Introduction
Doxorubicin is an anthracycline anticancer drug that
is clinically used to treat several types of hematological cancer
and solid tumors, including leukemia, breast, endometrial and
bladder cancer (1). However, the
strong anticancer effect of DOX is often accompanied by a range of
side effects, such as bone marrow suppression, stomatitis, fatigue
and alopecia; the most severe side effect is dose-dependent
cardiotoxicity, which limits the clinical use of DOX (2,3).
A previous study has reported that patients treated with a
cumulative dose of 400 mg/m2 DOX present an increased
risk of cardiotoxicity; this risk is increased by 26 and 48% at 550
and 700 mg/m2 compared with that before starting the DOX
treatment, respectively (4).
Myocardial oxidative stress, mitochondrial impairment,
intracellular calcium dysregulation and extracellular matrix
remodeling are the main molecular mechanisms underlying DOX-induced
cardiotoxicity (5). Reactive
oxygen species (ROS) produced by oxidative stress is a major
mechanism among those that have been elucidated to date,
accumulating in the myocardium, causing apoptosis and further
leading to cardiac dysfunction and eventually heart failure
(6). The development of drugs to
treat and prevent the cardiotoxicity induced by DOX has recently
attracted attention (7).
Dexrazoxane, the most effective agent for alleviating the
DOX-induced cardiotoxicity, is converted into a chelating agent in
cells and interferes with the formation of free radicals mediated
by iron; however, dexrazoxane may diminish the anticancer effects
and potentiate certain side effects of DOX (8). Therefore, it is necessary to find a
more effective strategy to treat DOX-induced cardiotoxicity, for
which oxidative stress may be a potential target.
The peptide Szeto-Schiller (SS)-31 (SS31;
H-D-Arg-Dmt- Lys-Phe-NH2) is an efficacious antioxidant that has
the capacity to reduce mitochondrial ROS and scavenge free radicals
(9). It has been reported that
SS31 is involved in the pathophysiological processes of a variety
of cardiovascular diseases, including protection of the myocardium
from ischemia-reperfusion (I/R) injury by reducing inflammation,
oxidative stress, apoptosis and fibrosis (10), reduction of proteomic alterations
in heart failure by preserving mitochondrial function (11), prevention of sepsis-induced
cardiac damage by suppressing the inflammatory response and
maintaining mitochondrial membrane potential (12), and amelioration of angiotensin
II-induced cardiomyopathy by decreasing the level of ROS (13). However, whether SS31 serves a
protective role in DOX-induced cardiotoxicity remains to be
elucidated.
The mitogen-activated protein kinase (MAPK) pathways
serve a crucial role in the regulation of apoptosis.
Extracellular-regulated kinase (ERK) 1/2, p38 MAPK and c-Jun
N-terminal kinase (JNK) are three major members of MAPKs and exert
antiapoptotic or proapoptotic effects in different cell types and
contexts (14). For example,
MAPK serves a protective role in I/R-induced cardiac myocyte
apoptosis and in isolated perfused hearts that have undergone
reperfusion injury, whereas p38 and JNK promote apoptosis in
cardiomyocytes subjected to I/R (15). Fasudil suppresses
isoproterenol-induced heart failure by inhibiting the activation of
JNK and the nuclear translocation of MAPK (16). Therefore, the present study
focused on the development of strategies to improve DOX-induced
cardiotoxicity with MAPK as a target.
The present aimed to determine the therapeutic
effects of SS31 on DOX-induced cardiotoxicity, and we hypothesized
that the administration of SS31 may provide a new insight into
therapeutic strategies for the treatment of DOX-induced
cardiotoxicity.
Materials and methods
Peptide synthesis and administration
The peptide SS31 (H-D-Arg-Dmt-Lys-Phe-NH2) was
synthesized by Shanghai Science Peptide Biological Technology Co.,
Ltd. The peptide crystal was dissolved in sterile double-distilled
water and diluted to 10, 20 and 50 μmol/l.
Cell culture and treatment
H9c2, a rat cardiomyocyte-derived cell line, was
acquired from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences. H9c2 cells were cultured in Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.) supplemented with 10% fetal bovine serum (Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin and streptomycin
(Wisent, Inc.) in a 95% air and 5% CO2 atmosphere at
37°C.
The H9c2 cell toxicity model was induced by DOX
(cat. no. CAS 25316-40-9; Target Molecule Corp.). The H9c2 cells at
80% confluence were preincubated with the SS31 peptide or vehicle
for 2 h at 37°C, followed by the addition of 1 μM DOX to the
medium for 24 h at 37°C (17).
Cells were designated into six groups: i) Control group; ii) SS31
(50 μmol/l) treatment group; iii) DOX treatment group; iv)
DOX and SS31 (10 μmol/l) cotreatment group; v) DOX and SS31
(20 μmol/l) cotreatment group; vi) DOX and SS31 (50
μmol/l) cotreatment group. The SS31 peptide was added to the
culture supernatant 2 h prior to DOX treatment. A selective agonist
of p38 MAPK P79350 (50 μM; Invitrogen; Thermo Fisher
Scientific, Inc.) was used to activate the p38 signaling pathway.
P79350 was added to the cell culture medium for 24 h at 37°C.
Crystal violet staining assay
Following treatment, H9c2 cells were seeded in
6-well plates at 2×105 cells/well, washed with PBS twice
to remove dead cells and fixed with 4% paraformaldehyde at room
temperature (RT) for 30 min. Subsequently, the cells were washed
again with PBS and stained with 0.1% crystal violet (Beyotime
Institute of Biotechnology) solution for 30 min at RT in the dark.
The dye was aspirated, and the cells were washed twice with PBS and
air-dried naturally at RT for image capture under light microscopy
(×200). Subsequently, the dye was solubilized with 33% acetic acid
solution for 10 min at RT, and the absorbance was quantified at 570
nm using a microplate reader.
Cell survival analysis
H9c2 cells were seeded in 6-well plates at
2×105 cells/well. Following DOX and peptide treatment,
the cells were digested with trypsin-EDTA solution for 3 min at
37°C and pipetting. Trypan blue staining was used to assess the
cell survival rates. Briefly, the cells were resuspended with 1 ml
PBS, and 100 μl of the cell suspension was added to 100
μl trypan blue solution and stained for 3 min. The number of
cells was counted in the four squares of the hemocytometer under a
Zeiss light microscope (magnification, ×100). The following formula
was used for the cell survival rate: Cell survival rate (%)=(no. of
living cells/no. of total cells) ×100%.
Analysis of cell viability
Cell viability was assessed using the CCK-8 Assay
kit (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. H9c2 cells were seeded in 96-well
plates at 1×104 cells/well and cultured to adherence,
followed by SS31 and DOX treatment as aforementioned. A total of 10
μl CCK-8 reagent was supplemented into each well, and the
cells were incubated in the dark for2 h at 37°C. The absorbance was
measured using a microplate reader at 450 nm, and the cell
viability was calculated based on the absorbance.
Lactate dehydrogenase (LDH)
determination
The levels of LDH were detected by the LDH Release
Assay kit (Beyotime Institute of Biotechnology). The reaction
solution was prepared according to the manufacturer's instructions.
The H9c2 cell supernatant (120 μl/well) was collected by
centrifugation at 400 × g for 5 min at RT and mixed with the
reaction solution (60 μl,/well), and the mixtures were added
into 96-well plates. The plates were wrapped in tin foil and
incubated for 30 min at RT on the shaker. Finally, the absorbance
was detected with a microplate reader at 490 nm wavelength.
Analysis of ROS production
The level of intracellular total ROS was assessed by
2′,7′-dichlorofluorescin diacetate (DCFH-DA) using a Reactive
Oxygen Species Assay kit (Beyotime Institute of Biotechnology).
Briefly, H9c2 cells were seeded in 6-well plates at
2×105 cells/well and treated as aforementioned when the
cells had grown to 80% confluence. DCFH-DA was diluted to 10
μM in serum-free DMEM and added into the medium (1 ml/well),
and the cells were incubated at 37°C away from light for 20 min.
Subsequently, the cells were washed with PBS thrice to remove the
residual DCFH-DA and examined in at ≥3 fields per sample under a
laser scanning confocal microscope (magnification, ×100). The
density of ROS fluorescence was examined by ImageJ software 1.26
(National Institutes of Health).
