Introduction
Insufficient blood supply to the myocardium, known
as acute myocardial injury, is a highly prevalent and fatal
condition that poses a significant risk to human well-being
(1). Therefore, the timely
restoration of blood flow (reperfusion) remains the foundation of
all current treatments to salvage ischemic myocardium (2). Research has indicated that
myocardial ischemia reperfusion injury (MI/RI) can trigger
different forms of regulated cell death (RCD), including apoptosis,
ferroptosis and autophagy-induced cell death (3). Growing evidence indicates that
autophagy could have both positive and negative effects on numerous
diseases (4). Likewise,
autophagy can have various impacts on MI/RI. While it can protect
the myocardium during ischemia, an excessive amount of autophagy
can harm the heart during reperfusion (5). The molecular mechanisms involved in
MI/RI are complex, with factors such as mitochondrial
abnormalities, oxidative stress and the generation of reactive
oxygen species (ROS) playing a role (6-8).
Rho Family GTPase 3 (RhoE), also referred to as the
Rnd3 protein, belongs to the Rho-GTPase group and controls the
movement of the actin cytoskeleton, cell cycle advancement and
programmed cell death (9). A
previous study revealed that RhoE influences inflammation following
myocardial infarction and promotes the recovery of the injured
heart (10). A previous study
revealed that RhoE could target and regulate gastric cancer
proliferation through chaperone-mediated autophagy (11). However, its role in myocardial
injury induced by MI/RI has not yet been investigated.
Berberine (BBR; PubChem ID: 2353) is an isoquinoline
alkaloid obtained from herbaceous plants of the Coptis genus
native to the Orient (12). A
previous study reported that BBR inhibits autophagy protecting the
myocardium from MI/RI (13).
However, the exact molecular mechanism by which BBR inhibits
autophagy and whether BBR inhibits autophagy through
RhoE/AMP-activated protein kinase (AMPK) remains unknown. BBR
demonstrated the ability to decrease apoptosis in myocardial cells
and enhance the compromised mechanical performance of the heart in
an MI/RI model (14). Thus, the
impact of BBR on mitigating MI/RI and its potential ability to
prevent injury involving the RhoE/AMPK, were examined.
In the present study, a hypoxia/reoxygenation (H/R)
injury model in H9c2 cells and a MI/RI injury model in mice were
employed to: i) Confirm whether MI/RI injury induces excessive
autophagy, which causes damage to the myocardium; ii) investigate
whether BBR inhibits excessive autophagy induced by MI/RI; iii)
investigate whether the RhoE/AMPK pathway mediates the inhibition
of myocardial excessive autophagy by BBR; and iv) determine whether
the cardioprotective benefits of BBR protective effects are linked
to the apoptosis inhibition, oxidative stress suppression, energy
metabolism enhancement and the maintenance of mitochondrial
function.
Materials and methods
Materials and animals
BBR (with a purity exceeding 98%) was provided from
Chengdu Must Bio-Technology Co., Ltd. Adenovirus pAd/RhoE-small
hairpin (sh)RNA was sourced from Shanghai GenePharma Co., Ltd.
Compound C and rapamycin (CC; cat. no. HY-13418A and Rap; cat. no.
HY-10219) were purchased from MedChemExpress. The antibodies
against NADH-ubiquinone oxidoreductase subunit B8 (NDUFB8; cat. no.
R383060), ubiquinol-cytochrome c reductase core protein 2
(UQCRC2; cat. no. R382096), Bcl-2-associated X protein (Bax; cat.
no. 380709), B-cell lymphoma 2 (Bcl-2; cat. no. 250198),
phosphorylated (p-)AMP-activated protein kinase (p-AMPK; cat. no.
381164) and microtubule-associated protein 1 light 3 (LC3; cat. no.
350140) were obtained from Chengdu Zen-Bioscience Co., Ltd.
(http://www.zen-bio.cn/). The antibodies
AMP-activated protein kinase (AMPK; cat. no. 10929-2-AP) and
anti-β-actin (cat. no. 66009-1-Ig) were purchased from Proteintech
Group, Inc. Anti-RhoE (cat. no. YN1227) and anti-P62 (cat. no.
YT7058) antibodies were supplied by ImmunoWay Biotechnology
Company. Mouse and rabbit secondary antibodies (cat. nos. 511103
and 511203, respectively) were provided by Chengdu Zen-Bioscience
Co., Ltd. 3-Methyladenine (3-MA; cat. no. HY-19312), an autophagic
inhibitor, was bought from MedChem Express. Dimethyl sulfoxide
(DMSO; cat. no. HY-Y0320) was obtained from MedChem Express.
A total of 30 healthy male C57BL/6 mice (6-8
weeks-old), weighing ~20 g, were supplied by the Animal Center of
Nanchang University (Nanchang, China). The experimental procedure
adhered to the guidelines of the National Institutes of Health
(NIH) and was authorized by the Animal Experimentation Ethics
Committee of The First Affiliated Hospital of Nanchang University
(approval no. CDYFY-IACUC-202209QR004). The mice were housed under
controlled conditions, including a temperature of 23±1°C, humidity
ranging between 40-50%, a 12-h light/dark cycle, and access to food
and water ad libitum.
The animal was euthanized in case-predefined humane
endpoints. These include: i) Weight loss: Rapid loss of 15-20% of
original body weight; ii) weakness: Unable to eat and drink on his
own, unable to stand for up to 24 h or unable to stand with extreme
reluctance; and iii) the animal exhibits depression and hypothermia
(<37°C) without anesthesia or sedation. No mice showed abnormal
signs of humanitarian endpoints throughout the experiment.
In vitro experiments
Cell culture
The H9c2 cell line was acquired from the Cell
Bank/Stem Cell Bank located in Beijing, China. The cells were
cultured in high-glucose Dulbecco's modified Eagle's medium
(H-DMEM; HyClone; Cytiva) with the addition of 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin,
and 100 μg/ml streptomycin (Wuhan Servicebio Technology Co.,
Ltd.). The cells were cultivated in a humid incubator at 37°C, a
humidity level of 95%, an oxygen concentration of 21%, and a
CO2 concentration of 5%.
Transfection of adenovirus and H/R
modeling
The transfection of adenovirus pAD/RhoE-shRNA
[multiplicity of infection (MOI): 80; target sense sequence: 5′-GCA
GCC ACT TAC ATA GAA T-3′; antisense sequence: 5′-ATT CTA TGT AAG
TGG CTG C-3′] into H9c2 cells was carried out in H-DMEM containing
10% FBS. The transfection efficiency was ~85% after 48 h under 95%
O2 and 5% CO2 at 37°C, and the following
experiments were conducted.
The cells were cultured in anoxia solution (1.0 mM
CaCl2, 20 mM HEPES, 10 mM KCl, 1.2 mM MgSO4,
98.5 mM NaCl, 0.9 mM NaH2PO4, 36 mM
NaHCO3 and 40 mM sodium lactate, at pH 6.8) for hypoxia
and in reoxygenation solution (1.0 mM CaCl2, 5.5 mM
glucose, 20 mM HEPES, 5 mM KCl, 1.2 mM MgSO4, 129.5 mM
NaCl, 0.9 mM NaH2PO4, 20 mM
NaHCO3, at pH 7.4) for reoxygenation. The H9c2 cells
were incubated in a sealed anoxic chamber at 37°C with a gas
mixture of 95% nitrogen and 5% carbon dioxide for 3 h in a Petri
dish. Following that, the gas mixture was altered to contain 95%
oxygen and 5% carbon dioxide for a duration of 2 h to cause H/R
damage (15,16).
Experimental grouping
H9c2 cells were assigned randomly into eight
different groups: i) control group; ii) H/R group: H9c2 cells
exposed to H/R injury; iii) BBR + H/R group: H9c2 cells were
treated with BBR at concentrations of 1.25, 2.5, 5, 10, 20 and 40
μM for 48 h before H/R injury; iv) BBR + pAd/RhoE-shRNA +
H/R group: H9c2 cells transfected with pAd/RhoE-shRNA for 48 h and
then pretreated with 20 μM BBR for an additional 48 h prior
to H/R injury; v) pAd/RhoE-shRNA + H/R group: H9c2 cells
transfected with pAd/RhoE-shRNA and incubated for 48 h prior to H/R
injury; vi) BBR + Rap group + H/R group: H9c2 cells pretreated with
20 μM BBR and 100 nM Rap for 48 h prior to H/R injury; vii)
BBR + 5 μM compound C + H/R group: H9c2 cells pretreated
with 20 μM BBR for 48 h and 100 nM Rap for 24 h before H/R
injury; and viii) 20 μM BBR + 5 mM 3-MA group: H9c2 cells
pretreated with 5 mM 3-MA for 24 h before H/R injury.
Cell viability and lactate
dehydrogenase (LDH) activity assay
Cell viability was evaluated by utilizing the Cell
Counting Kit-8 (CCK-8; Good Laboratory Practice Bioscience; cat.
no. GK10001). Each well was incubated with 10 μl of CCK-8
reagent for 1 h at 37°C, and the absorbance was measured at 450 nm.
LDH (Beyotime Institute of Biotechnology; cat. no. C0016)
concentrations were measured following the manufacturer's
guidelines. A total of 120 μl of supernatant were received
from each group and added to a new 96-well plate; then 60 μl
of LDH assay working solution was added to each well, incubated at
25°C (avoiding light) for 30 min, and the absorbance was measured
at 490 nm.
Measurement of oxidative stress
To evaluate intracellular ROS generation, a ROS
detection kit (Beyotime Institute of Biotechnology; cat. no.
S0033S) was utilized. H9c2 cell cultures were treated with DCFH-DA
at 37°C for a duration of 20 min without any exposure to light.
Inverted fluorescence microscopy (Olympus Corporation) was used to
observe the levels of ROS in the cell populations of different
experimental groups. Malondialdehyde (MDA), glutathione (GSH) and
glutathione disulfide (GSSG), levels were measured in the
supernatant of H9c2 cells in each group according to the
instructions provided in the kit (Beyotime Institute of
Biotechnology; cat. no. S0053). The GSH/GSSG ratio was
determined.
