Introduction
Pancreatic cancer is the seventh most common type of
cancer worldwide. However, with only 6% successfully treated cases,
it places fourth according to mortality rates (1). Delayed diagnosis and resistance to
chemotherapy are the main causes of such a poor outcome. Early
pancreatic cancer detection is limited by an asymptomatic disease
and the lack of reliable diagnostic biomarkers. Locally advanced
pancreatic tumors (30-40% of cases) can be surgically resected, and
the procedure is often combined with chemo- or radiotherapy.
However, it rarely leads to full tumor eradication (2). Metastatic pancreatic cancer has the
worst prognosis, as it cannot be surgically removed and is highly
resistant to traditional pancreatic cancer chemotherapeutic drugs,
such as gemcitabine. Although several novel combination therapies
(gemcitabine + nab-paclitaxel, gemcitabine + FOLFIRINOX) have been
proposed in recent years (3,4),
advanced pancreatic cancer still remains highly lethal. It is
estimated that by the year 2025, pancreatic cancer may even reach
the top three of the most lethal cancer types in Europe (5).
In 2012, a novel iron-dependent cell death form,
ferroptosis, was identified (6).
Cells that undergo ferroptosis die from excessive membrane lipid
peroxidation. Fenton reactions and enzymes that use iron as a
cofactor, such as lipoxygenases constantly generate lipid peroxides
as a part of a normal cellular homeostasis. To prevent membrane
damage, cells activate antioxidant enzyme glutathione peroxidase 4
(GPX4). The majority of ferroptosis inducers inhibit the activity
or expression of GPX4, which leads to an oxidative membrane damage
and eventual cell death. In recent years, ferroptosis has been
researched in the context of cardiovascular and neurodegenerative
diseases, as well as tissue injury and cancer (7). Of note, as cancer cells have more
soluble iron in their cytosol than their normal counterparts, they
require a milder oxidative stimulus to induce ferroptosis. Thus,
ferroptosis can be specifically targeted to malignant cells,
particularly in iron-addicted cancers (8). Apart from an upregulated iron
metabolism, other factors can also predetermine sensitivity to
ferroptosis. For example, in pancreatic cancer, susceptibility to
ferroptosis is associated with constitutive KRAS activation
(9). Indeed, the first known
ferroptosis inducers, erastin and RSL3, were originally discovered
as compounds selectively lethal to RAS-mutated cancer cells.
Moreover, it has been shown that therapy-resistant and metastatic
cancer cells are particularly sensitive to ferroptosis induction
(10,11). The vulnerability of metastatic
cancer to ferroptosis may be attributed to an increased
polyunsaturated fatty acid content in cell membranes, which renders
them an easy target of oxidation and elicits dependency on GPX4
(12,13). In addition, resistance to
ferroptosis inducers is coupled with metabolic reprograming and
glutamine/glucose dependency (14). Pancreatic cancer also falls into
a broad category of glutamine-dependent cancer (15).
The question why tumors metastasize has not yet been
fully answered. Metastasis does not appear to be predetermined
genetically, and although certain genetic mutations promote
metastasis, environmental factors play a major role (16). From an evolutionary perspective,
it can be hypothesized that cells increase their motility in order
to migrate towards a more favorable growth environment (for
example, more nutrient-rich). Such behavior is common for bacteria
and certain primitive eukaryotes (17,18). However, the atavistic point of
view does not necessarily fit complex systems, as tumors can
develop adaptive mechanisms. When starved, cancer cells use
alternative energy sources, micropinocytosis, metabolic symbiosis,
autophagy, increase nutrient supply via angiogenesis and vessel
co-option (19-25). Nevertheless, several studies
suggest that a dysregulated microenvironment, such as extensive
cell death, pH changes due to the Warburg effect, signals from
stromal and immune cells can induce and enhance metastasis
(26-30). From this point of view, nutrient
and growth factor deprivation are also metastatic triggers.
Moreover, metastasis itself can be viewed as a form of adaptation.
In recent years, efforts have been put forth into developing
therapies that collectively exploit metabolic vulnerabilities of
metastatic cancer. However, care should be taken to avoid an
adaptive response, which would elicit an opposing, cancer-promoting
result.
The present study unveils novel (to the best of our
knowledge) molecular mechanisms through which starvation mediates
resistance to ferroptosis in pancreatic cancer cells and proposes
ferroptosis modulation strategies with which to improve pancreatic
cancer treatment.
Materials and methods
Cell culture and treatments
In the present study, five human ductal
adenocarcinoma cell lines were used: Miapaca2, Panc-1, Su.86.86,
T3M4 and Capan-26. The Miapaca2 cell line was a kind gift from Dr
Vitalijus Karabanovas (Biomedical Physics Laboratory, National
Cancer Institute, Vilnius, Lithuania). The Panc-1, Su.86.86 and
T3M4 cells were gifts from Dr Arvydas Kanopka (Department of
Immunology and Cell Biology, Institute of Biotechnology, Life
Sciences Center, Vilnius, Lithuania). The Capan-26 cell line was
previously established in the authors' laboratory using an explant
method and characterized by assessing the doubling time, tumor and
stem cell marker expression, colony forming efficiency, mutations
of the KRAS and TP53 genes, karyotype and sensitivity
to drug treatment (31). To
summarize the main findings, Capan-26 expresses the pancreatic
cancer markers, CEA cell adhesion molecule 6 and carbohydrate
antigen 19-9, the epithelial marker, E-cadherin, as well as the
stem cell markers, CD44, octamer-binding transcription factor 4 and
zinc finger E-box binding homeobox 1. The cells successfully form
colonies in soft agar. Additionally, Capan-26 bears a deletion of
KRAS exon 3 and a Val172Phe point mutation V172F in
TP53 exon 5. It is a mixed aneuploid/polyploid population.
All cell lines tested negative for mycoplasma.
The Miapaca2, Panc-1, Su.86.86 and T3M4 cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.),
supplemented with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.)
and 1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific,
Inc.). The Capan-26 cells were cultured in Iscove's modified
Dulbecco's medium (Gibco Thermo Fisher Scientific, Inc.) with 10%
FBS and 1% penicillin/streptomycin. Cells were grown at 37°C in a
humidified atmosphere with 5% CO2. For all experiments,
cells were seeded at the following densities: Miapaca2,
8×104 cells/ml; Panc-1 and Su.86.86, 7×104
cells/ml; T3M4, 9×104 cells/ml. The Capan-26 cells were
split 3-4 times.