JC-1 mitochondrial membrane potential
determination
The JC-1 Mitochondrial Membrane Potential Assay kit
(Beyotime Institute of Biotechnology) was used to analyze
mitochondrial injury according to the manufacturer's instructions.
Briefly, H9c2 cells were seeded in 6-well plates at
2×105 cells/well, washed with PBS and incubated with
JC-1 solution for 10 min at 37°C. The cells were washed with the
dilution buffer and analyzed in ≥3 fields per sample under a laser
scanning confocal microscope (magnification, ×100).The density of
JC-1 fluorescence was examined by ImageJ software.
Western blotting
H9c2 cells were treated as aforementioned and lysed
by RIPA protein lysis buffer (Beyotime Institute of Biotechnology)
and 1% PMSF (Beyotime Institute of Biotechnology) to extract the
total protein. The protein concentrations were determined using the
Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology). The proteins (20 μg/lane) were separated by
10% SDS-PAGE and transferred to PVDF membranes (MilliporeSigma).
The membranes were blocked with skimmed milk (5%) for 2 h at RT and
incubated with the primary antibodies against PARP (1:1,000; cat.
no. 9542), cleaved caspase-3 (1:1,000; cat. no. 9661), bax
(1:1,000; cat. no. 2772), bcl-2 (1:1,000; cat. no. 4223),
phosphorylated (p-)p38 (1:1,000; cat. no. 4511), p38 (1:1,000; cat.
no. 8690), JNK (1:1,000; cat. no. 9255), p-JNK (1:1,000; cat. no.
9251), p-ERK (1:1,000; cat. no. 4376), ERK (1:1,000; cat. no.
4695), α/β-Tubulin (1:2,000; cat. no. 2148), β-actin (1:2,000; cat.
no. 4970) and GAPDH (1:2,000; cat. no. 2118) (all Cell Signaling
Technology, Inc.) at 4°C overnight. Notably, the internal control
antibody against α/β-tubulin produced a nonspecific faint band
(18,19). Subsequently, PVDF membranes were
washed three times for 10 min each time with TBS with 0.1% Tween-20
buffer and incubated with horseradish peroxidase-conjugated
anti-rabbit secondary antibodies (1:3,000; cat. no. 7074; Cell
Signaling Technology, Inc.) for 1 h at RT. The immunoreactive
protein bands were detected using an enhanced chemiluminescent
substrate (cat. no. SQ101; Epizyme, Inc.). The protein expression
levels were quantified according to their grey values determined
using ImageJ software.
In vivo experiment
All animal experiments were carried out in
accordance with the Guide for the Care and Use of Laboratory
(20) and approved by the
Institutional Animal Care and Use Committee of Nanjing Medical
University (approval no. IACUC-1903030; Nanjing, China). A total of
48 male C57BL/6 mice (6 weeks old; weight, 16-20 g) were obtained
from Shanghai SLAC laboratory animal corporation and maintained
under a 12:12-h light/dark cycle at 22-26°C with a relative
humidity of 40-50% and ad libitum food and water. Following
one week of adjustable feeding, the animals were randomly assigned
to the following groups: i) Vehicle; ii) SS31; iii) DOX; and iv)
DOX + SS31 treated animals. DOX was administrated by
intraperitoneal injection at 5 mg/kg weekly, and the final
cumulative dose was 20 mg/kg (21). SS31 (2.5 mg/kg) was injected into
the tail vein weekly, and the final cumulative dose was 10 mg/kg
(22). The mice that were not
treated with SS31 or DOX received the equal volumes of saline.
Following the treatment, the mice were maintained alive for one
week. The mice were anesthetized with 1.5% isoflurane inhalation;
the depth of anesthesia was evaluated by the immobility and the
absence of righting reflex, and echocardiography was used to detect
mouse cardiac function. Following euthanasia by carbon dioxide
asphyxia (30% chamber volume/min), the hearts were removed rapidly
and harvested to prepare paraffin sections for Masson and wheat
germ agglutinin (WGA) staining or to isolate the mitochondria. The
blood (1 ml) was collected from the abdominal aorta, and the serum
was obtained by centrifugation (1,200 × g, 20 min, 4°C).
Echocardiography analysis
For the evaluation of cardiac function, the mice
were anesthetized with 1.5% isoflurane, and echocardiography was
performed using a Vevo 2100 High Resolution Imaging System
(VisualSonics, Inc.). Cardiac contractile function was examined by
echocardiography in conscious, gently restrained mice using a Vevo
2100 system (MS400C probe). The main measured indicators included
ejection fraction (EF) and fractional shortening (FS). Other
echocardiographic parameters included left ventricular end-systolic
diameter (LVEDs), left ventricular end-diastolic diameter (LVEDd),
left ventricular end-systolic volume (LVESV) and left ventricular
end-diastolic volume (LVEDV). FS was calculated as follows: FS
(%)=[(LVEDd-LVEDs)/LVEDd] × 100; EF was calculated as follows: EF
(%)=[(LVEDV-LVESV)/LVEDV] × 100, where LVEDV=7 ×
LVEDd3/(2.4 + LVEDd) and LVESV= 7 ×
LVEDs3/(2.4 + LVEDs).
WGA and Masson staining
The hearts were harvested and fixed in 4% buffered
formaldehyde for 48 h at RT. After embedding in paraffin and
sectioning, 5-μm sections were stained with fluorescein
isothiocyanate-conjugated WGA (cat. no. L4895; Sigma-Aldrich; Merck
KGaA) staining according to the manufacturer's instructions.
Digital images (≥3 fields) were captured using a laser scanning
confocal microscope (magnification, ×400). A quantitative digital
image analysis system Image-Pro Plus 6.0 (Media Cybernetics, Inc.)
was used to measure the cross-sectional area of cardiomyocytes. To
assess cardiac fibrosis, the 5-μm sections were stained with
Masson's trichrome (cat. no. G1340; Beijing Solarbio Science &
Technology Co., Ltd.) according to the manufacturer's instructions.
Each stained section was observed under a microscope
(magnification, ×400), and ImageJ software was used to evaluate
histopathological damage.
LDH, superoxide dismutase (SOD),
malondialdehyde (MDA) and glutathione peroxidase (GSH-PX)
measurement
The serum LDH concentrations were measured using an
ELISA kit (cat. no. J2380; Promega Corporation) according the
manufacturer's instructions. A total of 40 mg heart tissues were
harvested to isolate the mitochondria. Briefly, tissues were
incised completely in 500 PBS and centrifuged at 800 × g for 5 min.
After centrifuge, 500 μl isolation reagent were added to
purify the mitochondria. The SOD, MDA and GSH-PX were determined
using commercially available kits (cat. nos. A001-3, A003-1 and
A005-1, respectively; Nanjing Jiancheng Bio Company) according to
the manufacturer's instructions by measuring absorbance at 450, 532
and 412 nm, respectively, with a microplate reader.
Statistical analysis
Data are presented as the mean ± standard deviation.
All experimental data were analyzed by GraphPad Prism 8 software
(GraphPad Software, Inc.). The differences amongst multiple groups
were analyzed by one-way ANOVA with a post hoc Bonferroni's
multiple comparisons test. Mouse survival was analyzed by the
log-rank test. P<0.05 was considered to indicate a statistically
significant difference.
Results
SS31 treatment attenuates DOX-induced
inhibition of H9c2 cell survival
To investigate the function of SS31, the present
study first treated H9c2 cells with SS31 to identify the location
of SS31. As presented in Fig.
S1A, SS31 entered the cells. The changes in the H9c2 cell
survival following treatment with various concentrations of DOX (0,
0.1, 0.25, 0.5, 1.0, 2.5, 5.0 and 10.0 μM) for various
durations (0, 6, 12, 24 and 36 h) were first assessed prior to
demonstrating the function of SS31 in DOX-induced cardiotoxicity.