Lyso tracker red staining
H9c2 cells were incubated with a working solution of
50 nM Lyso-Tracker Red (Beyotime Institute of Biotechnology; cat.
no. C1046) to perform Lyso tracker red staining. Incubation was
carried out at 37°C for 20 min while avoiding exposure to light.
The examination of the cells was performed using an inverted
fluorescence microscope (Olympus Corporation).
Assessment of caspase-3 activity
The activity of caspase-3 was evaluated in
accordance with the instructions provided in the caspase-3 activity
assay kit (cat. no. C1115) from Beyotime Institute of
Biotechnology. The combination of reaction buffer, cell group
homogenate, and caspase-3 substrate was thoroughly mixed and
subsequently added to 96-well plates. The mixture was incubated at
37°C for 2 h. Following incubation, the absorbance was measured at
a wavelength of 405 nm using a microplate reader (Thermo Fisher
Scientific, Inc.). The concentration of each group of proteins was
determined using the Bradford method. Finally, caspase-3 activity
was calculated.
Flow cytometry assay
Mitochondrial permeability transition (mPTP),
mitochondrial membrane potential (MMP), and apoptosis were measured
using the mPTP assay kit (cat. no. BB-48122), the JC-1 MMP assay
kit (cat. no. BB-4105), and the Annexin V-fluorescein
isothiocyanate (V-FITC) Apoptosis assay Kit (cat. no. BB-4101)
(BestBio; https://www.bestbio.com.cn/index.html), according to
the manufacturer's instruction, respectively. For mPTP detection,
the cell suspensions were incubated with BbcellProbe M61 as well as
a quencher for 15 min at 37°C in the dark, followed by
centrifugation at 1,000 × g at room temperature for 5 min and
washing. The level of mPTP was immediately determined using a
Cytomics FC 500 flow cytometer [excitation (Ex)=488 nm, emission
(Em)=558 nm] (Beckman Coulter, Inc.). MMP levels were determined by
incubating H9c2 cardiomyocytes with JC-1 dye at 37°C for 30 min in
the dark, followed by centrifugation, washing, and detection using
a Cytomics FC 500 flow cytometer [530/580 nm (red) and 485/530 nm
(green)]. For the apoptosis assay, cell suspensions were incubated
with 5 μl of membrane-bound protein V-FITC and 10 μl
of propidium iodide for 20 min at 4°C and the cells were analyzed
using a Cytomics FC 500 flow cytometer (Ex=488 nm; Em=578 nm).
NovoExpress (v.6.2; Agilent Technologies, Inc.) was used to analyze
the flow cytometric data.
Transmission electron microscopy
imaging
After treatment, cells were collected, fixed
(incubated in 2% glutaraldehyde at 25°C for 2 h), washed,
dehydrated, embedded, sectioned and stained (staining with 2%
uranyl acetate and 2.6% lead citrate at 37°C for 8 min). The
structure of autophagosome in H9c2 cells was examined using
transmission electron microscopy (TEM; Hitachi 7800; Hitachi,
Ltd.).
Western blot analysis
Total cellular proteins from H9c2 cells were
hydrolyzed in RIPA lysis buffer (Beyotime Institute of
Biotechnology). The concentration of proteins was determined using
the BCA protein assay kit (Good Laboratory Practice Bioscience).
Electrophoresis on a 10% or 12% SDS-PAGE gel was carried out to
separate 20 μg of proteins, which were then transferred onto
polyvinylidene fluoride membranes. After blocking, the membranes
were probed with primary antibodies (diluted to 1:1,000) against
RhoE, LC3B, P62, AMPK, p-AMPK, Bcl-2, Bax, NDUFB8, UQCRC2 and
β-actin for an overnight incubation at 4°C. Subsequently, the
membranes were exposed to secondary antibodies conjugated with
horseradish peroxidase (1:20,000) at room temperature for 1 h. The
internal reference for normalization was β-actin. Protein bands
were analyzed using ImageJ v1.5.3 software (National Institutes of
Health).
In vivo experiments
Mice were re-separated into four groups: i) sham
group, ii) sham + BBR group, iii) ischemia-reperfusion (I/R) group,
iv) I/R + BBR group. The sham + BBR group and I/R + BBR group were
intragastrically administered 40 mg/kg BBR for 3 weeks. Mice from
both the sham and I/R groups received a saline solution. Following
the induction of anesthesia using 3% isoflurane, mice were
positioned supine and maintained under 1.5% isoflurane.
Subsequently, a thoracotomy was performed at the fourth intercostal
space, opening the pericardium to reveal the heart. Closure of the
left anterior descending artery (LAD) was achieved with a 4-0 silk
suture, and a snare was created by passing a short polyethylene
tube through the suture ends. The snare was clamped against the
heart surface to establish ischemia and released for reperfusion.
Mice in the sham group underwent a procedure that was similar but
did not include clamping of the LAD. The hearts of the mice
experienced 60 min of ischemia followed by 24 h of reperfusion in
order to model MI/RI in vivo.
Following reperfusion, the Vevo2100 imaging system
(Visual Sonics, Inc.) was used to measure the left ventricular end
diastolic diameter, left ventricular end-systolic diameter, left
ventricular ejection fraction (LVEF), and left ventricular
fractional shortening (LVFS) of the left ventricle in mice under
anesthesia with 1.5% isoflurane, using 2-dimensional transthoracic
echocardiography.
After collecting ~0.5 ml of blood from each group of
anesthetized mice through cardiac puncture, serum was extracted and
prepared. Immediately after blood collection, the mice were
euthanized with a 30% vol/min volumetric CO2
displacement rate. The activities of serum LDH and creatine kinase
(CK)-MB were then assessed. The myocardial infarct area was
measured by triphenyl tetrazolium chloride staining, and the left
ventricle was routinely fixed and sliced into 8 μM-thick
sections, which were stained using Terminal Deoxynucleotidyl
Transferase mediated dUTP Nick-End Labeling (TUNEL) and
dihydroethidium (DHE) staining and visualized by light
microscopy.
Statistical analysis
GraphPad Prism (Dotmatics) was utilized to conduct
one-way analysis of variance (ANOVA) with Tukey post hoc analysis,
with results being displayed as the mean ± standard deviation.
P<0.05 was considered to indicate a statistically significant
difference.
Results
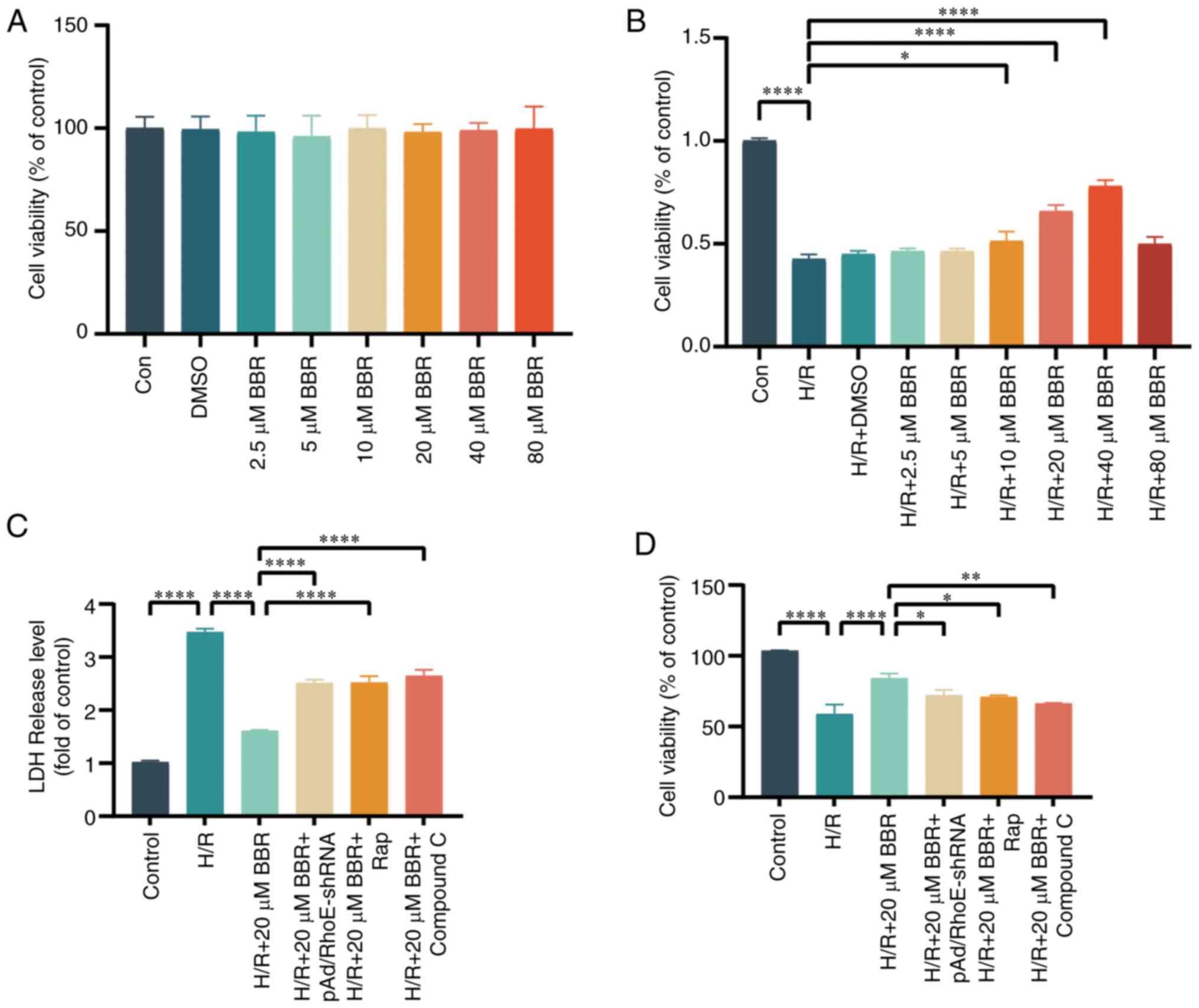
BBR protects H9c2 cells from injury
caused by H/R
An effective concentration of BBR was determined
using the CCK-8 test. The survival of H9c2 cells was mostly
unaffected after exposure to 0, 2.5, 5, 10, 20, 40, or 80 μM
BBR or 1‰ DMSO. BBR enhanced cell survival in a dose-dependent
manner but declined once it exceeded 80 μM. According to the
principle of drug dosing, the ideal therapeutic level of a
medication is typically above the level where it starts working and
below the maximum safe concentration. Consequently, a 20 μM
BBR concentration was utilized in following trials (Fig. 1A and B). A decrease in H9c2 cell
viability post-H/R injury confirmed the successful establishment of
the H/R injury model.
