Gliomas are the most common type of brain malignancy
in both children and adults. According to the World Health
Organization (WHO), glioma can be classified into the low-grade
(grades 1 and 2) and high-grade (grades 3 and 4) categories, based
on the degree of malignancy from lowest to highest (1-3).
The prognosis for gliomas remains poor despite the existence of
multiple treatment strategies, including surgery, radiation,
chemotherapy and targeted therapy. Specifically, glioblastoma (GBM)
multiforme (WHO grade 4) has a 5-year survival rate of only 5.5%
(4), which may be due to
chemoresistance, heterogeneity and infiltrative properties, making
the tumor difficult to remove completely (5). By contrast, low-grade gliomas (WHO
grades 1-2) have a relatively favorable prognosis, with an overall
survival of ~7 years (6).
Glioma tissues can consist not only of cancer cells
but can also contain various non-cancerous cell types, such as
resident microglia from the brain and monocytes (macrophages) from
the circulating bloodstream. In particular, macrophages and
microglia are highly heterogeneous and plastic, such that they
become cells of different phenotypes after in vitro
stimulation (7). Toll-like
receptor 4 (TLR4) ligands and IFN-γ stimulation typically result in
the pro-inflammatory M1 phenotype, whereas IL-4, IL-10 and IL-13
stimulation typically produce an anti-inflammatory M2 phenotype
(8). In addition, macrophages
can be selectively activated further and then subdivided into the
M2a [type II T-helper cell (Th2) response, type II inflammation,
pathogen killing and allergic response], M2b (Th2 activation,
immunomodulation) and M2c (immunomodulation, matrix deposition and
tissue remodeling) states (8,9).
These macrophage subpopulations differ in their receptor
expression, effector function, as well as cytokine and chemokine
expression profiles. However, this phenotype definition was
proposed based on data from mainly in vitro research,
meaning that they cannot be used to fully reflect the in
vivo situation of the different pathological conditions.
Metabolic reprogramming refers to the process by
which cells adjust their metabolic pathways and energy production
methods to adapt to environmental changes under specific
conditions. However, this process is not merely a simple metabolic
change. Instead, it typically involves profound systemic
adjustments aimed at meeting the specific physiological needs of
the cells (10,11). Metabolic reprogramming in tumors
will likely involve significant changes in the energy production
and metabolic pathways being activated. Changes that have been
previously reported include the Warburg effect, enhanced lipid
synthesis and abnormal amino acid metabolism (12,13). However, the majority of such
previous studies have mainly focused on tumor cells. Metabolic
reprogramming in other cell types that reside in the tumor
microenvironment, such as glioma-associated microglia/macrophages
(GAMs), should also be considered. Tumor cell metabolic
reprogramming can mediate macrophage phenotypic alterations through
various mechanisms, such as epigenetic modifications, leading to
altered macrophage metabolism and in turn tumor progression
(14). Studies over the past
decade have demonstrated that altered lipid metabolism in
tumor-associated macrophages (TAMs) can serve an important role in
tumor progression, though to the best of our knowledge, there have
been few similar studies on GAMs. Therefore, the present review
aims to systematically summarize the research progress on metabolic
reprogramming in GAMs. Based on existing studies on TAMs (15), hypotheses regarding the role of
GAMs in glioma are proposed, emphasizing their potential metabolic
similarities. In addition, the complex regulatory mechanisms
potentially driving these metabolic changes and their implications
for tumor progression and immune evasion are summarized. The
present review also explores potential therapeutic targets within
lipid metabolism, aiming to facilitate future strategies for
inhibiting glioma tumor growth by modulating GAM metabolism. The
novelty of the present review lies in its comprehensive focus on
the underexplored area of GAM lipid metabolism and integration of
recent findings to propose novel research directions and clinical
applications.
Similar to other rapidly proliferating cells, glioma
cells typically metabolize glucose into lactate even in the
presence of oxygen (the 'Warburg' effect). This allows tumor cells
to use glucose-derived carbon to synthesize essential cellular
components whilst simultaneously producing sufficient ATP to
support its substantial metabolic demands (16-18). In addition, glioma cells can
increase their own intracellular stores of fats, amino acids and
nucleotides through various pathways. These include extracellular
uptake, de novo synthesis and the delivery of carbon or
nitrogen via multiple routes (17,19).
The brain is a highly metabolically active organ
that relies on glucose as its major energy substrate. However,
lactate, ketone bodies, fatty acids (FAs) and amino acids can also
serve as its energy source (20-22). In addition, astrocytes, neurons
and microglia can all regulate the nutrient uptake processes of
each other (18). Specifically,
neurons can absorb lactate, cholesterol and FAs produced by
astrocytes, whilst astrocytes can take up glutamate produced by
neurons (20). Gliomas develop
in a complex and frequently hypoxic environment, which
significantly influences the metabolic decisions of glioma cells,
driving tumor growth, reproduction and invasion (23-26).
Microglia are macrophages that reside in the central
nervous system (CNS) and are distributed throughout the brain. They
serve as the key immune effector cell type in the CNS. GAMs
typically originate from two cell types, namely brain-resident
microglia (BRM) and bone marrow-derived monocytes (BMDM) (27). The debate over the origin of
microglia remains to the present day after it was first proposed by
del Rio-Hortega (28). It has
been suggested that increased microglial density after CNS injury
involves both BRM proliferation and active recruitment of BMDM
progenitors from the bloodstream (29-32). By contrast, it has also been
suggested that the increase in microglial density originates
primarily from the BRM (33).
The reason for this controversy may lie in the experimental
methodology used. To avoid the influence of the blood-brain
barrier, the method used to distinguish microglia from monocytes is
to first destroy the hematopoietic system of the recipient's bone
marrow with radiation and then transplant the labeled hematopoietic
stem cells into the recipient, before observing the infiltration of
the labeled monocytes into the tumor tissue of the brain. However,
irradiation can damage the blood-brain barrier in mice whilst
disrupting the immune system and non-specific infiltration of
immune cells into the brain, compromising the accuracy of the
experiment (34). This debate
continued until it was resolved when a chimeric animal was
generated by a form of heterologous symbiosis that required neither
irradiation nor transplantation. Both axotomy and neurodegeneration
models failed to recruit microglia from the circulation (33). In addition, similar results were
observed in a mouse model of experimental allergic
encephalomyelitis (35).
In high-grade gliomas, BMDM accounts for >85% of
GAMs, whereas BRM is predominantly distributed in peritumoral
tissues (36,37). In the past, the expression levels
of CD45 were typically used to differentiate between BRM (CD45 high
expression)-derived and BMDM (CD45 low expression)-derived GAMs
(38). However, different views
have emerged in recent years. A previous study has shown that
although the expression level of CD48 can distinguish BRM-GAMs from
BMDM-GAMs to a certain extent, the cell type-specific CD45
expression profiles of humans and mice are different. In addition,
the differentiation effect of CD45 is not precise, necessitating
the use of more sensitive and specific methods, such as RNA
sequencing and flow cytometry, to accurately distinguish between
BRM-derived and BMDM-derived GAMs (39).
A large-scale RNA sequencing analysis has previously
revealed the existence of BRM-derived GAMs and BMDM-derived GAMs
with distinct gene expression patterns. In particular,
subpopulations of GAMs from different origins may perform different
functions (36). Another
previous study found that transmembrane protein 119 (TMEM119) was
stably expressed only in BRM-derived GAMs. Subsequently,
RNA-sequencing was performed in this previous study based on the
expression profile of TMEM119 and differences in the transcript
fragments of BRM- and BMDM-derived GAMs were found. It was also
observed that the gene expression pattern of BRM may differ at
different stages of development, such that, as microglia mature,
the expression of their specifically expressed genes (such as
TMEM119, purinergic receptor P2Y12 and olfactomedin-like 3)
increases, but their proliferative capacity decreases (40). Using genealogical tracer
techniques and a mouse model of glioma, Bowman et al
(39) previously found that the
transcriptional profiles and epigenetic landscapes between the two
major subgroups of GAMs differed markedly, whereby CD49d was
proposed as a distinguishing marker. Furthermore, Müller et
al (41) previously
performed single-cell sequencing on clinical glioma specimens and
found that the levels of immunosuppressive cytokines, M2 activation
markers (IL-10 and TGF-βII), phagocytosis and tricarboxylic acid
(TCA) cycle activity were all upregulated in BMDM compared with
those in BRM.
Macrophages and microglia belong to the same
monocyte type. Therefore, they can have a high degree of diversity
and plasticity, allowing them to exhibit various phenotypes when
exposed to different in vitro stimuli (7). Stimulation with Toll-like receptor
(TLR)4 ligands and IFN-γ typically produces the pro-inflammatory M1
phenotype, whereas stimulation with IL-4, IL-10 and IL-13 produces
an anti-inflammatory M2 phenotype (8). In a previous study, RNA microarray
was applied to compare the expression profiles of microglia,
macrophages and control microglia obtained by CD11b
antibody-mediated magnetic beads sorting. The results showed that
~1,000 transcripts were differentially expressed in GAMs twice or
more compared with those in control microglia. This expression
pattern overlapped only partially with the reported gene profiles
of the M1, M2a, M2b and M2c phenotypes (42). It has also been shown that GAMs
can exhibit a different expression profile from the M1 and M2
phenotypes whilst highly expressing glycoprotein non-metastatic
melanoma protein B and secreted phosphoprotein 1.
