Introduction
Myocardial infarction (MI) remains the leading
cardiovascular disease worldwide and the number of people with
cardiovascular disease is expected to increase by 40.5% by 2030,
which imposes a substantial economic burden on society (1). While blood reperfusion strategies
effectively reduce the risk of death in patients with MI, the
restoration of blood and oxygen to regions of ischemic myocardium
can result in additional cardiac damage and complications known as
myocardial ischemia/reperfusion (I/R) injury (MIRI) (2). MIRI is characterized by
pathophysiological phenomena, such as excessive calcium influx,
reactive oxygen species (ROS) generation, impaired endothelial
function, disturbances in mitochondrial function and autophagy of
cardiomyocytes (3,4). Owing to the complexity of the
molecular mechanisms of MIRI, a single therapeutic regimen has not
shown satisfactory therapeutic outcomes, indicating that targeting
multiple pathophysiological features may lead to improved
therapeutic outcomes (5,6). Of note, many studies have shown
that blood reperfusion therapy combined with drugs can effectively
reduce MIRI and improve the heart pumping function (7-9).
Turmeric has been used for the treatment of cancer,
neurological disorder, chronic inflammation and cardiovascular
disease in traditional Chinese medicine (10,11). The plant has been found to
contain several active compounds, of which curcumin (Cur) is the
primary component (12). Cur
scavenges free radical activity and maintains cellular function
through various molecular mechanisms (13). There is increasing evidence that
Cur exhibits cardioprotective effects in cardiovascular disease
(14,15). A pharmacological study by Hong
et al (16) demonstrated
that a continuous 3-day Cur treatment (75 mg/kg/day) regimen
improves cardiac function and reduced MI size. Duan et al
(17) showed that the Cur-based
modulation of the JAK2/STAT3 pathway attenuates IRI, significantly
reducing infarct size and ROS levels. In addition to directly
modulating downstream molecular activity, Cur attenuates
mitochondrial dysfunction and oxidative stress mediated by MIRI
(18). Based on the
cardioprotective effects of Cur via inhibition of the different
modes of regulated cell death (13), it was hypothesized that Cur
attenuates MIRI through multiple pathways.
Ferroptosis is a newly defined form of
iron-dependent cell death characterized by ROS production and lipid
peroxidation (19). The
mechanisms by which ferroptosis occurs differ from those of
apoptosis, necrosis and autophagy. However, there is a complex
crosstalk between these processes (20,21). Ferroptosis is characterized by an
imbalance in iron metabolism, which induces production of large
amounts of ROS and lipid peroxides that lead to cell death
(22,23). Excessive iron deposition can lead
to pathological iron overload; excess iron in cardiomyocytes can
directly induce ferroptosis via accumulation of phospholipid
hydroperoxides in the cell membrane (24). Increasing evidence has suggested
that iron overload during MIRI is associated with the activation of
ferritin phagocytosis, in which autophagic vesicles release ferric
ions into labile iron pools by interacting with ferritin heavy
chain 1 (FTH1) via nuclear receptor coactivator 4 (NCOA4), directly
contributing to impaired iron metabolism in cardiomyocytes
(25,26). In addition, iron can catalyze ROS
production via the Fenton reaction and promote lipid peroxidation,
causing further oxidative injury to cells (19). In summary, autophagy-dependent
ferroptosis-mediated pathological iron deposition may exacerbate
MIRI, but the associated molecular mechanisms have not been
elucidated. Poly (ADP-ribose) polymerase family member 1 protects
against cardiac ischemic injury by decreasing forkhead box O3A
(FoxO3a) phosphorylation and sequestering it in the cytoplasm to
inhibit autophagy (27).
Therefore, it was hypothesized that Cur modulates localization of
FoxO3a in cardiomyocytes, thereby regulating autophagy and
attenuating ferroptosis.
