulas T,
Markovic D, Schultze JL, Holland EC, Synowitz M, Wolf SA and
Kettenmann H: Human glioblastoma-associated microglia/monocytes
express a distinct RNA profile compared to human control and murine
samples. Glia. 64:1416–1436. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young RM, Jamshidi A, Davis G and Sherman
JH: Current trends in the surgical management and treatment of
adult glioblastoma. Ann Transl Med. 3:1212015.PubMed/NCBI
|
|
4
|
Omuro A and DeAngelis LM: Glioblastoma and
other malignant gliomas: A clinical review. JAMA. 310:1842–1850.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gibb EA, Brown CJ and Lam WL: The
functional role of long non-coding RNA in human carcinomas. Mol
Cancer. 10:382011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tian Y, Zheng Y and Dong X: AGAP2-AS1
serves as an oncogenic lncRNA and prognostic biomarker in
glioblastoma multiforme. J Cell Biochem. 120:9056–9062. 2019.
View Article : Google Scholar
|
|
8
|
Zhou Y, Dai W, Wang H, Pan H and Wang Q:
Long non-coding RNA CASP5 promotes the malignant phenotypes of
human glioblastoma multiforme. Biochem Biophys Res Commun.
500:966–972. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu X, Yidayitula Y, Zhao H, Luo Y, Ma X
and Xu M: lncRNA LINC00152 promoted glioblastoma progression
through targeting the miR-107 expression. Environ Sci Pollut Res
Int. 25:17674–17681. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tan SK, Pastori C, Penas C, Komotar RJ,
Ivan ME, Wahlestedt C and Ayad NG: Serum long noncoding RNA HOTAIR
as a novel diagnostic and prognostic biomarker in glioblastoma
multiforme. Mol Cancer. 17:742018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hernando-Herraez I, Garcia-Perez R, Sharp
AJ and Marques-Bonet T: DNA methylation: Insights into human
evolution. PLoS Genet. 11:e10056612015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Forrest ME and Khalil AM: Review:
Regulation of the cancer epigenome by long non-coding RNAs. Cancer
Lett. 407:106–112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ghasemi A and Fallah S: Epigenetic
modification of MicroRNA-205 and its association with glioblastoma
multiform. Clin Lab. 63:1079–1088. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tabu K, Sasai K, Kimura T, Wang L,
Aoyanagi E, Kohsaka S, Tanino M, Nishihara H and Tanaka S: Promoter
hypomethylation regulates CD133 expression in human gliomas. Cell
Res. 18:1037–1046. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schulze M, Violonchi C, Swoboda S, Welz T,
Kerkhoff E, Hoja S, Brüggemann S, Simbürger J, Reinders J and
Riemenschneider MJ: RELN signaling modulates glioblastoma growth
and substrate-dependent migration. Brain Pathol. 28:695–709. 2018.
View Article : Google Scholar
|
|
16
|
Liu C, Fu H, Liu X, Lei Q, Zhang Y, She X,
Liu Q, Liu Q, Sun Y, Li G and Wu M: LINC00470 coordinates the
epigenetic regulation of ELFN2 to distract GBM cell autophagy. Mol
Ther. 26:2267–2281. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhi H, Ning S and Li X, Li Y, Wu W and Li
X: A novel reannotation strategy for dissecting DNA methylation
patterns of human long intergenic non-coding RNAs in cancers.
Nucleic Acids Res. 42:8258–8270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sandoval J, Heyn H, Moran S, Serra-Musach
J, Pujana MA, Bibikova M and Esteller M: Validation of a DNA
methylation microarray for 450,000 CpG sites in the human genome.