EGFR inhibitor, afatinib (Afa, 1 µM), FGFR
inhibitor, BGJ398 (BG, 1 µM), ferroptosis inducer, erastin
(Era, 0.4-20 µM), the ferroptosis inhibitor, ferrostatin-1
(Ferr-1, 1 µM), the dual mTORC1/mTORC2 inhibitor, INK128
(INK, 0.125 nM), the ERK1/2 inhibitor, SCH772984 (SCH, 1
µM), the FAK inhibitor, PF573228 (PF, 2 µM), the Src
inhibitor, saracatinib (Sar, 2 µM), the YAP antagonist,
super-TDU (TDU, 0.2 µM), the mammalian sterile 20-like
kinase (MST)1/2 inhibitor, XMU-MP-1 (XMU, 5 µM), the
glycogen synthase kinase (GSK)-3 inhibitor, tideglusib (TDG, 5
µM), and the IKK inhibitor, TPCA (5 µM), were
purchased from Selleck Chemicals. The tankyrase inhibitor, XAV939
(XAV, 5 µM), the JNK inhibitor, SP600125 (SP6, 5 µM),
and the Unc-51 like autophagy activating kinase 1 (ULK-1)
inhibitor, SBI-0206965 (SBI, 1 µM), were purchased from
MilliporeSigma. The mTORC1 inhibitor, rapamycin (Rap, 0.25 nM), was
obtained from Santa Cruz Biotechnology, Inc. The caspase inhibitor,
Z-VAD-FMK (ZVAD, 1 µM), was purchased from R&D Systems,
Inc. The RIPK1 inhibitor, necrostatin-1 (Nec-1, 1 µM), was
purchased from Alfa Aesar. All compounds were dissolved in dimethyl
sulfoxide (DMSO), stored at -20°C and diluted in growth medium to
their final concentrations immediately prior to use. An appropriate
volume of DMSO was used as a vehicle in control cells.
The EGF and bFGF (100 ng/ml) recombinant proteins
were purchased from Invitrogen and stored according to
manufacturer's instructions.
Cell viability assessment
Cell viability was evaluated using propidium iodide
(PI) staining. The day before treatment, the cells were seeded into
48-well plates (SigmaAldrich; Merck KGaA). Combined treatments were
administered sequentially: First, the cells were treated with
inhibitors for 1 h, and erastin was then added to the culture
medium. Overall, the cells were exposed to the treatments for 48 h.
The substratum-bound and detached cells were then collected,
centrifuged at 600 × g for 3 min at room temperature, suspended in
PBS and stained with 0.5 µg/ml PI for 5-10 min. The
proportion of cells with a permeabilized membrane was determined
using the Guava easyCyte 8HT flow cytometer (MilliporeSigma). If
the proportion of apoptotic (caspase-3/7-positive) cells was to be
evaluated simultaneously, the cells were also stained with
Caspase-3/7 Green detection reagent (Invitrogen; Thermo Fisher
Scientific, Inc.; 0.5 µM; 37°C for 30 min). The data were
analyzed using Flowing Software 2.5.1 (University of Turku, Turku,
Finland).
Detection of membrane lipid oxidation
using C11-BODIPY 581/591 staining
To detect cells with oxidized membrane lipids, the
cells were starved by culturing without FBS, without amino acids
L-glutamine, L-lysine and L-arginine or treated with Rap and
simultaneously exposed to erastin and/or Ferr-1, then stained with
0.5 µM C11 BODIPY 581/591 fluorescent probe (Invitrogen;
Thermo Fisher Scientific, Inc.) for 30 min at room temperature. The
cells were then collected, centrifuged at 600 × g for 3 min at room
temperature, resuspended in PBS and analyzed on the Guava easyCyte
8HT flow cytometer (MilliporeSigma). Data analysis was performed
using Flowing Software 2.5.1 (University of Turku).
Oxidized membrane lipids in Capan-26 cells were
visualized using confocal microscopy. Briefly, the cells were
seeded on glass coverslips, stained with 10 µM C11 BODIPY
581/591 for 30 min at 37°C, washed with PBS, mounted in Prolong
Gold Antifade reagent (Molecular Probes; Invitrogen; Thermo Fisher
Scientific, Inc.) and observed immediately using a confocal laser
scanning microscope (Eclipse TE2000-S; Nikon Corporation).
Fluorescence/confocal microscopy
For immunofluorescence experiments, the cells were
seeded on glass coverslips in 24-well plates (MilliporeSigma).
After 24 h, the cells were treated with the corresponding
compounds, washed twice with PBS and fixed with 4% paraformaldehyde
(15 min at room temperature). The cells were then washed three
times with 1% BSA (MilliporeSigma) in PBS and permeabilized with
0.2% of Triton X-100 (MilliporeSigma) in PBS (15 min at room
temperature). After washing, the non-specific binding sites were
blocked by incubating with 1% BSA in PBS at room temperature for 30
min. The coverslips were then stained with the following primary
antibodies for 1 h at 37°C: Rabbit anti-ERK1/2 (1:100; produced in
the authors' laboratory by rabbit immunization with a recombinant
protein), rabbit anti-phospho-JNK (1:400; cat. no. V7931; Promega
Corporation), rat anti-E-cadherin (1:400; cat. no. 13-1900; Thermo
Fisher Scientific, Inc.), followed by washing with 1% BSA in PBS
five times and 30 min of incubation at 37°C with Alexa Fluor™
594-conjugated anti-rabbit (H+L) or Alexa Fluor™ 594-conjugated
anti-rat (H+L) secondary antibodies (both 1:250; cat. nos. A32740
and A-11007, respectively; Thermo Fisher Scientific, Inc.). Cell
nuclei were stained with 300 nM 4′,6-diamidino-2-phenylindole
dihydrochloride (DAPI) dye (Thermo Fisher Scientific, Inc.) for 10
min at room temperature. After washing, the coverslips were mounted
in Prolong Gold antifade (Molecular Probes; Invitrogen) and
observed using a confocal laser scanning microscope (Eclipse
TE2000-S; Nikon Corporation). Fluorescence intensity in the cell
nuclei was quantified using ImageJ 1.52v software (National
Institutes of Health).
Western blot analysis
The cells were washed with PBS and lysed on ice in
EB++ lysis buffer (extraction buffer; 10 mM Tris-HCl, pH 7.4, 1 mM
Tris base, 50 mM NaCl, 50 mM NaF, 1% Triton X-100, 5 mM EDTA, 2 mM
Na3VO4 and 1 mM PMSF; pH 7,2-7,4). The cell
lysates were centrifuged 20,000 × g for 15 min at 4°C. The protein
concentration was quantified as previously described (32). Protein samples were subjected to
12% SDS-PAGE, transferred to polyvinylidene difluoride membranes
(Bio-Rad Laboratories, Inc.) by wet transfer and blocked in
blocking buffer containing 1% milk powder (45 min, room
temperature). The membranes were then incubated with primary mouse
anti-vimentin (1:2,000; cat. no. 550513; BD Pharmingen™) and mouse
anti-YAP1 (1:500; cat. no. sc-101199; Santa Cruz Biotechnology,
Inc.) antibodies for 2 h at room temperature. In addition, the
blots were probed with mouse anti-GAPDH antibody (1:1,000; cat. no.
AM4300; Thermo Fisher Scientific, Inc.) for the detection of GAPDH
as a loading control. After washing four times for 5 min at room
temperature with 0.1% Tween-20 (Carl Roth Gmbh & Co. Kg) in PBS
(PBS-T), membrane-bound primary antibodies were probed with
IRDye® 800CW Infrared dye conjugated secondary goat
anti-mouse antibody (1:10,000; cat. no. 926-32210; LI-COR
Biosciences) for 30 min at room temperature. After washing again
for four times with PBS-T and once with PBS, the membranes were
scanned on an Odyssey® Infrared Imaging System (LI-COR
Biosciences). Densitometric analysis was performed using ImageJ
1.52v software (National Institutes of Health).