As demonstrated in Fig. S1B and
C, a stable in vitro cardiotoxicity model was
established by 1-μM DOX treatment for 24 h. The results of
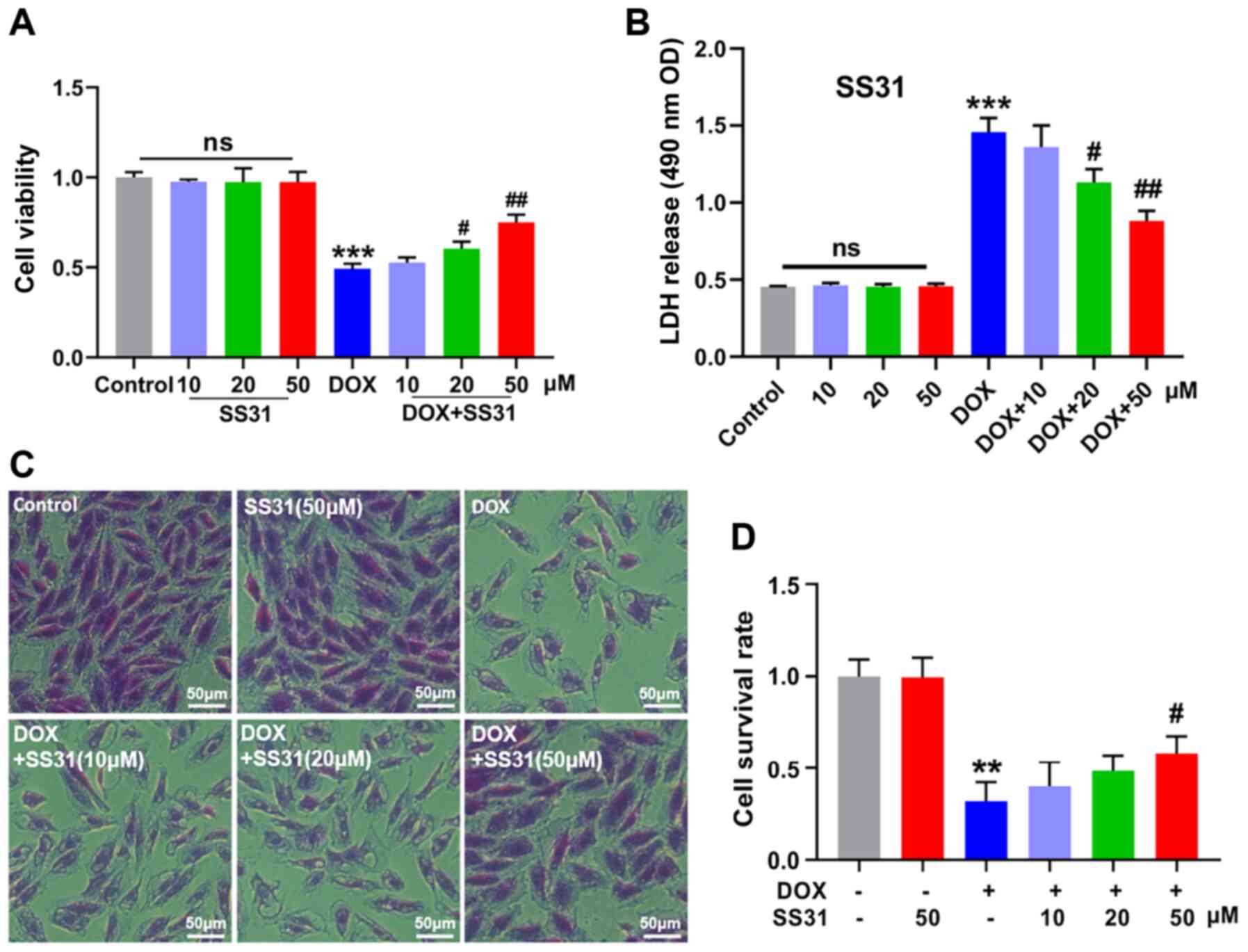
subsequent experiments demonstrated that SS31 alleviated
DOX-induced H9c2 cell damage and had no effect on H9c2 cells
without DOX treatment, as revealed by the cell viability and LDH
release assays (Fig. 1A and B).
In addition, crystal violet staining confirmed that SS31 (20 and 50
μM) improved the survival rate of DOX-treated H9c2 cells
(Fig. 1C and D). These results
indicated that SS31 may exert protection from DOX-induced H9c2 cell
damage.
SS31 attenuates DOX-induced apoptosis in
H9c2 cells
To further explore the function of SS31 in
DOX-induced myocardial injury, the expression levels of
apoptosis-associated proteins were evaluated in H9c2 cells.
Decreased PARP activation, cleaved caspase-3 and bax levels and
increased bcl-2 protein levels were detected in cells pretreated
with SS31 (20 and 50 μM) prior to DOX treatment compared
with those in cells only exposed to DOX (Fig. 2A-E). These results suggested that
SS31 attenuated DOX-induced apoptosis in H9c2 cells.
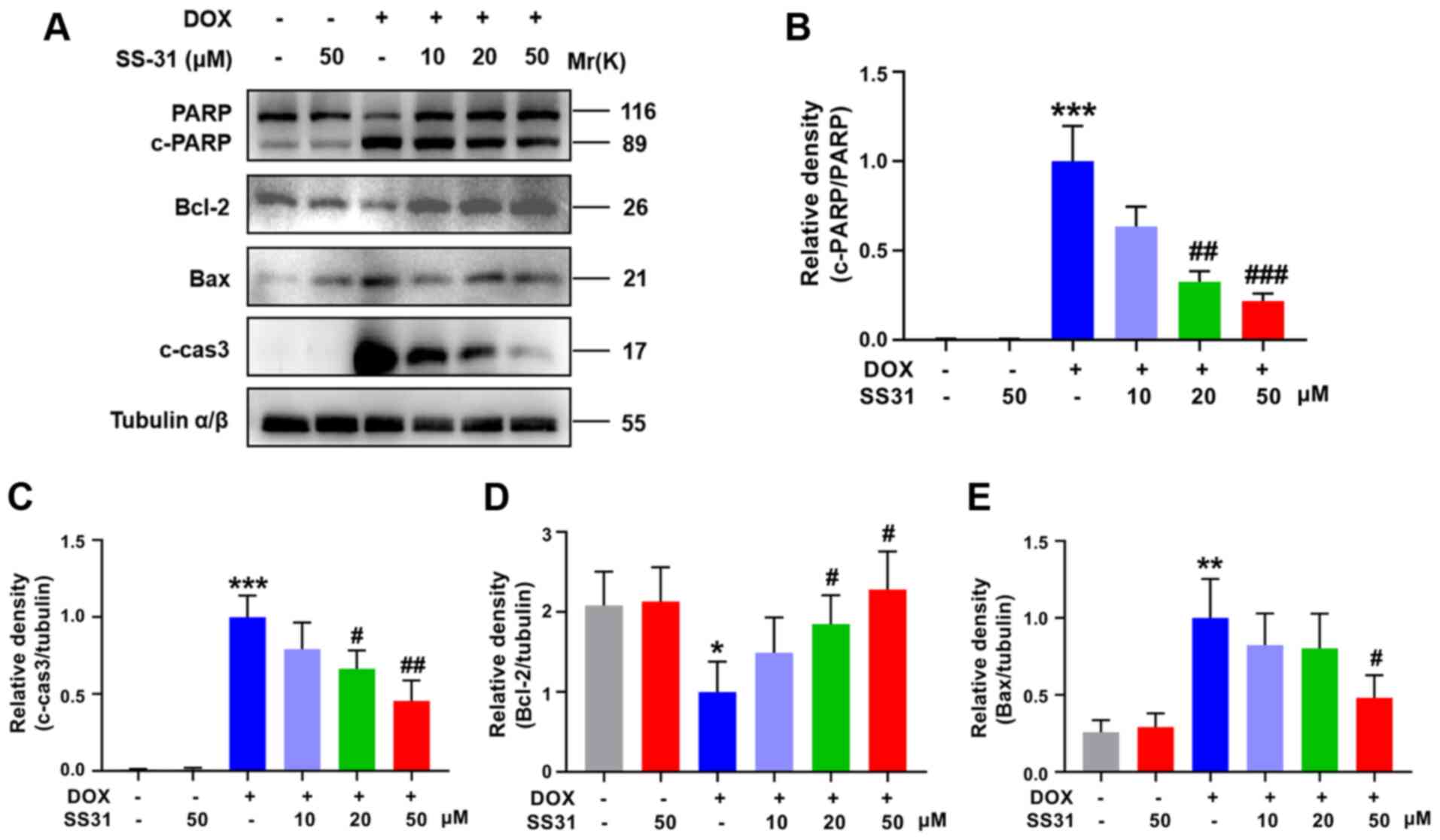 | Figure 2SS31 attenuates DOX-induced apoptosis
in H9c2 cells. (A) The levels of apoptosis-related proteins (PARP,
cleaved caspase-3, bax and bcl-2) were determined by western
blotting. (B-E) Quantitative analysis of the relative protein
levels of (B) c-PARP (C) cleaved caspase-3, (D) bcl-2 and (E) bax.
