Introduction
Acute lung injury (ALI) is the pulmonary
manifestations of an acute systemic inflammatory process that is
characterized by pulmonary infiltrates, hypoxemia and edema,
frequently resulting in significant morbidity and mortality
(1,2). At present, the mechanism underlying
ALI by driving an acute inflammatory response is not completely
understood. Over the last few decades, although a large number of
ongoing studies are evaluating the value of several new drugs, no
effective drug has been identified (3). Therefore, understanding the exact
mechanism and implicated factors may aid with identifying novel
targets and strategies for the clinical prevention and treatment of
ALI.
CD40 belongs to the tumor necrosis factor receptor
(TNFR) family and is involved in the onset and maintenance of
inflammation (4,5). CD40 is present in a variety of cell
types, including endothelial cells, vascular smooth muscle cells,
fibroblasts, macrophages and monocytes (6,7).
Ligation of CD40 (CD40L) leads to activation of several
inflammatory signaling pathways, including PI3K, MAPK, ERK1/2, IKK
and NF-κB, eventually manifesting as enhanced expression of
primarily proinflammatory genes and adhesion molecules (8). Therefore, CD40 may mediate the
transcriptional regulatory genes encoding inflammatory factors via
activation of NF-κB.
It was previously demonstrated that acute
inflammation caused by NF-κB activation was associated with ALI,
whereas lung injury was alleviated by NF-κB inhibition (9,10). The IKK complex, a crucial
activator of NF-κB, consists of three proteins: i) IKK-α; ii)
IKK-β; and iii) IKK-γ. The complex can be activated via IκB-α
phosphorylation and subsequent degradation. The activated IKK
complex phosphorylates the IκB protein, which results in
proteasomal degradation. Subsequently, the NF-κB subunits
translocate from the cytoplasm to the nucleus to regulate the
transcription of target genes (11,12). Eventually, various inflammatory
cytokines and proteins are stimulated, including IL-1β, TNF-α, IL-6
and cell adhesion molecules, such as intracellular adhesion
molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and
inducible nitric oxide synthase (iNOS), which can exacerbate the
inflammatory damage to varying degrees (13,14).
In addition, another important pathological feature
of ALI is infiltration and accumulation of polymorphonuclear
neutrophils (PMNs) in the interstitial and alveolar spaces of the
lung, which impairs respiratory function (15). Persistent and extreme PMN
infiltration may cause additional injury to the lung by promoting
the release of several toxic factors, including reactive oxygen
species (ROS), proinflammatory cytokines and procoagulant
molecules, further aggravating ALI (16). Therefore, suppressing PMN
infiltration may significantly decrease inflammation-induced damage
and provide protection against ALI.
Ginkgo biloba is a traditional Chinese
medicine that has been used for thousands of years, and its leaf
extracts have been reported to display various biological
properties, including cardioprotective, antineurovascular insult
and anticancer activities (17).
Our previous study reported that Ginkgolide C (GC), a flavonoid
monomer extracted from Ginkgo biloba leaf with a range of
biological activities, may protect against myocardial
ischemia/reperfusion injury via inhibiting CD40/NF-κB signaling and
late inflammatory responses (18). However, the current understanding
of the association between GC and ALI is limited and requires
further investigation.
Therefore, the present study aimed to investigate
the protective effects of GC against ALI in vivo and in
vitro, and to elucidate the potential underlying mechanism. The
results of the present study may indicate novel therapeutic
strategies and interventions for ALI. For example, the results
indicated that CD40 might serve as a valuable therapeutic target
for ALI.
Materials and methods
Materials and reagents
GC (PubChem CID: 161120) and lipopolysaccharide
(LPS) were purchased from Sigma-Aldrich (Merck KGaA). TNF-α, IL-1β,
IL-6 ELISA kits and anti-IKK-β antibody were purchased from Abcam.
Anti-CD40, anti-ICAM-1, anti-VCAM-1, anti-iNOS, anti-NF-κB p65,
anti-phosphorylated (p)-IκB-α, anti-IκB-α, anti-β-actin,
anti-histone and goat anti-mouse IgG-HRP antibodies were purchased
from Santa Cruz Biotechnology, Inc. Phenylmethanesulfonyl fluoride
and RIPA lysis buffer were purchased from Abcam. PVDF membranes,
the BCA protein concentration assay kit and the SDS-PAGE gel
preparation kit were obtained from Beyotime Institute of
Biotechnology. The ECL plus kit was purchased from Cusabio
Technology LLC.
Animals
A total of 48 male wild-type (WT) C57BL/6J mice
(age, 8-10 weeks; weight, 18-22 g; specific-pathogen-free grade)
were provided by the Experimental Animal Center of Shandong First
Medical University. A total of 32 CD40 gene conditional knockout
(CD40-cKO) male mice (age, 8-10 weeks; weight, 18-22 g;
specific-pathogen-free grade) were provided by Biocytogen. All
animals were housed at 20-25°C with 40-60% humidity, 12-h
light/dark cycles, and free access to fresh water and food.
Following lung tissue and blood collection, animals were sacrificed
via cervical dislocation. The present study was approved by the
Ethics Committee of Shandong Provincial Hospital Affiliated to
Shandong First Medical University (grant no. 2020-464).
ALI induction in WT and CD40-cKO
mice
A total of 40 mice were divided into five groups
(n=8 per group; Table I) as
follows: i) Control group, received PBS; ii) LPS group; iii) 12
mg/kg GC group, LPS mice received 12 mg/kg GC; iv) 24 mg/kg GC
group, LPS mice received 24 mg/kg GC; and v) 48 mg/kg GC group, LPS
mice received 48 mg/kg GC) (18). As previously described (19), mice were anaesthetized by the
intraperitoneal injection of pentobarbital sodium (50 mg/kg),
orally intubated with a sterile plastic catheter and subsequently
intratracheally administered LPS (5 mg/kg). At 24 h post-LPS
treatment, mice were sacrificed. Control mice were intratracheally
administered 50 µl PBS. In the GC groups, mice were
intraperitoneally administered with GC for 7 days prior to LPS
treatment. Prior to sacrifice, blood samples and lung tissues were
collected under anesthesia.
 | Table IAnimal groups included in the present
study. |
Table I
Animal groups included in the present
study.
| Experiment | Group
|
|---|
| Control | LPS | GC (12 mg/kg) | GC (24 mg/kg) | GC (48 mg/kg) |
|---|
| LPS-injured WT
mice | WT mice | WT mice | WT mice | WT mice | WT mice |
| LPS-injured
alveolar epithelial cells of WT mice | WT mice | WT mice | WT mice | WT mice | WT mice |
| LPS-injured
CD40-cKO mice | WT mice | CD40-cKO mice | CD40-cKO mice | CD40-cKO mice | CD40-cKO mice |
| LPS-injured
alveolar epithelial cells of CD40-cKO mice | WT mice | CD40-cKO mice | CD40-cKO mice | CD40-cKO mice | CD40-cKO mice |
Histopathological assessment and PMN
infiltration analysis
Following fixation in 10% neutral-buffered formalin,
lung tissues were embedded in paraffin using an automated
processor. After paraffin embedding, lung tissues were treated with
a series of graded alcohols and xylene at 45°C for 10 min. Sections
(6-µm thick) were stained with hematoxylin and eosin at 37°C
for 15 min. Histopathological examination was performed using a
BX51 light microscope (Olympus Corporation; magnification, ×200).
The mean number of PMNs was recorded in three randomly selected
high-power fields.