Wound healing assay
The cells were seeded in 24-well plates
(MilliporeSigma) and cultured until they reached confluency. In one
group, a scratch was made using a sterile 10 µl pipette tip,
and the cells were washed with PBS and supplemented with fresh
medium without FBS. In the other group, cells were FBS-starved for
an additional 4 days, and a scratch was then made. In both cases,
images of the same three fields were captured using an Eclipse
TE2000-S microscope (Nikon Corporation; magnification, ×10) at 0
and 48 h after scratching. The width of the healed wound was
calculated using the ImageJ 1.52v plugin MRI Wound Healing Tool
(National Institutes of Health).
Glutathione (GSH) assay
GSH levels were determined in the cells using
Ellman's reagent (5,5-dithio-bis-(2-nitrobenzoic acid) (DTNB;
MilliporeSigma). The cells were collected, centrifuged at 400 × g
for 4 min at room temperature and resuspended in PBS. Subsequently,
one-tenth of the cell suspension was used for cell counting using a
Guava easy-Cyte 8HT flow cytometer (MilliporeSigma). The remaining
cells were centrifuged again at the same conditions as in the
previous step and resuspended in 20 µl working buffer (100
mM Tris-HCl; 1 mM EDTA). To precipitate proteins, 20 µl 5%
trichloroacetic acid were added, and the samples were incubated on
ice at 4°C for 15 min. Acid was neutralized with 5 µl 2 M
Tris base. The samples were then centrifuged at 12,000 × g for 5
min at room temperature, and 1.5 mM of DTNB was added to the
supernatant. The absorbance was measured at 412 nm with Varioskan
Flash Multimode Reader (Thermo Fisher Scientific, Inc.). The GSH
level was calculated according to the calibration curve prepared
with pure GSH (Carl Roth Gmbh & Co. Kg) and normalized to the
cell number in each sample.
Statistical analysis
Microsoft Office Excel (v16.0) software was used for
statistical analysis. The data are presented as the mean ± standard
deviation from at least three independent assays, each one at least
in duplicate. The unpaired Student's t-test was used to compare two
groups. Multiple comparisons were performed using Tukey post-hoc
test, following one-way ANOVA. P<0.05 was considered to indicate
a statistically significant difference.
Results
Starved pancreatic cancer cells react
differently to ferroptosis induction
In eukaryotic cells, the main sensor of
environmental conditions is mTOR (33). As mTOR signaling mediates both
adaptation to nutrient/growth factor deprivation and oxidative
stress, the interplay between pharmacological mTOR inhibition and
sensitivity to erastin-induced ferroptosis was first analyzed. Cell
viability following erastin treatment was examined in a panel of
pancreatic cancer cell lines cultured in standard medium in the
absence of FBS, L-glutamine, L-lysine and L-arginine (33) and pseudo-starved by exposing them
to the mTOR inhibitors, Rap and INK (Fig. 1A-D). Starvation elicited
contrasting responses in different cell lines. In the
mesenchymal-like Miapaca2 cells, FBS, amino acid starvation and
rapamycin treatment prevented cell death. By contrast, in the
epithelial-like Panc-1 and Su.86.86 cells, ferroptosis was strongly
induced. Pseudo-starvation induced by rapamycin exerted the most
prominent effect, whereas amino acid starvation slightly decreased
the viability of the control cells, although this was not due to
ferroptosis. The dual mTORC1/mTORC2 inhibitor, INK, on the other
hand, did not affect sensitivity to ferroptosis, apart from the
Panc-1 cells. Lipid peroxidation detection by C11 BODIPY staining
was also performed. Unless otherwise stated, for the Miapaca2 and
Panc-1 cell lines, the proportion of the oxidized population was
quantified (Fig. S1). For the
Su.86.86 and T3M4 cells, the medium fluorescence intensity was
analyzed. C11 BODIPY staining supported the cell viability results
(Fig. 1E-H). In the T3M4 cells,
FBS withdrawal increased the proportion of oxidized membrane
lipids, as well as cell death following erastin treatment. However,
Ferr-1 did not prevent erastin-induced T3M4 cell death and neither
did the inhibitors of apoptosis, necroptosis or autophagy (Fig. S2A). Erastin also failed to
elevate the proportion of apoptotic (caspase-3/7+) cells
(Fig. S2B). Such results
indicate a partial change in the cell death type following erastin
treatment and overall stronger ROS-adaptive mechanisms in T3M4
cells. Taken together, these data suggest the differential
regulation of the erastin-induced ferroptosis of starved pancreatic
cancer cells.
 | Figure 1Starved pancreatic cancer cells react
differently to ferroptosis induction. (A-D) Viability assessment of
(A) Miapaca2, (B) Panc-1, (C) Su.86.86 and (D) T3M4 cells cultured
without FBS, without the amino acids L-glutamine, L-lysine and
L-arginine or pseudo-starved using treatment with the mTOR
inhibitors rapamycin (0.25 nM) and INK (0.125 nM). (E-H)
Measurement of lipid peroxidation of (E) Miapaca2, (F) Panc-1, (G)
Su.86.86 and (H) T3M4 cells cultured without FBS, L-glutamine,
L-lysine and L-arginine and pseudo-starved using treatment with
rapamycin (0.25 nM). Ferroptosis was induced by erastin and
inhibited by Ferr-1. The data are presented as the mean ± SD; n=3.
Cells cultured under standard conditions were used as the control.
*P<0.05, **P<0.01 and
***P<0.001, vs. control. -AA, cells, cultured without
L-glutamine, L-lysine and L-arginine; Rap, rapamycin; INK, INK128;
Ferr-1, ferrostatin-1. |
Starvation regulates ferroptosis via
ERK1/2
The inability of the selective mTORC1/mTORC2
inhibitor, INK, to induce changes in pancreatic cancer cell
viability in contrast to the mTORC1 inhibitor, rapamycin, strongly
suggests the involvement of mTORC2-mediated feedback loops. One of
the possible mTORC2 targets is ERK1/2. Although Soares et al
(34) demonstrated that
prolonged incubation with rapamycin did not activate ERK1/2 in
pancreatic cancer cells, ERK1/2 translocated to the nucleus in
cells starved for a short period of time and/or treated with
erastin (Figs. 2 and S3). Rapamycin and erastin
synergistically promoted ERK1/2 translocation to the nucleus in the
Miapaca2, Panc-1, Su.86.86 and T3M4 cells. ERK1/2 also translocated
to the nucleus of FBS and amino acid-deprived control Panc-1 cells
(not treated with erastin); however, as erastin itself enhanced
ERK1/2 translocation, there was no significant difference in the
fluorescence intensity in the nucleus of the erastin-treated
control and starved cells. In the control Miapaca2 cells, FBS and
amino acid starvation also resulted in ERK1/2 translocation;
however, erastin did not enhance this effect. To further elucidate
the role of ERK1/2 in ferroptosis, this kinase was inhibited with
its inhibitor, SCH, and it was then examined whether sensitivity to
erastin was affected in starved pancreatic cancer cells (Fig. 3A-D). The results suggested that
ERK1/2 inhibition increased the viability of rapamycin-treated and
FBS-deprived Panc-1 and Su.86.86 cells. Moreover, SCH failed to
rescue the starved cells from lipid oxidation following exposure to
a higher erastin concentration (Figs. 3E-H and S4). In the Miapaca2 cells, SCH
treatment did not reverse starvation-induced erastin resistance
either. Collectively, these data indicate that ERK1/2 primes
starved Panc-1, Su.86.86 and T3M4 cells for erastin-induced
ferroptosis.