*P<0.05, **P<0.01 and ***P<0.001 vs. control; #P<0.05,
##P<0.01 and ###P<0.001 vs. DOX. DOX,
doxorubicin; SS31, Szeto-Schiller 31 peptide; PARP,
poly(ADP-ribose) polymerase; c-, cleaved; cas3, caspase-3. |
SS31 attenuates DOX-induced mitochondrial
oxidative stress injury in H9c2 cells
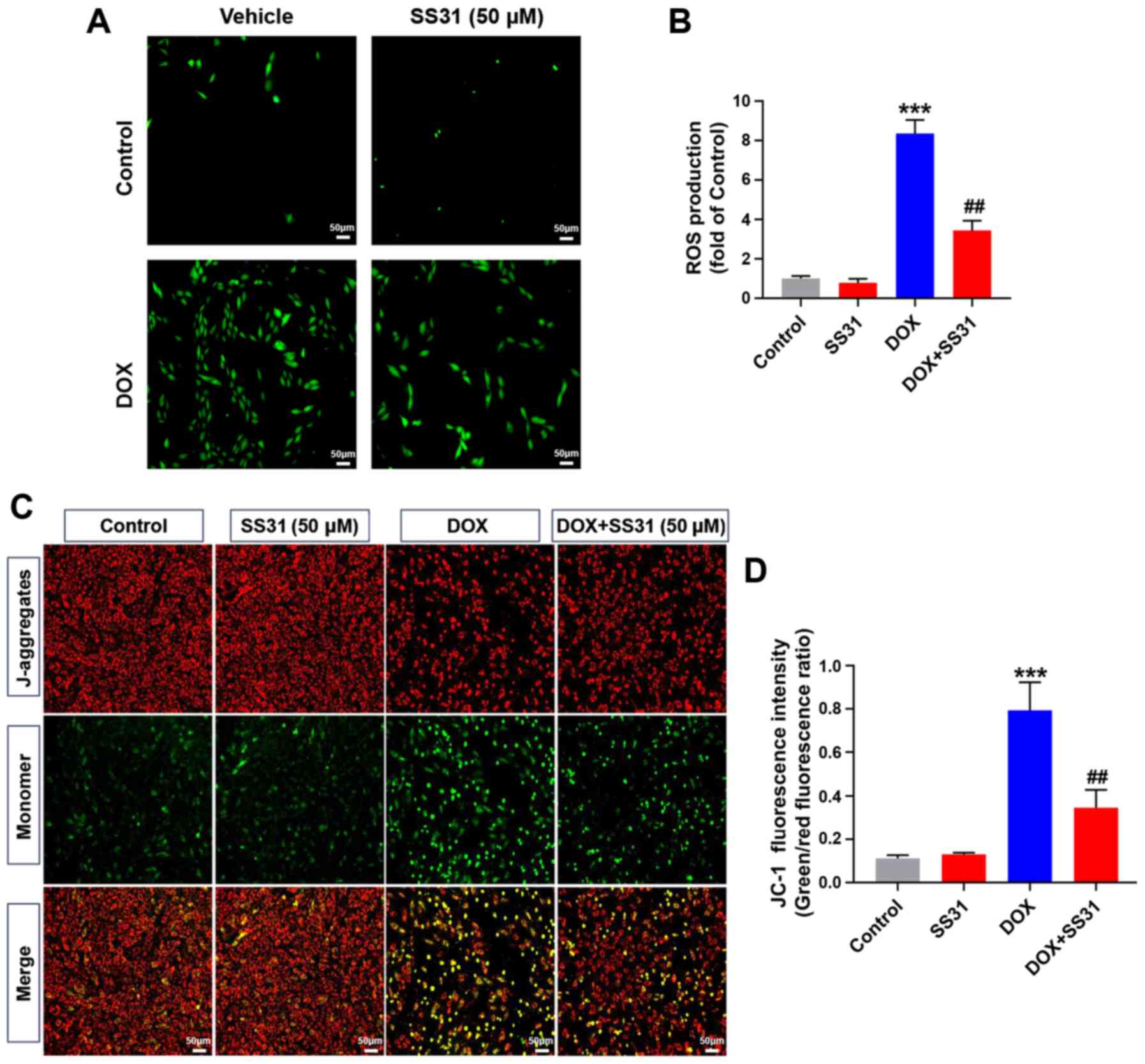
Since the accumulation of intracellular total ROS is
the main biological event of DOX-induced myocardial injury
(6), the total ROS accumulation
in H9c2 cells was examined by DCFH-DA staining. The results
demonstrated that 50 μM SS31 significantly decreased the
level of ROS following DOX treatment in H9c2 cells compared with
that in the cells treated with DOX alone (Fig. 3A and B). In addition, 50
μM SS31 significantly alleviated the decrease of
mitochondrial membrane potential induced by DOX, as evidenced by
the lower ratio of JC-1 monomer to aggregate in the SS31-pretreated
cells compared with that in the cells treated with DOX alone
(Fig. 3C and D).
Effects of SS31 on DOX-induced cardiac
injury in vivo
To assess the effects of SS31 on DOX-induced cardiac
injury in vivo, a mouse model of cardiotoxicity was
constructed by an intraperitoneal injection of 20 mg/kg DOX and a
tail vein injection of 10 mg/kg SS31 (Fig. 4A). The results of the in
vivo experiments demonstrated that SS31 protected the mouse
heart from DOX-induced injury, as evidenced by improved cardiac
function determined by echocardiography (Fig. 4B and C) and a lower level of
cardiac fibrosis (Fig. 4D and E)
in mice treated with SS31 and DOX compared with those in the DOX
group. The cardiomyocyte area was evaluated by WGA staining, and
the results revealed a significant decrease of the cardiomyocyte
area in the DOX group compared with the control group; cotreatment
with SS31 reversed this effect (Fig.
4F and G). In addition, DOX-induced cardiac injury was
significantly improved in the SS31 intervention group, which
exhibited a decrease in the serum LDH level compared with that in
mice treated with DOX alone (Fig.
4H). Oxidative stress markers were detected to evaluate the
mitochondrial oxidative stress injury; the results demonstrated
that SS31 attenuated the DOX-induced mitochondrial oxidative stress
injury, as indicated by the differences in the MDA, SOD and GSH-PX
content between the DOX and DOX + SS31 groups (Fig. 4I-K). As presented in Fig. 4L, treatment with SS31 did not
affect the survival of mice following DOX injection. These results
indicated that SS31 exerted a cardioprotective effect on
DOX-induced cardiac injury without improving survival in
vivo.