Evaluation of lung injury
The severity of lung injury was scored according to
the alveolar wall thickness, the amount of cellular infiltration
and the level of hemorrhaging, as previously described with minor
modifications (20). Lung injury
scores were graded on a scale of 0-8 as follows: 0, no damage; 2,
mild damage; 4, moderate damage; 6, severe damage; and 8, extremely
severe damage.
Determination of myeloperoxidase (MPO)
activity
MPO activity in lung tissues was measured using an
MPO kit (cat. no. 20190618; Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocol. MPO activity
was expressed as U/g of protein.
Transmission electron microscopy
Lung tissues were fixed with glutaraldehyde buffered
fixative (3%; pH 7.2) at 25°C for 2-3 days. Tissues were embedded
in Spon 812 (SPI-Chem, Inc.; Structure Probe, Inc.) at 60°C for 48
h. Sections (thickness, 60-80 nm) were stained with 2% uranium
acetate at 25°C for 10 min and lead citrate at 25°C for 5 min.
Following washing with PBS, sections were analyzed using a
JEM-2000EX transmission electron microscope (JEOL, Ltd.;
magnification, ×17,000) in three randomly selected fields of
view.
Measurement of lung wet-to-dry (W/D)
weight ratio
Lung W/D weight ratio was calculated to assess the
extent of lung edema/water accumulation. The wet lung weight was
measured immediately after isolation from the mice. Following
drying at 60°C for 24 h, the dry lung weight was measured.
Hydroxyproline (Hyp) assay
Hyp activity was determined using a Hyp Colorimetric
Assay kit (BioVision, Inc.) according to the manufacturer's
protocol.
Immunohistochemistry
To evaluate ICAM-1, VCAM-1 and iNOS expression in
lung tissue, immunohistochemistry was performed. Lung tissues were
fixed in 4% paraformaldehyde at 4°C for 2 h and embedded in
paraffin at 60°C for 2 h. Tissues were then blocked with 10% BSA
(Sigma-Aldrich, Inc.) at 25°C for 30 min. Subsequently, the
sections (thickness, 3-5 µm) were incubated with anti-ICAM-1
(cat. no. p05362), anti-VCAM-1 (cat. no. p29533) and anti-iNOS
(cat. no. p35228) antibodies (all 1:800) overnight at 4°C.
Following primary antibody incubation, the samples were incubated
with an anti-rabbit IgG secondary antibody (cat. no. 3678s;
1:1,000) at 25°C for 30 min. The sections were stained with 0.05%
DAB at 25°C for 5 min. Immunohistological analysis was performed
using a fluorescence microscope (magnification, ×1,000). The
optical density (OD) of the positively stained area was quantified
using Image-Pro Plus software (version 6; Media Cybernetics
Inc.).
Culture of primary alveolar epithelial
cells
Following sacrifice, mouse alveolar epithelial cells
were separated from the lungs of mice in each group. First, 20 ml
PBS was used to flush blood from the lung vasculature.
Subsequently, a 1-2-mm section of lung tissue was excised and cut
into 1-mm3 pieces on ice. To disperse and culture fresh
tissue, tissues were cultured in DMEM (3 ml; Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 20% fetal calf serum (Gibco;
Thermo Fisher Scientific, Inc.) in 25 ml uncoated culture flasks
with 5% CO2 at 37°C for 48-52 h. Tissue explants were
removed and cells were maintained in DMEM (3 ml) supplemented with
20% fetal calf serum. After monolayers formed, cells were purified
(1×105/ml) and cultured in DMEM supplemented with 10%
fetal calf serum. Following culture for 3 days, cells were used for
subsequent experiments. Alveolar epithelial cell morphology and
proliferation were observed using an inverted light microscope
(magnification, ×200) to determine cell viability and purity (data
not shown). Alveolar epithelial cell structure was observed by
transmission electron microscopy using a JEM-2000EX transmission
electron microscope (magnification, ×17,000) according to the
aforementioned protocol.
Experimental protocol in vitro
Cells were randomly divided into five groups (n=8
per group; Table I) as follows:
i) Control group, cells were cultured in DMEM; ii) LPS group, cells
treated with 1 µg/ml LPS for 4 h; iii) 1 µM GC group,
cells pre-incubated with 1 µM GC for 24 h prior to LPS
treatment; iv) 10 µM GC group, cells pre-incubated with 10
µM GC for 24 h prior to LPS treatment; and v) 100 µM
GC group, cells pre-incubated with 100 µM GC for 24 h prior
to LPS treatment.
Cell viability assay
MTT colorimetric assays were performed to assess
alveolar epithelial cell viability. Following treatment with 1
µg/ml LPS for 4 h, cells were incubated with 5 mg/ml MTT at
37°C for 4 h. Subsequently, 100 µl DMSO was added for 15 min
to dissolve the formazan crystals. OD was determined at a
wavelength of 490 nm using a microplate reader (Thermo Fisher
Scientific, Inc.). The results are presented as a percentage of the
OD in the control group.
Determination of cytokines
Blood samples and cell medium were collected
separately. TNF-α (cat. no. 208348), IL-1β (cat. no. 197742) and
IL-6 (cat. no. ab222503) levels were measured using mouse ELISA
kits according to the manufacturer's protocol.
Western blotting
Cytoplasmic and nuclear proteins were extracted from
cells using a Nuclear and Cytoplasmic Protein Extraction kit
(Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Protein concentrations were determined
using a BCA assay. Western blotting was performed as previously
described with minor modifications (18). Proteins (50 µg) were
separated via 10% SDS-PAGE and transferred to PVDF membranes (20 V;
100 mA) overnight. Following blocking with 5% skimmed milk at 37°C
for 4 h, the membranes were incubated with primary antibodies (all
1:1,000) targeted against: CD40 (cat. no. sc-59047), ICAM-1 (cat.
no. sc-8439), VCAM-1 (cat. no. sc-13160), iNOS (cat. no. sc-7271),
NF-κB p65 (cat. no. sc-166748), p-IκB-α (cat. no. sc-8404), IκB-α
(cat. no. sc-1643) and IKK-β (cat. no. ab32135) at 4°C for 24 h.
Subsequently, the membranes were incubated with HRP-conjugated
secondary antibodies (cat. no. sc-2005; 1:800) at 25°C for 12 h.
Protein bands were visualized using an ECL Plus kit and a Gel
Imaging System (Thermo Fisher Scientific, Inc.). Protein expression
was semi-quantified using Quantity One software (version 4.0;
Bio-Rad Laboratories, Inc.). Histone and β-actin were used as the
loading controls for nuclear and cytoplasmic proteins,
respectively.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 5.0; GraphPad Software, Inc.). Data are
presented as the mean ± SD of three independent experiments.
Comparisons among groups were analyzed using one-way ANOVA followed
by Bonferroni's post hoc test. P<0.05 was considered to indicate
a statistically significant difference.
Results
GC alleviates histopathological
alterations, lung injury score, PMN infiltration and MPO activity
in LPS-injured WT mice
The control group did not display lung injury, as
indicated by simple columnar epithelium and cuboidal epithelium
respiratory bronchioles, and normal wall structure in the pulmonary
alveoli (Fig. 1A-a). By
contrast, severe pathological alterations were observed in the LPS
group, including cellular inflammatory infiltration, hemorrhage,
pulmonary edema and thickening of the alveolar walls with
disorganized alveolar structure (Fig. 1A-b). However, pretreatment with
12, 24 or 48 mg/kg GC notably ameliorated LPS-induced
histopathological alterations (Fig.