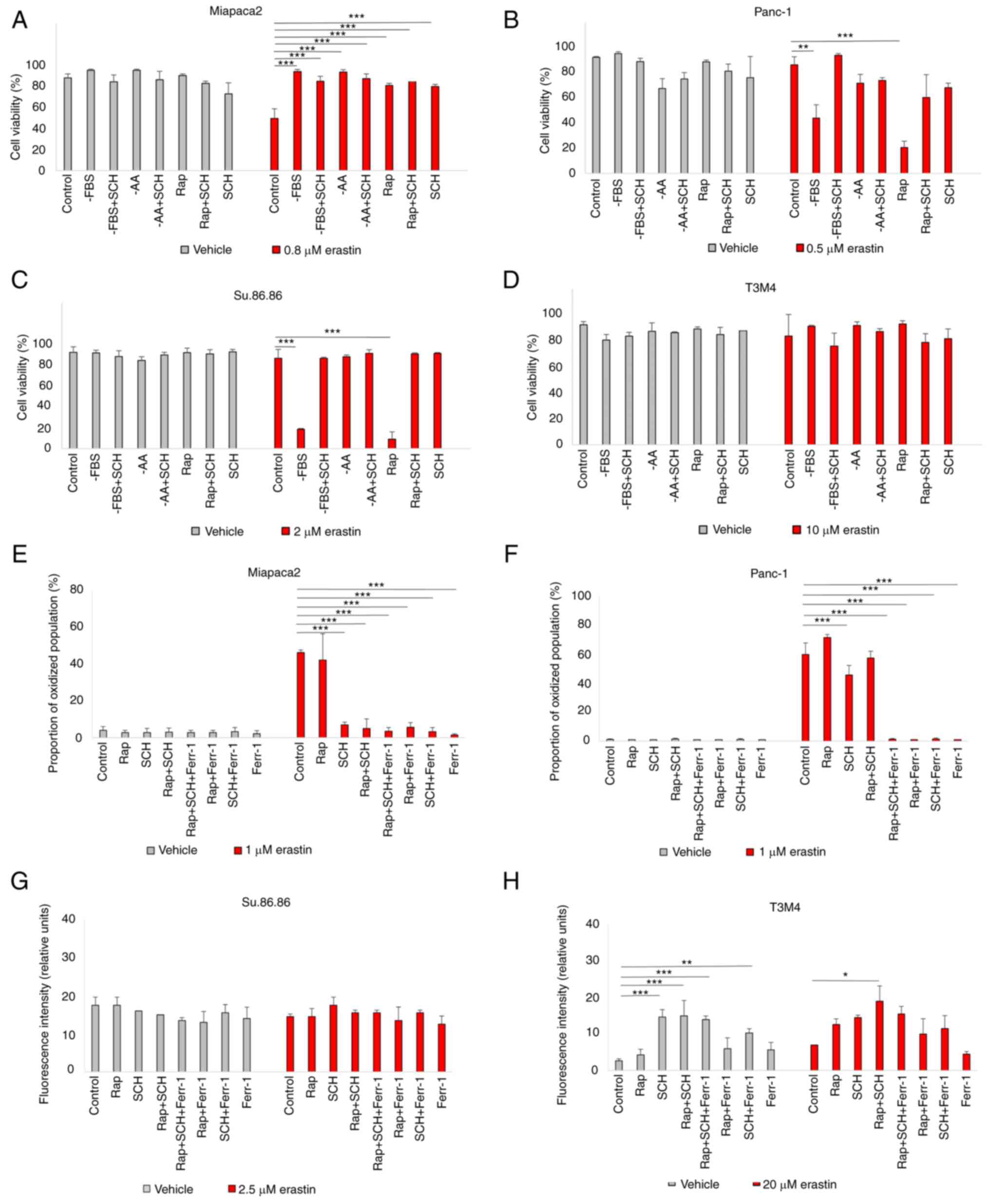 | Figure 3Starvation regulates ferroptosis via
ERK1/2. (A-D) Cell viability assessment of (A) Miapaca2, (B)
Panc-1, (C) Su.86.86 and (D) T3M4 cells cultured without FBS,
without amino acids L-glutamine, L-lysine and L-arginine,
pseudo-starved by treating with Rap and treated with the ERK1/2
inhibitor, SCH (1 µM). (E-H) Measurement of lipid
peroxidation of pseudo-starved (E) Miapaca2, (F) Panc-1, (G)
Su.86.86 and (H) T3M4 cells. Ferroptosis was induced by erastin and
inhibited by Ferr-1. The data are presented as the mean ± SD; n=3.
Cells cultured under standard conditions were used as the control.
*P<0.05, **P<0.01 and
***P<0.001, vs. control. -AA, cells, cultured without
L-glutamine, L-lysine and L-arginine; Rap, rapamycin; SCH,
SCH772984; Ferr-1, ferrostatin-1. |
Starvation regulates ferroptosis via
JNK
The c-Jun transcription factor is another regulator
of anti-oxidative stress responses and is activated by JNK. In the
present study, phospho-JNK immunofluorescence revealed that erastin
treatment activated JNK only in Miapaca2 cells (Fig. 4A). However, starvation alone
induced JNK phosphorylation in all four cell lines. JNK activation
was prominent following FBS and amino acid starvation and with
rapamycin treatment in the Miapaca2 and Su.86.86 cells, following
FBS withdrawal and rapamycin treatment in Panc-1 cells and
following FBS and amino acid starvation in T3M4 cells. Of note,
treatment with the JNK inhibitor, SP6, prevented the ferroptosis of
cells exposed to rapamycin (Fig.
4B-E). JNK inhibition reduced cell viability under standard
conditions only in Panc-1 cells, although its activation was not
observed in immunofluorescence experiments, which suggests delayed
JNK activation in these cells. These data highlight a novel role
for JNK in oxidative stress responses: In starved pancreatic cancer
cells, JNK activation elevates erastin-induced ferroptosis.
 | Figure 4Starvation regulates ferroptosis via
JNK. (A) Confocal microscopy images of phospho-JNK staining in
(pseudo)starved Miapaca2, Panc-1, Su.86.86 and T3M4 cells, with or
without erastin treatment. Arrowheads indicate phospho-JNK
accumulation in the cell membrane. Scale bar, 20 μm. (B-E) Cell
viability assessment of (B) Miapaca2, (C) Panc-1, (D) Su.86.86 and
(E) T3M4 cells, cultured without FBS, without amino acids
L-glutamine, L-lysine and L-arginine, pseudo-starved by treating
with rapamycin and treated with the JNK inhibitor, SP6 (5
µM). Ferroptosis was induced by erastin and inhibited by
Ferr-1. The data are presented as the mean ± SD; n=3. Cells
cultured under standard conditions were used as the control.