Silent information regulator 1 (Sirt1), a class III
histone deacetylase (28), is
involved in regulation of cell energy metabolism, senescence and
the transcription of oxidative stress-associated factors (29). In addition, Sirt1 can exhibit
chromatin-modifying activity and maintenance of gluconeogenesis,
fatty acid oxidation, oxidative phosphorylation and other processes
depends on the regulation of Sirt1 (29,30). Thus, Sirt1 regulates multiple
molecular mechanisms to maintain cardiac function.
Phosphatidylinositol 3-kinase (PI3K)/AKT pathway is activated by
Sirt1 to promote endothelial histiocyte migration and
proliferation, which may be achieved by promoting phosphorylation
of endothelial nitric oxide synthase (eNOS) (31). In the heart, Sirt1 can block the
accumulation of FoxO1 via the PI3K/AKT pathway, decreasing ROS
levels and cardiomyocyte apoptosis in the diabetic heart and
ameliorating metabolic abnormalities. Nevertheless, the molecular
mechanism between Sirt1 and MIRI remains unclear. Furthermore, the
ability of Cur to regulate downstream molecules via Sirt1 to
alleviate MIRI requires further investigation.
Here, a H9c2 cell anoxia/reoxygenation (A/R) and a
rat MIRI model were used to verify whether MIRI induces
autophagy-dependent ferroptosis and whether Cur attenuates MIRI by
attenuating autophagy-dependent ferroptosis, to investigate whether
the cardioprotective effect of Cur is related to the
Sirt1/AKT/FoxO3a pathway.
Materials and methods
Materials
Cur (purity ≥98%, batch no. DC0279-0005) was
purchased from Dester Technology Co., Ltd. and Akt inhibitor
triciribine (API-2; cat. no. GC15392) was purchased from GLPBIO
Technology LLC. In addition, 3-methyladenine (3-MA, cat. no.
HY-19312), an inhibitor of autophagy, was procured from
MedChemExpress. Small interfering RNA (siRNA) specific to Sirt1,
along with non-specific siRNA (scrambled control), were synthesized
by RIBOBIO Co., Ltd. The primary antibodies targeting Bcl-2 (cat.
no. 381702), Bax (cat. no. R22708), Sirt1 (cat. no. R25721), FTH1
(cat. no. R23306), phosphorylated (p-) FoxO3a (cat. no. R24347),
FoxO3a (cat. no. 381451) and microtubule-associated protein 1 light
chain 3β (LC3II; cat. no. 381544) as well as anti-rabbit (cat. no.
550076) and anti-mouse (cat. no. 550047) secondary antibodies
conjugated with horseradish peroxidase (HRP) were sourced from
ZEN-BIOSCIENCE Biotechnology Co., Ltd. Primary antibodies against
caspase 3 (cat. no. AF7022) and P62 (cat. no. AF5384) were
purchased from Affinity Biosciences Co., Ltd. Primary antibodies
targeting β-actin (cat. no. 20536-1-AP) and PCNA (cat. no.
60097-1-Ig) were sourced from Proteintech Inc. Finally, primary
antibodies targeting NCOA4 (cat. no. A5695) were sourced from
ABclonal Technology Co., Ltd. and antibodies against p-AKT (cat.
no. 9271) and AKT (cat. no. 9272) were sourced from Cell Signaling
Technology, Inc.
Sprague Dawley (SD) rats and H9c2
cells
A total of 18 adult male SD rats (weight, 250±20 g;
age, 8 weeks) were sourced from Tianqin Biotechnology Co. The
experimental protocols were performed following principles
established by the National Institutes of Health regarding the
treatment and utilization of laboratory animals (32). The research protocol was approved
by the Ethics Committee of the First Affiliated Hospital of
Nanchang University (approval no. CDYFY-IACUC-202211QR010).
H9c2 cells were sourced from the Cell Bank or Stem
Cell Bank at the Chinese Academy of Sciences. The cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.),
supplemented with 10% FBS (HyClone; Cytiva), in a 37°C incubator
containing 5% CO2 and 21% O2 at 95%
humidity.