Epigenetics. 6:692–702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Guintivano J, Aryee MJ and Kaminsky ZA: A
cell epigenotype specific model for the correction of brain
cellular heterogeneity bias and its application to age, brain
region and major depression. Epigenetics. 8:290–302. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41(Database Issue):
D991–D995. 2013. View Article : Google Scholar
|
|
21
|
Yang X, Gao L and Zhang S: Comparative
pan-cancer DNA methylation analysis reveals cancer common and
specific patterns. Brief Bioinform. 18:761–773. 2017.
|
|
22
|
Harrow J, Frankish A, Gonzalez JM,
Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa
A, Searle S, et al: GENCODE: The reference human genome annotation
for the ENCODE project. Genome Res. 22:1760–1774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li JH, Liu S, Zhou H, Qu LH and Yang JH:
starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA
interaction networks from large-scale CLIP-Seq data. Nucleic Acids
Res. 42(Database Issue): D92–D97. 2014. View Article : Google Scholar
|
|
24
|
Paraskevopoulou MD, Vlachos IS, Karagkouni
D, Georgakilas G, Kanellos I, Vergoulis T, Zagganas K, Tsanakas P,
Floros E, Dalamagas T and Hatzigeorgiou AG: DIANA-LncBase v2:
Indexing microRNA targets on non-coding transcripts. Nucleic Acids
Res. 44(D1): D231–D238. 2016. View Article : Google Scholar :
|
|
25
|
Chou CH, Shrestha S, Yang CD, Chang NW,
Lin YL, Liao KW, Huang WC, Sun TH, Tu SJ, Lee WH, et al: miRTarBase
update 2018: A resource for experimentally validated
microRNA-target interactions. Nucleic Acids Res. 46(D1): D296–D302.
2018. View Article : Google Scholar :
|
|
26
|
Huang Z, Shi J, Gao Y, Cui C, Zhang S, Li
J, Zhou Y and Cui Q: HMDD v3.0: A database for experimentally
supported human microRNA-disease associations. Nucleic Acids Res.
47(D1): D1013–D1017. 2019. View Article : Google Scholar
|
|
27
|
Liu X, Wang S, Meng F, Wang J, Zhang Y,
Dai E, Yu X, Li X and Jiang W: SM2miR: A database of the
experimentally validated small molecules' effects on microRNA
expression. Bioinformatics. 29:409–411. 2013. View Article : Google Scholar
|
|
28
|
Wishart DS, Feunang YD, Guo AC, Lo EJ,
Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al:
DrugBank 5.0: A major update to the DrugBank database for 2018.
Nucleic Acids Res. 46(D1): D1074–D1082. 2018. View Article : Google Scholar :
|
|
29
|
Barbarino JM, Whirl-Carrillo M, Altman RB
and Klein TE: PharmGKB: A worldwide resource for pharmacogenomic
information. Wiley Interdiscip Rev Syst Biol Med. 10:e14172018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar
|
|
31
|
Law CW, Chen Y, Shi W and Smyth GK: voom:
Precision weights unlock linear model analysis tools for RNA-seq
read counts. Genome Biol. 15:R292014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yosipof A and Senderowitz H: k-Nearest
neighbors optimization-based outlier removal. J Comput Chem.
36:493–506. 2015. View Article : Google Scholar
|
|
33
|
Zhi H, Li X, Wang P, Gao Y, Gao B, Zhou D,
Zhang Y, Guo M, Yue M, Shen W, et al: Lnc2Meth: A manually curated
database of regulatory relationships between long non-coding RNAs
and DNA methylation associated with human disease. Nucleic Acids
Res. 46(D1): D133–D138. 2018. View Article : Google Scholar :
|
|
34
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ferreira JA: The Benjamini-Hochberg method
in the case of discrete test statistics. Int J Biostat. 3:Article
11. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu J, Li Y, Lu J, Pan T, Ding N, Wang Z,
Shao T, Zhang J, Wang L and Li X: The mRNA related ceRNA-ceRNA
landscape and significance across 20 major cancer types. Nucleic
Acids Res. 43:8169–8182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dong Z, Zhang A, Liu S, Lu F, Guo Y, Zhang
G, Xu F, Shi Y, Shen S, Liang J and Guo W: Aberrant
methylation-mediated silencing of lncRNA MEG3 functions as a ceRNA
in esophageal cancer. Mol Cancer Res. 15:800–810. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liao Q, Xiao H, Bu D, Xie C, Miao R, Luo
H, Zhao G, Yu K, Zhao H, Skogerbø G, et al: ncFANs: A web server
for functional annotation of long non-coding RNAs. Nucleic Acids
Res. 39(Web Server Issue): W118–W124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao Z, Bai J, Wu A, Wang Y, Zhang J, Wang
Z, Li Y, Xu J and Li X: Co-lncRNA: Investigating the lncRNA
combinatorial effects in GO annotations and KEGG pathways based on
human RNA-Seq data. Database (Oxford). 2015:pii: bav082. 2015.