*P<0.05, **P<0.01 and
***P<0.001, vs. control. -AA, cells, cultured without
L-glutamine, L-lysine and L-arginine; Rap, rapamycin; SP6,
SP600125; Ferr-1, ferrostatin-1. |
Pancreatic cancer cells transition
between different mesenchymal states during FBS starvation
The results of the present study indicated that
starvation elicited opposing effects in Miapaca2 cells and in the
other tested cell lines, Panc-1, Su.86.86 and T3M4. When starved,
the Miapaca2 cells acquired resistance to erastin, rather than
sensitivity. Notably, starvation provoked changes in the Miapaca2
cell growth pattern and morphology: After growing without FBS for 2
days, the originally mesenchymal-like Miapaca2 cells formed
islands, characteristic of epithelial cells; however, after a 5-day
starvation period, they become spindle-shaped (Fig. 5A). Therefore, it was hypothesized
that these morphological changes may indicate transitioning between
epithelial and mesenchymal cell states. Indeed, wound healing
assays revealed that during the first 2 days of starvation, the
cells were about as twice less motile as during days 4-6 of
starvation (Fig. 5B and C).
Western blot analysis was used to examine the expression of the
mesenchymal markers, vimentin and YAP1, which confirmed that the
expression of vimentin and YAP1 was the lowest on day 2 of FBS
starvation, and the highest on day 5 (Figs. 5D and E, and S5A and B). No similar changes in
plasticity were observed in the Panc-1, Su.86.86 and T3M4 cell
lines, which are originally more epithelial-like in nature
(Fig. S6). However, the
Capan-26 cell line, which was established and characterized as an
epithelial pancreatic cancer cell line in the authors' laboratory,
exhibited similarities to the Miapaca2 cells. Indeed, after 3 days
of FBS starvation, the cells at the edge of the islands acquired a
mesenchymal shape (Fig. 5F) and
lost membrane E-cadherin expression (Fig. 5G). However, vimentin expression
did not increase during starvation, possibly because only a small
population of cells was affected (Fig. S5C). With respect to ferroptosis,
the Miapaca2 cells on day 2 of FBS starvation (most
epithelial-like) were about twice less sensitive to erastin than on
day 5 (most mesenchymal-like) (Fig.
6A). The Capan-26 cells were overall very resistant to erastin,
although erastin treatment following a 3-day starvation period
induced lipid oxidation (Fig. 6B and
C). However, Ferr-1 failed to reduce membrane oxidation. C11
BODIPY microscopy clearly identified oxidized lipids in the
membranes of mesenchymal-shaped cells at the edges of starved cell
islands, compared with a non-starved control (Fig. 6D). Together, these data indicate
that in Miapaca-2 and Capan-26 cells, starvation promotes
transitioning between mesenchymal and epithelial states, with the
mesenchymal state being more sensitive to ferroptosis induction
than the epithelial state.
Modulation of epithelial-to-mesenchymal
transition (EMT) can be used to increase pancreatic cancer cell
sensitivity to ferroptosis
The aforementioned results encouraged the
exploration of the possibilities of ferroptosis modulation in
EMT-prone mesenchymal-like pancreatic cancer cells. A combinatorial
cell viability analysis of erastin and EMT-targeting compounds in
FBS-starved and non-starved cells was performed. Firstly, the
EMT-inducing growth factors, EGF and bFGF, sensitized the Miapaca2
cells to erastin under standard conditions, and had no significant
effect on cells cultured in the absence of FBS for 2 days (most
epithelial-like state) (Fig.
7A). The sensitivity-inducing effect was completely abolished
by treating the cells with the EGFR and FGFR inhibitors, afatinib
and BGJ398, respectively. Secondly, two anti-EMT agents, the Src
inhibitor, Sar, and the functionally similar FAK inhibitor, PF,
were tested for their effects on ferroptosis. Sar completely
rescued the Miapaca2 cells from erastin-induced cell death, alone
or in combination with PF, although PF did not affect cell
viability (Fig. 7B). Finally,
other compounds, which are not conventional EMT-targeted drugs, but
modulate EMT-related signaling pathways, were also tested in the
Miapaca2, Panc-1, Su.86.86 and T3M4 cells: NF-κB (IKK inhibitor,
TPCA), Wnt (tankyrase inhibitor, XAV, and GSK-3 inhibitor, TDG) and
Hippo (MST 1/2 inhibitor, XMU) (Fig.
7C). Wnt and Hippo signaling inhibition increased cell
sensitivity to erastin, in most cases at a high erastin
concentration. The combination of erastin and TDG exerted the most
prominent cytotoxic effect under standard conditions in all cell
lines tested, even in T3M4 cells, which are overall resistant to
erastin. These data indicate that pancreatic cancer cell
sensitivity to ferroptosis can be modulated and promoted using
EMT-targeting compounds.
 | Figure 7EMT modulation increases pancreatic
cancer cell sensitivity to ferroptosis. (A) Miapaca2 cell viability
following combined treatment with erastin and EMT-inducing growth
factors, EGF and FGF (100 ng/ml). Afa and BG inhibited EGFR and
FGFR, respectively. (B) Miapaca2 cell viability following
simultaneous exposure to erastin and the EMT inhibitors, Sar and
PF. (C) Combinatorial cell viability analysis of other
EMT-targeting compounds and erastin in different pancreatic cancer
cell lines. Cells cultured under standard conditions were used as
the control.*P<0.05, **P<0.01 and
***P<0.001 vs. control (untreated) cells. EMT,
epithelial-to-mesenchymal transition; Era, erastin; FGF/bFGF; basic
fibroblast growth factor; Afa, afatinib; Ferr-1, ferrostatin-1; BG,
BGJ398; Sar, saracatinib; PF, PF573228; TPCA, TPCA-1; XAV, XAV939;
XMU, XMU-MP-1; TDG, tideglusib. |
Discussion
Oxidative stress lies in the nature of pancreatic
cancer, as known triggers of pancreatic carcinogenesis, such as
alcohol consumption and inflammation, generate ROS and promote
malignant lesions in the pancreas (35,36). Generally, there are two main
strategies to kill cells which thrive on accumulating ROS: To
deplete ROS or to elevate oxidative stress above the bearable
threshold, i.e., anti-oxidant or pro-oxidant cancer therapies.
Although both have been tested for pancreatic cancer, the present
study focused on the latter. In the present study, new strategies
with which to improve pancreatic cancer treatment were identified
by modulating ferroptosis, a unique cell death type based on
membrane lipid oxidation due to iron overload. Ferroptosis is
morphologically and biochemically distinct from apoptosis,
necroptosis or autophagy, and has gained considerable attention
since it was first identified. One of the most notable recent
findings is that ferroptosis induction inhibits pancreatic cancer
resistance to gemcitabine, a first-line pancreatic cancer drug
(37).
Sensitivity to ferroptosis is dependent on metabolic
rewiring, a common trait of pancreatic cancer, which can be
exemplified by glucose and glutamine dependence. The
cystine-glutamate antiporter system xc− mediates
cellular cystine import by exchanging one molecule of cystine for
glutamate. In the cell, cystine is rapidly reduced to cysteine and
used for glutathione biosynthesis. Cystine reduction involves
nicotinamide-adenine dinucleotide phosphate (NADPH), which is
generated from glucose via the pentose phosphate pathway. NADPH is
also used to reduce oxidized glutathione. Cells overexpressing
solute carrier family 7 member 11, a transporter component of the
xc− system, tend to be more resistant to ferroptosis
inducers due to elevated levels of reduced glutathione and GPX4
activity. However, to meet energy demands and supply sufficient
amounts of glutamate for the tricarboxylic acid cycle, cells need
to enhance glutamate import, commonly in the form of glutamine. In
this manner, ferroptosis resistance is coupled to glutamine and
glucose dependency (14).