Animal experiments
All rats had unrestricted access to water and food,
a temperature range of 22-24°C, humidity of 40-60%, and a 12/12-h
light/dark cycle. To simulate MIRI, SD rats were anesthetized with
2.5% isoflurane gas inhalation and 100 mg/kg ketamine
intraperitoneal injection. The rats were tracheally intubated and
connected to a ventilator to maintain respiration. To maintain body
temperature, anesthetized SD rats were positioned on a continuously
heated plate. The hair on the left chest was removed and the chest
cavity opened at the fourth intercostal space to expose the heart.
A 7-0 silk suture was used to ligate the left anterior descending
coronary artery and the ligature wire was removed after 30 min
ischemia, followed by 2 h of reperfusion after chest closure. Any
residual intrathoracic air was removed before closing the chest
with a 4-0 wire silk suture.
A total of 18 SD rats were randomly assigned to one
of three groups (n=6/group): Sham, SD rats were fed routinely and
underwent open chest surgery without ligation; I/R, SD rats were
subjected to continuous intraperitoneal injection of saline before
induction of MIRI and I/R + Cur, Cur (50 mg/kg) was injected
continuously intraperitoneally for 4 weeks before induction of MIRI
(13). Rats were checked daily
for weight, health and behavior. The following conditions were
defined as humane endpoints: i) Weight loss >20%; ii) inability
to eat, drink or stand; iii) depression (immobility, sniffing,
trembling, scratching) and body temperature <37°C in
unanesthetized or sedated animals and iv) abnormal central nervous
system responses and inability to effectively control pain (foot
retraction and licking and abdominal retraction). The total
duration of the experiment was 4 weeks and no humane endpoints were
observed in all animals. All rats were subjected to blood sampling
and echocardiographic assessment at the end of the experiment. Rats
were euthanized by CO2 asphyxiation at a flow rate of
60% of luminal volume/min according to the American Veterinary
Medical Association guidelines for animal euthanasia (33), and death was confirmed based on
respiratory arrest and loss of muscle tone.
Echocardiographic measurement
SD rats were anesthetized with isoflurane inhalation
(induction and maintenance concentration of isoflurane were 2.5%)
with 1 l/min O2. An echocardiographic probe (V6, 23 MHz
linear transducer, VINNO) was placed anteriorly on the left chest
wall to assess left ventricular ejection fraction (LVEF) and left
ventricular shortening fraction (LVFS). For each parameter, three
individual measurements were performed and the mean value was
subsequently computed (34).
Determination of lactate dehydrogenase
(LDH) and creatine kinase isoenzyme (CK-MB) levels
SD rats were anesthetized with isoflurane inhalation
(induction and maintenance, 2.5%) with 1 l/min O2,
connected to a ventilator and fixed on a continuously heated plate.
After MIRI induction, heart function was assessed using an
echocardiography system (V6, 23 MHz linear transducer, VINNO).
Then, 0.5 ml blood samples were taken by cardiac puncture and
collected in a heparinized tube. Rats were euthanized and their
hearts were rapidly harvested, cleaned with PBS and stored in
liquid nitrogen (-210°C) for subsequent experiments (13). The blood samples were stored
overnight at 4°C and centrifuged for 15 min to obtain serum (13,400
× g, 4°C). LDH and CK-MB levels were determined using assay kits
according to the manufacturer's instructions (cat. nos. E006-1-1;
A020-2-2; both Nanjing Jiancheng Bioengineering Institute).
Evans blue and triphenyl tetrazolium
chloride (TTC) staining
SD rats were anesthetized with isoflurane inhalation
(induction and maintenance; 2.5%) with 1 l/min O2,
connected to a ventilator and fixed on a continuously heated plate.
The chest was opened in the left anterior thoracic region between
the 4 and 5th intercostal spaces and the left anterior descending
flow was blocked by ligating a 7-0 silk thread and injecting Evans
blue dye (Beijing Solarbio Science & Technology Co., Ltd.; cat.
no. E8010) from the apex of the heart until cardiac arrest. The
hearts were removed and frozen at -80°C for 30 min, sliced (2 mm
thickness) and incubated in 2% TTC solution (Merck KGaA; cat. no.