The effects of 20 μM BBR were significantly
counteracted by pAD/RhoE-shRNA, 100 nM Rap (a substance that
activates autophagy), and 5 μM compound C (a blocker of
AMPK). The results indicated that BBR could protect H9c2 cells from
H/R injury (Fig. 1C and D).
BBR inhibits autophagy by H/R-induced in
H9c2 cells
The levels of RhoE and autophagy indicators were
examined, such as P62 and the ratio of LC3-II to LC3-I, to evaluate
the impact of BBR on the expression of RhoE and autophagy during
H/R injury. Upon exposure to H/R, BBR enhanced the expression of
RhoE and P62 while reducing the LC3-II/LC3-I ratio. Nevertheless,
prior administration of pAd/RhoE-shRNA could reverse the expression
of the aforementioned proteins (Fig.
2A-D).
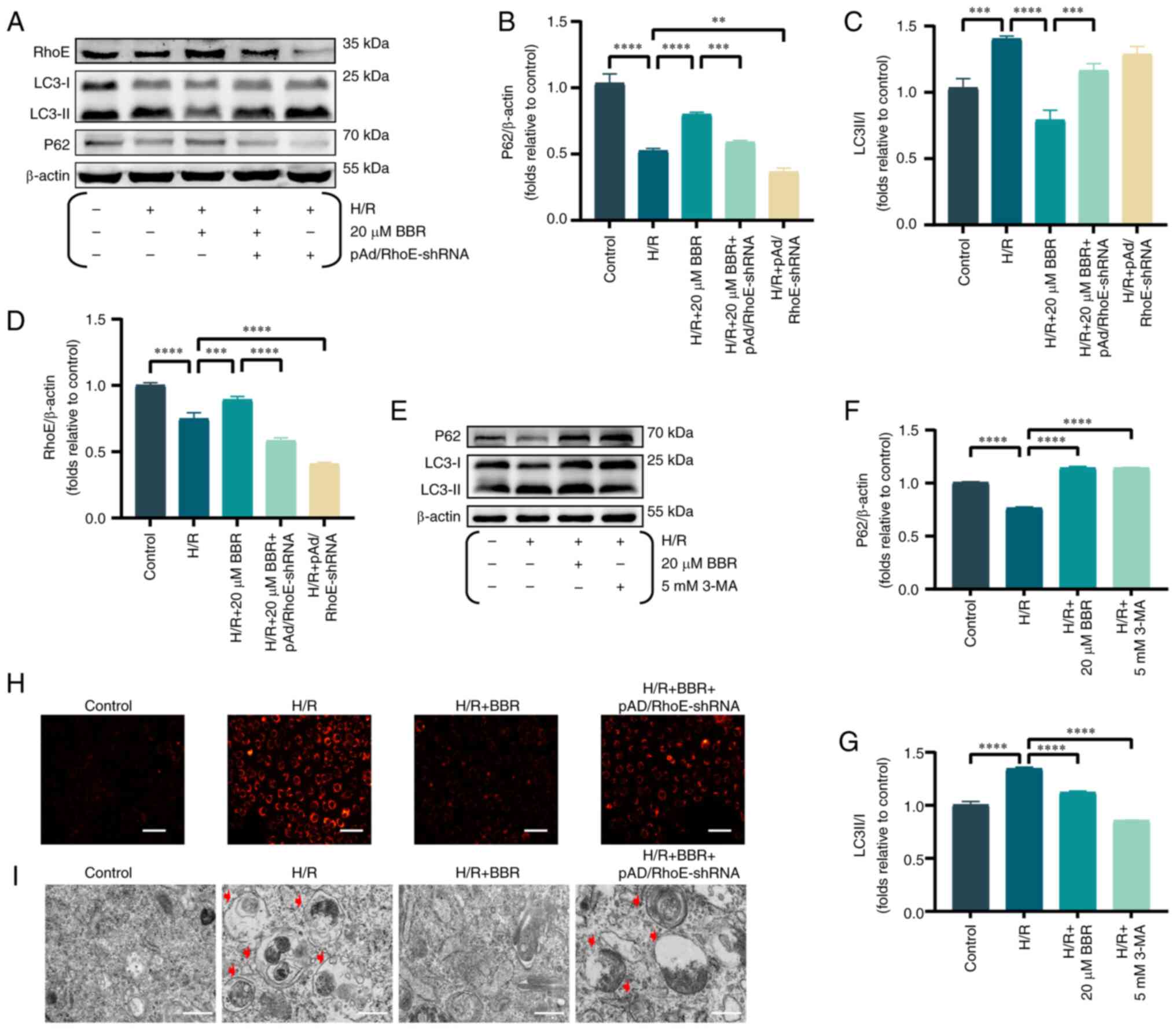 | Figure 2BBR inhibits autophagy by H/R-induced
in H9c2 cells. (A) Western blot detection of RhoE protein, LC3
protein and P62 protein expression in H/R-induced cells after
pretreatment with BBR, BBR + pAd/RhoE-shRNA and pAd/RhoE-shRNA.
(B-D) Histogram of RhoE protein, LC3 protein and P62 protein
expression. (E) Western blot detection of LC3 protein and P62
protein expression in H/R-induced cells after pretreatment with BBR
and 3-MA. (F and G) Histogram of LC3 and P62 protein expression.
(H) LysoTracker Red DND-99-stained images of H9c2 cells
(magnification, ×200; scale bar, 50 μm). (I) Transmission
electron microscopy images of H9c2 cells (magnification, ×6,000;
scale bar, 1 μm). Data are expressed as the mean ± SD (n=3).
**P<0.01, ***P<0.001 and
****P<0.0001. BBR, berberine H/R,
hypoxia/reoxygenation; RhoE, Rho family GTPase 3; LC3,
microtubule-associated protein 1 light 3; P62, Sequestosome 1; Ad,
adenovirus; sh-, small hairpin; 3-MA, 3-Methyladenine. |
The aforementioned effects of 20 μM BBR were
simulated by adding 5 mM 3-MA (an autophagy inhibitor). The results
showed that BBR had the same effect as the autophagy inhibitor 3-MA
(Fig. 2E-G).
LysoTracker Red DND-99-stained H9c2 cells exhibited
increased fluorescence intensity in the H/R group compared with the
control group, suggesting that H/R induced autophagosome formation
by decreasing lysosomal pH and enhancing autophagy. By contrast,
BBR reduced the strong fluorescence in the H/R group and suppressed
the formation of autophagosomes. pAd/RhoE-shRNA transfection
ameliorated the aforementioned changes (Fig. 2H).
Transmission electron microscopy results revealed a
rise in the number of autophagic vesicles in H9c2 cells within the
H/R group. Additionally, pretreatment with BBR resulted in a
reduction of autophagic vesicles, thereby inhibiting the autophagic
process. The aforementioned Changes mentioned above could be
reversed by pAd/RhoE-shRNA (Fig.
2I).
BBR inhibits excessive autophagy by H/R
induced in H9c2 cells via the RhoE/AMPK pathway
Western blot analysis of AMPK protein
phosphorylation was conducted to further examine the inhibitory
impact of RhoE-mediated BBR on autophagy in the H/R injury model.
BBR pretreatment significantly increased the levels of AMPK
phosphorylation. However, pAd/RhoE-shRNA transfection only
partially reversed the effects of BBR (Fig. 3A and B). The expression of
p-AMPK, AMPK, P62 and LC3 were analyzed in H9c2 cells, both with
and without compound C (an AMPK inhibitor), to analyze the effects
of the RhoE/AMPK pathway on BBR's protection against H/R damage.
BBR enhanced the expression of P62 and the p-AMPK/AMPK ratio, while
reducing the LC3-II/LC3-I ratio. Treatment with compound C
nullified the impacts of BBR, indicating that the RhoE/AMPK
pathway, which is associated with BBR, hindered the excessive
autophagy induced by H/R (Fig.
3C-F).