1A-c-e). Moreover, pathological lung injury scores were
significantly increased in the LPS group compared with the control
group (Fig. 1B). However, 12, 24
or 48 mg/kg GC significantly reversed LPS-induced high lung injury
scores. Additionally, prominent infiltration by leukocytes,
primarily PMNs, in the interstitial and alveolar spaces of the
lungs is one of the most important pathological hallmarks of ALI
(21). In the present study, a
significant increase in the number of PMNs was observed in the LPS
group compared with the control group, suggesting increased
inflammatory infiltration in the lungs (Fig. 1C). Furthermore, MPO activity was
measured to evaluate the level of neutrophilic infiltration
(Fig. 1D). MPO activity was very
low (0.56±0.32 U/g protein) in the control group, but was
significantly increased to 5.95±0.60 U/g protein in the LPS group.
The results revealed that pretreatment with GC significantly
inhibited LPS-induced MPO activity (12 mg/kg, 4.79±0.43 U/g
protein; 24 mg/kg, 2.69±0.43 U/g protein; and 48 mg/kg, 1.63±0.31
U/g protein, respectively).
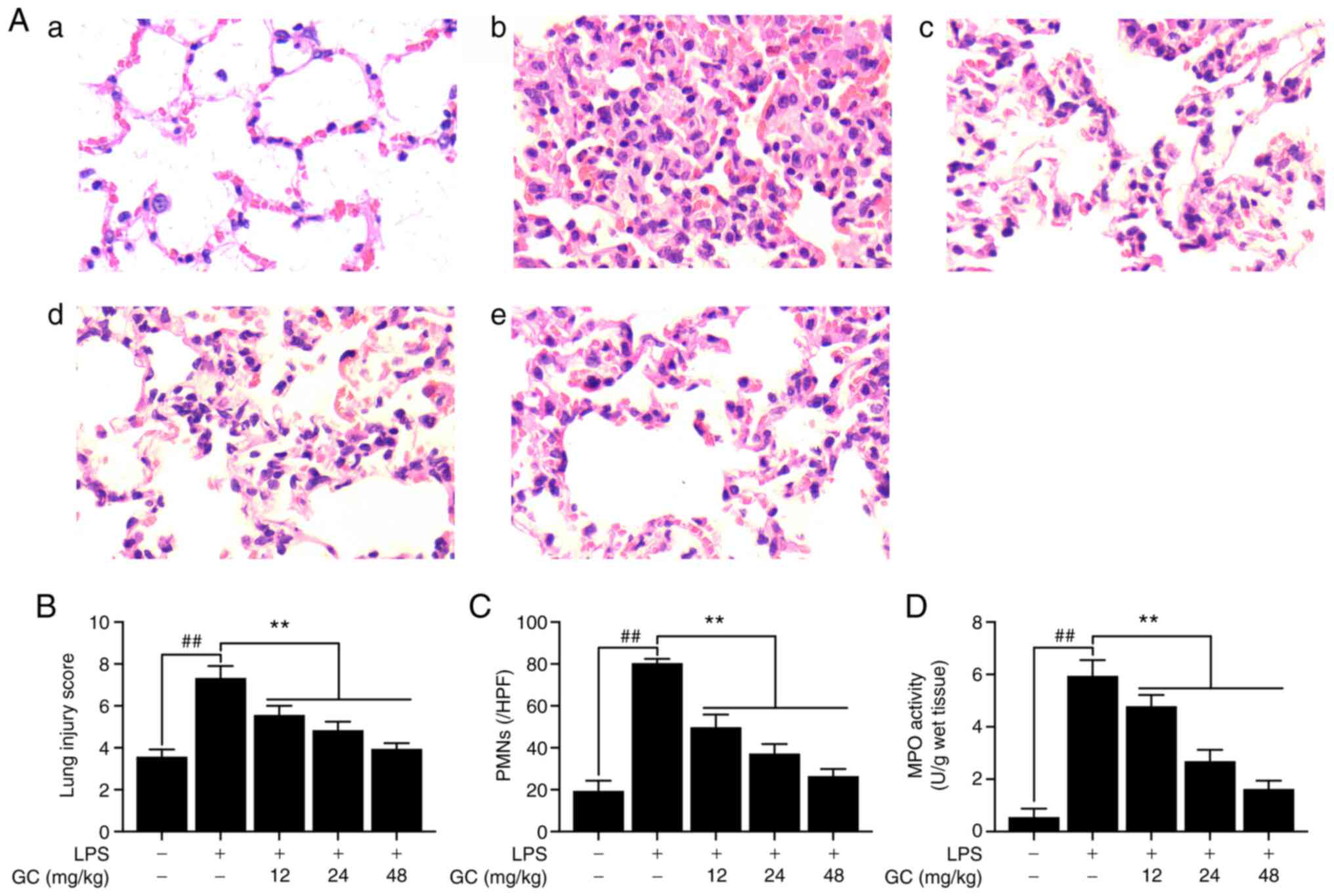 | Figure 1Effects of GC on histopathological
alterations, lung injury score, PMN count and MPO activity of lung
tissue in LPS-injured wild-type mice. (A) Histopathological
morphology was assessed by performing hematoxylin and eosin
staining (magnification, ×200) in the (a) control group, (b) LPS
group, (c) 12 mg/kg GC group, (d) 24 mg/kg GC group and (e) 48
mg/kg GC group. Histological images were captured from three
randomly selected fields of view. Effects of GC on (B) lung injury
score, (C) PMN count and (D) MPO activity. Data are presented as
the mean ± SD (n=8). ##P<0.01 vs. control;
**P<0.01 vs. LPS. GC, Ginkgolide C; PMN,
polymorphonuclear neutrophil; MPO, myeloperoxidase; LPS,
lipopolysaccharide; HPF, high-power field. |
GC improves the ultrastructural
characteristics of the lung and reduces the W/D weight ratio, Hyp
activity and expression of CD40 in LPS-injured WT mice
The results demonstrated that there was no
endothelial cell edema and the vascular basement membrane was
intact in the control group (Fig.
2A-a). However, pathological alterations were obvious in the
LPS group (Fig. 2A-b),
including: i) Vacuolization, degeneration and necrosis in type I
and II alveolar epithelial cells; ii) extensive shedding of
microvilli and empty lamellar bodies; and iii) obvious PMN
infiltration, vascular endothelial cell edema and basement membrane
rupture in the alveolar spaces. Pretreatment with 12, 24 or 48
mg/kg GC notably reversed LPS-induced vascular endothelial cell
damage and PMN infiltration (Fig.
2A-c-e). The lung W/D weight ratio was calculated to evaluate
the degree of pulmonary edema in LPS-induced ALI. The W/D weight
ratio in the LPS group was significantly higher compared with the
control group (Fig. 2B). After
pretreatment with 12, 24 or 48 mg/kg GC, lung permeability was
significantly decreased and alveolar epithelial barrier damage was
markedly reversed compared with the LPS group (Fig. 2B). Hyp activity in lung tissues
reflects the proportion of tissue with collagen fibers (22). Hyp content of lung tissues in the
control group was 0.91±0.34 µg/mg lung, which was
significantly increased to 1.51±0.05 µg/mg lung in the LPS
group (Fig. 2C). Hyp content in
the 12, 24 and 48 mg/kg GC groups was significantly reduced to
2.11±0.13 (P<0.05), 2.70±0.15 (P<0.01) and 3.26±0.32
µg/mg lung (P<0.01), respectively, compared with the LPS
group. Subsequently the expression of CD40 in lung tissue was
measured. CD40 expression in the LPS group was significantly higher
compared with the control group (P<0.01; Fig. 2D). However, 12, 24 and 48 mg/kg
GC significantly reduced LPS-induced CD40 expression levels
(P<0.01).