Pancreatic cancer falls into a broad category of
glutamine-dependent cancers, although cells use different enzymes
to metabolize glutamine due to KRAS mediated reprogramming
(15). Thus, depleting cells
from nutrients, not exclusively glucose or glutamine, may have a
therapeutic benefit in pancreatic cancer, which is sensitive to
ferroptosis induction. This hypothesis led to the present
study.
The findings of the present study indicate that
treatments that induce or mimic starvation (growth factor and amino
acid withdrawal, together with exposure to rapamycin) elicit
contrasting effects in different pancreatic cancer cell lines.
Generally, Miapaca2 cells acquired resistance to ferroptosis, while
Panc-1, Su.86.86 and T3M4 cells became more sensitive. Some
previous findings shed light on oxidative stress resistance in
cells, encountering starvation. For example, it has previously been
reported that growth factor starvation induces a quiescent
phenotype and NF-κB activation in prostate cancer cells and
protects them from ROS-induced cell death, but not specifically
ferroptosis (38). Additionally,
recently, Lee et al (39)
demonstrated that energy stress inhibits ferroptosis in cancer
cells through AMPK activation and the acceleration of fatty acid
biosynthesis. However, in some cells encountering growth factor
deprivation, AMPK activation requires gradual ROS accumulation
(40). In contrast to the Panc-1
and Su.86.86 cells, the same time course of FBS and amino acid
starvation increased the amount of reduced glutathione in Miapaca2
cells, indicating slower antioxidant responses (Fig. S7), which explains, at least in
part, the differences in sensitivity. It has also been observed
that amino acid starvation exerted an anti-ferroptotic effect on
Miapaca2 cells. Sato et al (41) demonstrated that the deprivation
of lysine, arginine and other amino acids increased cystine import
and the expression of the components of cystine-glutamate
antiporter xc−, which could protect cells against
ferroptosis. Even more pronounced effects to erastin sensitivity
were observed in the present study following pseudo-starvation
induced by treating cells with rapamycin, but not with the dual
mTORC1/mTORC2 inhibitor INK128. This demonstrates the involvement
of mTORC2-mediated feedback loops, as prolonged incubation with
rapamycin inhibits S6K and thus, indirectly activates its
downstream target Rictor and consequently mTORC2. Gu et al
(42) indicated that active
mTORC2 diminished cystine import, while the knockout or
pharmacological inhibition of this protein exerted an opposite
effect. This mechanism supports the current observations in Panc-1,
Su.86.86 and T3M4 cells, in which rapamycin enhances erastin
cytotoxicity. ERK1/2 is an established downstream target of
rapamycin-induced mTORC2 feedback signaling. The results confirmed
ERK1/2 translocation to the cell nucleus after rapamycin and
erastin combined treatment in Miapaca2, Panc-1, Su.86.86 and T3M4
cells. Pharmacological ERK1/2 inhibition protected pancreatic
cancer cells from erastin-induced ferroptosis. In addition,
starvation-induced resistance to erastin in pancreatic cancer cells
was mediated by another kinase, JNK. Indeed, JNK was also activated
in pseudo-starved and erastin-treated pancreatic cancer cells. Of
note, the findings of the present study highlight a dual role of
JNK in oxidative stress response regulation. In Panc-1 cells under
standard conditions, JNK inhibition enhanced ROS-induced cell
death; however, starvation combined with JNK inhibition protected
these cells from erastin cytotoxicity. The antioxidant properties
of the JNK inhibitor, SP6, were also observed in Miapaca2 cells
even at nutrient-rich conditions. Although these results appear
paradoxical, some insights on a pro-oxidant JNK role have been
proposed; however, mainly in the context of the regulation of
mitochondria function in normal cell apoptosis (43-45). With respect to ferroptosis, it
has been shown that the activation of JNK and p38 elevates the
expression of NADPH oxidase 4 and enhances the ferroptosis of
pancreatic islet cells (46).
Moreover, Yang et al (47) recently reported that JNK
downregulated GPX4 and promoted the ferroptosis of colorectal
cancer cells (46). However,
none of these mechanisms is directly linked to cell metabolism. To
our knowledge, the present study is the first to report the
starvation-induced anti-ferroptotic role of JNK.
Nutrient withdrawal mimics conditions that cancer
cells face in larger tumors before neovascularization. Poor
perfusion is a common trait of pancreatic cancer tumors and it
impairs nutrient access to the deeper tumor layers (48). It is known that serum starvation
enhances metastatic properties of cancer cells (49). The results of the present study
demonstrated that FBS starvation induced changes in cell morphology
and epithelial and mesenchymal marker expression and/or increased
motility of two pancreatic cancer cell lines, Miapaca2 and
Capan-26. These changes were not observed in the pancreatic cancer
cell lines, Panc-1, Su.86 86 and T3M4. While starved, the Miapaca2
cells transitioned between mesenchymal-like (original) to
epithelial-like to mesenchymal states. The epithelial-like state
proved to be the most resistant to erastin. One possible
explanation may be that, in this case, mesenchymal properties and
possibly cell sensitivity are mediated by an autocrine stimulation
of unknown growth factors, which elicit their effect only when
their concentration is below the threshold; i.e., the concept of
dependence receptors (50). From
an evolutionary perspective, it may be hypothesized that gaining
more epithelial properties and enhancing cell-cell junctions could
help stromal cells to form a shield and protect epithelial cancer
(stem) cells from oxidative damage in pancreatic tumors.
Increased mesenchymal properties under FBS
starvation conditions sensitized the Miapaca2 cells to
erastin-induced ferroptosis and was associated with an increased
proportion of oxidized membrane lipids in Capan-26 cells. With this
in mind, several EMT-targeting compounds were evaluated for their
effects on cell sensitivity to erastin. As was expected, the
EMT-promoting growth factors, EGF and FGF, enhanced Miapaca2 cell
sensitivity to erastin, whereas blocking their receptors diminished
this effect. Furthermore, the classical anti-EMT drug Src
inhibitor, Sar, but not functionally related FAK inhibitor, PF,
prevented erastin-induced cell death. Lastly, the inhibitors of the
Wnt and Hippo signaling pathways (XAV and XMU, respectively)
enhanced erastin cytotoxicity. In some cases, a biphasic inhibitory
effect was observed: In Panc-1, Su.86.86 and T3M4 cells, the
tankyrase inhibitor, XAV, prevented cell death at a low erastin
concentration, but enhanced its cytotoxic effect at higher erastin
concentrations. The GSK-3 inhibitor, tideglusib, combined with
erastin, exerted the most prominent cytotoxic effect in all cell
lines tested, even in T3M4 cells, which are overall resistant to
erastin. From a mechanistic perspective, by inhibiting GSK-3,
tideglusib indirectly activates β-catenin, which can promote EMT by
blocking cell-cell junctions (51).