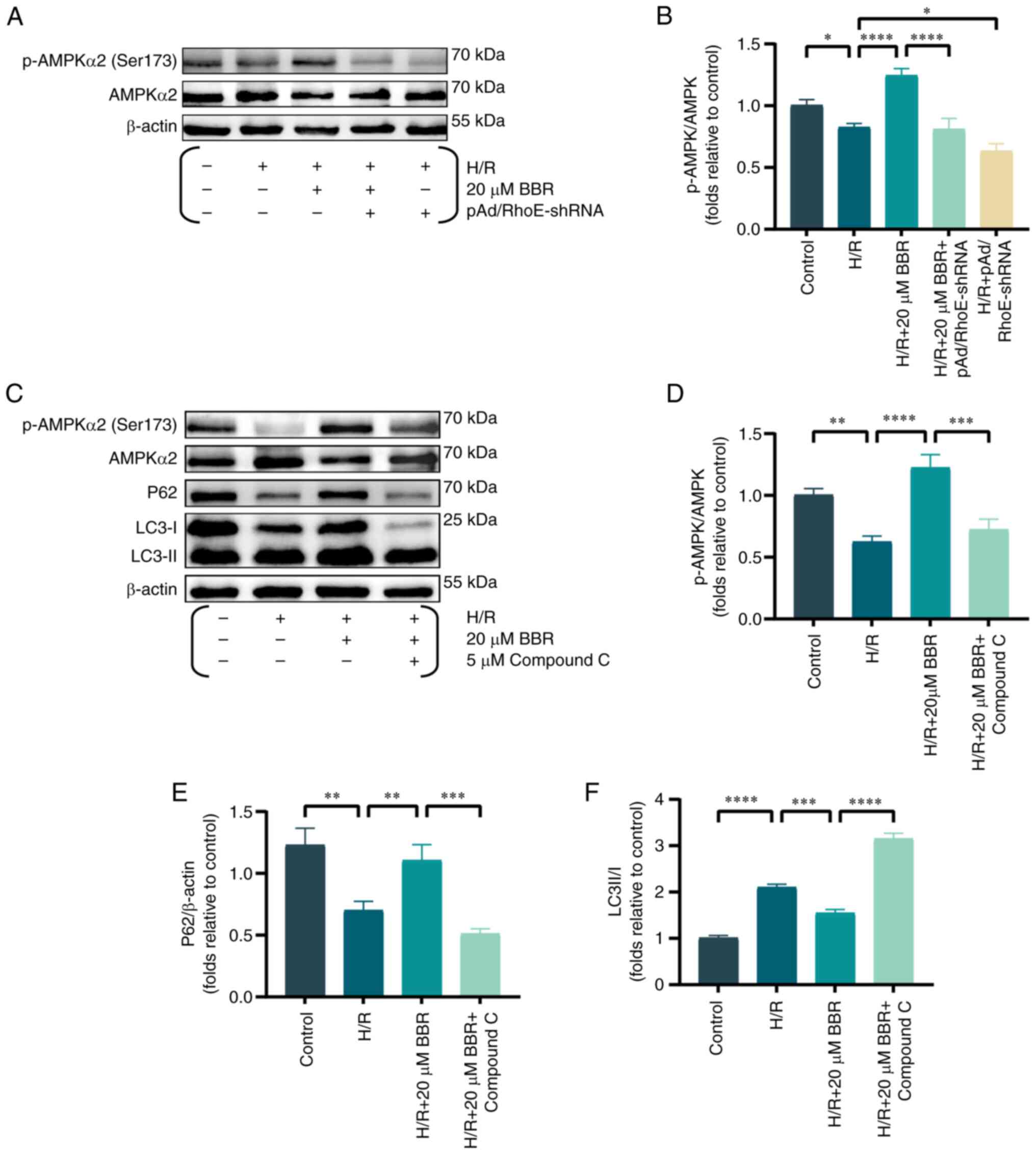 | Figure 3BBR inhibits excessive autophagy by
H/R induced in H9c2 cells via the RhoE/Ampk pathway. (A) Western
blot detection of p-AMPK protein and AMPK protein expression in
H/R-induced cells after pretreatment with BBR, BBR + pAd/RhoE-shRNA
and pAd/RhoE-shRNA. (B) Histogram of p-AMPK and AMPK protein
ratios. (C) Western blot detection of p-AMPK, AMPK, LC3 and P62
protein expression in H/R-induced cells after pretreatment with BBR
and BBR + Compound C. (D-F) Histogram of p-AMPK and AMPK protein
ratios, LC3 and P62 protein expression. Data are expressed as the
mean ± SD (n=3). *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. BBR,
berberine; H/R, hypoxia/reoxygenation; RhoE, Rho family GTPase 3;
p-, phosphorylated; AMPK, AMP-activated protein kinase; Ad,
adenovirus; sh, small hairpin; LC3, microtubule-associated protein
1 light 3; P62, Sequestosome 1. |
BBR improves mitochondrial function in
H9c2 cells after H/R injury
Mitochondria are recognized as the focal point of
cellular energy metabolism, while the AMPK pathway serves as an
energy detector that mirrors the state of the mitochondria.
Previous studies verified a strong connection between mitochondrial
impairment and MI/RI (17,18).
The energy supply of cardiomyocytes is maintained by
the crucial involvement of mitochondrial electron transfer chain
complexes I and III (19).
Consequently, the protein quantity of Ubiquinone Oxidoreductase
Subunit B8 (NDUFB 8) (component of complex I) and Cytochrome b-c1
complex subunit 2, mitochondrial (UQCRC 2) component of complex III
protein were analysed. BBR pretreatment rescued the downregulation
of both levels in H/R-injured H9c2 cells compared with controls
(Fig. 4A-C).
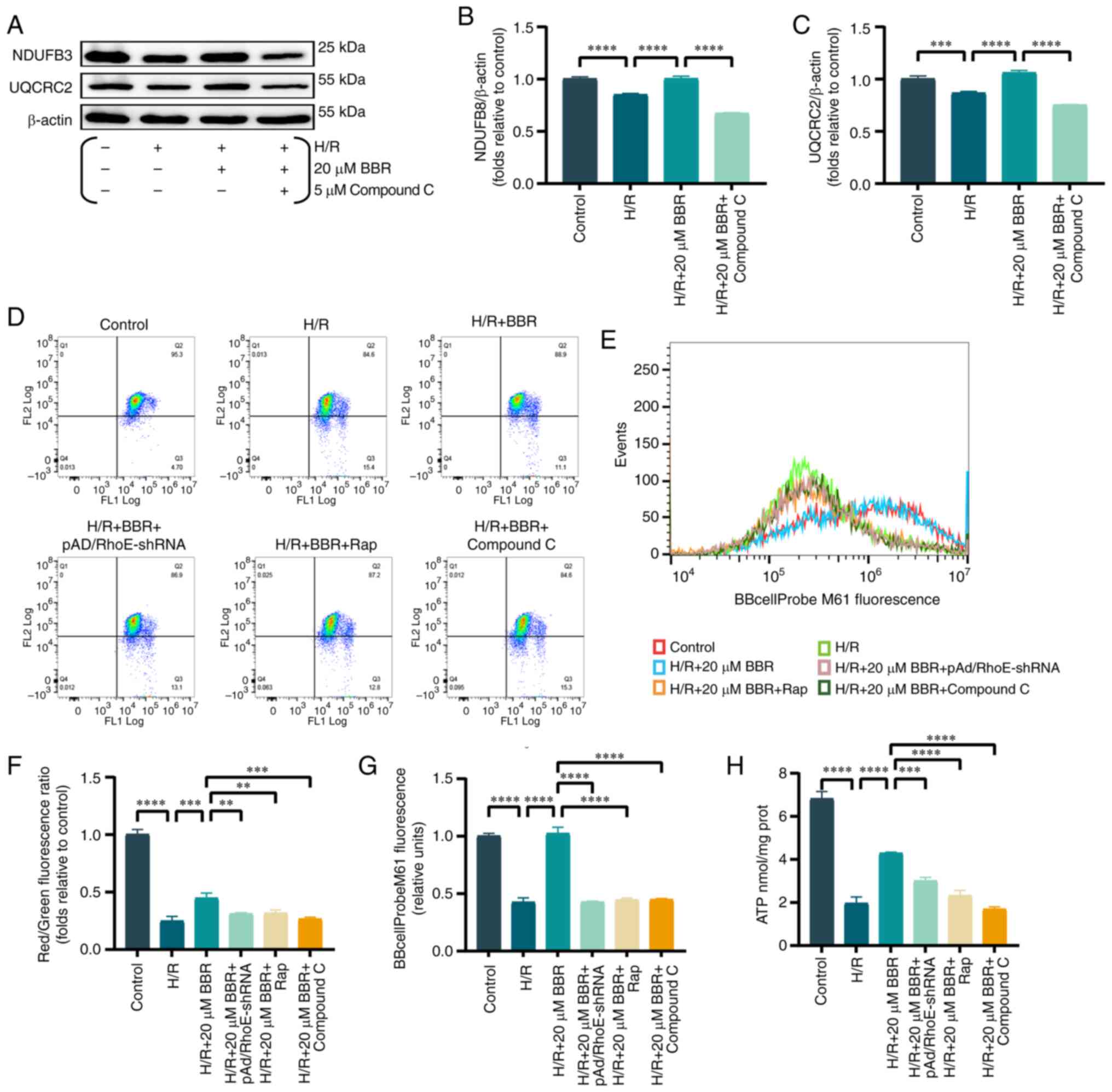 | Figure 4BBR improves mitochondrial function
in H9c2 cells after H/R injury. (A) Western blot detection of NDUFB
8 and UQCRC 2 protein expression in H/R-induced cells after
pretreatment with BBR and BBR + Compound C. (B and C) Histogram of
NDUFB 8 and UQCRC 2 protein expression. (D) MMP levels detected by
JC-1 in H9c2 cells by red/green fluorescence ratio. (E) Histogram
of red/green fluorescence ratio. (F) Histogram of mPTP flow
cytometry results. (G) Fluorescent probe BBcellProbe M61 indicating
mPTP opening was detected by flow cytometry. (H) Histogram of ATP.
Data are expressed as the mean ± SD (n=3). **P<0.01,
***P<0.001, ****P<0.0001. BBR,
berberine; H/R, hypoxia/reoxygenation; NDUFB 8, Ubiquinone
Oxidoreductase Subunit B8; UQCRC 2, Cytochrome b-c1 complex subunit
2, mitochondrial; MMP, mitochondrial membrane potential; mPTP,
mitochondrial permeability transition; Ad, adenovirus. |
Similarly, MMP and abnormal opening of the mPTP are
important judges of mitochondrial function. The mPTP assay and the
MMP results revealed that the green fluorescence and the ratio of
red/green fluorescence were significantly increased/decreased in
the H/R group compared with the control group. Conversely, these
effects were reversed in the H/R + BBR group. Notably, the
protective effects of BBR pretreatment were eliminated in cells
transfected with pAd/RhoE-shRNA and treated with Rap and compound C
(Fig. 4D-G).
A reduction in ATP levels, which is the primary
source of cellular energy, was observed following H/R damage.
However, BBR pretreatment partially restored ATP levels.
pAd/RhoE-shRNA, Rap and compound C reversed the aforementioned
effects (Fig. 4H).
The findings indicated that H/R has the potential to
hinder mitochondrial function, while BBR has the ability to
preserve mitochondrial function by suppressing excessive autophagy
via the RhoE/AMPK pathway.