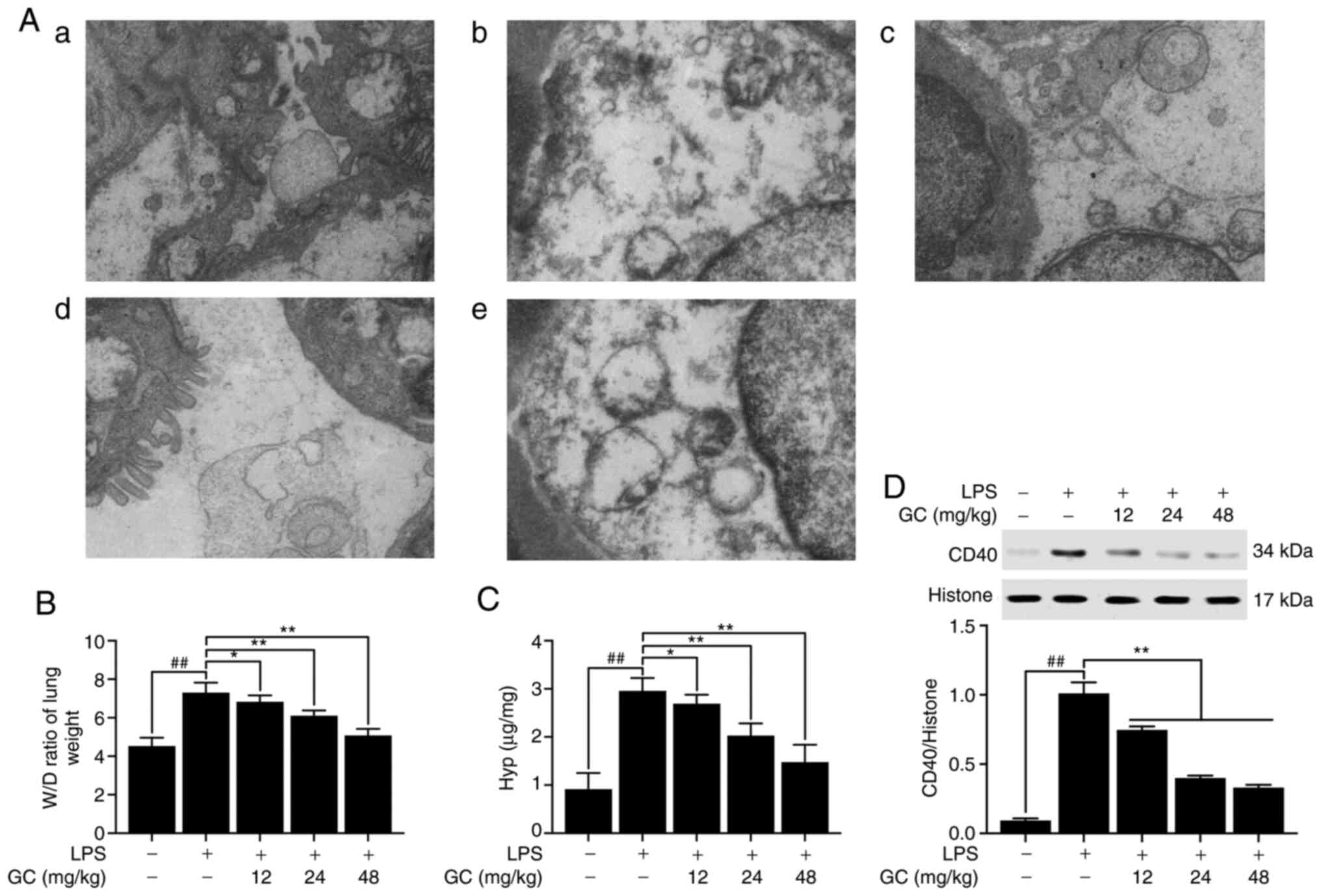 | Figure 2Effects of GC on ultrastructural
characteristics, W/D weight ratio, Hyp activity and CD40 expression
of lung tissue in LPS-injured wild-type mice. (A) Representative
transmission electron microscopy observation (magnification,
×17,000) of lung injury in the (a) control group, (b) LPS group,
(c) 12 mg/kg GC group, (d) 24 mg/kg GC group and (e) 48 mg/kg GC
group. Effects of GC on (B) W/D weight ratio, (C) Hyp activity and
(D) CD40 expression. Data are presented as the mean ± SD (n=8).
##P<0.01 vs. control; *P<0.05 and
**P<0.01 vs. LPS. GC, Ginkgolide C; W/D, wet-to-dry;
Hyp, hydroxyproline; LPS, lipopolysaccharide. |
GC prevents ICAM-1, VCAM-1 and iNOS
upregulation, and lowers inflammatory cytokine serum levels in
LPS-injured WT mice
ICAM-1, VCAM-1 and iNOS expression levels in the LPS
group were significantly increased by 1.90-, 2.58- and 2.29-fold,
respectively, compared with the control group (P<0.01; Fig. 3). Moreover, the levels of TNF-α,
IL-1β and IL-6 were significantly increased by 4.73-, 6.57- and
138.08-fold, respectively, in the LPS group compared with the
control group (P<0.01; Table
II). However, 12, 24 and 48 mg/kg GC significantly
downregulated ICAM-1, VCAM-1 and iNOS expression levels, and the
levels of TNF-α, IL-1β and IL-6 compared with the LPS group
(P<0.01).
| Table IIEffects of GC on serum inflammatory
cytokines in LPS-injured wild-type mice. |
Table II
Effects of GC on serum inflammatory
cytokines in LPS-injured wild-type mice.
| Group | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-6 (pg/ml) |
|---|
| Control | 10.74±3.24 | 3.20±0.59 | 0.57±0.25 |
| LPS | 50.82±6.55a | 20.99±2.48a | 78.93±5.85a |
| GC (12 mg/kg) | 36.70±6.09b | 14.51±0.65b | 55.10±4.30b |
| GC (24 mg/kg) | 31.84±4.13b | 9.98±1.35b | 41.54±3.92b |
| GC (48 mg/kg) | 17.18±5.08b | 7.48±1.63b | 30.41±6.85b |
GC protects against cell injury,
downregulates inflammatory factor expression and inhibits
activation of the NF-κB signaling pathway in LPS-injured alveolar
epithelial cells of WT mice
Compared with the control group, cell viability in
the LPS group was significantly reduced to 58.71±4.78% (P<0.01;
Fig. 4A). Treatment with 1, 10
or 100 µM GC significantly increased cell viability in
LPS-treated alveolar epithelial cells (73.11±5.94, 80.04±3.84 and
86.44±2.29%, respectively; all P<0.01). CD40 expression was low
in the control group, but LPS treatment significantly increased the
expression of CD40 compared with the control group (P<0.01;
Fig. 4B). In addition, treatment
with LPS significantly increased the expression levels of ICAM-1
(8.50-fold), VCAM-1 (13.82-fold) and iNOS (16.94-fold) compared
with the control group (P<0.01; Fig. 4C-E). Pretreatment with 1, 10 or
100 µM GC significantly decreased the expression levels of
CD40 (by 27.46, 61.70 and 71.53%, respectively), ICAM-1 (by 28.15,
53.78 and 76.89%, respectively), VCAM-1 (by 23.83, 56.60 and
78.72%, respectively) and iNOS (by 11.07, 49.82 and 76.38%,
respectively) compared with the LPS group (all P<0.01). No
significant differences in the expression levels of total IκB-α
were observed among the groups (Fig.