View Article : Google Scholar
|
|
41
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Y, Xu Y, Feng L, Li F, Sun Z, Wu T,
Shi X, Li J and Li X: Comprehensive characterization of lncRNA-mRNA
related ceRNA network across 12 major cancers. Oncotarget.
7:64148–64167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang W, Qu Y, Yang Q, Ma X, Meng Q, Xu J,
Liu X and Wang S: D-lnc: A comprehensive database and analytical
platform to dissect the modification of drugs on lncRNA expression.
RNA Biol. 16:1586–1591. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bett JVS, Batistella EÂ, Melo G, Munhoz
EA, Silva CAB, Guerra ENDS, Porporatti AL and De Luca Canto G:
Prevalence of oral mucosal disorders during pregnancy: A systematic
review and meta-analysis. J Oral Pathol Med. 48:270–277. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shen H and Laird PW: Interplay between the
cancer genome and epigenome. Cell. 153:38–55. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cadieux B, Ching TT, VandenBerg SR and
Costello JF: Genome-wide hypomethylation in human glioblastomas
associated with specific copy number alteration,
methylenetet-rahydrofolate reductase allele status, and increased
proliferation. Cancer Res. 66:8469–8476. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lai RK, Chen Y, Guan X, Nousome D, Sharma
C, Canoll P, Bruce J, Sloan AE, Cortes E, Vonsattel JP, et al:
Genome-wide methylation analyses in glioblastoma multiforme. PLoS
One. 9:e893762014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Inbar-Feigenberg M, Choufani S, Butcher
DT, Roifman M and Weksberg R: Basic concepts of epigenetics. Fertil
Steril. 99:607–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jia H, Osak M, Bogu GK, Stanton LW,
Johnson R and Lipovich L: Genome-wide computational identification
and manual annotation of human long noncoding RNA genes. RNA.
16:1478–1487. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Xiao W, Cao Y, Long H, Luo Z, Li S, Deng
N, Wang J, Lu X, Wang T, Ning S, et al: Genome-wide DNA methylation
patterns analysis of noncoding RNAs in temporal lobe epilepsy
patients. Mol Neurobiol. 55:793–803. 2018. View Article : Google Scholar
|
|
51
|
Li Y, Zhang Y, Li S, Lu J, Chen J, Wang Y,
Li Y, Xu J and Li X: Genome-wide DNA methylome analysis reveals
epigenetically dysregulated non-coding RNAs in human breast cancer.
Sci Rep. 5:87902015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cabili MN, Trapnell C, Goff L, Koziol M,
Tazon-Vega B, Regev A and Rinn JL: Integrative annotation of human
large intergenic noncoding RNAs reveals global properties and
specific subclasses. Genes Dev. 25:1915–1927. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liao Q, Liu C, Yuan X, Kang S, Miao R,
Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al: Large-scale prediction
of long non-coding RNA functions in a coding-non-coding gene
co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ren J, Yang Y, Xue J, Xi Z, Hu L, Pan SJ
and Sun Q: Long noncoding RNA SNHG7 promotes the progression and
growth of glioblastoma via inhibition of miR-5095. Biochem Biophys
Res Commun. 496:712–718. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zheng R, Yao Q, Ren C, Liu Y, Yang H, Xie
G, Du S, Yang K and Yuan Y: Upregulation of long noncoding RNA
small nucleolar RNA host gene 18 promotes radioresistance of glioma
by repressing semaphorin 5A. Int J Radiat Oncol Biol Phys.
96:877–887. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wu P, Tang Y, Fang X, Xie C, Zeng J, Wang
W and Zhao S: Metformin suppresses hypopharyngeal cancer growth by
epigenetically silencing long non-coding RNA SNHG7 in FaDu cells.