According to previous histological investigations
that focused on single cells, the characteristics of GAMs are not
limited to only M1 and M2 phenotypes. Instead, a wide range of
variations have been noted. At present, no one superior typing
method has been found compared with M1/M2. Since the majority of
the relevant studies have continued to concentrate on M1/M2,
discussion of data related to this topic will also center around
M1/M2. GAMs express a number of markers that characterize the M1 or
M2 phenotype (7). It has been
previously shown that glioma-derived macrophage colony-stimulating
factor (CSF) can induce microglia and macrophages to shift into an
M2 phenotype, thereby promoting tumor growth (43). Similarly, mTOR and CSF-1 was
found to inhibit microglia transformation into the M1 phenotype
(43,44). Dopamine, microRNA (miR)-142-3p,
prolyl 4-hydroxylase subunit α 1 downregulation and anti-programmed
cell death protein 1 also showed similar anti-tumor effects
(45-48). Based on these previous
aforementioned studies, it has been proposed that targeted therapy
aiming at converting the M2 phenotype into the M1 phenotype is a
potential therapeutic strategy to inhibit glioma growth. However,
other previous studies have also shown that M1-specific markers or
associated pathways (IL1-β) are positively associated with glioma
growth (49). It has also been
indicated that sterile α and HEAT/armadillo motif can inhibit
glioma progression by inducing the M2 polarization of GAMs
(50).
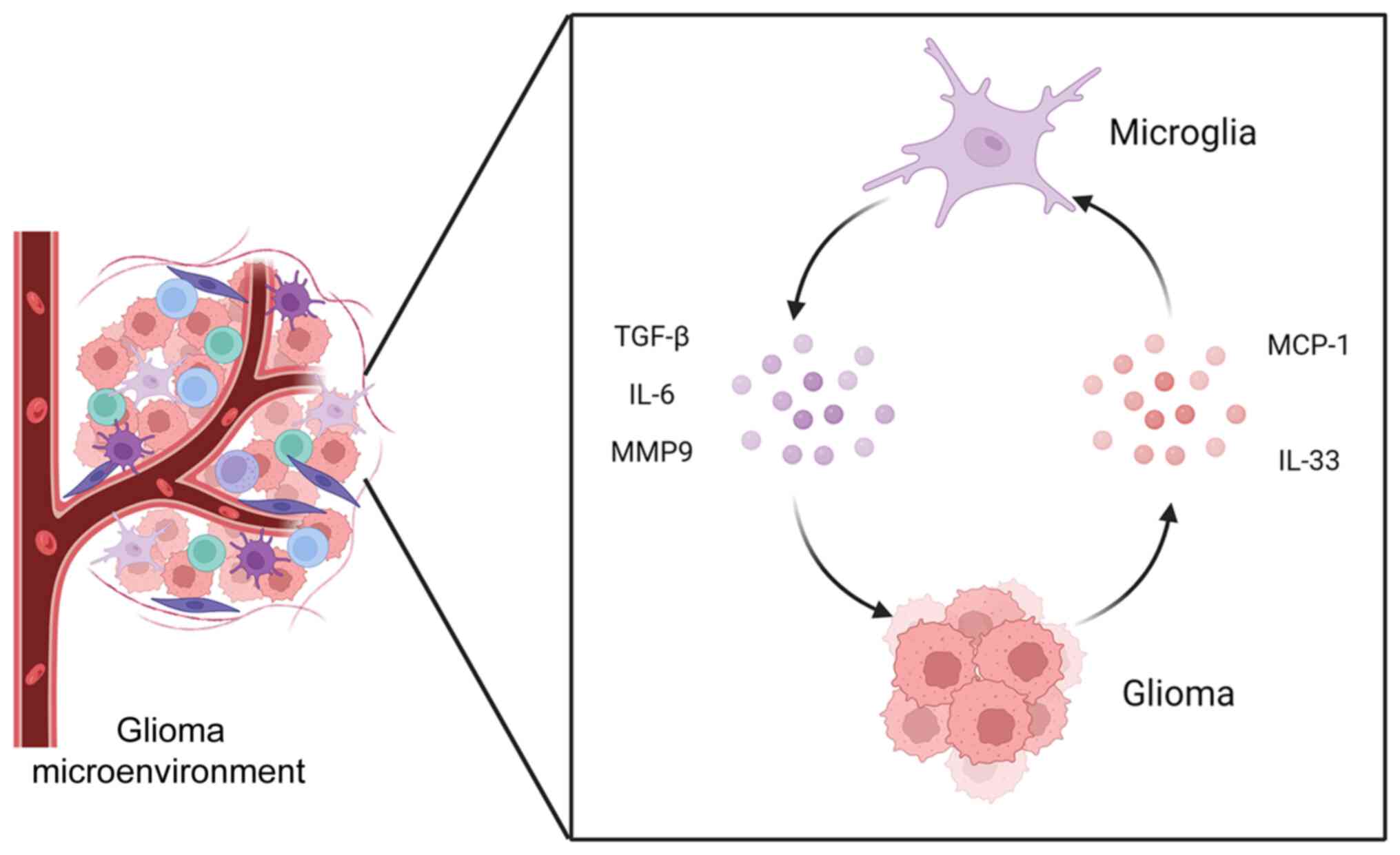
In the glioma microenvironment, microglia act
through two main mechanisms. Microglia first become active upon
glioma stimulation, producing cytokines, growth factors and MMPs to
promote tumor growth and invasion (51). Subsequently, tumor cells secrete
chemotactic agents and chemokines to recruit another population of
microglia for activation, creating a continuous cycle (7,52,53). It has been previously shown that
several common chemokines and receptors are upregulated in gliomas,
including monocyte chemoattractant protein-1 (MCP-1),
granulocyte-macrophage (GM)-CSF and fractalkine (54). MCP-1 has been considered to serve
a key role in recruiting microglia to gliomas, where IL-33 may also
be involved (Fig. 1) (51,54,55). In addition, microglia can have an
important effect on angiogenesis, an effect associated with VEGF,
which stimulates angiogenesis and promotes tumor growth (8). It has also been shown that
inflammation is a key factor in brain tumor progression.
Inflammation leads to the production of chemokines, such as
C-X-C-motif chemokine ligand (CXCL)12, CXCL18 and reactive oxygen
species (ROS), which promote tumor development by damaging DNA,
proteins and lipids (56).
Another previous study showed that the programmed cell death 10
protein serves an important role in the CXCL2/C-X-C chemokine
receptor type 2 signaling pathway (57).
Lipid metabolic reprogramming is a major feature of
tumorigenesis and progression, serving a crucial role in GAMs.
Initially, during tumor development, GAMs exhibit an M1 phenotype.
However, as the tumor progresses, GAMs predominantly show the M2
phenotype. This metabolic reprogramming has been indicated to be
regulated by hypoxia-inducible factor 1α and its downstream
components (58-64). However, due to the limited
availability of pertinent studies on gliomas, this section will
discuss lipid metabolic reprogramming in TAMs of other tumor types.
From these insights, the potential impact of GAMs of gliomas will
be speculated.
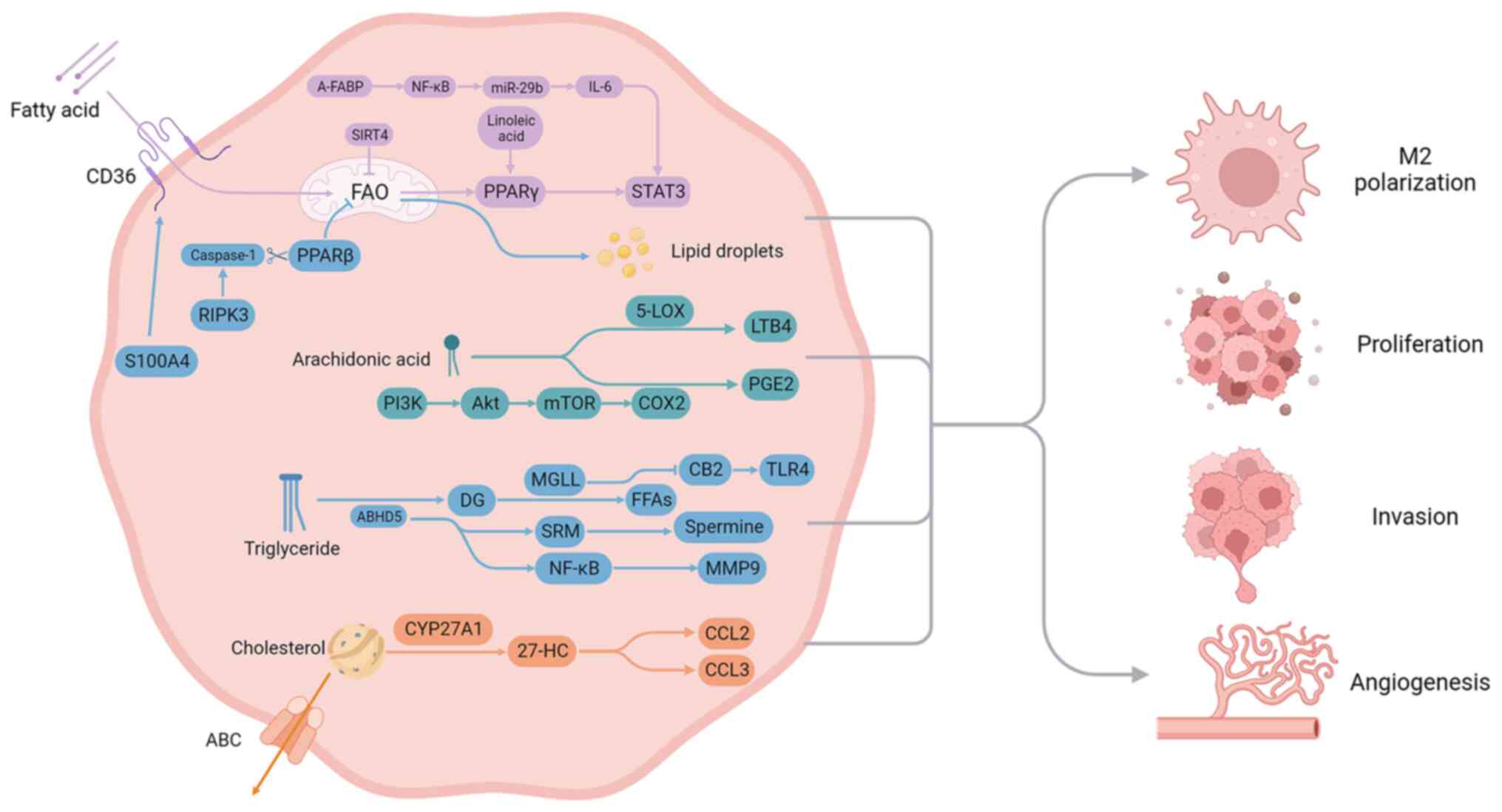
Altered FA metabolism in tumor cells increases lipid
accumulation in the TAM, which in turn promotes TAM activation and
polarization (65). It has been
previously found that M2 polarization is associated with FA
oxidation (FAO). The scavenger receptor CD36 is highly expressed in
TAMs, through which they take up and accumulate lipids (66). Results from a previous in
vivo experiment corroborated this finding, where TAMs from
tumor-bearing mice were found to have a higher lipid content
compared with macrophages from tumor-free mice (67). High levels of FAO can promote
mitochondrial oxidative phosphorylation and downstream signaling,
accompanied by activation of the TCA cycle, which in turn promotes
the M2 polarization of TAMs (15,60,66,68-70). Other previous studies have also
shown that the metabolic efficiency of FAO serves an important role
in regulating the polarization of TAMs, whereby β-oxidation is
closely associated with the phenotype of TAMs (60,70). The peroxisome
proliferator-activated receptor (PPAR) system regulates FAO and
significantly influences the metabolic reprogramming of TAMs and
their polarization towards the M2 phenotype (71,72). Specifically, the PPAR system
enhances FAO metabolic efficiency mediated by STAT6 and PPARγ
coactivator-1β (73,74).