4F). However, compared with the control group, the expression
levels of p-IκB-α were significantly increased by 5.02-fold in the
LPS group (P<0.01). Compared with the control group, IKK-β
expression was significantly increased by 7.78-fold in the LPS
group (P<0.01), which was significantly reversed by pretreatment
with 1, 10 or 100 µM GC (P<0.01; Fig. 4G). Moreover, the results
demonstrated that the level of NF-κB p65 was relatively high in the
cytoplasm but low the in nucleus in the control group (Fig. 4H and I). However, the
translocation of p65 from the cytosol into the nucleus was
significant in the LPS group compared with the control group
(P<0.01). LPS-induced effects on NF-κB p65 were significantly
reversed by pretreatment with 1, 10 or 100 µM GC
(P<0.01). The levels of TNF-α, IL-1β and IL-6 in the control
group were 13.65±2.48, 34.85±2.30 and 50.28±7.87 pg/ml,
respectively (Table III). The
levels of TNF-α, IL-1β and IL-6 in the LPS group were significantly
increased to 89.61±7.04 pg/ml (6.56-fold), 709.67±56.24 pg/ml
(20.36-fold) and 655.95±19.12 pg/ml (13.05-fold), respectively,
compared with the control group (all P<0.01). Pretreatment with
1, 10 or 100 µM GC significantly decreased the levels of
TNF-α, IL-1β and IL-6 compared with the LPS group (P<0.01).
 | Figure 4Effects of GC on cell viability and
CD40, ICAM-1, VCAM-1, iNOS, p-IκB-α, IKK-β and NF-κB p65 expression
levels in LPS-injured alveolar epithelial cells of wild-type mice.
(A) Effects of GC on cell viability. Effects of GC on the protein
expression levels of (B) CD40, (C) ICAM-1, (D) VCAM-1, (E) iNOS,
(F) p-IκB-α/total IκB-α, (G) IKK-β, (H) cytoplasmic NF-κB p65 and
(I) nuclear NF-κB p65. Data are presented as the mean ± SD of three
independent experiments. ##P<0.01, vs. control;
**P<0.01 vs. LPS. GC, Ginkgolide C; ICAM-1,
intracellular adhesion molecule-1; VCAM-1, vascular cell adhesion
molecule-1; iNOS, inducible nitric oxide synthase; p,
phosphorylated; LPS, lipopolysaccharide. |
| Table IIIEffects of GC on inflammatory
cytokines levels in the cell medium of LPS-injured alveolar
epithelial cells of wild-type mice. |
Table III
Effects of GC on inflammatory
cytokines levels in the cell medium of LPS-injured alveolar
epithelial cells of wild-type mice.
| Group | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-6 (pg/ml) |
|---|
| Control | 13.65±2.48 | 34.85±2.30 | 50.28±7.87 |
| LPS | 89.61±7.04a |
709.67±56.24a |
655.95±19.12a |
| GC (1
µM) | 61.76±4.68b |
278.99±37.31b |
389.36±55.80b |
| GC (10
µM) | 40.56±6.32b |
232.06±35.37b |
292.07±42.87b |
| GC (100
µM) | 34.89±3.21b |
182.19±27.55b |
209.68±15.26b |
GC does not improve LPS-induced injury or
inhibit the expression of inflammatory factors in LPS-injured
CD40-cKO mice
To further verify the key roles of CD40 in the
anti-ALI effects of GC, CD40-cKO mice were employed to establish an
in vivo LPS-induced ALI mouse model. Compared with the
control group, LPS treatment markedly aggravated histopathological
alterations (Fig. 5A-2), lung
injury scores (Fig. 5B), PMN
infiltration (Fig. 5C) and MPO
activity (Fig. 5D), destroyed
the ultrastructural characteristics of lung tissue (Fig. 6A-2), elevated the W/D weight
ratio (Fig. 6B) and Hyp activity
(Fig. 6C). Moreover, the results
presented in Fig. 6D
demonstrated that CD40 gene was successfully and stably silenced in
the lung tissues of mice, except in the control group. Moreover,
LPS treatment significantly induced inflammatory reactions in
CD40-cKO mice compared with the control group (Fig. 7 and Table IV). Notably, 12, 24 and 48 mg/kg
GC failed to ameliorate LPS-induced alterations following CD40 gene
silencing.
 | Figure 5Effects of GC on histopathological
alterations, lung injury score, PMN count and MPO activity of lung
tissue in LPS-injured CD40-cKO mice. (A) Histopathological
morphology was assessed by performing hematoxylin and eosin
staining (magnification, ×200) in the (a) control group, (b) LPS
group, (c) 12 mg/kg GC group, (d) 24 mg/kg GC group and (e) 48
mg/kg GC group. Histological images were captured from three
randomly selected fields of view. Effects of GC on (B) lung injury
score, (C) PMN count and (D) MPO activity. Wild-type mice were used
as the control group. Data are presented as the mean ± SD (n=8).
##P<0.01 vs. control. GC, Ginkgolide C; PMN,
polymorphonuclear neutrophil; MPO, myeloperoxidase; cKO,
conditional knockout; LPS, lipopolysaccharide; HPF, high-power
field; NS, not significant. |
 | Figure 6Effects of GC on ultrastructural
characteristics, W/D weight ratio, Hyp activity and CD40 expression
of lung tissue in LPS-injured CD40-cKO mice. (A) Representative
transmission electron microscopy observation (magnification,
×17,000) of lung injury in the (a) control group, (b) LPS group,
(c) 12 mg/kg GC group, (d) 24 mg/kg GC group and (e) 48 mg/kg GC
group. Effects of GC on (B) W/D weight ratio, (C) Hyp activity and
(D) CD40 expression. Wild-type mice were used as the control group.
Data are presented as the mean ± SD (n=8). ##P<0.01
vs. control. GC, Ginkgolide C; W/D, wet-to-dry; Hyp,
hydroxyproline; cKO, conditional knockout; LPS, lipopolysaccharide;
NS, not significant. |
 | Figure 7Effects of GC on ICAM-1, VCAM-1 and
iNOS expression in LPS-injured CD40-cKO mice. Immunohistochemistry
was performed to evaluate ICAM-1, VCAM-1 and iNOS expression in
lung tissue. Magnification, ×1,000. Wild-type mice were used as the
control group. ##P<0.01 vs. control. GC, Ginkgolide
C; ICAM-1, intracellular adhesion molecule-1; VCAM-1, vascular cell
adhesion molecule-1; iNOS, inducible nitric oxide synthase; LPS,
lipopolysaccharide; cKO, conditional knockout; NS., not
significant. |
| Table IVEffects of GC on serum inflammatory
cytokines in LPS-injured CD40-cKO mice. |
Table IV
Effects of GC on serum inflammatory
cytokines in LPS-injured CD40-cKO mice.
| Group | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-6 (pg/ml) |
|---|
| Control | 8.99±0.70 | 3.86±0.52 | 0.44±0.28 |
| LPS | 50.14±9.31a | 20.96±2.00a | 58.19±6.01a |
| GC (12 mg/kg) | 54.23±6.39 | 19.56±1.62 | 62.24±5.64 |
| GC (24 mg/kg) | 45.05±5.80 | 19.91±2.26 | 61.87±5.76 |
| GC (48 mg/kg) | 48.30±7.22 | 21.13±2.31 | 53.08±4.10 |
GC exerts no effect on cell injury,
expression of inflammatory factors or activation of the NF-κB
signaling pathway in LPS-injured alveolar epithelial cells of
CD40-cKO mice
CD40 was not expressed following CD40 gene
conditional knockout, except in the control group (Fig. 8B). Following CD40 gene silencing,
cell viability was significantly reduced, and the expression levels
of CD40, ICAM-1, VCAM-1, iNOS and IKK-β, the phosphorylation of
IκB-α, the translocation of NF-κB and the levels of inflammatory
cytokines were significantly enhanced in the LPS group compared
with the control group (P<0.01; Fig. 8 and Table V). As expected, the in
vitro results were similar to the in vivo results; GC
did not alter the viability or reverse injury in LPS-treated
alveolar epithelial cells following CD40 gene silencing.