In recent years, cancer starvation therapies have
gained considerable levels of attention. Common strategies with
which to starve tumors include treatment with antiangiogenic
compounds, vascular blood supply disruption, direct decomposition
of intratumoral nutrients and treatment with agents that induce
pseudo-starvation, such as inhibitors of growth factor receptors
and the mTOR pathway (52-55). The combination of starvation and
pro-oxidant therapies has proven to be effective both in
vitro and in vivo (56). The present study demonstrated
that in pancreatic cancer cells, starvation mediated sensitivity to
ferroptosis via the ERK1/2 and JNK kinases and by inducing the
transition between epithelial and mesenchymal states. Therefore,
there may be some new avenues for further research, both in the
basic and the clinical sciences, regarding therapies that combine
kinase inhibitors, ferroptosis-inducing agents and common
anti-proliferative drugs.
Supplementary Data
Availability of data and materials
All data generated or analyzed during this study is
included in this published article or are available from the
corresponding author on reasonable request.
Authors' contributions
EZ conceptualized the study. EZ and JC designed the
experiments. EZ carried out the experiments. EZ and JC wrote the
manuscript and confirm the authenticity of all the raw data. Both
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors acknowledge financial support for
publishing the manuscript from Dr Violeta Jonusiene (Institute of
Biosciences, Vilnius University Life Sciences Centre, Vilnius,
Lithuania). The authors would also thank Dr Mindaugas Valius
(Proteomics Centre, Institute of Biochemistry, Vilnius University
Life Sciences Centre, Vilnius, Lithuania) for the laboratory
equipment and materials.
Funding
No funding was received.
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
FBS
|
fetal bovine serum
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spadi R, Brusa F, Ponzetti A, Chiappino I,
Birocco N, Ciuffreda L and Satolli MA: Current therapeutic
strategies for advanced pancreatic cancer: A review for clinicians.
World J Clin Oncol. 7:27–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Petrillo A, Pappalardo A, Calabrese F,
Tirino G, Pompella L, Ventriglia J, Laterza MM, Caterino M, Sforza
V, Iranzo V, et al: First line nab-paclitaxel plus gemcitabine in
elderly metastatic pancreatic patients: A good choice beyond age. J
Gastroint Oncol. 10:910–917. 2019. View Article : Google Scholar
|
|
4
|
Sarabi M, Mais L, Oussaid N, Desseigne F,
Guibert P and De La Fouchardiere C: Use of gemcitabine as a
second-line treatment following chemotherapy with folfirinox for
metastatic pancreatic adenocarcinoma. Oncol Lett. 13:4917–4924.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ferlay J, Partensky C and Bray F: More
deaths from pancreatic cancer than breast cancer in the EU by 2017.
Acta Oncol. 55:1158–1160. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Han C, Liu Y, Dai R, Ismail N, Su W and Li
B: Ferroptosis and its potential role in human diseases. Front
Pharmacol. 11:2392020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Basuli D, Tesfay L, Deng Z, Paul B,
Yamamoto Y, Ning G, Xian W, Mckeon F, Lynch M, Crum CP, et al: Iron
addiction: A novel therapeutic target in ovarian cancer. Oncogene.
36:4089–4099. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eling N, Reuter L, Hazin J, Hamacher-Brady
A and Brady NR: Identification of artesunate as a specific
activator of ferroptosis in pancreatic cancer cells. Oncoscience.
2:517–532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li B, Yang L, Peng X, Fan Q, Wei S, Yang
S, Li X, Jin H, Wu B, Huang M, et al: Emerging mechanisms and
applications of ferroptosis in the treatment of resistant cancers.
Biomed Pharmacother. 130:1107102020. View Article : Google Scholar
|
|
11
|
Gagliardi M, Saverio V, Monzani R, Ferrari
E, Piacentini M and Corazzari M: Ferroptosis: A new unexpected
chance to treat metastatic melanoma? Cell Cycle. 19:2411–2425.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu H, Schreiber SL and Stockwell BR:
Targeting dependency on the GPX4 lipid peroxide repair pathway for
cancer therapy. Biochemistry. 57:2059–2060. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Viswanathan VS, Ryan MJ, Dhruv HD, Gill S,
Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada
K, Aguirre AJ, et al: Dependency of a therapy-resistant state of
cancer cells on a lipid peroxidase pathway. Nature. 547:453–457.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar :
|
|
15
|
Son J, Lyssiotis CA, Ying H, Wang X, Hua
S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et
al: Glutamine supports pancreatic cancer growth through a
KRAS-regulated metabolic pathway. Nature. 496:101–105. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Q, Zhang H, Jiang X, Qian C, Liu Z and
Luo D: Factors involved in cancer metastasis: A better
understanding to 'seed and soil' hypothesis. Mol Cancer.
16:1762017. View Article : Google Scholar
|
|
17
|
Ni B, Ghosh B, Paldy FS, Colin R, Heimerl
T and Sourjik V: Evolutionary remodeling of bacterial motility
checkpoint control. Cell Rep. 18:866–877. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gancedo JM: Control of pseudohyphae
formation in Saccharomyces cerevisiae. FEMS Microbiol Rev.
25:107–123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Keenan MM and Chi JT: Alternative fuels
for cancer cells. Cancer J. 21:49–55. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Commisso C, Davidson SM, Soydaner-Azeloglu
RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin
JA, Thompson CB, et al: Macropinocytosis of protein is an amino
acid supply route in Ras-transformed cells. Nature. 497:633–637.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Martinez-Outschoorn UE, Lisanti MP and
Sotgia F: Catabolic cancer-associated fibroblasts transfer energy
and biomass to anabolic cancer cells, fueling tumor growth. Semin
Cancer Biol. 25:47–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yang S, Wang X, Contino G, Liesa M, Sahin
E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al:
Pancreatic cancers require autophagy for tumor growth. Genes Devel.
25:717–729. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lugano R, Ramachandran M and Dimberg A:
Tumor angiogenesis: Causes, consequences, challenges and
opportunities. Cell Mol Life Sci. 77:1745–1770. 2020. View Article : Google Scholar :
|
|
24
|
Prabhakar NR and Semenza GL: Adaptive and
maladaptive cardiorespiratory responses to continuous and
intermittent hypoxia mediated by hypoxia-inducible factors 1-2.
Physiol Rev. 92:967–1003. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kuczynski EA, Vermeulen PB, Pezzella F,
Kerbel RS and Reynolds AR: Vessel co-option in cancer. Nat Rev Clin
Oncol. 16:469–493. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Neophytou CM, Panagi M, Stylianopoulos T
and Papageorgis P: The role of tumor microenvironment in cancer
metastasis: Molecular mechanisms and therapeutic opportunities.
Cancers (Basel). 13:20532021. View Article : Google Scholar
|
|
27
|
Janssen LME, Ramsay EE, Logsdon CD and
Overwijk WW: The immune system in cancer metastasis: Friend or foe?
J Immunotherapy Cancer. 5:792017. View Article : Google Scholar
|
|
28
|
Wang RA, Lu YY and Fan DM: Reasons for
cancer metastasis: A holistic perspective. Mol Clin Oncol.