However, other potentially noteworthy pathways have
not been intensively studied. In particular, IFN-γ, GM-CSF and
lipopolysaccharide (LPS) are factors that can induce M1
polarization (75). Previous
studies have shown that there may be associations between the
aforementioned factors and the FAO (76,77). In addition, proposals of
regulating FAO by targeting IFN-γ, GM-CSF and LPS to in turn
achieve a desired anti-tumor effect have been made (78-81).
PPARβ/δ can promote TAM polarization toward M2,
tumor invasion and angiogenesis. A previous lipidomic analysis of
ovarian cancer ascites has revealed that high concentrations of
polyunsaturated FAs (PUFAs), particularly linoleic acid, can
function as potent PPARβ/δ agonists in macrophages, thereby
promoting the M2 polarization of TAMs (85). Sirtuin 4 (SIRT4) is a member of
the SIRT family that can regulate cell proliferation and
metabolism. It has been previously shown that upon downregulation
of SIRT4 in human hepatocellular carcinoma, TAMs can activate the
FAO/PPARβ/δ-STAT3 signaling pathway, which leads to M2 polarization
(86,87).
A series of studies have shown that the intact
structural PPAR system is required for the regulation of FAO.
Caspase-1 activation generates a 41-kDa PPARγ fragment by cleaving
PPARγ on Asp64. This fragment can then enter the mitochondria and
inhibit medium-chain acyl-CoA dehydrogenase activity, reducing the
efficiency of FAO and leading to lipid droplet accumulation, which
in turn promotes M2 polarization (88-90). In addition, receptor-interacting
protein kinase 3 (RIPK3) is another key factor mediating macrophage
necrosis. It has been shown that in human and mouse hepatocellular
carcinoma tissues, downregulation of RIPK3 can inhibit
caspase-1-mediated PPAR cleavage, promote FAO, polarize TAMs toward
the M2 phenotype and enhance tumor immunosuppression (89).
In addition, the FA binding protein (FABP) family
serves another important role in FA metabolism, where its
intracellular localization is involved in glioma progression
(91). It has been previously
shown that epidermal FABP is significantly overexpressed in mouse
mammary carcinoma TAMs, which promotes the production of IFN-β by
modulating lipid droplets, thereby recruiting immune effector cells
and inhibiting tumor progression (92,93). By contrast, adipocyte/macrophage
FABP is highly expressed in mouse and human breast cancer TAMs,
where it promotes breast cancer cell proliferation and metastasis
through the NF-κB/miR-29b/IL-6/STAT3 pathway (92,94). This suggests the different roles
of different subtypes of FABP in cancer, where some types can
promote tumor growth and metastasis, whilst others have oncolytic
effects.
CD36 is a scavenger receptor that mediates lipid
uptake, immune recognition, inflammation, molecular adhesion and
apoptosis. This protein is a transmembrane glycoprotein and can
bind to a variety of ligands, including FAs, to exert its effects
(95). TAMs highly express CD36
and extensively utilize FAO for their energy supply. This process
promotes mitochondrial oxidative phosphorylation and the production
of ROS, leading to the activation of STAT6 and modulation of TAM
polarization (66). S100A4 is
another well-established pre-metastatic oncoprotein that is
primarily expressed by macrophages in the tumor microenvironment.
S100A4 has been shown to enhance CD36-mediated FA uptake through
the PPARγ pathway, thereby promoting and polarizing TAMs towards
the M2 phenotype (96).
Overall, the aforementioned previous observations
have demonstrated that FA metabolism serves a role in promoting M2
polarization in TAMs to a certain degree. Therefore, it can be
hypothesized that a comparable phenomenon may be present in GAMs.
However, it must be emphasized that the existing body of
experimental evidence on GAMs is insufficient to substantiate such
a hypothesis. Further studies in this area are required for further
advancement.
Arachidonic acid (AA) is a membrane phospholipid
produced by phospholipase A2 and is released into the cytosol.
Known enzymes involved in AA metabolism include cytochrome P450,
cyclooxygenase (COX) and lipoxygenase (LOX), which breaks AA down
into hydroxyeicosatraenoic acids, prostaglandins and leukotrienes,
respectively (97). In addition,
AA or phospholipid metabolism in TAM mainly regulates the immune
escape and proliferation of tumor cells.
A previous study has shown that TAMs can increase
the expression of COX2 and prostaglandin E2 (PGE2) through the
PI3K/Akt/mTOR pathway, leading to tamoxifen resistance and enhanced
endocrine resistance in breast cancer (98). Meanwhile, PGE2 stimulates
angiogenesis, suppresses immune function, promotes cancer cell
migration and inhibits CD80 expression on tumor-associated
phagocytes, thereby promoting cancer progression (99,100). TAM-derived osteopontin binds to
α9β1 integrins, which upregulates COX2 expression, then increases
the expression of PGE2 and MMP9 and accelerates angiogenesis
(101). Another study showed
that blocking the microsomal prostaglandin E synthase-1 and COX2
promoted TAM polarization toward M2 in colon cancer, thereby
inhibiting tumor progression (102). Furthermore, it has also been
previously shown that 5-LOX serves an important role in TAMs. In a
metastatic lung cancer model, 5-LOX-expressing macrophages were
observed to promote tumor cell proliferation by upregulating
leukotriene B4 expression, whereas 5-LOX-suppressed macrophages
exhibited reduced tumor proliferation (103,104). Similarly, reduced 5-LOX
expression in human breast cancer TAMs can lead to decreased
leukotriene synthesis and reduced effector T-cell recruitment,
thereby promoting tumor progression (105).
Triglycerides are produced by the esterification of
three hydroxyl groups on glycerol with three long-chain FA
molecules and are involved in anabolism and catabolism. Anabolism
is mainly regulated by diacylglycerol O-acyltransferases and
monoacylglycerol O-acyltransferases, whilst catabolism is mainly
regulated by hormone-sensitive lipase, abhydrolase
domain-containing (ABHD)5, adipose triglyceride lipase and
monoglyceride lipase (MGLL) (106,107). It has been shown that TAM can
affect tumor development by regulating triglyceride metabolizing
enzymes, where ABHD and MGLL serve a key role in this process.
ABHD is a key enzyme in triglyceride catabolism
whilst also being able to inhibit autophagy and apoptosis in tumor
cells (108). A previous study
found that ABHD5 can promote the expression of spermine synthase
(SRM) in TAMs of human and mouse colon cancer tissues. Spermine
promotes apoptosis in tumor cells. Single-cell sequencing results
also showed that high expression of ABHD5 in TAMs can promote tumor
growth. Therefore, targeting the ABHD5/SRM/spermine axis in TAM may
serve as a potential therapeutic strategy for colon cancer
(109). Furthermore, it has
been shown that ABHD5 in TAMs can increase MMP9 expression through
the NF-κB pathway, thereby promoting the lung metastasis of
colorectal cancer (110).
MGLL is another important component of the
triglyceride catabolic pathway, which hydrolyzes triglycerides into
free FAs. It has been previously found in mouse models of colon and
breast cancer that MGLL deficiency can cause lipid accumulation in
TAMs and promotes endocannabinoid receptor-2/TLR4 activation in
TAMs, which enhances immunosuppression and promotes tumor
progression (111).
Cholesterol is an important component of biological
membranes. It regulates cell membrane fluidity and participates in
various signaling pathways as a solubilizer of other lipids.
Cholesterol metabolic reprogramming in TAMs has been previously
shown to serve an important role in tumor development through TAM
activation and recruitment, whilst promoting M2 polarization. It
has been indicated that cholesterol metabolic reprogramming in TAMs
mainly focuses on the alteration of the cholesterol efflux pathway.