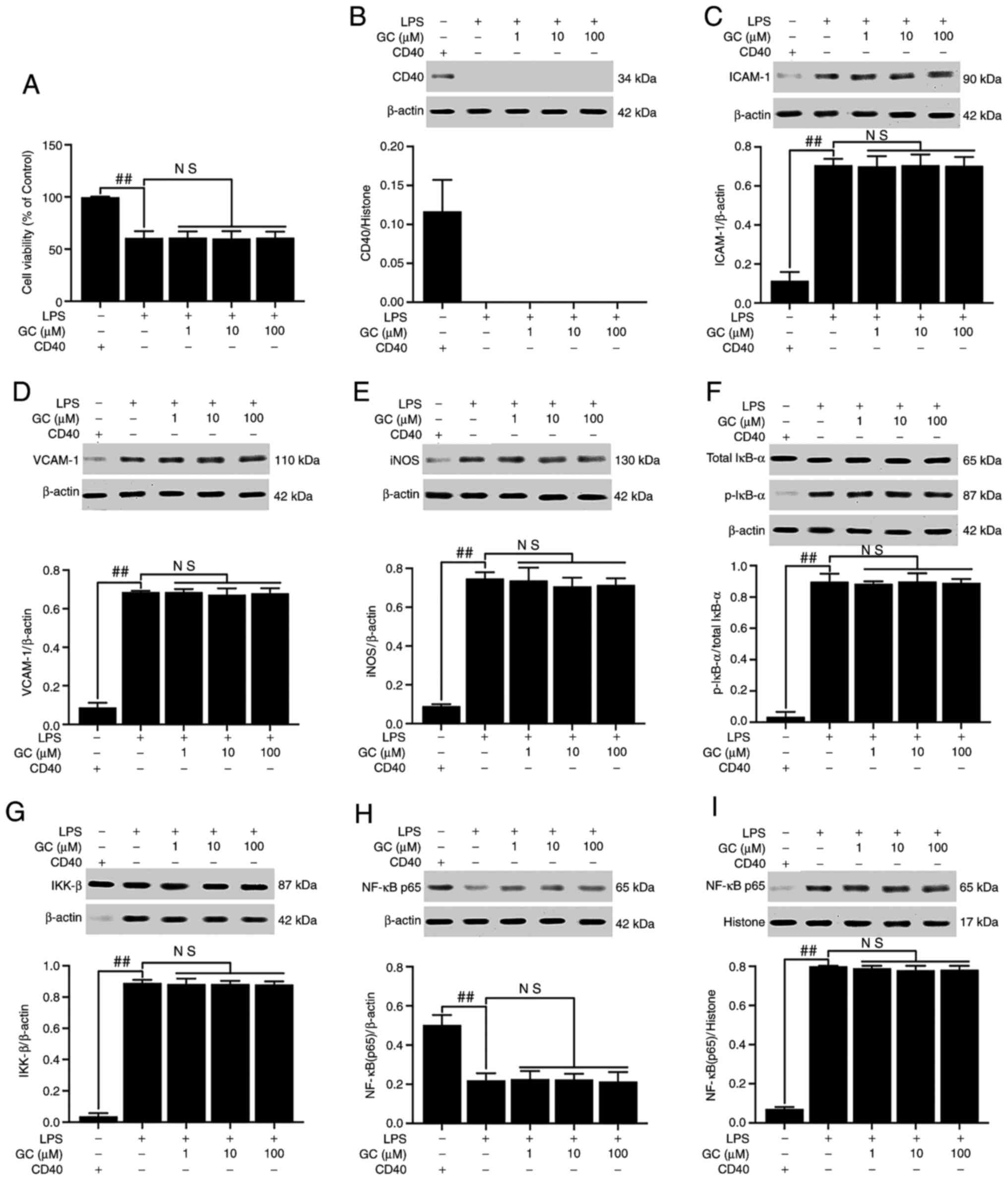 | Figure 8Effects of GC on cell viability and
CD40, ICAM-1, VCAM-1, iNOS, p-IκB-α, IKK-β and NF-κB p65 expression
levels in LPS-injured alveolar epithelial cells of CD40-cKO mice.
(A) Effects of GC on cell viability. Effects of GC on the protein
expression levels of (B) CD40, (C) ICAM-1, (D) VCAM-1, (E) iNOS,
(F) p-IκB-α/total IκB-α, (G) IKK-β, (H) cytoplasmic NF-κB p65 and
(I) nuclear NF-κB p65. Alveolar epithelial cells from wild-type
mice were used as the control group. Data are presented as the mean
± SD of three independent experiments. ##P<0.01 vs.
control. GC, Ginkgolide C; ICAM-1, intracellular adhesion
molecule-1; VCAM-1, vascular cell adhesion molecule-1; iNOS,
inducible nitric oxide synthase; p, phosphorylated; LPS,
lipopolysaccharide; cKO, conditional knockout; NS, not
significant. |
| Table VEffects of GC on inflammatory
cytokine levels in the cell medium of LPS-injured alveolar
epithelial cells of CD40-cKO mice. |
Table V
Effects of GC on inflammatory
cytokine levels in the cell medium of LPS-injured alveolar
epithelial cells of CD40-cKO mice.
| Group | TNF-α (pg/ml) | IL-1β (pg/ml) | IL-6 (pg/ml) |
|---|
| Control | 13.84±2.55 | 30.38±6.04 | 60.02±14.72 |
| LPS | 85.56±9.57a |
709.65±72.75a |
741.05±104.58a |
| GC (1
µM) | 81.91±7.44 | 729.46±59.26 | 655.20±96.86 |
| GC (10
µM) | 82.22±8.90 | 680.37±69.73 | 616.25±88.45 |
| GC (100
µM) | 87.30±10.30 | 725.60±67.53 | 691.05±108.84 |
Discussion
LPS-induced excessive lung inflammation and
destruction of the structure and function of alveolar epithelial
cells are important pathological processes of ALI, which commonly
result in significant morbidity or even death (23). Augmentation of inflammatory
responses in the lung tissue stimulates the production of numerous
cytotoxic substances, including various proinflammatory cytokines,
ROS and granular enzymes, thereby affecting the development and
severity of LPS-induced ALI (24). Therefore, inhibiting inflammation
may serve as a promising and effective therapy for ALI. To the best
of our knowledge, the present study was the first to suggest the
key role of GC in the inflammatory pathological process of ALI via
inhibition of the CD40/NF-κB signaling pathway in vivo and
in vitro.