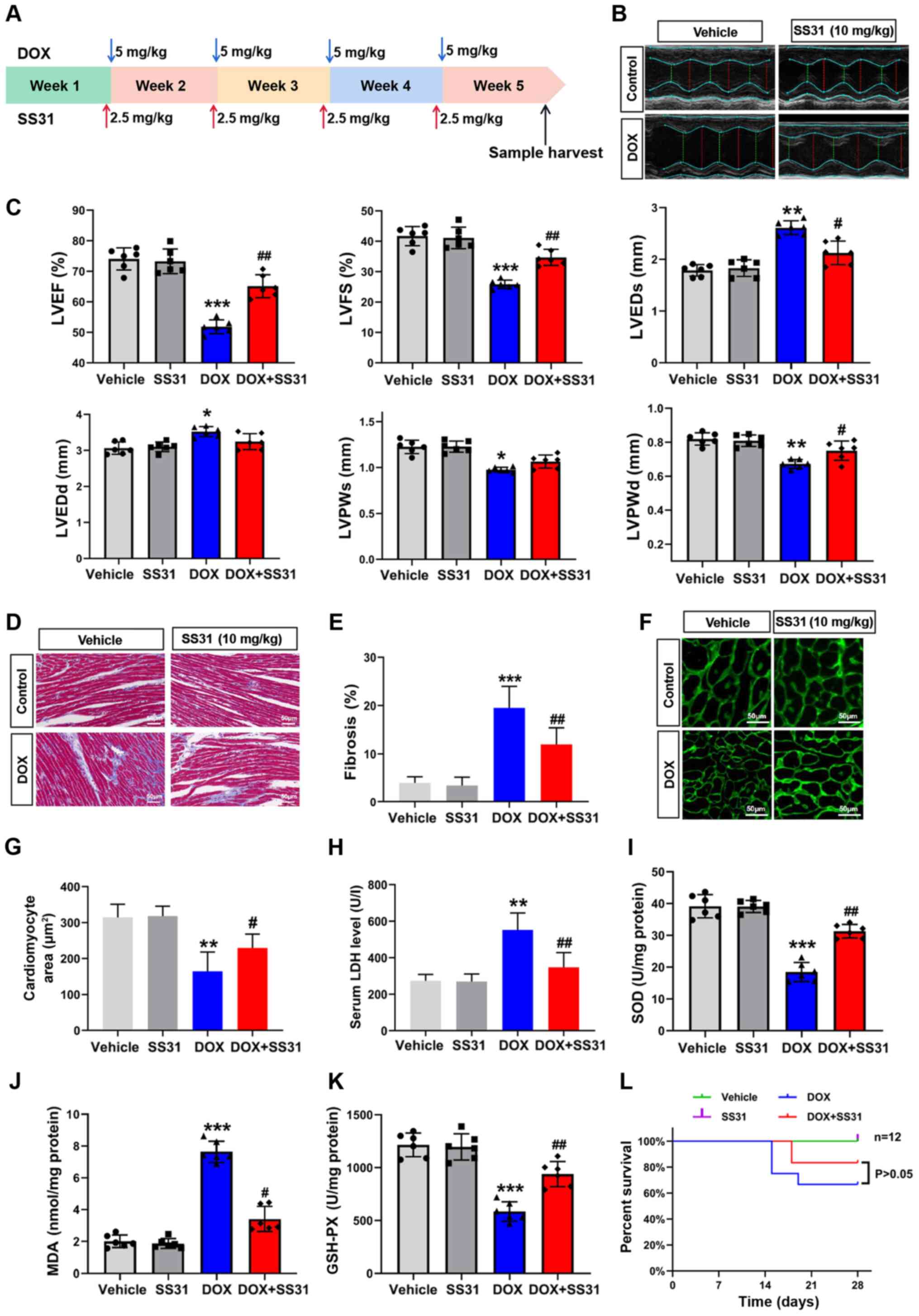 | Figure 4Effects of SS31 on DOX-induced
cardiac injury in vivo. (A) Timeline of the experimental
procedure for the DOX-induced mouse cardiotoxicity model. (B)
Representative photographs of the echocardiography analysis. (C)
Quantified data of the echocardiography analysis. (D) Masson
trichrome staining. Magnification, ×400. (E) Quantitative analysis
of fibrosis in the Masson-stained sections. (F) Representative
photographs of wheat germ agglutinin staining. Magnification, ×400.
(G) Quantitative analysis of the cardiomyocyte area. (H) Serum LDH
levels. n=6 mice/group. (I-K) SOD, MDA and GSH-PX levels were
evaluated in mouse heart tissue samples. n=6 mice/group. (L)
Survival of mice following DOX-induced cardiac injury. Day 0 refers
to the first DOX injection. *P<0.05, **P<0.01 and
***P<0.001 vs. vehicle; #P<0.05 and ##P<0.01
vs. DOX. DOX, doxorubicin; SS31, Szeto-Schiller 31 peptide; LDH,
lactate dehydrogenase; SOD, superoxide dismutase; MDA,
malondialdehyde; GSH-PX, glutathione peroxidase; EF, ejection
fraction; FS, fractional shortening; LVEDs, left ventricular
end-systolic diameter; LVEDd, left ventricular end-diastolic
diameter; LVESV, left ventricular end-systolic volume; LVEDV, left
ventricular end-diastolic volume. |
SS31 inhibits the activation of p38 MAPK
signaling in DOX-treated H9c2 cells
The present study further aimed to determine the
mechanism underlying the effects of SS31 in ameliorating
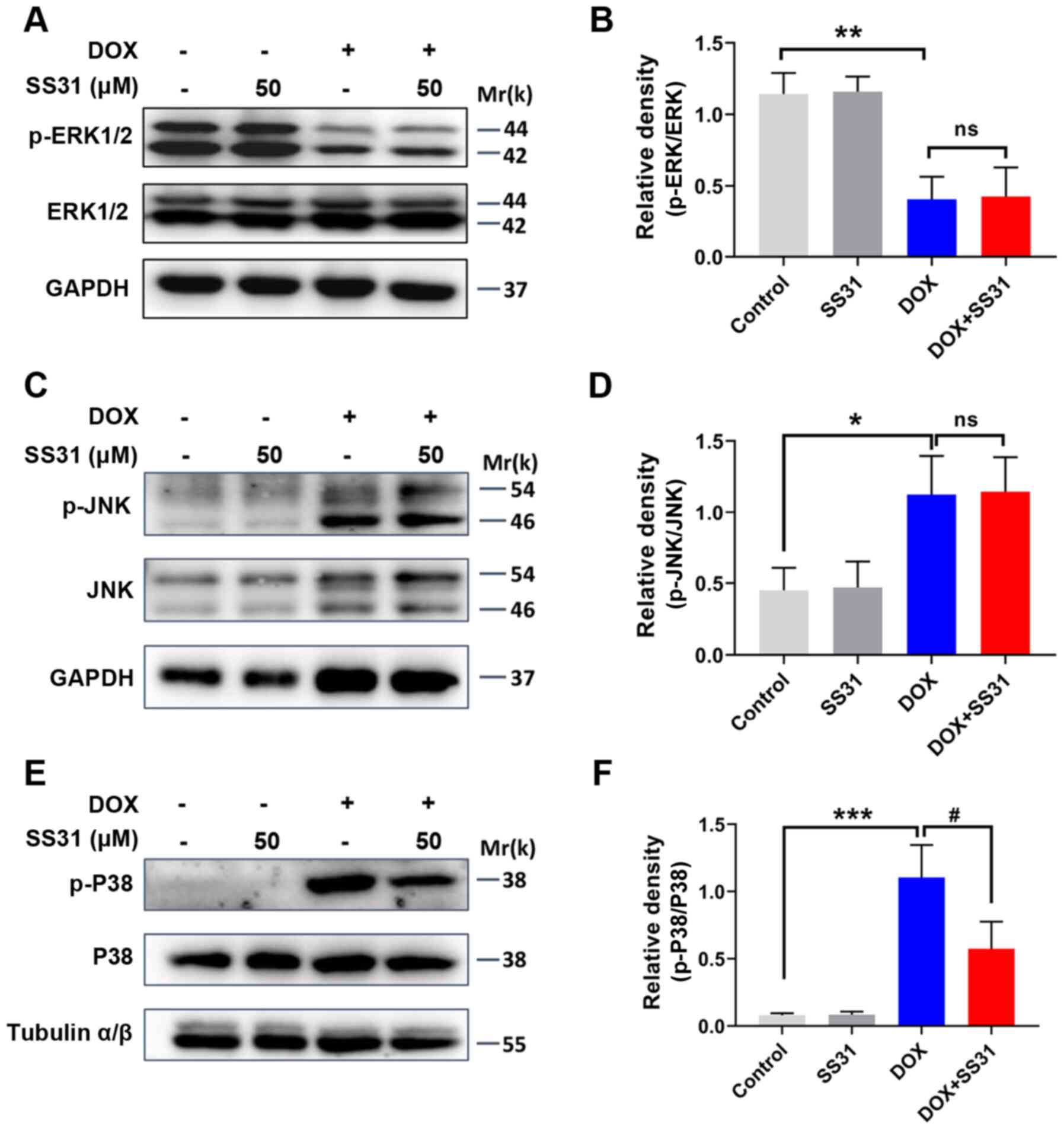
DOX-induced cardiotoxicity in H9c2 cells. The effects of SS31 on
the expression and phosphorylation of ERK1/2, JNK and p38 were
examined in DOX-treated H9c2 cells. SS31 treatment had no
significant effects on the levels of p-ERK1/2 and p-JNK, and the
total levels of ERK1/2 and JNK were also unchanged (Fig. 5A-D). However, the DOX-induced
upregulation of p-p38 was significantly inhibited by pretreatment
with SS31 in H9c2 cells (Fig. 5E and
F). Thus, these results revealed that SS31 inhibited the
activation of the p38 MAPK signaling pathway to attenuate the ROS
accumulation and apoptosis in H9c2 cells.