Front Pharmacol. 10:1432019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang D, Zheng J, Liu X, Xue Y, Liu L, Ma
J, He Q, Li Z, Cai H and Liu Y: Knockdown of USF1 inhibits the
vasculogenic mimicry of glioma cells via stimulating
SNHG16/miR-212-3p and linc00667/miR-429 axis. Mol Ther Nucleic
Acids. 14:465–482. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lu YF, Cai XL, Li ZZ, Lv J, Xiang YA, Chen
JJ, Chen WJ, Sun WY, Liu XM and Chen JB: lncRNA SNHG16 functions as
an oncogene by sponging miR-4518 and Up-regulating PRMT5 expression
in glioma. Cell Physiol Biochem. 45:1975–1985. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang BY, Meng Q, Sun Y, Gao L and Yang JX:
Long non-coding RNA SNHG16 contributes to glioma malignancy by
competitively binding miR-20a-5p with E2F1. J Biol Regul Homeost
Agents. 32:251–261. 2018.PubMed/NCBI
|
|
60
|
Wu ZB, Qiu C, Zhang AL, Cai L, Lin SJ, Yao
Y, Tang QS, Xu M, Hua W, Chu YW, et al: Glioma-associated antigen
HEATR1 induces functional cytotoxic T lymphocytes in patients with
glioma. J Immunol Res. 2014:1314942014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Drappatz J, Wong ET, Schiff D, Kesari S,
Batchelor TT, Doherty L, Lafrankie DC, Ramakrishna N, Weiss S,
Smith ST, et al: A pilot safety study of lenalidomide and
radiotherapy for patients with newly diagnosed glioblastoma
multiforme. Int J Radiat Oncol Biol Phys. 73:222–227. 2009.
View Article : Google Scholar
|
|
62
|
Arcella A, Oliva MA, Staffieri S, Sanchez
M, Madonna M, Riozzi B, Esposito V, Giangaspero F and Frati L:
Effects of aloe emodin on U87MG glioblastoma cell growth: In vitro
and in vivo study. Environ Toxicol. 33:1160–1167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Tang B, He WL, Zheng C, Cheang TY, Zhang
XF, Wu H and Yang HL: Marine fungal metabolite 1386A alters the
microRNA profile in MCF-7 breast cancer cells. Mol Med Rep.
5:610–618. 2012.
|
|
64
|
Tunca B, Tezcan G, Cecener G, Egeli U, Ak
S, Malyer H, Tumen G and Bilir A: Olea europaea leaf extract alters
microRNA expression in human glioblastoma cells. J Cancer Res Clin
Oncol. 138:1831–1844. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Večeřa M, Šána J, Bútová R, Reguli Š,
Hermanová M, Křen L, Lipina R, Smrčka M and Slabý O: Dysregulation
of long non-coding RNAs in glioblastoma multiforme and their study
through use of modern molecular-genetic approaches. Klin Onkol.
31(Suppl 1): S168–S170. 2018.
|
|
66
|
Wang Y, Kong X, Guo Y, Wang R and Ma W:
Continuous dose-intense temozolomide and cisplatin in recurrent
glioblastoma patients. Medicine (Baltimore). 96:e62612017.
View Article : Google Scholar
|
|
67
|
Sang L, Wang XM, Xu DY and Zhao WJ:
Bioinformatics analysis of aberrantly methylated-differentially
expressed genes and pathways in hepatocellular carcinoma. World J
Gastroenterol. 24:2605–2616. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liang R, Zhi Y, Zheng G, Zhang B, Zhu H
and Wang M: Analysis of long non-coding RNAs in glioblastoma for
prognosis prediction using weighted gene co-expression network
analysis, Cox regression, and L1-LASSO penalization. Onco Targets
Ther. 12:157–168. 2018. View Article : Google Scholar
|
|
69
|
Forloni M, Gupta R, Nagarajan A, Sun LS,
Dong Y, Pirazzoli V, Toki M, Wurtz A, Melnick MA, Kobayashi S, et
al: Oncogenic EGFR represses the TET1 DNA demethylase to induce
silencing of tumor suppressors in cancer cells. Cell Rep.
16:457–471. 2016. View Article : Google Scholar : PubMed/NCBI
|