GC, a terpene lactone component of Ginkgo
biloba leaves, displays a broad range of biological and
pharmacological functions against various diseases, including
memory loss and cognitive disorders, arrhythmias and ischemic heart
disease, cancer, diabetes and thromboses (25,26). Our previous study focused on the
cardioprotective effects of GC via suppression of CD40-NF-κB
signaling pathway-activated inflammation (18); however, whether GC exerts a
beneficial effect in ALI is not completely understood. To the best
of our knowledge, the present study demonstrated for the first time
that pretreatment with GC at 12, 24 or 48 mg/kg (per day for 7
days) markedly alleviated LPS-induced ALI, as evidenced by notable
improvements in histopathological alterations, reductions in lung
injury scores and restored lung ultrastructure. The significant
alterations induced by GC may be partially explained by the fact
that GC exerted a prominent protective effect against LPS-induced
ALI.
It was previously reported that activated PMNs were
a key event in the development of inflammation during ALI (27). These communications include
paracrine cross talk between PMNs and alveolar epithelial cells. In
the present study, LPS treatment results in widespread PMN
infiltration, interstitial edema in the alveolar space and septum,
notable thickening of the alveolar membrane and disruption of the
normal alveolar structure. MPO, which is also considered as a
specific marker of ALI and a risk factor for long-term mortality,
is secreted by PMNs (28). The
present study demonstrated that administration of 12, 24 or 48
mg/kg GC to WT mice significantly alleviated LPS-induced PMN
infiltration and MPO activity. In addition, compared with the
control group, significantly higher W/D weight ratios of the lung
and Hyp content levels were observed in the LPS group, suggesting
gradual aggravation of lung fluid leakage during the acute phase of
lung injury. However, the W/D weight ratio and Hyp content were
significantly decreased following treatment with 12, 24 and 48
mg/kg GC compared with the LPS group.
CD40 belongs to the TNFR superfamily and is
universally expressed on the surface of resting and activated
vascular endothelial cells and platelets (29). CD40L serves as a CD40 ligand and
is primarily expressed on endothelial and epithelial cells, B
cells, neutrophils and macrophages. The CD40-CD40L system serves a
key role in various clinical settings, including inflammation,
thrombosis, cancer and autoimmune diseases (30). More importantly, PMNs can express
CD40, and CD40-CD40L binding rapidly primes PMNs, ultimately
inducing PMN-mediated toxicity against alveolar epithelial cells
(31). Although it is clear that
CD40 induction of proinflammatory activity contributes to chronic
inflammation in various clinical settings, the signaling pathway
mediating its effects in ALI has only been partially described. In
the present study, compared with the control group, CD40 expression
in WT mice was significantly increased following LPS exposure, and
GC pretreatment significantly downregulated CD40 expression in a
concentration-dependent manner. Therefore, suppressing CD40
expression by GC may alleviate inflammation, thereby attenuating
the severity of ALI.
CD40 signaling is closely associated with the
non-canonical NF-κB signaling pathway, followed by strongly
triggered downstream signaling events (32,33). Under normal conditions,
CD40-induced NF-κB activation involves the phosphorylation of IκB,
which results in proteasome-mediated degradation. After the
ubiquitylation and degradation of phosphorylated IκB, nuclear
translocation of NF-κB is observed, which regulates the
transcription of target genes (34). The present study demonstrated
that compared with the control group, LPS exposure in alveolar
epithelial cells from WT mice significantly increased the
phosphorylation of IκB and translocation of NF-κB p65, indicating
increased transcription of NF-κB and strict regulation of NF-κB
signaling at the level of IκB phosphorylation. The IKK complex
(IKK-α, -β and -γ) is activated by phosphorylation (primarily of
IKK-α or IKK-β) within their activation loops, either by upstream
kinases or via autophosphorylation. Then, phosphorylated IκB-α
combines with the activated complex, causing its ubiquitin-mediated
degradation and release of the NF-κB subunits (35,36). The present study demonstrated
that IKK-β expression was significantly increased by LPS exposure
compared with the control group. Moreover, the results indicated
that IKK-β expression was significantly downregulation by GC in
LPS-induced alveolar epithelial cells. Therefore, the
CD40/IKK/NF-κB signaling pathway may serve as an inflammatory
target in LPS-induced ALI.
A previous study indicated that activation of NF-κB
occurs during the very early stages of ALI, and then sequentially
regulates the expression of downstream inflammatory factors
(37). Accordingly,
immunostaining assays, ELISAs and western blotting were performed
in the present study to verify whether LPS triggered a severe
inflammatory reaction induced by NF-κB-dependent gene
transcription. The results demonstrated that LPS aggravated ALI by
promoting the production of proinflammatory factors (TNF-α, IL-1β
and IL-6) and the expression of inflammatory proteins (ICAM-1,
VCAM-1 and iNOS) via an NF-κB-dependent signaling pathway, both
in vivo and in vitro. However, pretreatment with GC
effectively reversed LPS-induced effects. The results suggested
that GC served a protective role against LPS-induced ALI via
inhibiting inflammation induced by activation of the NF-κB
signaling pathway.
Based on the aforementioned results, it was inferred
that CD40 served a key role in stimulating LPS-induced inflammation
in ALI. Therefore, CD40-cKO mice and their alveolar epithelial
cells were further examined to investigate this hypothesis in
vivo and in vitro. The protective effects of GC against
LPS-induced ALI were no longer observed following CD40 silencing,
confirming that GC improved LPS-induced ALI by inhibiting the NF-κB
signaling pathway via CD40 (Fig.
9). Therefore, further elucidating how CD40 regulates
LPS-induced inflammation may identify promising intervention
targets for ALI.
Taken together, the results of the present study
demonstrated that GC exerted a protective effect against
LPS-induced ALI via inhibiting CD40/NF-κB signaling
pathway-dependent inflammation. Therefore, GC may serve as an
effective therapeutic agent for of ALI, and CD40 may be considered
as a novel potential therapeutic target for ALI.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
RZ, NG, TG, GY, QW and BZ performed the experiments.
RZ, NG and NH designed the present study. TG, GY, QW and BZ
analyzed the data. RZ drafted and edited the manuscript. RZ and NH
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Shandong Provincial Hospital Affiliated to Shandong
First Medical University (grant no. 2020-464).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Mowery NT, Terzian WT and Nelson AC: Acute
lung injury. Curr Probl Surg. 57:1007772020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuldanek SA, Kelher M and Silliman CC:
Risk factors, management and prevention of transfusion-related
acute lung injury: A comprehensive update. Expert Rev Hematol.
12:773–785. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Semple JW, McVey MJ, Kim M, Rebetz J,
Kuebler WM and Kapur R: Targeting transfusion-related acute lung
injury: The journey from basic science to novel therapies. Crit
Care Med. 46:e452–e458. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leite LFB, Máximo TA, Mosca T and Forte
WCN: CD40 ligand deficiency. Allergol Immunopathol (Madr).
48:409–413. 2020. View Article : Google Scholar
|
|
5
|
França TT, Barreiros LA, Al-Ramadi BK,
Ochs HD, Cabral-Marques O and Condino-Neto A: CD40 ligand
deficiency: Treatment strategies and novel therapeutic
perspectives. Expert Rev Clin Immunol. 15:529–540. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dasgupta S, Dasgupta S and Bandyopadhyay
M: Regulatory B cells in infection, inflammation, and autoimmunity.
Cell Immunol. 352:1040762020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Subauste CS: The CD40-ATP-P2X7
receptor pathway: Cell to cell cross-talk to promote inflammation
and programmed cell death of endothelial cells. Front Immunol.