Therefore, targeting cholesterol efflux may be a potential method
of controlling or treating cancer (112,113).
An ATP-binding cassette transporter (ABC) is a type
of ATP-powered pump that consists of two transmembrane structural
domains and two ATP-binding domains on the cytoplasmic side. ABC
proteins scavenge surplus cholesterol within cells and regulate the
balance of cholesterol to maintain homeostasis (84). Various cancers have been found to
have elevated cholesterol levels (114). A recent study discovered that
Apolipoprotein A (ApoA1) can enhance the removal of cholesterol
from GAMs, decreasing intracellular cholesterol levels; this
process was found to activate CD8+ T cells, enhancing anti-tumor
immunity in a mouse GBM model (115). In another study, it was
discovered that ABC-mediated cholesterol efflux from TAM membranes
facilitated M2 polarization in a mouse model of metastatic ovarian
cancer; this in turn led to IL-4-associated immunosuppression and
invasive metastasis, whilst also inhibiting the IFN-γ-induced
antitumor effects (116).
Studies on bladder cancer and melanoma mouse models have also
revealed a similar phenomenon, whereby ABCG1 can facilitate the
removal of cholesterol to control the balance of cholesterol within
cells. The absence of ABCG1 in mice led to activation of the NF-κB
pathway and a transformation of macrophages from M2 to M1,
resulting in enhanced direct cytotoxic effects on tumors, which
hinders the growth of malignancies (114). However, further studies are
required to clarify the effects of cholesterol metabolism in a cell
type-specific context.
27-Hydroxycholesterol (27-HC) is a major metabolite
of cholesterol that is catalyzed by its cytochrome P450 oxidase
(CYP27A1). CYP27A1 is highly expressed in M2 macrophages and
activates M2 polarization, thereby promoting tumor progression
(117). It has been previously
found that CYP27A1 is highly expressed in mouse breast cancer TAMs,
whilst the 27-HC catabolic enzyme CYP27B1 is not expressed at high
levels in breast cancer cells, a setup that results in the
accumulation of 27-HC in the tumor cells. This accumulation of
27-HC in turn promotes the proliferation of the tumor cells and
facilitates the expression of several chemokines by the TAMs,
including chemokine (C-C motif) ligand (CCL)2 and CCL3, which then
recruit monocytes to the tumor site to promote tumor progression
(118).
The lipid metabolic reprogramming in TAMs and its
regulatory mechanism for tumor progression are shown in Fig. 2.
The role of SCFAs in macrophages in inflammation has
been extensively studied, but their role in TAMs has remained
elusive. Therefore, this section will focus on their role in
inflammation and, by extension, their role in tumor regulation.
During the inflammatory response, SCFAs can mediate
both pro-inflammatory and anti-inflammatory effects. This
phenomenon may be due to the expression and local concentration
profiles of the different SCFA receptors and SCFAs themselves,
respectively. It has been previously shown that in macrophages,
SCFAs (namely butyrate) can bind to and activate free FA receptor
(FFAR)3 to downregulate the levels of proinflammatory factors
(including inducible nitric oxide synthase, TNF, MCP-1 and IL-6),
thereby exerting anti-inflammatory effects (119,120). In addition, during airway
inflammation, SCFAs can downregulate IL-8 expression by targeting
FFAR2 and FFAR3 in macrophages, thereby exerting an
anti-inflammatory effect and improving patient symptoms (121). These results suggest that SCFAs
can exert potent anti-inflammatory effects that are realized
through FFAR2 and FFAR3. Therefore, inhibitors of FFAR2/3 may
mediate both proinflammatory and anti-tumor effects, to inhibit
tumor progression. However, other studies have found that SCFAs can
exert proinflammatory effects. It has been previously found that
when FFAR2/3 is activated, it further activates mTOR, PI3K and MAPK
signaling pathways downstream to mediate proinflammatory effects
(122,123). In addition, SCFAs (acetate)
were found to upregulate the production of proinflammatory
cytokines and chemokines, such as CXCL1/2 and IL-6, by activating
FFAR3/FFAR2 and ERK1/2 downstream. These aforementioned studies
suggest that SCFAs can have opposite roles in inflammation. This
phenomenon is associated with the local concentration of SCFAs.
It has been demonstrated that SCFAs can also
regulate inflammation through the binding of hydroxycarboxylic acid
receptor 2 (GPR109A), a butyrate receptor present in intestinal
epithelial cells and immune cells that serves an important role in
inflammation and immunity. Stimulation with IFN-γ has been shown to
upregulate GPR109A expression in macrophages (124). GPR109A activation is involved
in IL-8 and IL-10 production downstream, which affects regulatory T
cells to reduce inflammation (125-128). Therefore, GPR109A may exert
anti-inflammatory and immunomodulatory effects. However, to the
best of our knowledge, there are relatively few relevant studies in
TAMs, meaning that the relationship between GPR109A and tumors
requires further exploration.
SCFAs can not only exert anti-inflammatory effects
through signaling but also participate in inflammatory regulation
by inhibiting histone deacetylase (HDAC). In macrophages, SCFAs
(propionate and butyrate) have been observed to exert
anti-inflammatory effects by inhibiting the TNF and NF-κB signaling
pathways, in addition to inhibiting HDAC and promoting IL-10
production (129-131). However, it remains elusive
which HDAC is inhibited and further studies are warranted.
To conclude, SCFAs serve a role in controlling
inflammation in macrophages mainly through two primary modes of
behavior. One method likely involves attaching to G protein-coupled
receptors (such as FFAR2/3 and GPR109A) to trigger signaling
pathways further downstream. Another method may involve the
inhibition of HDAC once it has entered the cell, resulting in
anti-inflammatory effects. Therefore, it would be of benefit to
study whether a similar mechanism exists in GAMs in future studies,
where SCFAs can potentially exert anti-inflammatory effects in
addition to promoting tumor growth.
Omega-3 FAs are a family of long-chain PUFAs that
also includes α-linolenic acid, eicosapentaenoic acid (EPA) and
docosahexaenoic acid (DHA). G-protein coupled receptor 120 (GPR120)
is a G-protein coupled receptor that is involved in the regulation
of metabolic, endocrine and immune functions. GPR120 can be
activated by long-chain PUFAs (132). It has been previously shown
that GPR120 is highly expressed in adipose tissue and
proinflammatory macrophages in mice fed a high saturated fat diet,
where the use of fish oil containing DHA and EPA can exert
anti-inflammatory effects through GPR120 (133). In addition, mice fed an omega-3
diet had a significant reduction in the number of M2-like TAMs and
expression of M2-associated cytokines, chemokines and growth
factors in tumor tissues, compared with those in mice fed on an
omega-6 diet (134). Data from
another previous study corroborated this finding, as DHA was found
to combine with ethanolamine to generate DHEA in breast cancer
cells, reducing the secretion of CCL5 and affecting TAM recruitment
and tumor progression. In addition, omega-3 FAs have been found to
inhibit prostate cancer progression through a variety of
mechanisms, including inhibition of COX2-mediated PGE2 formation,
LOX activity, TLRs, formation of pro-resolvin metabolites,
activation of PPARγ and inhibition of NF-κB (135).
Traditional views suggest that M1-like TAMs prefer
glycolysis as an energy source, whilst M2-like TAMs favor FAO
(66,136). Therefore, regulating FAO may
offer a strategy to inhibit tumor progression.
S100A4 is a well-known pre-metastatic oncoprotein
that is primarily expressed by macrophages in the tumor
microenvironment. S100A4 can enhance CD36-mediated FA uptake
through the PPARγ pathway, promoting the polarization of TAMs
towards the M2 phenotype (96).
Previous studies have shown that injecting S100A4-knockout
macrophages can significantly reduce tumor growth in mice (71). Similarly, using VT1021 to block
CD36 lipid uptake was found to inhibit the M2 polarization of TAMs,
thereby suppressing cancer (137,138).
The caspase-1/PPARγ/FAO axis is another crucial
target for cancer therapy. A study has previously found that the
caspase-1 inhibitor (Tyr-Val-Ala-Asp) can inhibit breast cancer
progression by blocking caspase-1-mediated PPARγ cleavage (90). In addition, RIPK3 deficiency was
found to inhibit caspase-1-mediated cleavage of PPARα and PPARγ in
TAMs, leading to increased FAO and promoting hepatocellular
carcinoma. This process can be inhibited by using the RIPK3 agonist
decitabine (90). It has also
been shown that lipid metabolism can be reprogrammed and TAM
polarization reversed by blocking FAO using etomoxir (66,139).
Rofecoxib, a specific COX-2 inhibitor, has been
documented to restore the adhesion and antitumor activity of TAMs
(140). Celecoxib was also
observed to exert similar effects (141). Another study previously showed
that a selective COX-2 inhibitor, LM-1685, significantly reduced
the level of arginase 1 in M2 macrophages, thereby inhibiting tumor
progression (142).
Zileuton, a 5-LOX inhibitor, was found to decrease
MMP7 expression whilst reducing TAM migration and infiltration
(143,144).
A recombinant tumor lysing adenovirus carrying ApoA1
was previously designed to overexpress ApoA1 in the tumor
microenvironment. This led to an increase in cholesterol removal
from GAMs and a significant decrease in cholesterol levels within
GAMs. As a result, GAMs were able to regain their ability to engulf
and present antigens, which enhanced the effectiveness of
CD8+ T cells in eliminating GBM. Furthermore, this
treatment also induced a long-lasting immune response (115). In another study, ATR101, an
inhibitor of ABC, was found to inhibit the M2 polarization of TAM
by inhibiting cholesterol efflux from TAM, leading to cholesterol
accumulation in cells (145).