p38 agonist reverses the effects of
SS31
To further verify that SS31 protected H9c2 cells
from DOX-induced injury by regulating the p38 MAPK signaling
pathway, a selective agonist of p38 MAPK, P79350, was used to
activate the p38 signaling pathway. As demonstrated in Fig. 6A, compared with the SS31 + DOX
group, p38 was activated when the cells were treated with P79350. A
comprehensive experiment was subsequently performed. The p38
agonist reversed the inhibitory effects of SS31 on the cell damage,
as evidenced by the decreased cell viability, cell survival rates
and increased LDH release in DOX-induced cells cotreated with
P79350 and SS31 compared with those in DOX-induced cells treated
with SS31 alone (Fig. 6C-F). In
addition, the mitochondrial ROS content analysis results
demonstrated that P79350 completely rescued effects of SS31 on ROS
(Fig. 6G and H). The levels of
apoptosis-associated proteins were also assessed, and the results
revealed that the levels of cleaved PARP and cleaved caspase-3 were
increased in the DOX, SS31 and P79350 cotreatment group compared
with those in the DOX and SS31 group, suggesting that the p38
agonist reversed the effects of SS31 (Fig. 6I-K).
 | Figure 6p38 agonist reverses the effects of
SS31 in DOX-treated H9c2 cells. (A) H9c2 cells were treated with
P79350 to activate the p38 signaling pathway. (B) Quantification of
the western blot data. (C) H9c2 cell viability was detected
following treatment with DOX, SS31 and P79350. (D) LDH release was
detected in H9c2 cells. (E) Crystal violet staining of living
cells. Magnification, ×200. (F) Quantitative analysis of the
crystal violet assay. (G) ROS contents were detected in H9c2 cells
following treatment with DOX, SS31 and P79350. (H) Quantitative
analysis of ROS contents. (I) The effects of P79350 on the levels
of apoptosis-associated proteins in H9c2 cells treated with DOX and
SS31. (J) Quantification of the PARP western blot bands. (K)
Quantification of the c-cas3 western blot bands. **P<0.01 and
***P<0.001 vs. control; ##P<0.01 vs. DOX;
&P<0.05 and &&P<0.01 vs. DOX + SS31. p-,
phosphorylated; DOX, doxorubicin; SS31, Szeto-Schiller 31 peptide;
LDH, lactate dehydrogenase; ROS, reactive oxygen species; PARP,
poly(ADP-ribose) polymerase; OD, optical density; c-, cleaved;
cas3, caspase-3. |
Discussion
DOX is an efficient broad-spectrum antitumor drug
and has been used to treat various types of cancer as a basic
chemotherapy agent in clinical practice (1). However, DOX is a double-edged
sword, as it also induces acute and chronic cardiac injury during
or after treatment (23).
Oxidative stress and apoptosis of cardiomyocytes serve important
roles in DOX-induced cardiac injury (24,25). In the present study, SS31 was
demonstrated to exert a protective effect against DOX-induced
cardiotoxicity by scavenging ROS and reducing myocardial apoptosis
through the inhibition of the p38 MAPK signaling pathway. In
addition, the administration of SS31 improved cardiac function and
limited the extent of myocardial fibrosis in a mouse model of
cardiotoxicity caused by DOX. These results suggested that SS31 may
be a candidate drug for the treatment of DOX-induced
cardiotoxicity.
To determine the myocardial protective effects of
SS31 against DOX-induced cardiotoxicity, in vitro
experiments were first performed in the present study. In previous
studies, the concentration and time of the DOX in vitro
model treatment were different compared with those used in the
present study (26,27). For example, treatment with 0.5
μM DOX for 24 h significantly diminishes the protein
expression levels of COX5A in H9c2 cells (27). MicroRNA-140-5p aggravates 5
μM DOX-induced cardiotoxicity by promoting myocardial
oxidative stress via targeting Nrf2 and Sirt2 in H9c2 cells
(26). The present study used
H9c2 cells to construct a stable in vitro cardiotoxicity
model by 24-h treatment with 1 μM DOX. A previous study has
reported that 200 nM DOX induces cell cycle arrest (28). However, other studies have
demonstrated that 1 μM DOX induces ROS accumulation and
apoptosis in cardiomyocytes (17,29). DOX has been reported to induce
apoptosis through a number of mechanisms, including its effects on
topoisomerase II, excessive production of ceramide and production
of free radicals and ROS (30).
In the present study, 1 μM DOX induced ROS accumulation and
apoptosis in cardiomyocytes. These results were consistent with the
aforementioned studies and indicated that DOX induced apoptosis in
H9c2 cells. Subsequently, the results of the present study
demonstrated that pretreatment with SS31 improved the inhibition of
cell viability induced by DOX. This effect was
concentration-dependent; in addition 50 μM SS31 did not
exert any toxic effects on H9c2 cells. Consistent with these
results, crystal violet staining confirmed that SS31 improve the
survival rate of DOX-treated H9c2 cells. These results suggested
that SS31 may exert myocardial protection.
Although a number of mechanisms are involved in
DOX-induced cardiotoxicity, oxidative stress, defined as an
imbalance between the production of ROS and antioxidant reactions,
is recognized as the primary mechanism (6). DOX preferentially localizes to the
anionic phospholipid cardiolipin on the mitochondrial inner
membrane, where it causes organelle toxicity by producing
superabundant ROS (31).
Supraphysiologic ROS not only contributes to mitochondrially injury
via inactivation of iron-sulfur proteins and stimulation of lipid
peroxidation, but also impairs macromolecules in the cells, such as
nucleic acids, proteins and phospholipids, thus leading to the
structural collapse of organelles and apoptosis (32,33). Previous studies have reported
that SS31 protects cells from oxidative stress by restoring
mitochondrial function (34).
SS31 exhibits potent protective effects in numerous diseases, such
as diabetic nephropathy (35),
Alzheimer's disease (36),
subarachnoid hemorrhage (37)
and Huntington's disease (38).
SS31 has also been demonstrated to exhibit high efficacy in several
types of cardiovascular diseases, including myocardial I/R injury,
heart failure and hypertensive cardiomyopathy (9). In the present study, compared with
the DOX group, SS31 reduced the release of LDH. In addition, 50
μM SS31 markedly decreased the levels of ROS and maintained
the mitochondrial membrane potential following DOX treatment in
H9c2 cells compared with those in the cells treated with DOX alone.
These results were consistent with the aforementioned studies and
further confirmed that the antioxidation of SS31 may serve crucial
roles in the protection of DOX-induced H9c2 cytotoxicity.
DOX has been reported to induce cardiomyocyte
apoptosis by activating extrinsic and intrinsic apoptotic
signaling, causing tissue injury (39,40). Additionally, alleviating
myocardial apoptosis potently suppresses the toxic effect of DOX on
myocytes (41,42). Consistent with previous studies,
the results of the present study demonstrated that
apoptosis-related proteins were activated following DOX treatment,
suggesting that DOX indeed induced apoptosis in H9c2 cells, whereas
SS31 reduced the levels of proapoptotic proteins PARP, cleaved
caspase-3 and bax and enhanced the expression levels of the
antiapoptotic protein bcl-2 compared with those in cells treated
with DOX alone. Therefore, the antiapoptotic effect of SS31 may
also play a key role in protecting DOX-induced cardiotoxicity.