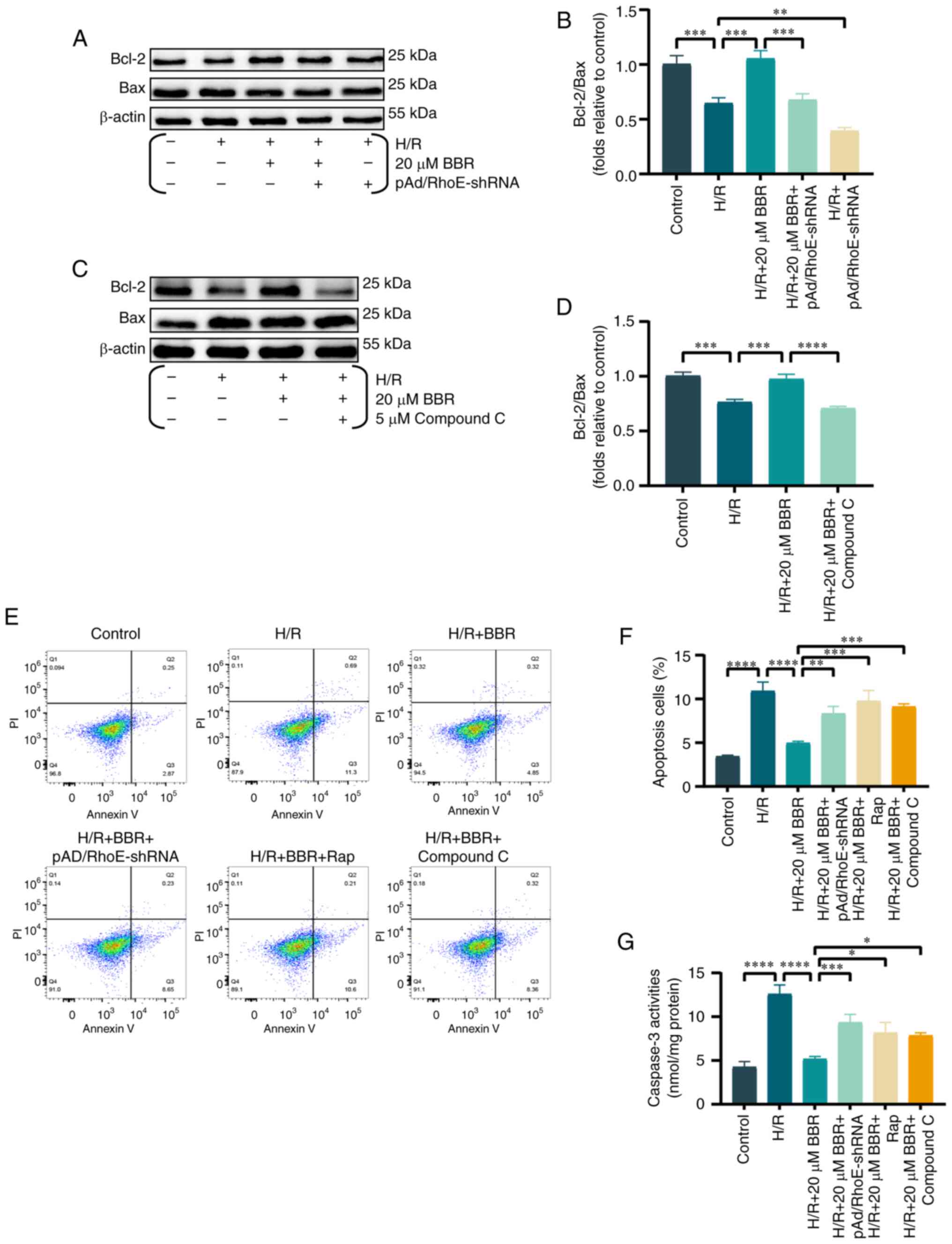
BBR attenuates apoptosis in H9c2 cells
after H/R injury through the RhoE/AMPK pathways
Prior research indicated the significant involvement
of apoptosis along with heightened autophagy in MI/RI injury
(20). During the investigation
into the suppression of excessive autophagy by BBR in H/R-damaged
H9c2 cells, corresponding alterations were observed in apoptosis
indicators. It was found that the ratio of Bcl-2/Bax ratio was
decreased in H9c2 cells when exposed to H/R. However, the effect
was prevented by pretreatment with BBR. The addition of
pAd/RhoE-shRNA or compound C reversed these changes (Fig. 5A-D).
Similarly, apoptosis by flow cytometry was assayed
and it was revealed that BBR inhibited H/R-mediated apoptosis and
that pAd/RhoE-shRNA, Rap and compound c still abrogated the
protective effect of BBR. The results suggested that BBR also
inhibits apoptosis through the RhoE/AMPK pathway (Fig. 5E and F).
Caspase-3 levels were analyzed to study the effect
of BBR on decreasing apoptosis in H9c2 cells following H/R injury
via the RhoE/AMPK pathways (21), as caspase-3 is considered the
main caspase involved in apoptosis execution. In the H/R group,
there was an increase in caspase-3 activity, which was
significantly reduced by BBR. However, pAd/RhoE-shRNA, Rap and
compound C pretreatment all ameliorated the protective effects of
BBR (Fig. 5G).
BBR inhibits oxidative stress by H/R
induced in H9c2 cells
The generation of cellular ROS, and the levels of
GSH, GSSG and MDA were investigated to illustrate the connection
between the suppression of excessive autophagy and oxidative stress
in H/R injury. BBR decreased the production within cells,
replenished GSH levels and the ratio of GSH to GSSG, and lowered
MDA and GSSG levels. The addition of pAd/RhoE-shRNA, Rap, or
compound C prevented the protective effect of BBR against H/R
injury in H9c2 cells (Fig.
6A-E). These results suggested that the inhibition of oxidative
stress by BBR was RhoE/AMPK pathway-dependent and associated with
the suppression of excessive autophagy.
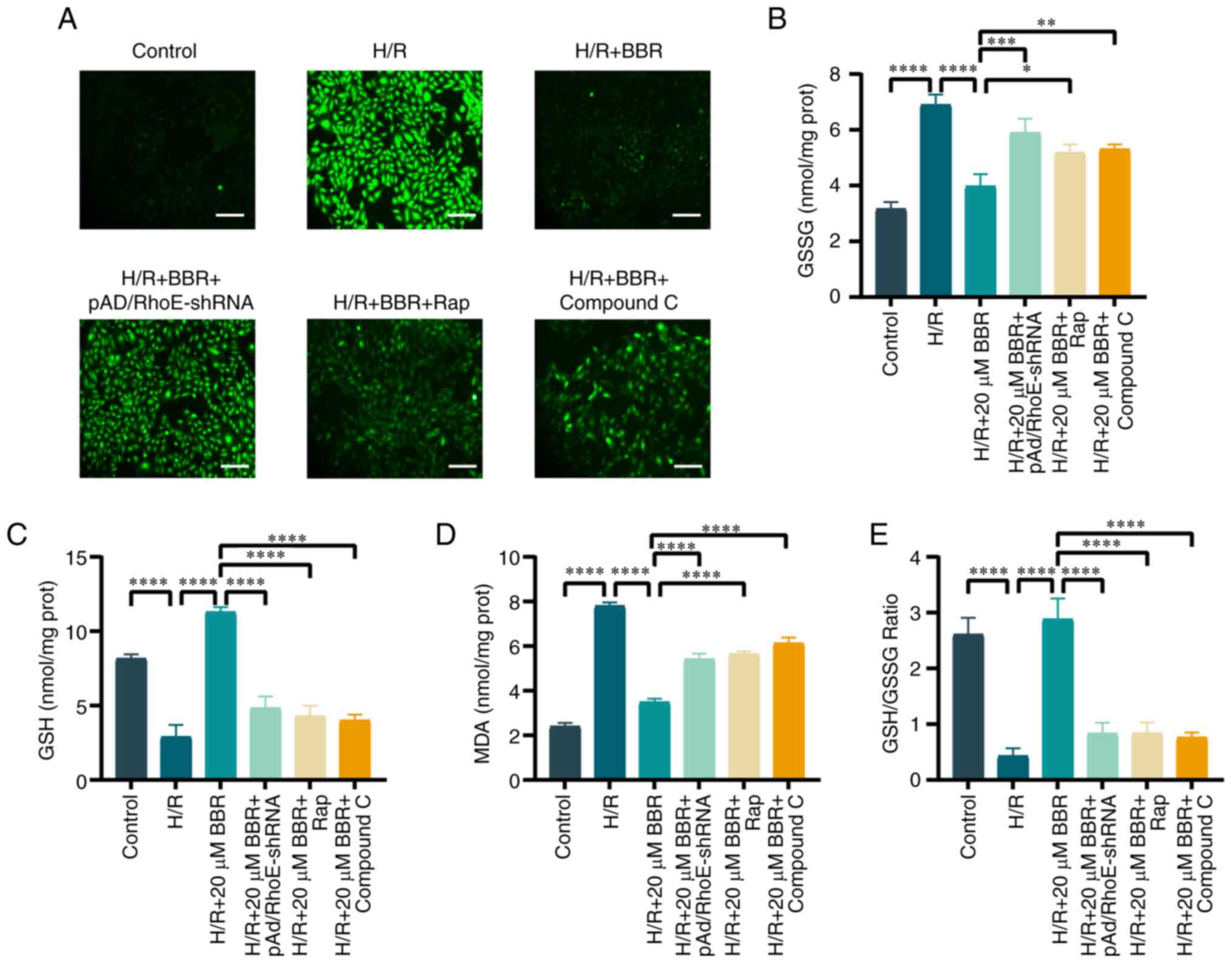 | Figure 6BBR inhibits oxidative stress by H/R
induced in H9c2 cells. (A) DCFH-DA stained images for detection of
ROS (magnification, ×200; scale bar, 50 μm). (B) Histogram
of GSH. (C) Histogram of GSSG. (D) Histogram of GSH and GSSG
ratios. (E) Histogram of MDA. Data are expressed as the mean ± SD
(n=3). *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001. BBR,
berberine; H/R, hypoxia/reoxygenation; ROS, reactive oxygen
species; GSH, glutathione; GSSG, glutathione disulfide; MDA,