Therapies that have been experimentally proven to be
feasible through metabolic modulation are listed in Table I.
However, to the best of our knowledge, few therapies
targeting the metabolic pathway of GAMs are available at present
and the aforementioned drugs have not yet been tested in clinical
trials in patients with glioma or animal models of glioma. It
remains speculative whether similar drugs may be effective in GAMs.
Further research is needed to explore these possibilities.
GAMs play an important role in the tumor
microenvironment, influencing glioma growth, invasion and
angiogenesis through specific signaling molecules like MCP-1,
GM-CSF and VEGF. They exhibit high plasticity, allowing them to
differentiate into various phenotypes under different stimuli,
adapting their metabolic pathways to support tumor progression.
While recent studies have highlighted significant
alterations in lipid metabolism within TAMs, literature on lipid
metabolic reprogramming in GAMs remains scarce. More in-depth
studies focusing on GAMs are essential to understanding their
unique metabolic adaptations and roles in glioma. Targeted
therapies modulating lipid metabolism in GAMs hold promise for
inhibiting tumor progression. Inhibiting FAO and targeting pathways
involving COX-2 and PGE2 have shown potential in preclinical
studies. Developing drugs specifically modulating GAM metabolic
pathways may provide more effective treatment options for
glioma.
Future research should focus on specific drug
development for GAMs, targeting lipid metabolism and other pathways
unique to these cells. Testing potential metabolic modulation drugs
in preclinical or clinical trials is urgently needed to evaluate
their efficacy and safety. Considering the complexity of tumor
metabolic pathways, multi-targeted therapeutic strategies may
enhance therapeutic outcomes by disrupting the tumor's metabolic
network comprehensively.
In addition, identifying and validating biomarkers
for monitoring metabolic reprogramming and therapeutic response is
crucial. Biomarkers will help optimize treatment regimens, improve
efficacy and provide critical information for personalized therapy.
Further research into GAM metabolic reprogramming mechanisms will
provide a foundation for developing novel therapeutic approaches,
potentially in combination with existing treatments like
immunotherapy and chemotherapy.
By addressing these areas, future research can
advance the understanding of GAM metabolism, leading to effective
therapies for glioma, ultimately improving patient outcomes.
Not applicable.
Writing-original draft preparation: YM and YH;
writing-review and editing: FH and KS; visualization: YM and YH;
supervision: FH and KS. All authors have read and agreed to the
published version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
This research was funded by the National Natural Science
Foundation of China (grant no. 82203683), the National Key Research
and Development Program of China (grant no. 2023YFC2510001) and the
Chinese Society of Clinical Oncology Foundation-Zai Lab Cancer
Treatment Research Foundation (grant no. Y-zai2021/qn-0217).
|
1
|
Laug D, Glasgow SM and Deneen B: A glial
blueprint for gliomagenesis. Nat Rev Neurosci. 19:393–403. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J, Leavenworth JW, Hjelmeland AB,
Smith R, Patel N, Borg B, Si Y and King PH: Deletion of the RNA
regulator HuR in tumor-associated microglia and macrophages
stimulates anti-tumor immunity and attenuates glioma growth. Glia.
67:2424–2439. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jiang Y, Marinescu VD, Xie Y, Jarvius M,
Maturi NP, Haglund C, Olofsson S, Lindberg N, Olofsson T,
Leijonmarck C, et al: Glioblastoma cell malignancy and drug
sensitivity are affected by the cell of origin. Cell Rep.
18:977–990. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ostrom QT, Gittleman H, Liao P,
Vecchione-Koval T, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: Primary brain and other central nervous
system tumors diagnosed in the United States in 2010-2014. Neuro
Oncol. 19(Suppl 5): V1–V88. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Uddin MS, Mamun AA, Alghamdi BS, Tewari D,
Jeandet P, Sarwar MS and Ashraf GM: Epigenetics of glioblastoma
multiforme: From molecular mechanisms to therapeutic approaches.
Semin Cancer Biol. 83:100–120. 2022. View Article : Google Scholar
|
|
6
|
Claus EB, Walsh KM, Wiencke JK, Molinaro
AM, Wiemels JL, Schildkraut JM, Bondy ML, Berger M, Jenkins R and
Wrensch M: Survival and low-grade glioma: The emergence of genetic
information. Neurosurg Focus. 38:E62015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hambardzumyan D, Gutmann DH and Kettenmann
H: The role of microglia and macrophages in glioma maintenance and
progression. Nat Neurosci. 19:20–27. 2016. View Article : Google Scholar :
|
|
8
|
Mantovani A, Sozzani S, Locati M, Allavena
P and Sica A: Macrophage polarization: Tumor-associated macrophages
as a paradigm for polarized M2 mononuclear phagocytes. Trends
Immunol. 23:549–555. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N,
Yi P, Tang L, Pan Q, Rao S, et al: The cancer metabolic
reprogramming and immune response. Mol Cancer. 20:282021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang K, Wang X, Song C, He Z, Wang R, Xu
Y, Jiang G, Wan Y, Mei J and Mao W: The role of lipid metabolic
reprogramming in tumor microenvironment. Theranostics.
13:1774–1808. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fernandez LP, Gomez de Cedron M and
Ramirez de Molina A: Alterations of lipid metabolism in cancer:
Implications in prognosis and treatment. Front Oncol.
10:5774202020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen JQ and Russo J: Dysregulation of
glucose transport, glycolysis, TCA cycle and glutaminolysis by
oncogenes and tumor suppressors in cancer cells. Biochim Biophys
Acta. 1826:370–384. 2012.PubMed/NCBI
|
|
15
|
Xiang Y and Miao H: Lipid metabolism in
tumor-associated macrophages. Adv Exp Med Biol. 1316:87–101. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vander Heiden MG and DeBerardinis RJ:
Understanding the intersections between metabolism and cancer
biology. Cell. 168:657–669. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Venneti S and Thompson CB: Metabolic
reprogramming in brain tumors. Annu Rev Pathol. 12:515–545. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bi J, Chowdhry S, Wu S, Zhang W, Masui K
and Mischel PS: Altered cellular metabolism in gliomas-an emerging
landscape of actionable co-dependency targets. Nat Rev Cancer.
20:57–70. 2020. View Article : Google Scholar
|
|
19
|
Pavlova NN and Thompson CB: The emerging
hallmarks of cancer metabolism. Cell Metab. 23:27–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Belanger M, Allaman I and Magistretti PJ:
Brain energy metabolism: Focus on astrocyte-neuron metabolic
cooperation. Cell Metab. 14:724–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Magistretti PJ and Allaman I: A cellular
perspective on brain energy metabolism and functional imaging.
Neuron. 86:883–901. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zielke HR, Zielke CL and Baab PJ: Direct
measurement of oxidative metabolism in the living brain by
microdialysis: A review. J Neurochem. 109(Suppl 1): S24–S29. 2009.
View Article : Google Scholar
|
|
23
|
Kaur B, Khwaja FW, Severson EA, Matheny
SL, Brat DJ and Van Meir EG: Hypoxia and the
hypoxia-inducible-factor pathway in glioma growth and angiogenesis.
Neuro Oncol. 7:134–153. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kayama T, Yoshimoto T, Fujimoto S and
Sakurai Y: Intratumoral oxygen pressure in malignant brain tumor. J
Neurosurg. 74:55–59. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kucharzewska P, Christianson HC and
Belting M: Global profiling of metabolic adaptation to hypoxic
stress in human glioblastoma cells. PLoS One. 10:e01167402015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Z, Bao S, Wu Q, Wang H, Eyler C,
Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al:
Hypoxia-Inducible factors regulate tumorigenic capacity of glioma
stem cells. Cancer Cell. 15:501–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ricard C, Tchoghandjian A, Luche H, Grenot
P, Figarella-Branger D, Rougon G, Malissen M and Debarbieux F:
Phenotypic dynamics of microglial and monocyte-derived cells in
glioblastoma-bearing mice. Sci Rep. 6:263812016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
del Rio-Hortega P: The third element of
the nerve centers. Bulletin of the Spanish Society of Biology.
9:69–129. 1919.In Spanish.
|
|
29
|
Simard AR and Rivest S: Bone marrow stem
cells have the ability to populate the entire central nervous
system into fully differentiated parenchymal microglia. FASEB J.
18:998–1000. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Priller J, Flugel A, Wehner T, Boentert M,
Haas CA, Prinz M, Fernández-Klett F, Prass K, Bechmann I, de Boer
BA, et al: Targeting gene-modified hematopoietic cells to the
central nervous system: Use of green fluorescent protein uncovers
microglial engraftment. Nat Med. 7:1356–1361. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Flugel A, Bradl M, Kreutzberg GW and
Graeber MB: Transformation of donor-derived bone marrow precursors
into host microglia during autoimmune CNS inflammation and during
the retrograde response to axotomy. J Neurosci Res. 66:74–82. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hickey WF and Kimura H: Perivascular
microglial cells of the CNS are bone marrow-derived and present
antigen in vivo. Science. 239:290–292. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Massengale M, Wagers AJ, Vogel H and
Weissman IL: Hematopoietic cells maintain hematopoietic fates upon
entering the brain. J Exp Med. 201:1579–1589. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
De Leo A, Ugolini A and Veglia F: Myeloid
cells in glioblastoma microenvironment. Cells. 10:182020.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ajami B, Bennett JL, Krieger C, McNagny KM
and Rossi FM: Infiltrating monocytes trigger EAE progression, but
do not contribute to the resident microglia pool. Nat Neurosci.