The in vitro experiments in the present study
demonstrated the antioxidant and antiapoptotic functions of SS31 in
DOX-treated H9c2 cells. The results of the in vivo
experiments further confirmed the cardiotoxicity of DOX by
histopathological analysis of mouse tissues, revealing notable
fibrosis in the myocardial tissues of DOX-treated animals.
Administration of SS31 partially reversed that detrimental changes,
indicating that SS31 exerted a potential cardioprotective effect
in vivo. Thus, the present study provided evidence that SS31
may protect the heart from DOX-induced toxicity in vitro and
in vivo.
The present study further assessed the underlying
mechanism of the effects of SS31 in ameliorating DOX-induced
toxicity in H9c2 cells. As a member of the MAPK family, p38 MAPK is
susceptible to various stimuli, such as oxidative stress,
inflammatory cytokines, growth factors or high glucose levels. The
canonical pathway of p38 MAPK activation is phosphorylation on a
threonine and a tyrosine residue of the activation loop mediated by
MAP2K (43). In cardiomyocytes,
a previous study has demonstrated that the phosphorylation of p38
is triggered by hypoxia and exogenous H2O2
treatment (44). In the present
study, DOX treatment significantly increased the phosphorylation of
p38 MAPK compared with that in the control cells, whereas
administration of SS31 inhibited this trend in H9c2 cells. Notably,
SS31 treatment exerted no significant effects on the expression and
phosphorylation levels of ERK1/2 and JNK. A previous study has
reported that SS31 peptide eliminates high glucose-induced
mitochondrial oxidative stress by regulating the p38 MAPK signal
pathway, which may be a novel therapeutic strategy to prevent
hyperglycemia-induced neuronal perturbation (45). Another study has demonstrated
that SS31 reduces the effects of sepsis-induced heart injury by
inhibiting the p38 MAPK signaling pathway (12). The results of these studies were
consistent with those of the present study. Therefore, SS31 may be
a potent antioxidant peptide with a capacity for reducing ROS and
inhibiting apoptosis, which may be associated with the inactivation
of the p38 MAPK signaling pathway.
A previous study has mainly focused on the mechanism
of DOX-induced cardiotoxicity and reported that high levels of
mitochondrial ROS production are required for DOX-induced cardiac
damage, and that the p38 MAPK signaling pathway is involved in
DOX-induced cardiotoxicity (46). However, the aforementioned study
has failed to provide the mechanism by which SS31 affects
DOX-induced cardiomyocyte apoptosis. The present study demonstrated
that SS31 may ameliorate DOX-induced ROS production and apoptosis
in cardiomyocytes by inhibiting the activation of the p38 MAPK
signaling pathway. To the best of the author's knowledge, this is
the first report on the effects of SS31 on mitochondrial function
describing the underlying mechanism to date.
Although the present study illustrated the
anticardiotoxic effects of SS31 in DOX-induced injury models in
vitro and in vivo, the study had certain limitations. It
is necessary to evaluate the function of SS31 in other types of
cells derived from heart, such as primary neonatal rat myocardial
cells and AC16 cardiomyocytes. In addition, whether different types
of modification affect the function of SS31 remains to be further
verified. Therefore, our future studies will involve evaluating the
effects of SS31 in multiple types of cardiomyocytes and performing
modifications of SS31 to verify its cardioprotective function.
In summary, the results of the present study
demonstrated that the mitochondria-targeted antioxidant peptide
SS31 suppressed the generation of ROS and apoptosis by inhibiting
the phosphorylation of p38 MAPK in DOX-treated H9c2 cells. In
vivo, cotreatment with SS31 improved cardiac function and
suppressed the occurrence of myocardial fibrosis induced by DOX
compared with those in mice treated with DOX alone. Therefore, the
results of the present study may provide a potential candidate
molecule for the treatment of DOX-induced cardiotoxicity.
Supplementary Data
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ and MF performed the experiments and wrote the
manuscript. XW and HZ performed the cell experiments and
participated in drafting the manuscript. JD performed a part of the
in vivo study and participated in drafting the manuscript.
LQ and ZC conceived, designed and supervised the project, and
revised the manuscript. LZ and MF confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were carried out in
accordance with the Guide for the Care and Use of Laboratory and
approved by the Institutional Animal Care and Use Committee of
Nanjing Medical University (approval no. IACUC-1903030; Nanjing,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Martins-Teixeira MB and Carvalho I:
Antitumour anthracyclines: Progress and perspectives. ChemMedChem.
15:933–948. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ji X, Ding W, Xu T, Zheng X, Zhang J, Liu
M, Liu G and Wang J: MicroRNA-31-5p attenuates doxorubicin-induced
cardiotoxicity via quaking and circular RNA Pan3. J Mol Cell
Cardiol. 140:56–67. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kalyanaraman B: Teaching the basics of the
mechanism of doxorubicin-induced cardiotoxicity: Have we been
barking up the wrong tree? Redox Biol. 29:1013942020. View Article : Google Scholar
|
|
4
|
Li DL and Hill JA: Cardiomyocyte autophagy
and cancer chemotherapy. J Mol Cell Cardiol. 71:54–61. 2014.
View Article : Google Scholar
|
|
5
|
Osataphan N, Phrommintikul A, Chattipakorn
SC and Chattipakorn N: Effects of doxorubicin-induced
cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial
function: Insights for future interventions. J Cell Mol Med.
24:6534–6557. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: From molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang AJ, Zhang J, Xiao M, Wang S, Wang BJ,
Guo Y, Tang Y and Gu J: Molecular mechanisms of doxorubicin-induced
cardiotoxicity: Novel roles of sirtuin 1-mediated signaling
pathways. Cell Mol Life Sci. Jan 13–2021.Epub ahead of print.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tahover E, Segal A, Isacson R, Rosengarten
O, Grenader T, Gips M, Cherny N, Heching NI, Mesika L, Catane R and
Gabizon A: Dexrazoxane added to doxorubicin-based adjuvant
chemotherapy of breast cancer: A retrospective cohort study with a
comparative analysis of toxicity and survival. Anticancer Drugs.
28:787–794. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ajith TA and Jayakumar TG:
Mitochondria-targeted agents: Future perspectives of mitochondrial
pharmaceutics in cardiovascular diseases. World J Cardiol.
6:1091–1099. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee FY, Shao PL, Wallace CG, Chua S, Sung
PH, Ko SF, Chai HT, Chung SY, Chen KH, Lu HI, et al: Combined
therapy with SS31 and mitochondria mitigates myocardial
ischemia-reperfusion injury in rats. Int J Mol Sci. 19:27822018.
View Article : Google Scholar :
|
|
11
|
Dai DF, Hsieh EJ, Chen T, Menendez LG,
Basisty NB, Tsai L, Beyer RP, Crispin DA, Shulman NJ, Szeto HH, et
al: Global proteomics and pathway analysis of
pressure-overload-induced heart failure and its attenuation by
mitochondrial-targeted peptides. Circ Heart Fail. 6:1067–1076.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Y, Yang W, Sun X, Xie L, Yang Y, Sang
M and Jiao R: SS31 ameliorates sepsis-induced heart injury by
inhibiting oxidative stress and inflammation. Inflammation.
42:2170–2180. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dai DF, Chen T, Szeto H, Nieves-Cintron M,
Kutyavin V, Santana LF and Rabinovitch PS: Mitochondrial targeted
antioxidant Peptide ameliorates hypertensive cardiomyopathy. J Am
Coll Cardiol. 58:73–82. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yue J and Lopez JM: Understanding MAPK
signaling pathways in apoptosis. Int J Mol Sci. 21:23462020.
View Article : Google Scholar :
|
|
15
|
Yue TL, Wang C, Gu JL, Ma XL, Kumar S, Lee
JC, Feuerstein GZ, Thomas H, Maleeff B and Ohlstein EH: Inhibition
of extracellular signal-regulated kinase enhances
ischemia/reoxygenation-induced apoptosis in cultured cardiac
myocytes and exaggerates reperfusion injury in isolated perfused
heart. Circ Res. 86:692–699. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang N, Guan P, Zhang JP, Li YQ, Chang YZ,
Shi ZH, Wang FY and Chu L: Fasudil hydrochloride hydrate, a
Rho-kinase inhibitor, suppresses isoproterenol-induced heart
failure in rats via JNK and ERK1/2 pathways. J Cell Biochem.