10:29582019. View Article : Google Scholar
|
|
8
|
Fujihara C, Kanai Y, Masumoto R, Kitagaki
J, Matsumoto M, Yamada S, Kajikawa T and Murakami S: Fibroblast
growth factor-2 inhibits CD40-mediated periodontal inflammation. J
Cell Physiol. 234:7149–7160. 2019. View Article : Google Scholar
|
|
9
|
Seigner J, Basilio J, Resch U and de
Martin R: CD40L and TNF both activate the classical NF-κB pathway,
which is not required for the CD40L induced alternative pathway in
endothelial cells. Biochem Biophys Res Commun. 495:1389–1394. 2018.
View Article : Google Scholar
|
|
10
|
Woolaver RA, Wang X, Dollin Y, Xie P, Wang
JH and Chen Z: TRAF2 deficiency in B cells impairs CD40-Induced
isotype switching that can be rescued by restoring NF-κB1
activation. J Immunol. 201:3421–3430. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mulero MC, Huxford T and Ghosh G:
NF-kappaB, IkappaB, and IKK: Integral components of immune system
signaling. Adv Exp Med Biol. 1172:207–226. 2019. View Article : Google Scholar
|
|
12
|
Durand JK and Baldwin AS: Targeting IKK
and NF-κB for therapy. Adv Protein Chem Struct Biol. 107:77–115.
2017. View Article : Google Scholar
|
|
13
|
Pordanjani SM and Hosseinimehr SJ: The
role of NF-κB inhibitors in cell response to radiation. Curr Med
Chem. 23:3951–3963. 2016. View Article : Google Scholar
|
|
14
|
Scott O and Roifman CM: NF-κB pathway and
the goldilocks principle: Lessons from human disorders of immunity
and inflammation. J Allergy Clin Immunol. 143:1688–1701. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kellner M, Noonepalle S, Lu Q, Srivastava
A, Zemskov E and Black SM: ROS signaling in the pathogenesis of
acute lung injury (ALI) and acute respiratory distress syndrome
(ARDS). Adv Exp Med Biol. 967:105–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ward PA, Fattahi F and Bosmann M: New
insights into molecular mechanisms of immune complex-induced injury
in lung. Front Immunol. 7:862016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fang J, Wang Z, Wang P and Wang M:
Extraction, structure and bioactivities of the polysaccharides from
Ginkgo biloba: A review. Int J Biol Macromol. 62:1897–1905. 2020.
View Article : Google Scholar
|
|
18
|
Zhang R, Han D, Li Z, Shen C, Zhang Y, Li
J, Yan G, Li S, Hu B, Li J and Liu P: Ginkgolide C alleviates
myocardial ischemia/reperfusion-induced inflammatory injury via
inhibition of CD40-NF-κB pathway. Front Pharmacol. 9:1092018.
View Article : Google Scholar
|
|
19
|
Su X, Wang L, Song Y and Bai C: Inhibition
of inflammatory responses by ambroxol, a mucolytic agent, in a
murine model of acute lung injury induced by lipopolysaccharide.
Intensive Care Med. 30:133–140. 2004. View Article : Google Scholar
|
|
20
|
McGuigan RM, Mullenix P, Norlund LL, Ward
D, Walts M and Azarow K: Acute lung injury using oleic acid in the
laboratory rat: Establishment of a working model and evidence
against free radicals in the acute phase. Curr Surg. 60:412–417.
2003. View Article : Google Scholar
|
|
21
|
Chopra M, Reuben JS and Sharma AC: Acute
lung injury: Apoptosis and signaling mechanisms. Exp Biol Med
(Maywood). 234:361–371. 2009. View Article : Google Scholar
|
|
22
|
Zhao F, Shi D, Li T, Li L and Zhao M:
Silymarin attenuates paraquat-induced lung injury via Nrf2-mediated
pathway in vivo and in vitro. Clin Exp Pharmacol Physiol.
42:988–998. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Toumpanakis D, Vassilakopoulou V, Sigala
I, Zacharatos P, Vraila I, Karavana V, Theocharis S and
Vassilakopoulos T: The role of Src and ERK1/2 kinases in
inspiratory resistive breathing induced acute lung injury and
inflammation. Respir Res. 18:2092017. View Article : Google Scholar
|
|
24
|
Gouda MM and Bhandary YP: Acute lung
injury: IL-17A-mediated inflammatory pathway and its regulation by
curcumin. Inflammation. 42:1160–1169. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Omidkhoda SF, Razavi BM and Hosseinzadeh
H: Protective effects of Ginkgo biloba L. against natural toxins,
chemical toxicities, and radiation: A comprehensive review.
Phytother Res. 33:2821–2840. 2019. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dubey AK, Shankar PR, Upadhyaya D and
Deshpande VY: Ginkgo biloba-an appraisal. Kathmandu Univ Med J
(KUMJ). 2:225–229. 2004.
|
|
27
|
Boxio R, Wartelle J, Nawrocki-Raby B,
Lagrange B, Malleret L, Hirche T, Taggart C, Pacheco Y, Devouassoux
G and Bentaher A: Neutrophil elastase cleaves epithelial cadherin
in acutely injured lung epithelium. Respir Res. 17:1292016.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao H, Chen H, Xiaoyin M, Yang G, Hu Y,
Xie K and Yu Y: Autophagy activation improves lung injury and
inflammation in sepsis. Inflammation. 42:426–439. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Masuda H, Mori M, Umehara K, Furihata T,
Uchida T, Uzawa A and Kuwabara S: Soluble CD40 ligand disrupts the
blood-brain barrier and exacerbates inflammation in experimental
autoimmune encephalomyelitis. J Neuroimmunol. 316:117–120. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Takada YK, Yu J, Shimoda M and Takada Y:
Integrin binding to the trimeric interface of CD40L plays a
critical role in CD40/CD40L signaling. J Immunol. 203:1383–1391.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu ZL, Hu J, Xiao XF, Peng Y, Zhao SP,
Xiao XZ and Yang MS: The CD40 rs1883832 polymorphism affects sepsis
susceptibility and sCD40L levels. Biomed Res Int.
2018:74973142018.
|
|
32
|
Bishop GA, Stunz LL and Hostager BS: TRAF3
as a multifaceted regulator of B lymphocyte survival and
activation. Front Immunol. 9:21612018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yu N, Lambert S, Bornstein J, Nair RP,
Enerbäck C and Elder JT: The Act1 D10N missense variant impairs
CD40 signaling in human B-cells. Genes Immun. 20:23–31. 2019.
View Article : Google Scholar
|
|
34
|
Pan WZ, Du J, Zhang LY and Ma JH: The
roles of NF-κB in the development of lung injury after one-lung
ventilation. Eur Rev Med Pharmacol Sci. 22:7414–7422.
2018.PubMed/NCBI
|
|
35
|
Ulivi V, Giannoni P, Gentili C, Cancedda R
and Descalzi F: p38/NF-κB-dependent expression of COX-2 during
differentiation and inflammatory response of chondrocytes. J Cell
Biochem. 104:1393–1406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yin HC, Liu XY, Liu PM, Zhang H, Liang P,
Wang ZL and She MP: Effect of mm-LDL on NF-κB activation in
endothelial cell. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 23:312–316.
2001.In Chinese.
|
|
37
|
Asaad NY and Sadek GS: Pulmonary
cryptosporidiosis: Role of COX2 and NF-κB. APMIS. 114:682–689.
2006. View Article : Google Scholar : PubMed/NCBI
|
