14:1142–1249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen Z, Feng X, Herting CJ, Garcia VA, Nie
K, Pong WW, Rasmussen R, Dwivedi B, Seby S, Wolf SA, et al:
Cellular and molecular identity of tumor-associated macrophages in
glioblastoma. Cancer Res. 77:2266–2278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Landry AP, Balas M, Alli S, Spears J and
Zador Z: Distinct regional ontogeny and activation of tumor
associated macrophages in human glioblastoma. Sci Rep.
10:195422020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Xu C, Xiao M, Li X, Xin L, Song J, Zhan Q,
Wang C, Zhang Q, Yuan X, Tan Y and Fang C: Origin, activation, and
targeted therapy of glioma-associated macrophages. Front Immunol.
13:9749962022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bowman RL, Klemm F, Akkari L, Pyonteck SM,
Sevenich L, Quail DF, Dhara S, Simpson K, Gardner EE,
Iacobuzio-Donahue CA, et al: Macrophage ontogeny underlies
differences in tumor-specific education in brain malignancies. Cell
Rep. 17:2445–2459. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bennett ML, Bennett FC, Liddelow SA, Ajami
B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A,
Tucker A, et al: New tools for studying microglia in the mouse and
human CNS. Proc Natl Acad Sci USA. 113:E1738–E1746. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Müller S, Kohanbash G, Liu SJ, Alvarado B,
Carrera D, Bhaduri A, Watchmaker PB, Yagnik G, Di Lullo E,
Malatesta M, et al: Single-cell profiling of human gliomas reveals
macrophage ontogeny as a basis for regional differences in
macrophage activation in the tumor microenvironment. Genome Biol.
18:2342017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Szulzewsky F, Pelz A, Feng X, Synowitz M,
Markovic D, Langmann T, Holtman I R, Wang X, Eggen BJ, Boddeke HW,
et al: Glioma-Associated microglia/macrophages display an
expression profile different from M1 and M2 polarization and highly
express Gpnmb and Spp1. PLoS One. 10:e01166442015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pyonteck SM, Akkari L, Schuhmacher AJ,
Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT,
Teijeiro V, et al: CSF-1R inhibition alters macrophage polarization
and blocks glioma progression. Nat Med. 19:1264–1272. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lisi L, Laudati E, Navarra P and Dello
Russo C: The mTOR kinase inhibitors polarize glioma-activated
microglia to express a M1 phenotype. J Neuroinflammation.
11:1252014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin T, Wang C, Chen X, Duan C, Zhang X,
Zhang J, Chai H, Tang T, Chen H, Yue J, et al: Dopamine induces
growth inhibition and vascular normalization through reprogramming
M2-polarized macrophages in rat C6 glioma. Toxicol Appl Pharmacol.
286:112–123. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xu S, Wei J, Wang F, Kong LY, Ling XY,
Nduom E, Gabrusiewicz K, Doucette T, Yang Y, Yaghi NK, et al:
Effect of miR-142-3p on the M2 macrophage and therapeutic efficacy
against murine glioblastoma. J Natl Cancer Inst. 106:dju1622014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Q, Zhang J, Fang S, Wang J, Han X,
Liu F and Jin G: P4HA1 down-regulation inhibits glioma invasiveness
by promoting M1 microglia polarization. Onco Targets Ther.
14:1771–1782. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rao G, Latha K, Ott M, Sabbagh A,
Marisetty A, Ling X, Zamler D, Doucette TA, Yang Y, Kong LY, et al:
Anti-PD-1 Induces M1 polarization in the glioma microenvironment
and exerts therapeutic efficacy in the absence of CD8 Cytotoxic T
cells. Clin Cancer Res. 26:4699–4712. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Feng X, Szulzewsky F, Yerevanian A, Chen
Z, Heinzmann D, Rasmussen RD, Alvarez-Garcia V, Kim Y, Wang B,
Tamagno I, et al: Loss of CX3CR1 increases accumulation of
inflammatory monocytes and promotes gliomagenesis. Oncotarget.
6:15077–15094. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhou C, Li T, Dong Q, Liang H and Xu L:
SARM suppresses glioma progression in GL261 glioma cells and
regulates microglial polarization. Cell Biol Int. 46:1927–1936.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wei J, Gabrusiewicz K and Heimberger A:
The controversial role of microglia in malignant gliomas. Clin Dev
Immunol. 2013:2852462013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gutmann DH and Kettenmann H:
Microglia/Brain macrophages as central drivers of brain tumor
pathobiology. Neuron. 104:442–449. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zeppellini A, Galimberti S, Leone BE,
Pacifico C, Riva F, Cicchiello F, Capici S, Maggioni C, Sala L and
Cazzaniga ME: Comparison of tumor microenvironment in primary and
paired metastatic ER+/HER2-breast cancers: Results of a pilot
study. BMC Cancer. 21:2602021. View Article : Google Scholar
|
|
54
|
Lailler C, Louandre C, Morisse MC,
Lhossein T, Godin C, Lottin M, Constans JM, Chauffert B, Galmiche A
and Saidak Z: ERK1/2 signaling regulates the immune
microenvironment and macrophage recruitment in glioblastoma. Biosci
Rep. 39:BSR201914332019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
De Boeck A, Ahn BY, D'Mello C, Lun X,
Menon SV, Alshehri MM, Szulzewsky F, Shen Y, Khan L, Dang NH, et
al: Glioma-derived IL-33 orchestrates an inflammatory brain tumor
microenvironment that accelerates glioma progression. Nat Commun.
11:49972020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Greten FR and Grivennikov SI: Inflammation
and cancer: Triggers, mechanisms, and consequences. Immunity.
51:27–41. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang Q, Wang J, Yao X, Wu S, Tian W, Gan
C, Wan X, You C, Hu F, Zhang S, et al: Programmed Cell Death 10
Mediated CXCL2-CXCR2 signaling in regulating tumor-associated
microglia/macrophages recruitment in glioblastoma. Front Immunol.
12:6370532021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Anagnostakis F and Piperi C: Targeting
options of tumor-associated macrophages (TAM) activity in gliomas.
Curr Neuropharmacol. 21:457–470. 2023. View Article : Google Scholar :
|
|
59
|
Sun L, Zhang H and Gao P: Metabolic
reprogramming and epigenetic modifications on the path to cancer.
Protein Cell. 13:877–919. 2022. View Article : Google Scholar :
|
|
60
|
Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W,
Huang X, Yan H, He J and Cai Z: Metabolic reprogramming in
macrophage responses. Biomark Res. 9:12021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Wang Y, Wang D, Yang L and Zhang Y:
Metabolic reprogramming in the immunosuppression of
tumor-associated macrophages. Chin Med J (Engl). 135:2405–2416.
2022.PubMed/NCBI
|
|
62
|
Muri J and Kopf M: Redox regulation of
immunometabolism. Nat Rev Immunol. 21:363–381. 2021. View Article : Google Scholar
|
|
63
|
Blouin CC, Pagé EL, Soucy GM and Richard
DE: Hypoxic gene activation by lipopolysaccharide in macrophages:
Implication of hypoxia-inducible factor 1alpha. Blood.
103:1124–1130. 2004. View Article : Google Scholar
|
|
64
|
Van den Bossche J, Baardman J, Otto NA,
van der Velden S, Neele AE, van den Berg SM, Luque-Martin R, Chen
HJ, Boshuizen MC, Ahmed M, et al: Mitochondrial dysfunction
prevents repolarization of inflammatory macrophages. Cell Rep.
17:684–696. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Liu S, Liu J, Ma Q, Cao L, Fattah RJ, Yu
Z, Bugge TH, Finkel T and Leppla SH: Solid tumor therapy by
selectively targeting stromal endothelial cells. Proc Natl Acad Sci
USA. 113:E4079–E4087. 2016.PubMed/NCBI
|
|
66
|
Su P, Wang Q, Bi E, Ma X, Liu L, Yang M,
Qian J and Yi Q: enhanced lipid accumulation and metabolism are
required for the differentiation and activation of tumor-associated
macrophages. Cancer Res. 80:1438–1450. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Luo Q, Zheng NS, Jiang L, Wang T, Zhang P,
Liu Y, Zheng P, Wang W, Xie G, Chen L, et al: Lipid accumulation in
macrophages confers protumorigenic polarization and immunity in
gastric cancer. Cancer Sci. 111:4000–4011. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Huang SC, Everts B, Ivanova Y, O'Sullivan
D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY,
O'Neill CM, et al: Cell-intrinsic lysosomal lipolysis is essential
for alternative activation of macrophages. Nat Immunol. 15:846–855.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Teng Y, Xu L, Li W, Liu P, Tian L and Liu
M: Targeting reactive oxygen species and fat acid oxidation for the
modulation of tumor-associated macrophages: A narrative review.
Front Immunol. 14:12244432023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kumar S, Mittal S, Gupta P, Singh M,
Chaluvally-Raghavan P and Pradeep S: Metabolic reprogramming in
tumor-associated macrophages in the ovarian tumor microenvironment.
Cancers (Basel). 14:52242022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu S, Zhang H, Li Y, Zhang Y, Bian Y,
Zeng Y, Yao X, Wan J, Chen X, Li J, et al: S100A4 enhances protumor
macrophage polarization by control of PPAR-γ-dependent induction of
fatty acid oxidation. J Immunother Cancer. 9:e0025482021.
View Article : Google Scholar
|
|
72
|
Zhou D, Ji L and Chen Y: TSPO Modulates
IL-4-Induced Microglia/Macrophage M2 Polarization via PPAR-gamma
Pathway. J Mol Neurosci. 70:542–549. 2020. View Article : Google Scholar
|
|
73
|
Dubey S, Ghosh S, Goswami D, Ghatak D and
De R: Immunometabolic attributes and mitochondria-associated
signaling of Tumor-Associated Macrophages in tumor microenvironment
modulate cancer progression. Biochem Pharmacol. 208:1153692023.