3:1199–1202. 2015. View Article : Google Scholar
|
|
29
|
Trauzold A, Siegmund D, Schniewind B,
Sipos B, Egberts J, Zorenkov D, Emme D, Röder C, Kalthoff H and
Wajant H: TRAIL promotes metastasis of human pancreatic ductal
adenocarcinoma. Oncogene. 25:7434–7439. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiao D, Cai Z, Choksi S, Ma D, Choe M,
Kwon HN, Baik JY, Rowan BG, Liu C and Liu ZG: Necroptosis of tumor
cells leads to tumor necrosis and promotes tumor metastasis. Cell
Res. 28:868–870. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zalyte E, Dedonyte V, Kurlinkus B,
Sileikis A, Schemmer P and Valius M: Establishment and
characterization of a new pancreatic ductal adenocarcinoma cell
line capan-26. Anticancer Res. 41:1401–1406. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ger M, Žalytė E, Kaupinis A, Kurlinkus B,
Petrulionis M, Šileikis A, Strupas K and Valius M: Primary
pancreatic ductal adenocarcinoma cell cultures represent the
features of native tumours. Biologija. 65:20–33. 2019. View Article : Google Scholar
|
|
33
|
Rabanal-Ruiz Y, Otten EG and Korolchuk VI:
mTORC1 as the main gateway to autophagy. Essays Biochem.
61:565–584. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith
J and Rozengurt E: Different patterns of Akt and ERK feedback
activation in response to rapamycin, active-site mTOR inhibitors
and metformin in pancreatic cancer cells. PLoS One. 8:e572892013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liou GY, Döppler H, DelGiorno KE, Zhang L,
Leitges M, Crawford HC, Murphy MP and Storz P: Mutant kras-induced
mitochondrial oxidative stress in acinar cells upregulates EGFR
signaling to drive formation of pancreatic precancerous lesions.
Cell Rep. 14:2325–2336. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Palmieri VO, Grattagliano I and Palasciano
G: Ethanol induces secretion of oxidized proteins by pancreatic
acinar cells. Cell Biol Toxicol. 23:459–464. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu S, Zhang Q, Sun X, Zeh HJ III, Lotze
MT, Kang R and Tang D: HSPA5 regulates ferroptotic cell death in
cancer cells. Cancer Res. 77:2064–2077. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
White EZ, Pennant NM, Carter JR, Hawsawi
O, Odero-Marah V and Hinton CV: Serum deprivation initiates
adaptation and survival to oxidative stress in prostate cancer
cells. Sci Rep. 10:125052020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee H, Zandkarimi F, Zhang Y, Meena JK,
Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al:
Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat
Cell Biol. 22:225–234. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wu CA, Chao Y, Shiah SG and Lin WW:
Nutrient deprivation induces the warburg effect through
ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase.
Biochimica et biophysica Acta. 1833:1147–1156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sato H, Nomura S, Maebara K, Sato K, Tamba
M and Bannai S: Transcriptional control of cystine/glutamate
transporter gene by amino acid deprivation. Biochem Biophysical Res
Communicati. 325:109–116. 2004. View Article : Google Scholar
|
|
42
|
Gu Y, Albuquerque CP, Braas D, Zhang W,
Villa GR, Bi J, Ikegami S, Masui K, Gini B, Yang H, et al: mTORC2
regulates amino acid metabolism in cancer by phosphorylation of the
cystine-glutamate antiporter xCT. Mol Cell. 67:128–138 e127. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hanawa N, Shinohara M, Saberi B, Gaarde
WA, Han D and Kaplowitz N: Role of JNK translocation to
mitochondria leading to inhibition of mitochondria bioenergetics in
acetaminophen-induced liver injury. J Biol Chem. 283:13565–13577.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Win S, Than TA, Le BH, Garcia-Ruiz C,
Fernandez-Checa JC and Kaplowitz N: Sab (Sh3bp5) dependence of JNK
mediated inhibition of mitochondrial respiration in palmitic acid
induced hepatocyte lipotoxicity. J Hepatol. 62:1367–1374. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lee YH, Govinda B, Kim JC, Kim TI, Lee NH,
Lee JC, Yi HK and Jhee EC: Oxidative stress resistance through
blocking Hsp60 translocation followed by SAPK/JNK inhibition in
aged human diploid fibroblasts. Cell Biochem Funct. 27:35–39. 2009.
View Article : Google Scholar
|
|
46
|
Li XY and Leung PS: Erastin-induced
ferroptosis is a regulator for the growth and function of human
pancreatic islet-like cell clusters. Cell Regen. 9:162020.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang Y, Lin Z, Han Z, Wu Z, Hua J, Zhong
R, Zhao R, Ran H, Qu K, Huang H, et al: miR-539 activates the
SAPK/JNK signaling pathway to promote ferropotosis in colorectal
cancer by directly targeting TIPE. Cell Death Dis. 7:2722021.
View Article : Google Scholar
|
|
48
|
Kamphorst JJ, Nofal M, Commisso C, Hackett
SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA,
Bar-Sagi D, et al: Human pancreatic cancer tumors are nutrient poor
and tumor cells actively scavenge extracellular protein. Cancer
Res. 75:544–553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tong H, Yin H, Hossain MA, Wang Y, Wu F,
Dong X, Gao S, Zhan K and He W: Starvation-induced autophagy
promotes the invasion and migration of human bladder cancer cells
via TGF-β1/Smad3-mediated epithelial-mesenchymal transition
activation. J Cell Biochem. 120:5118–5127. 2019. View Article : Google Scholar
|
|
50
|
Stone TW: Dependence and guidance
receptors-DCC and Neogenin-in partial EMT and the actions of serine
proteases. Front Oncol. 10:942020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kim WK, Kwon Y, Jang M, Park M, Kim J, Cho
S, Jang DG, Lee WB, Jung SH, Choi HJ, et al: β-catenin activation
down-regulates cell-cell junction-related genes and induces
epithelial-to-mesenchymal transition in colorectal cancers. Sci
Rep. 9:184402019. View Article : Google Scholar
|
|
52
|
Coppock JD, Vermeer PD, Vermeer DW, Lee
KM, Miskimins WK, Spanos WC and Lee JH: mTOR inhibition as an
adjuvant therapy in a metastatic model of HPV+ HNSCC. Oncotarget.
7:24228–24241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chan DLH, Segelov E, Wong RS, Smith A,
Herbertson RA, Li BT, Tebbutt N, Price T and Pavlakis N: Epidermal
growth factor receptor (EGFR) inhibitors for metastatic colorectal
cancer. Cochrane Database Syst Rev. 6:CD0070472017.PubMed/NCBI
|
|
54
|
Demkova L and Kucerova L: Role of the
HGF/c-MET tyrosine kinase inhibitors in metastasic melanoma. Mol
Cancer. 17:262018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu S, Chen Z, Zeng X, Chen X and Gu Z:
Advances in nanomedicine for cancer starvation therapy.
Theranostics. 9:8026–8047. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
D'Aronzo M, Vinciguerra M, Mazza T,
Panebianco C, Saracino C, Pereira SP, Graziano P and Pazienza V:
Fasting cycles potentiate the efficacy of gemcitabine treatment in
in vitro and in vivo pancreatic cancer models. Oncotarget.
6:18545–18557. 2015. View Article : Google Scholar : PubMed/NCBI
|

