malondialdehyde; Ad, adenovirus. |
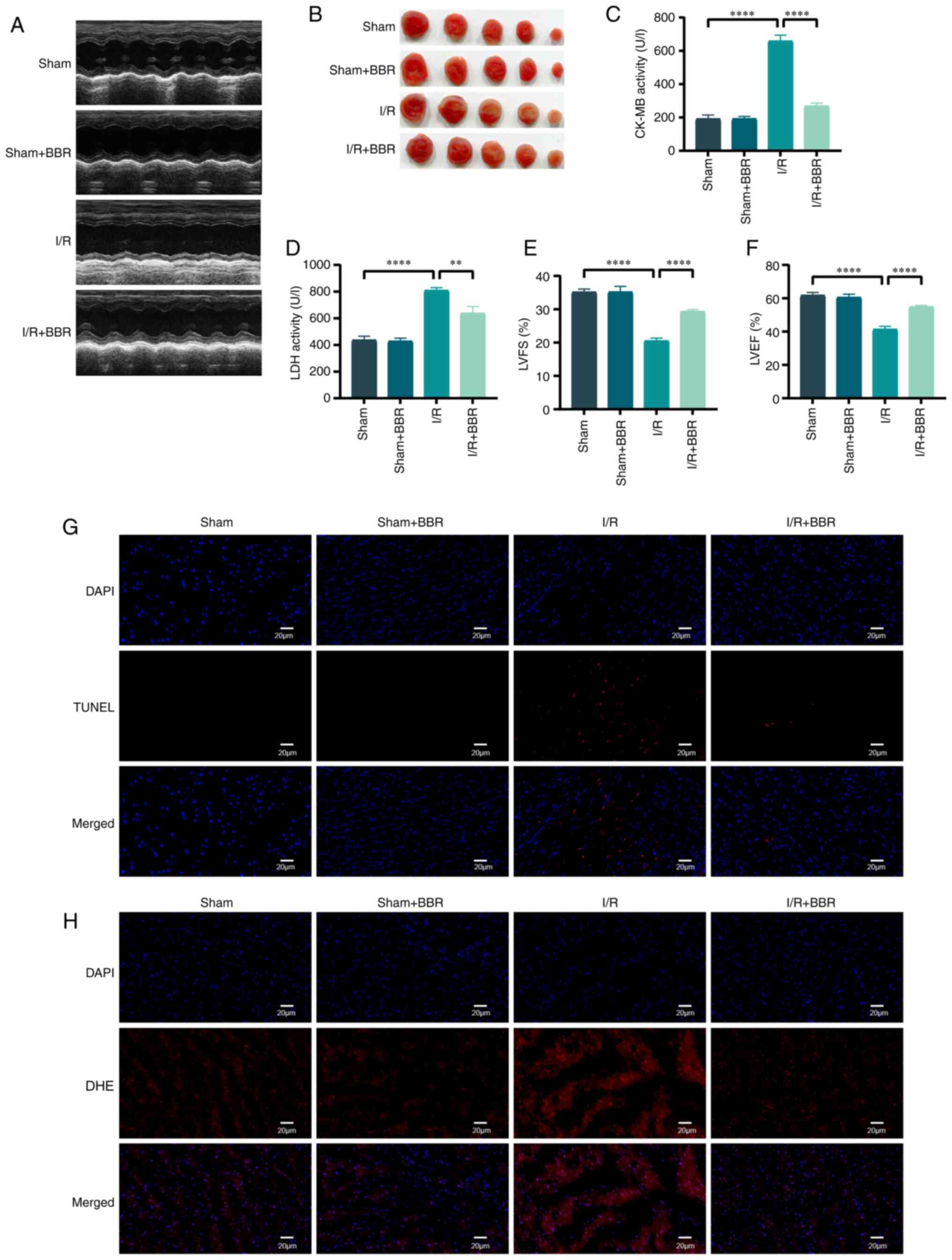
BBR protects mouse myocardium from I/R
damage
Male C57 BL/6 mice were utilized to simulate H/R
injury in order to validate the protective impact of BBR in
vivo. Mice afflicted with MI/RI injury exhibited significant
elevation in serum LDH and CK-MB levels. The assessment of LV
function in mice relied heavily on echocardiography. Post I/R
injury, cardiac function suffered a severe decline manifested by
decreased LVEF and LVFS. The administration of BBR (40 mg/kg)
effectively restored these anomalous functional and enzymatic
markers in I/R-induced injured mice (Fig. 7A and C-F). The infarct area was
also notably increased, whereas the infarct area of I/R
injury-induced mice treated with BBR was notably reduced (Fig. 7B). After I/R injury, TUNEL
staining revealed numerous distinct TUNEL-positive cardiomyocytes
and DHE staining demonstrated an increase in DHE intensity.
TUNEL-positive cardiomyocytes and DHE-positive cells decreased
after BBR treatment (Fig. 7G and
H). By contrast, none of the aforementioned indexes changed
significantly in the sham group after treatment with BBR (Fig. 7A-H).
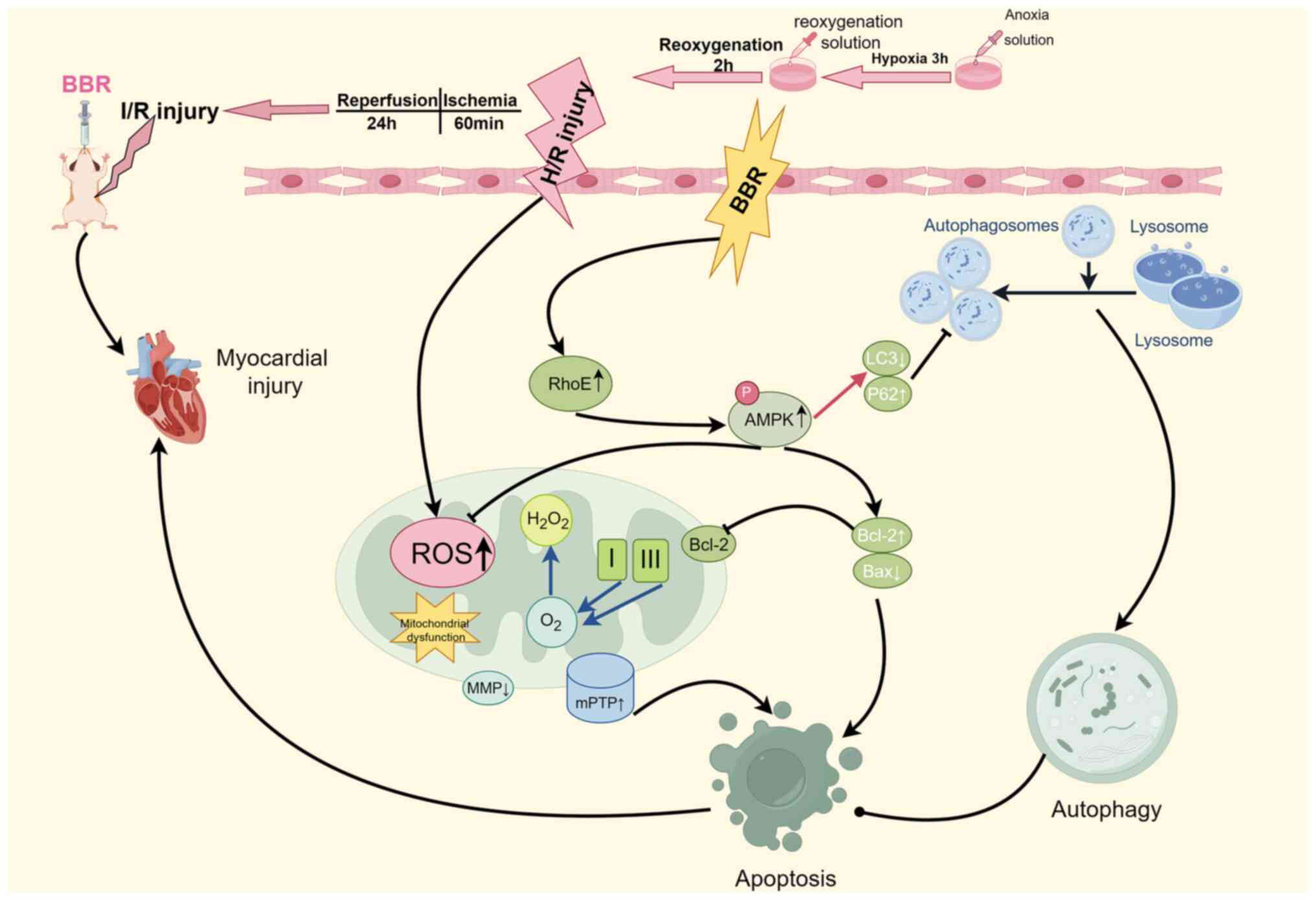
Discussion
Autophagy serves as a cellular mechanism for
survival, although uncontrolled autophagy can result in the demise
of cells (22). Autophagy is
often considered a double-edged sword in MI/RI. Moderate autophagy
is beneficial for cardiomyocyte resistance to ischemia-reperfusion
(23), but uncontrolled
autophagy results in cardiomyocyte death (24). This moderate autophagy is usually
called protective autophagy, while this uncontrolled autophagy is
called excessive autophagy. In the present study, excessive
autophagy during MI/RI severely damaged cardiomyocytes. However, to
date, there are no studies on whether the cardioprotective effects
of BBR are RhoE/AMPK pathway-dependent and which RCDs are involved
in the protective process. The innovation of the present study lies
in the fact that the myocardial protective effect of BBR was
dependent on the RhoE/AMPK pathway, and various enzymatic and
functional indices suggested that the apoptosis and autophagy
mechanisms of RCD were involved in the pathologic process of MI/RI
through. The H9c2 cell line was used in the present study because
it can well mimic the response of primary cellular cardiomyocytes
to hypoxia, and its energy metabolism pattern is similar to that of
primary cardiomyocytes. However, the use of a single cell line is
also a potential limitation to the present study.