View Article : Google Scholar
|
|
74
|
Puthenveetil A and Dubey S: Metabolic
reprograming of tumor-associated macrophages. Ann Transl Med.
8:10302020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Shapouri-Moghaddam A, Mohammadian S,
Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi
A, Afshari JT and Sahebkar A: Macrophage plasticity, polarization,
and function in health and disease. J Cell Physiol. 233:6425–6440.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lee LY, Oldham WM, He H, Wang R, Mulhern
R, Handy DE and Loscalzo J: Interferon-ү impairs human coronary
artery endothelial glucose metabolism by tryptophan catabolism and
activates fatty acid oxidation. Circulation. 144:1612–1628. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Friedmann Angeli JP, Xavier da Silva TN
and Schilling B: CD8+ T cells PUF(A)ing the flames of
cancer ferroptotic cell death. Cancer Cell. 40:346–348. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yerrapragada MR and Mampallil D:
Interferon-γ detection in point of care diagnostics: Short review.
Talanta. 245:1234282022. View Article : Google Scholar
|
|
79
|
Lin CY, Chen WL, Huang YC, Lim CL and Yang
CH: Gum Arabic in combination with IFN-γ promotes the M1
polarization in macrophage. Int J Biol Macromol. 209:506–512. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Abdi K, Laky K, Abshari M, Hill EM, Lantz
L, Singh NJ and Long EO: Dendritic cells Trigger IFN-γ secretion by
NK cells independent of IL-12 and IL-18. Eur J Immunol.
52:1431–1440. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhao X, Peng T, Cao X, Hou Y, Li R, Han T,
Fan Z, Zhao M, Chang Y, Chen H, et al: In vivo G-CSF treatment
activates the GR-SOCS1 axis to suppress IFN-y secretion by natural
killer cells. Cell Rep. 40:1113422022. View Article : Google Scholar
|
|
82
|
Chawla A: Control of Macrophage Activation
and Function by PPARs. Circ Res. 106:1559–1569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Christofides A, Konstantinidou E, Jani C
and Boussiotis VA: The role of peroxisome proliferator-activated
receptors (PPAR) in immune responses. Metabolism. 114:1543382021.
View Article : Google Scholar
|
|
84
|
Qiao X, Hu Z, Xiong F, Yang Y, Peng C,
Wang D and Li X: Lipid metabolism reprogramming in tumor-associated
macrophages and implications for therapy. Lipids Health Dis.
22:452023. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Schumann T, Adhikary T, Wortmann A,
Finkernagel F, Lieber S, Schnitzer E, Legrand N, Schober Y, Nockher
WA, Toth PM, et al: Deregulation of PPARβ/δ target genes in
tumor-associated macrophages by fatty acid ligands in the ovarian
cancer microenvironment. Oncotarget. 6:13416–13433. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Fernandez-Marcos PJ and Serrano M: Sirt4:
The glutamine gatekeeper. Cancer Cell. 23:427–428. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li Z, Li H, Zhao ZB, Zhu W, Feng PP, Zhu
XW and Gong JP: SIRT4 silencing in tumor-associated macrophages
promotes HCC development via PPARδ signalling-mediated alternative
activation of macrophages. J Exp Clin Cancer Res. 38:4692019.
View Article : Google Scholar
|
|
88
|
Mojsilovic SS, Mojsilovic S, Villar VH and
Santibanez JF: The metabolic features of tumor-associated
macrophages: Opportunities for immunotherapy? Anal Cell Pathol
(Amst). 2021:55230552021.PubMed/NCBI
|
|
89
|
Wu L, Zhang X, Zheng L, Zhao H, Yan G,
Zhang Q, Zhou Y, Lei J, Zhang J, Wang J, et al: RIPK3 orchestrates
fatty acid metabolism in tumor-associated macrophages and
hepatocarcinogenesis. Cancer Immunol Res. 8:710–721. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Niu Z, Shi Q, Zhang W, Shu Y, Yang N, Chen
B, Wang Q, Zhao X, Chen J, Cheng N, et al: Caspase-1 cleaves PPARγ
for potentiating the pro-tumor action of TAMs. Nat Commun.
8:7662017. View Article : Google Scholar
|
|
91
|
McKillop LH, Girardi CA and Thompson KJ:
Role of fatty acid binding proteins (FABPs) in cancer development
and progression. Cell Signal. 62:1093362019. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhang Y, Sun Y, Rao E, Yan F, Li Q, Zhang
Y, Silverstein KA, Liu S, Sauter E, Cleary MP and Li B: Fatty
Acid-Binding Protein E-FABP restricts tumor growth by promoting
IFN-β responses in tumor-associated macrophages. Cancer Res.
74:2986–2998. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Furuhashi M and Hotamisligil GS: Fatty
acid-binding proteins: role in metabolic diseases and potential as
drug targets. Nat Rev Drug Discov. 7:489–503. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Hao J, Yan F, Zhang Y, Triplett A, Zhang
Y, Schultz DA, Sun Y, Zeng J, Silverstein KAT, Zheng Q, et al:
Expression of adipocyte/macrophage fatty acid-binding protein in
tumor-associated macrophages promotes breast cancer progression.
Cancer Res. 78:2343–2355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wang J and Li Y: CD36 tango in cancer:
Signaling pathways and functions. Theranostics. 9:4893–4908. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Nath A and Chan C: Genetic alterations in
fatty acid transport and metabolism genes are associated with
metastatic progression and poor prognosis of human cancers. Sci
Rep. 6:186692016. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Harizi H, Corcuff JB and Gualde N:
Arachidonic-acid-derived eicosanoids: Roles in biology and
immunopathology. Trends Mol Med. 14:461–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Qin Q, Ji H, Li D, Zhang H, Zhang Z and
Zhang Q: Tumor-associated macrophages increase COX-2 expression
promoting endocrine resistance in breast cancer via the
PI3K/Akt/mTOR pathway. Neoplasma. 68:938–946. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Rabold K, Netea MG, Adema GJ and
Netea-Maier RT: Cellular metabolism of tumor-associated
macrophages-functional impact and consequences. FEBS Lett.
591:3022–3041. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Olesch C, Sha W, Angioni C, Sha LK, Açaf
E, Patrignani P, Jakobsson PJ, Radeke HH, Grösch S, Geisslinger G,
et al: MPGES-1-derived PGE2 suppresses CD80 expression on
tumor-associated phagocytes to inhibit anti-tumor immune responses
in breast cancer. Oncotarget. 6:10284–10296. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kale S, Raja R, Thorat D, Soundararajan G,
Patil TV and Kundu GC: Osteopontin signaling upregulates
cyclooxygenase-2 expression in tumor-associated macrophages leading
to enhanced angiogenesis and melanoma growth via alpha9beta1
integrin. Oncogene. 33:2295–2306. 2014. View Article : Google Scholar
|
|
102
|
Nakanishi Y, Nakatsuji M, Seno H, Ishizu
S, Akitake-Kawano R, Kanda K, Ueo T, Komekado H, Kawada M, Minami M
and Chiba T: COX-2 inhibition alters the phenotype of
tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse
polyps. Carcinogenesis. 32:1333–1339. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nosaka T, Baba T, Naito T, et al:
Leukotriene B 4 generated by alveolar macrophages drive
hepatocellular carcinoma lung metastasis. Hepatology. 66:85A.
2017.
|
|
104
|
Hall Z, Ament Z, Wilson CH, Burkhart DL,
Ashmore T, Koulman A, Littlewood T, Evan GI and Griffin JL: Myc
expression drives aberrant lipid metabolism in lung cancer. Cancer
Res. 76:4608–4618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Ringleb J, Strack E, Angioni C,
Geisslinger G, Steinhilber D, Weigert A and Brüne B: Apoptotic
cancer cells suppress 5-lipoxygenase in tumor-associated
macrophages. J Immunol. 200:857–868. 2018. View Article : Google Scholar
|
|
106
|
Cheng C, Geng F, Cheng X and Guo D: Lipid
metabolism reprogramming and its potential targets in cancer.
Cancer Commun (Lond). 38:272018.PubMed/NCBI
|
|
107
|
Ou J, Miao H, Ma Y, Guo F, Deng J, Wei X,
Zhou J, Xie G, Shi H, Xue B, et al: Loss of Abhd5 promotes
colorectal tumor development and progression by inducing aerobic
glycolysis and epithelial-mesenchymal transition. Cell Rep.
9:1798–1811. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Yen CL, Nelson DW and Yen MI: Intestinal
triacylglycerol synthesis in fat absorption and systemic energy
metabolism. J Lipid Res. 56:489–501. 2015. View Article : Google Scholar :
|
|
109
|
Miao H, Ou J, Peng Y, Zhang X, Chen Y, Hao
L, Xie G, Wang Z, Pang X, Ruan Z, et al: Macrophage ABHD5 promotes
colorectal cancer growth by suppressing spermidine production by
SRM. Nat Commun. 7:117162016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Shang S, Ji X, Zhang L, Chen J, Li C, Shi
R, Xiang W, Kang X, Zhang D, Yang F, et al: Macrophage ABHD5
Suppresses NFκB-Dependent matrix metalloproteinase expression and
cancer metastasis. Cancer Res. 79:5513–5526. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Xiang W, Shi R, Kang X, Zhang X, Chen P,
Zhang L, Hou A, Wang R, Zhao Y, Zhao K, et al: Monoacylglycerol
lipase regulates cannabinoid receptor 2-dependent macrophage
activation and cancer progression. Nat Commun. 9:25742018.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
King RJ, Singh PK and Mehla K: The
cholesterol pathway: Impact on immunity and cancer. Trends Immunol.