112:1920–1929. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L,
Reiter RJ, Qiao S and Yuan J: AMPK/PGC1a activation by melatonin
attenuates acute doxorubicin cardiotoxicity via alleviating
mitochondrial oxidative damage and apoptosis. Free Radic Biol Med.
129:59–72. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Krais JJ and Johnson N: Ectopic RNF168
expression promotes break-induced replication-like DNA synthesis at
stalled replication forks. Nucleic Acids Res. 48:4298–4308. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fan J, Shen W, Lee SR, Mathai AE, Zhang R,
Xu G and Gillies MC: Targeting the Notch and TGF-ß signaling
pathways to prevent retinal fibrosis in vitro and in vivo.
Theranostics. 10:7956–7973. 2020. View Article : Google Scholar :
|
|
20
|
National Research Council (US): Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington, DC: 2011
|
|
21
|
Oh J, Lee BS, Lim G, Lim H, Lee CJ, Park
S, Lee SH, Chung JH and Kang SM: Atorvastatin protects
cardiomyocyte from doxorubicin toxicity by modulating survivin
expression through FOXO1 inhibition. J Mol Cell Cardiol.
138:244–255. 2020. View Article : Google Scholar
|
|
22
|
Zhang L, Wang X, Feng M, Zhang H, Xu J,
Ding J, Cheng Z and Qian L: Peptidomics analysis reveals peptide
PDCryab1 inhibits doxorubicin-induced cardiotoxicity. Oxid Med Cell
Longev. 2020:71824282020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang L, Tu Y, Lu J, Wang P, Guo Z, Wang
Q, Guo K, Lan R, Li H and Liu P: Dkk1 exacerbates
doxorubicin-induced cardiotoxicity by inhibiting the Wnt/ß-catenin
signaling pathway. J Cell Sci. 132:cs2284782019. View Article : Google Scholar
|
|
24
|
Rochette L, Guenancia C, Gudjoncik A,
Hachet O, Zeller M, Cottin Y and Vergely C:
Anthracyclines/trastuzumab: New aspects of cardiotoxicity and
molecular mechanisms. Trends Pharmacol Sci. 36:326–348. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hantson P: Mechanisms of toxic
cardiomyopathy. Clin Toxicol (Phila). 57:1–9. 2019. View Article : Google Scholar
|
|
26
|
Zhao L, Qi Y, Xu L, Tao X, Han X, Yin L
and Peng J: MicroRNA-140-5p aggravates doxorubicin-induced
cardiotoxicity by promoting myocardial oxidative stress via
targeting Nrf2 and Sirt2. Redox Biol. 15:284–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang P, Chen Z, Lu D, Wu Y, Fan M, Qian J
and Ge J: Overexpression of COX5A protects H9c2 cells against
doxorubicin-induced cardiotoxicity. Biochem Biophys Res Commun.
524:43–49. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kim HS, Lee YS and Kim DK: Doxorubicin
exerts cytotoxic effects through cell cycle arrest and Fas-mediated
cell death. Pharmacology. 84:300–309. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu J, Gao H, Wu C, Xu QM, Lu JJ and Chen
X: Diethyl blechnic, a novel natural product isolated from salvia
miltiorrhiza bunge, inhibits doxorubicin-induced apoptosis by
inhibiting ROS and activating JNK1/2. Int J Mol Sci. 19:18092018.
View Article : Google Scholar :
|
|
30
|
Varela-Lopez A, Battino M, Navarro-Hortal
MD, Giampieri F, Forbes-Hernandez TY, Romero-Marquez JM, Collado R
and Quiles JL: An update on the mechanisms related to cell death
and toxicity of doxorubicin and the protective role of nutrients.
Food Chem Toxicol. 134:1108342019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gorini S, De Angelis A, Berrino L, Malara
N, Rosano G and Ferraro E: Chemotherapeutic drugs and mitochondrial
dysfunction: Focus on doxorubicin, trastuzumab, and sunitinib. Oxid
Med Cell Longev. 2018:75827302018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Mitochondrial oxidative stress: Implications for cell death. Annu
Rev Pharmacol Toxicol. 47:143–183. 2007. View Article : Google Scholar
|
|
33
|
Wallace KB, Sardao VA and Oliveira PJ:
Mitochondrial determinants of doxorubicin-induced cardiomyopathy.
Circ Res. 126:926–941. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Mitchell W, Ng EA, Tamucci JD, Boyd KJ,
Sathappa M, Coscia A, Pan M, Han X, Eddy NA, May ER, et al: The
mitochondria-targeted peptide SS-31 binds lipid bilayers and
modulates surface electrostatics as a key component of its
mechanism of action. J Biol Chem. 295:7452–7469. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hou Y, Shi Y, Han B, Liu X, Qiao X, Qi Y
and Wang L: The antioxidant peptide SS31 prevents oxidative stress,
downregu- lates CD36 and improves renal function in diabetic
nephropathy. Nephrol Dial Transplant. 33:1908–1918. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Reddy PH, Manczak M, Yin X and Reddy AP:
Synergistic protective effects of mitochondrial division inhibitor
1 and mito- chondria-targeted small peptide SS31 in Alzheimer's
disease. J Alzheimers Dis. 62:1549–1565. 2018. View Article : Google Scholar
|
|
37
|
Zhou J, Li Z, Chen Z and Yang K:
Protective effect of mitochondria-targeted antioxidant SS31 on
early brain injury following subarachnoid hemorrhage in rats. Zhong
Nan Da Xue Xue Bao Yi Xue Ban. 42:1003–1009. 2017.In Chinese.
PubMed/NCBI
|
|
38
|
Yin X, Manczak M and Reddy PH:
Mitochondria-targeted molecules MitoQ and SS31 reduce mutant
huntingtin-induced mitochondrial toxicity and synaptic damage in
Huntington's disease. Hum Mol Genet. 25:1739–1753. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ma H, Chen S, Xiong H, Wang M, Hang W, Zhu
X, Zheng Y, Ge B, Li R and Cui H: Astaxanthin from Haematococcus
pluvialis ameliorates the chemotherapeutic drug (doxorubicin)
induced liver injury through the Keap1/Nrf2/HO-1 pathway in mice.
Food Funct. 11:4659–4671. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Vu M, Kassouf N, Ofili R, Lund T, Bell C
and Appiah S: Doxorubicin selectively induces apoptosis through the
inhibition of a novel isoform of Bcl2 in acute myeloid leukaemia
MOLM13 cells with reduced Beclin 1 expression. Int J Oncol.
57:113–121. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Faridvand Y, Haddadi P, Vahedian V, Nozari
S, Nejabati HR, Pezeshkian M, Afrasiabi A, Safaie N, Jodati A and
Nouri M: Human amnion membrane proteins prevent doxorubicin-
induced oxidative stress injury and apoptosis in rat H9c2
cardiomyocytes. Cardiovasc Toxicol. 20:370–379. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu YZ, Zhang L, Wu ZX, Shan TT and Xiong
C: Berberine ameliorates doxorubicin-induced cardiotoxicity via a
SIRT1/p66Shc-mediated pathway. Oxid Med Cell Longev.
2019:21503942019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kulisz A, Chen N, Chandel NS, Shao Z and
Schumacker PT: Mitochondrial ROS initiate phosphorylation of p38
MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell
Mol Physiol. 282:L1324–L1329. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cao M, Jiang J, Du Y and Yan P:
Mitochondria-targeted antioxidant attenuates high glucose-induced
P38 MAPK pathway activation in human neuroblastoma cells. Mol Med
Rep. 5:929–934. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guo Z, Tang N, Liu FY, Yang Z, Ma SQ, An
P, Wu HM, Fan D and Tang QZ: TLR9 deficiency alleviates
doxorubicin- induced cardiotoxicity via the regulation of
autophagy. J Cell Mol Med. 24:10913–10923. 2020. View Article : Google Scholar : PubMed/NCBI
|