BBR has been extensively utilized in the management
of heart conditions (25).
Numerous studies have reported that BBR improves MI/RI through
various mechanisms, such as its antioxidant, anti-apoptotic,
anti-inflammatory and endoplasmic reticulum stress properties
(25-30). However, whether BBR can target
multiple RCDs and whether the RhoE/AMPK pathway is involved is
unclear. In a previous investigation, it was found that TSN
pretreatment successfully reduced H9c2 cardiomyocyte damage in an
H/R injury model by inhibiting apoptosis and ferroptosis (16). Therefore, drugs capable of
targeting multiple RCDs are expected to be the best treatment for
MI/RI. In summary, it was hypothesized that BBR may also attenuate
MI/RI by inhibiting different RCDs. It was found that along with
RhoE/AMPK pathway activation, BBR significantly inhibited
MI/RI-induced excessive autophagy. Notably, reducing RhoE
expression, inhibiting the AMPK pathway or suppressing excessive
autophagy greatly diminished the cardioprotective effects and
inhibited excessive autophagy, providing evidence that the
RhoE/AMPK pathway and the inhibition of excessive autophagy play a
crucial role in the anti-MI/RI effects of BBR.
RhoE belongs to the RND subfamily in the RHO family
(31). Earlier investigations
have indicated that RhoE may function as a chaperone-mediated
autophagy substrate implicated in controlling the proliferation of
gastric cancer cells (11).
Prior research by the authors revealed that RhoE regulates
inflammation following myocardial infarction and facilitates the
healing of the damaged heart (10). In the present study, it was
revealed that RhoE not only participates in the protective effect
of BBR on cardiomyocytes in MI/RI but it also has an important role
in inhibiting excessive autophagy.
It has been previously indicated that the AMPK
complex consists of three subunits: A catalytic α-subunit and two
regulatory subunits, β and γ (32). Numerous prior investigations
demonstrated that AMPK stimulation had the potential to enhance
autophagy (33,34). However, recent research indicated
that AMPK can impede autophagy in a photo-oxidative damage model,
thereby providing substantial protection to photoreceptors against
photo-oxidative damage (35). In
addition, data from a study on heart failure suggested that AMPK
could improve cardiac function by attenuating autophagy during the
development of chronic heart failure, which may be related to
mTORC2 activation and downstream effects (36). In the experiments performed in
the present study, BBR triggered AMPK and protected cardiomyocytes
by suppressing excessive autophagy in MI/RI. Remarkably, the
introduction of the AMPK inhibitor compound C resulted in the
restoration of certain autophagic flux. Numerous prior
investigations similarly demonstrated that the inhibition of
autophagy induced by AMPK activation might encompass the regulation
of various signaling pathways, including the suppression of nuclear
factor (NF)-κB signaling, the phosphorylation of ULK 1, or the
inhibition of endoplasmic reticulum stress signaling (37,38). The inhibition of autophagy
dependent on AMPK onset is also reliant on a particular form of
stimulus or trauma and is intricately linked to the cellular energy
status (39). Nevertheless, the
specific molecular pathways still require further
investigation.
As markers of autophagic flux, LC3 and P62 are often
used to evaluate the level of autophagy. Activated LC3-I can be
transferred to Atg3, which then binds phosphatidylethanolamine to
carboxyglycine to produce treated LC3B-II (40-42). P62/SQSTM1 attaches to
polyubiquitylated proteins and forms clumps via its
ubiquitin-binding structural domain while also binding to LC3B-II
through its LC3 interacting region to facilitate the breakdown of
ubiquitylated protein clumps in autophagic lysosomes (43-45). In the present study, it was
identified that P62 and LC3 displayed synchronized alterations,
indicating that BBR preconditioning safeguards cardiomyocytes
against MI/RI by restraining excessive autophagy. The inhibitory
effect of BBR on autophagy in cardiomyocytes in MI/RI was nullified
by pAd/RhoE-shRNA transfection or treatment with Rap, or compound
C. TEM and the LysoTracker Red DND-99-stained observations
demonstrated similar results.
Previously, various forms of cellular demise were
considered to be unrelated to one another. However, advancements in
molecular biology have led to a growing examination of their
interconnected communication (46,47). Interactions between different
autophagy-associated and apoptosis-associated proteins have been
identified, and crosstalk between the two modes of cell death
occurs at various stages of MI/RI development (48,49). The experiments of the present
study revealed that BBR inhibited excessive autophagy while
significantly reducing H/R-induced apoptosis in H9c2 cells,
decreasing caspase-3 activity, and inducing an increase in the
Bcl-2/Bax ratio. Of note, the effect of BBR on apoptosis-related
indicators was prevented not only by the inclusion of
pAd/RhoE-shRNA transfection or compound C treatment, but also by
the inclusion of the autophagy stimulator Rap. These findings
indicated that BBR has the ability to protect cardiomyocytes from
MI/RI by suppressing excessive autophagy via the RhoE/AMPK pathway.
Additionally, it can also impact apoptosis through the RhoE/AMPK
pathway to prevent cardiomyocytes from MI/RI. The two modes of cell
death were observed to be in correlated crosstalk. However, the
specific proteins through which the crosstalk process is
accomplished need to be further investigated.
Prior research has indicated that irregularities in
MMP and the opening of mPTP are significant contributors to MI/RI
(50,51). The opening of mPTP can decrease
MMP, release of cytochrome c, decrease in ATP synthesis, and
ultimately cause the death of cardiomyocytes (52).The results of the present study
suggested that BBR inhibits oxidative stress, maintains
mitochondrial function, and protects the myocardium from MI/RI.
AMPK has additionally been discovered to be linked with energy
metabolism, facilitating the oxidation of fatty acids and
tricarboxylic acid cycling (53,54). The present study revealed that
pre-treatment with BBR activated AMPKα2 and boosted energy
production. Cardiomyocytes received an adequate energy supply, and
resistance to MI/RI was enhanced. In summary, it was identified
that MI/RI induces severe damage to cardiomyocytes by activating
excessive autophagy. However, BBR can counteract this process by
inhibiting autophagy, preserving mitochondrial function, enhancing
energy supply, maintaining redox homeostasis, and reducing
apoptosis via the RhoE/AMPK pathway. These effects ultimately
protect the myocardium from MI/RI (Fig. 8).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author on reasonable request.
Authors' contributions
SL, JL and HH conceptualized, designed and
administrated the present study. FH, TH and YQ performed cell
experiments, animal experiments, data analysis and interpretation.
ZZ and WH performed other cell experiments. SL and JL confirm the
authenticity of all the raw data. All authors wrote the manuscript.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Animal experiments followed the guidelines of the
National Institutes of Health and were authorized by the Animal
Experimentation Ethics Committee of the First Affiliated Hospital
of Nanchang University (Nanchang, China) (approval. no.
CDYFY-IACUC-202209QR004).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grand nos. 82160073 and 82360057), the Jiangxi
Provincial Natural Science Foundation (grant. nos. 20224ACB206002
and 20232BAB206009) and the Young Research and Cultivation Fund of
the First Affiliated Hospital of Nanchang University (grant. no.
YFYPY202128).
References
|
1
|
Heusch G: Myocardial ischaemia-reperfusion
injury and cardioprotection in perspective. Nat Rev Cardiol.
17:773–789. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang K, Li Y, Qiang T, Chen J and Wang X:
Role of epigenetic regulation in myocardial ischemia/reperfusion
injury. Pharmacol Res. 170:1057432021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deng J: Advanced research on the regulated
necrosis mechanism in myocardial ischemia-reperfusion injury. Int J
Cardiol. 334:97–101. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018. View Article : Google Scholar
|
|
5
|
Dong Y, Chen H, Gao J, Liu Y, Li J and
Wang J: Molecular machinery and interplay of apoptosis and
autophagy in coronary heart disease. J Mol Cell Cardiol. 136:27–41.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu Y, Wang M, Chen R and Sun X, Sun G and
Sun X: Gypenoside XVII protects against myocardial ischemia and
reperfusion injury by inhibiting ER stress-induced mitochondrial
injury. J Ginseng Res. 45:642–653. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tie R, Ji L, Nan Y, Wang W, Liang X, Tian
F, Xing W, Zhu M, Li R and Zhang H: Achyranthes bidentata
polypeptides reduces oxidative stress and exerts protective effects
against myocardial ischemic/reperfusion injury in rats. Int J Mol
Sci. 14:19792–19804. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pisarenko O, Shulzhenko V, Studneva I,
Pelogeykina Y, Timoshin A, Anesia R, Valet P, Parini A and
Kunduzova O: Structural apelin analogues: Mitochondrial ROS
inhibition and cardiometabolic protection in myocardial ischaemia
reperfusion injury. Br J Pharmacol. 172:2933–2945. 2015. View Article : Google Scholar :
|
|
9
|
Jie W, Andrade KC, Lin X, Yang X, Yue X
and Chang J: Pathophysiological functions of Rnd3/RhoE. Compr
Physiol. 6:169–186. 2015. View Article : Google Scholar
|
|
10
|
Dai Y, Song J, Li W, Yang T, Yue X, Lin X,
Yang X, Luo W, Guo J, Wang X, et al: RhoE fine-tunes inflammatory
response in myocardial infarction. Circulation. 139:1185–1198.
2019. View Article : Google Scholar :
|
|
11
|
Zhou J, Yang J, Fan X, Hu S, Zhou F, Dong
J, Zhang S, Shang Y, Jiang X, Guo H, et al: Chaperone-mediated
autophagy regulates proliferation by targeting RND3 in gastric
cancer. Autophagy. 12:515–528. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fang X, Wu H, Wei J, Miao R, Zhang Y and
Tian J: Research progress on the pharmacological effects of
berberine targeting mitochondria. Front Endocrinol (Lausanne).