43:78–92. 2022. View Article : Google Scholar :
|
|
113
|
van der Vorst EPC, Theodorou K, Wu Y,
Hoeksema MA, Goossens P, Bursill CA, Aliyev T, Huitema LFA, Tas SW,
Wolfs IMJ, et al: High-Density lipoproteins exert pro-inflammatory
effects on macrophages via passive cholesterol depletion and
PKC-NF-κB/STAT1-IRF1 signaling. Cell Metab. 25:197–207. 2017.
View Article : Google Scholar
|
|
114
|
Sag D, Cekic C, Wu R, Linden J and Hedrick
CC: The cholesterol transporter ABCG1 links cholesterol homeostasis
and tumour immunity. Nat Commun. 6:63542015. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang S, Yan W, Kong L, Zuo S, Wu J, Zhu C,
Huang H, He B, Dong J and Wei J: Oncolytic viruses engineered to
enforce cholesterol efflux restore tumor-associated macrophage
phagocytosis and anti-tumor immunity in glioblastoma. Nat Commun.
14:43672023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Goossens P, Rodriguez-Vita J, Etzerodt A,
Masse M, Rastoin O, Gouirand V, Ulas T, Papantonopoulou O, Van Eck
M, Auphan-Anezin N, et al: Membrane cholesterol efflux drives
tumor-associated macrophage reprogramming and tumor progression.
Cell Metab. 29:1376–1389.e4. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Nelson ER, Wardell SE, Jasper JS, Park S,
Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V,
et al: 27-Hydroxycholesterol links hypercholesterolemia and breast
cancer pathophysiology. Science. 342:1094–1098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Shi SZ, Lee EJ, Lin YJ, Chen L, Zheng HY,
He XQ, Peng JY, Noonepalle SK, Shull AY, Pei FC, et al: Recruitment
of monocytes and epigenetic silencing of intratumoral CYP7B1
primarily contribute to the accumulation of 27-hydroxycholesterol
in breast cancer. Am J Cancer Res. 9:2194–2208. 2019.PubMed/NCBI
|
|
119
|
Ohira H, Fujioka Y, Katagiri C, Mamoto R,
Aoyama-Ishikawa M, Amako K, Izumi Y, Nishiumi S, Yoshida M, Usami M
and Ikeda M: Butyrate attenuates inflammation and lipolysis
generated by the interaction of adipocytes and macrophages. J
Atheroscler Thromb. 20:425–442. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yao Y, Cai X, Fei W, Ye Y, Zhao M and
Zheng C: The role of short-chain fatty acids in immunity,
inflammation and metabolism. Crit Rev Food Sci Nutr. 62:1–12. 2022.
View Article : Google Scholar
|
|
121
|
Masui R, Sasaki M, Funaki Y, Ogasawara N,
Mizuno M, Iida A, Izawa S, Kondo Y, Ito Y, Tamura Y, et al: G
protein-coupled receptor 43 moderates gut inflammation through
cytokine regulation from mononuclear cells. Inflamm Bowel Dis.
19:2848–2856. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Seljeset S and Siehler S:
Receptor-specific regulation of ERK1/2 activation by members of the
'free fatty acid receptor' family. J Recept Signal Transduct Res.
32:196–201. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Tan J, McKenzie C, Potamitis M, Thorburn
AN, Mackay CR and Macia L: The role of short-chain fatty acids in
health and disease. Adv Immunol. 121:91–119. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Schaub A, Fütterer A and Pfeffer K:
PUMA-G, an IFN-gamma-inducible gene in macrophages is a novel
member of the seven transmembrane spanning receptor superfamily.
Eur J Immunol. 31:3714–3725. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Singh N, Gurav A, Sivaprakasam S, Brady E,
Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et
al: Activation of Gpr109a, receptor for niacin and the commensal
metabolite butyrate, suppresses colonic inflammation and
carcinogenesis. Immunity. 40:128–139. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Karunaratne TB, Okereke C, Seamon M,
Purohit S, Wakade C and Sharma A: Niacin and Butyrate:
Nutraceuticals targeting dysbiosis and intestinal permeability in
parkinson's disease. Nutrients. 13:282020. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bach Knudsen KE, Laerke HN, Hedemann MS,
Nielsen TS, Ingerslev AK, Gundelund Nielsen DS, Theil PK, Purup S,
Hald S, Schioldan AG, et al: Impact of diet-modulated butyrate
production on intestinal barrier function and inflammation.
Nutrients. 10:14992018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Chai JT, Digby JE and Choudhury RP:
GPR109A and vascular inflammation. Curr Atheroscler Rep.
15:3252013. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Aoyama M, Kotani J and Usami M: Butyrate
and propionate induced activated or non-activated neutrophil
apoptosis via HDAC inhibitor activity but without activating
GPR-41/GPR-43 pathways. Nutrition. 26:653–661. 2010. View Article : Google Scholar
|
|
130
|
Chang PV, Hao L, Offermanns S and
Medzhitov R: The microbial metabolite butyrate regulates intestinal
macrophage function via histone deacetylase inhibition. Proc Natl
Acad Sci USA. 111:2247–2252. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Usami M, Kishimoto K, Ohata A, Miyoshi M,
Aoyama M, Fueda Y and Kotani J: Butyrate and trichostatin A
attenuate nuclear factor kappaB activation and tumor necrosis
factor α secretion and increase prostaglandin E2 secretion in human
peripheral blood mononuclear cells. Nutr Res. 28:321–328. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Hara T, Hirasawa A, Ichimura A, Kimura I
and Tsujimoto G: Free fatty acid receptors FFAR1 and GPR120 as
novel therapeutic targets for metabolic disorders. J Pharm Sci.
100:3594–3601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Oh DY, Talukdar S, Bae EJ, Imamura T,
Morinaga H, Fan W, Li P, Lu WJ, Watkins SM and Olefsky JM: GPR120
is an omega-3 fatty acid receptor mediating potent
anti-inflammatory and insulin-sensitizing effects. Cell.
142:687–698. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Liang P, Henning SM, Guan J, Grogan T,
Elashoff D, Olefsky JM, Cohen P and Aronson WJ: Role of Host GPR120
in mediating dietary omega-3 fatty acid inhibition of prostate
cancer. J Natl Cancer Inst. 111:52–59. 2019. View Article : Google Scholar :
|
|
135
|
Liang P, Henning SM, Schokrpur S, Wu L,
Doan N, Said J, Grogan T, Elashoff D, Cohen P and Aronson WJ:
Effect of dietary omega-3 fatty acids on tumor-associated
macrophages and prostate cancer progression. Prostate.
76:1293–1302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zhu L, Zhao Q, Yang T, Ding W and Zhao Y:
Cellular metabolism and macrophage functional polarization. Int Rev
Immunol. 34:82–100. 2015. View Article : Google Scholar
|
|
137
|
Wu H, Han Y, Rodriguez Sillke Y, Deng H,
Siddiqui S, Treese C, Schmidt F, Friedrich M, Keye J, Wan J, et al:
Lipid droplet-dependent fatty acid metabolism controls the immune
suppressive phenotype of tumor-associated macrophages. EMBO Mol
Med. 11:e106982019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Ahluwalia M, Battiste J, Bockorny B,
Bullock A, Patel MR, Wen P, Shepard D, Vaickus L, Vincent M,
Vincent M, et al: Clinical efficacy and biomarker assessment of
VT1021, A CD36/CD47 dual-targeting agent, in recurrent
glioblastoma. Neuro-Oncology. 23:50. 2021. View Article : Google Scholar
|
|
139
|
Zhang Q, Wang H, Mao C, Sun M, Dominah G,
Chen L and Zhuang Z: Fatty acid oxidation contributes to IL-1β
secretion in M2 macrophages and promotes macrophage-mediated tumor
cell migration. Mol Immunol. 94:27–35. 2018. View Article : Google Scholar
|
|
140
|
Lang S, Tiwari S, Andratschke M, Loehr I,
Lauffer L, Bergmann C, Mack B, Lebeau A, Moosmann A, Whiteside TL
and Zeidler R: Immune restoration in head and neck cancer patients
after in vivo COX-2 inhibition. Cancer Immunol Immunother.
56:1645–1652. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zheng X, Mansouri S, Krager A, Grimminger
F, Seeger W, Pullamsetti SS, Wheelock CE and Savai R: Metabolism in
tumour-associated macrophages: A quid pro quo with the tumour
microenvironment. Eur Respir Rev. 29:2001342020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Eruslanov E, Daurkin I, Ortiz J, Vieweg J
and Kusmartsev S: Tumor-mediated induction of myeloid-derived
suppressor cells and M2-polarized macrophages by altering
intracellular PGE2 catabolism in myeloid cells. J Leukoc
Biol. 88:839–848. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Wen Z, Liu H, Li M, Li B, Gao W, Shao Q,
Fan B, Zhao F, Wang Q, Xie Q, et al: Increased metabolites of
5-lipoxygenase from hypoxic ovarian cancer cells promote
tumor-associated macrophage infiltration. Oncogene. 34:1241–1252.
2015. View Article : Google Scholar
|
|
144
|
Nosaka T, Baba T, Tanabe Y, Sasaki S,
Nishimura T, Imamura Y, Yurino H, Hashimoto S, Arita M, Nakamoto Y
and Mukaida N: Alveolar macrophages drive hepatocellular carcinoma
lung metastasis by generating leukotriene B4. J Immunol.
200:1839–1852. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Hoppstadter J, Dembek A, Horing M, Schymik
HS, Dahlem C, Sultan A, Wirth N, Al-Fityan S, Diesel B, Gasparoni
G, et al: Dysregulation of cholesterol homeostasis in human lung
cancer tissue and tumour-associated macrophages. Ebiomedicine.
72:1035782021. View Article : Google Scholar : PubMed/NCBI
|