13:9821452022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu X, Liu Z, Yu XY, Xu S and Luo J:
Autophagy and cardiac diseases: Therapeutic potential of natural
products. Med Res Rev. 41:314–341. 2021. View Article : Google Scholar
|
|
14
|
Yu L, Li F, Zhao G, Yang Y, Jin Z, Zhai M,
Yu W, Zhao L, Chen W, Duan W and Yu S: Protective effect of
berberine against myocardial ischemia reperfusion injury: Role of
Notch1/Hes1-PTEN/Akt signaling. Apoptosis. 20:796–810. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang H, Lai S, Luo Y, Wan Q, Wu Q, Wan L,
Qi W and Liu J: Nutritional preconditioning of apigenin alleviates
myocardial ischemia/reperfusion injury via the mitochondrial
pathway mediated by Notch1/Hes1. Oxid Med Cell Longev.
2019:79730982019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu T, Zou HX, Le SY, Wang YR, Qiao YM,
Yuan Y, Liu JC, Lai SQ and Huang H: Tanshinone IIA confers
protection against myocardial ischemia/reperfusion injury by
inhibiting ferroptosis and apoptosis via VDAC1. Int J Mol Med.
52:1092023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cai CC, Zhu JH, Ye LX, Dai YY, Fang MC, Hu
YY, Pan SL, Chen S, Li PJ, Fu XQ and Lin ZL: Glycine protects
against hypoxic-ischemic brain injury by regulating
mitochondriamediated autophagy via the AMPK pathway. Oxid Med Cell
Longev. 2019:42485292019. View Article : Google Scholar
|
|
18
|
Chaudhary KR, Batchu SN, Das D, Suresh MR,
Falck JR, Graves JP, Zeldin DC and Seubert JM: Role of B-type
natriuretic peptide in epoxyeicosatrienoic acid-mediated improved
post-ischaemic recovery of heart contractile function. Cardiovasc
Res. 83:362–370. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Monzel AS, Enriquez JA and Picard M:
Multifaceted mitochondria: Moving mitochondrial science beyond
function and dysfunction. Nat Metab. 5:546–562. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deng X, Ye F, Zeng L, Luo W, Tu S, Wang X
and Zhang Z: Dexmedetomidine mitigates myocardial
ischemia/reperfusion-induced mitochondrial apoptosis through
targeting lncRNA HCP5. Am J Chin Med. 50:1529–1551. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Diao X, Wang J, Zhu H and He B:
Overexpression of programmed cell death 5 in a mouse model of
ovalbumin-induced allergic asthma. BMC Pulm Med. 16:1492016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Willis MS, Min JN, Wang S, McDonough H,
Lockyer P, Wadosky KM and Patterson C: Carboxyl terminus of
Hsp70-interacting protein (CHIP) is required to modulate cardiac
hypertrophy and attenuate autophagy during exercise. Cell Biochem
Funct. 31:724–735. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Campos JC, Queliconi BB, Bozi LHM, Bechara
LRG, Dourado PMM, Andres AM, Jannig PR, Gomes KMS, Zambelli VO,
Rocha-Resende C, et al: Exercise reestablishes autophagic flux and
mitochondrial quality control in heart failure. Autophagy.
13:1304–1317. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bitirim CV, Ozer ZB, Aydos D, Genc K,
Demirsoy S, Akcali KC and Turan B: Cardioprotective effect of
extracellular vesicles derived from ticagrelor-pretreated
cardiomyocyte on hyperglycemic cardiomyocytes through alleviation
of oxidative and endoplasmic reticulum stress. Sci Rep.
12:56512022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fu L, Chen W, Guo W, Wang J, Tian Y, Shi
D, Zhang X, Qiu H, Xiao X, Kang T, et al: Berberine targets
AP-2/hTERT, NF-κB/COX-2, HIF-1α/VEGF and cytochrome-c/caspase
signaling to suppress human cancer cell growth. PLoS One.
8:e692402013. View Article : Google Scholar
|
|
26
|
Liu DQ, Chen SP, Sun J, Wang XM, Chen N,
Zhou YQ, Tian YK and Ye DW: Berberine protects against
ischemia-reperfusion injury: A review of evidence from animal
models and clinical studies. Pharmacol Res. 148:1043852019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao L, Li H, Gao Q, Xu J, Zhu Y, Zhai M,
Zhang P, Shen N, Di Y, Wang J, et al: Berberine attenuates cerebral
ischemia-reperfusion injury induced neuronal apoptosis by
down-regulating the CNPY2 signaling pathway. Front Pharmacol.
12:6096932021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhu JR, Lu HD, Guo C, Fang WR, Zhao HD,
Zhou JS, Wang F, Zhao YL, Li YM, Zhang YD, et al: Berberine
attenuates ischemia-reperfusion injury through inhibiting HMGB1
release and NF-κB nuclear translocation. Acta Pharmacol Sin.
39:1706–1715. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang J, Yan H, Li S and Zhang M: Berberine
ameliorates MCAO induced cerebral ischemia/reperfusion injury via
activation of the BDNF-TrkB-PI3K/Akt signaling pathway. Neurochem
Res. 43:702–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chen C, Lin Q, Zhu XY, Xia J, Mao T, Chi
T, Wan J, Lu JJ, Li Y, Cui J, et al: Pre-clinical evidence:
Berberine as a promising cardioprotective candidate for myocardial
ischemia/reperfusion injury, a systematic review, and
meta-analysis. Front Cardiovasc Med. 8:6463062021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Endzhievskaya S, Hsu CK, Yang HS, Huang
HY, Lin YC, Hong YK, Lee JYW, Onoufriadis A, Takeichi T, Yu-Yun Lee
J, et al: Loss of RhoE function in dermatofibroma promotes
disorganized dermal fibroblast extracellular matrix and increased
integrin activation. J Invest Dermatol. 143:1487–1497.e9. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Herzig S and Shaw RJ: AMPK: Guardian of
metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol.
19:121–135. 2018. View Article : Google Scholar :
|
|
33
|
Zhuang A, Chai P, Wang S, Zuo S, Yu J, Jia
S, Ge S, Jia R, Zhou Y, Shi W, et al: Metformin promotes histone
deacetylation of optineurin and suppresses tumour growth through
autophagy inhibition in ocular melanoma. Clin Transl Med.
12:e6602022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kim TW, Cheon C and Ko SG: SH003 activates
autophagic cell death by activating ATF4 and inhibiting G9a under
hypoxia in gastric cancer cells. Cell Death Dis. 11:7172020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li YL, Zhang TZ, Han LK, He C, Pan YR, Fan
B and Li GY: The AMPK-dependent inhibition of autophagy plays a
crucial role in protecting photoreceptor from photooxidative
injury. J Photochem Photobiol B. 245:1127352023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y, Wang Y, Zou M, Chen C, Chen Y, Xue
R, Dong Y and Liu C: AMPK blunts chronic heart failure by
inhibiting autophagy. Biosci Rep. 38:BSR201709822018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lu G, Wu Z, Shang J, Xie Z, Chen C and
Zhang C: The effects of metformin on autophagy. Biomed
Pharmacother. 137:1112862021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nwadike C, Williamson LE, Gallagher LE,
Guan JL and Chan EYW: AMPK inhibits ULK1-dependent autophagosome
formation and lysosomal acidification via distinct mechanisms. Mol
Cell Biol. 38:e00023–18. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
He H, Wang L, Qiao Y, Yang B, Yin D and He
M: Epigallocatechin-3-gallate pretreatment alleviates
doxorubicin-induced ferroptosis and cardiotoxicity by upregulating
AMPKα2 and activating adaptive autophagy. Redox Biol.
48:1021852021. View Article : Google Scholar
|
|
40
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014. View Article : Google Scholar :
|
|
41
|
Glick D, Barth S and Macleod KF:
Autophagy: Cellular and molecular mechanisms. J Pathol. 221:3–12.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lamark T, Svenning S and Johansen T:
Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays
Biochem. 61:609–624. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jeong SJ, Zhang X, Rodriguez-Velez A,
Evans TD and Razani B: p62/SQSTM1 and selective autophagy in
cardiometabolic diseases. Antioxid Redox Signal. 31:458–471. 2019.
View Article : Google Scholar :
|
|
45
|
Deng Z, Lim J, Wang Q, Purtell K, Wu S,
Palomo GM, Tan H, Manfredi G, Zhao Y, Peng J, et al:
ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy
and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy.
16:917–931. 2020. View Article : Google Scholar :
|
|
46
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Su Z, Yang Z, Xu Y, Chen Y and Yu Q:
Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol
Cancer. 14:482015. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fairlie WD, Tran S and Lee EF: Crosstalk
between apoptosis and autophagy signaling pathways. Int Rev Cell
Mol Biol. 352:115–158. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cong L, Bai Y and Guo Z: The crosstalk
among autophagy, apoptosis, and pyroptosis in cardiovascular
disease. Front Cardiovasc Med. 9:9974692022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhao WK, Zhou Y, Xu TT and Wu Q:
Ferroptosis: Opportunities and challenges in myocardial
ischemia-reperfusion injury. Oxid Med Cell Longev.
2021:99296872021.PubMed/NCBI
|
|
51
|
Zhang CX, Cheng Y, Liu DZ, Liu M, Cui H,
Zhang BL, Mei QB and Zhou SY: Mitochondria-targeted cyclosporin A
delivery system to treat myocardial ischemia reperfusion injury of
rats. J Nanobiotechnology. 17:182019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bugger H and Pfeil K: Mitochondrial ROS in
myocardial ischemia reperfusion and remodeling. Biochim Biophys
Acta Mol Basis Dis. 1866:1657682020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhang Y, Wang Y, Xu J, Tian F, Hu S, Chen
Y and Fu Z: Melatonin attenuates myocardial ischemia-reperfusion
injury via improving mitochondrial fusion/mitophagy and activating
the AMPK-OPA1 signaling pathways. J Pineal Res. 66:e125422019.
View Article : Google Scholar
|
|
54
|
Cai Z, Li CF, Han F, Liu C, Zhang A, Hsu
CC, Peng D, Zhang X, Jin G, Rezaeian AH, et al: Phosphorylation of
PDHA by AMPK Drives TCA Cycle to promote cancer metastasis. Mol
Cell. 80:263–278.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|