298-96-4) at 37°C for 30 min in the dark. The images were captured
using a digital camera (×5; EOS M200; Canon; n=3/group) and
analyzed using the ImageJ software (version 1.8.0; National
Institutes of Health).
Hematoxylin and eosin (HE) and TUNEL
staining
Isolated rat hearts were fixed in 10%
paraformaldehyde for 24 h at room temperature and embedded in
paraffin. To determine the extent of cardiomyocyte apoptosis and
necrosis, sections (5 μm) were prepared and subjected to HE
(cat. no. G1005) and TUNEL staining (both Wuhan Servicebio
Technology Co., Ltd.; cat. no. G1507) kits according to the
manufacturer's instructions (n=3/group). Sections were stained with
0.50 hematoxylin for 10 and 0.05% eosin for 2 min at room
temperature. TdT incubation buffer (2 μl recombinant TdT
enzyme, 5 μl biotin-dUTP labeling mix, 50 μl
equilibration buffer) was added at 37°C for 1 h and sections were
washed 3 times with PBS, 0.5% streptavidin-HRP buffer was incubated
at 37°C for 30 min and then washed 3 times with PBS, 5% DAB
chromogenic solution was added at room temperature for 30 min and
then washed 3 times with PBS, and 0.5% hematoxylin staining
solution was stained at room temperature for 5 min. Percentage of
TUNEL-positive nuclei=number of TUNEL-positive nuclei (brown
staining) in 3 different fields of view/total nuclei ×100%. The
images were captured from three fields per slide using a light
microscope (BX53, Olympus Corporation) at a magnification of
×400.
Cell transfection
siRNA against Sirt1 (si-Sirt1) and the non-targeting
control siRNA (si-NC; cat. no. siN0000001-1-5; both Guangzhou
RiboBio Co., Ltd.; Table SI)
were introduced into H9c2 cells. Transfection was performed using
jetPRIME (Polyplus-transfection SA; cat. no. 101000006) according
to the manufacturer's protocol. A total of 5 μl si-RNA (20
μM), 200 μl jetPRIME buffer, 5 μl jetPRIME
reagent were mixed for 20 min at room temperature, followed by the
addition of the above complexes to 2 ml of culture DMEM (Final
siRNA concentration was 50 nM), and transfected for 8 h at 37°C.
Following transfection, cells were allowed to recover for 24 h
before subsequent experimentation. Transfection efficiency was
determined via reverse transcription-quantitative (RT-q) PCR and
western blotting.
Cell treatment
H9c2 cell A/R model was established as previously
described (35). In brief, H9c2
cells were treated with an anoxia medium (CaCl2 1.0,
HEPES 20.0, KCl 10.0, MgSO4 1.2, NaCl 98.5,
NaH2PO4 0.9, NaHcO3 6.0, sodium
lactate 40.0 mM, pH 6.8) for 4 h under oxygen-deficient conditions
(95% N2, 5% CO2; 37°C). Subsequently, the
anoxia medium was replaced with a reoxygenation medium
(CaCl2 1.0, glucose 5.5, HEPES 20.0, KCl 5.0, MgSO4 1.2
mM, NaCl 129.5, NaH2PO4 0.9 and
NaHcO3 20.0 mM, pH 7.4) and incubated for 4 h at 37°C in
an air-tight reoxygenation chamber containing 5% CO2 and
95% O2.
The experimental groups were as follows: i) control,
untreated cells; ii) A/R, cells subjected to A/R injury; iii) Cur
(2.5, 5.0, 10.0, 20.0 and 40.0 μM) + A/R, prior to A/R
injury, cells were pretreated with Cur at 37°C for 48 h; iv) Cur
(10 μM) + A/R, prior to A/R injury, cells were pretreated
with 10 μM Cur at 37°C for 48 h; v) 3-MA (5 mM)+ A/R, prior
to A/R injury, cells were treated with 5 mM 3-MA at 37°C for 24 h;
vi) Cur (10 μM) + si-NC +A/R group: prior to A/R injury,
cells were transfected with si-NC and pretreated with 10 μM
Cur at 37°C for 48 h; vii) Cur (10 μM) + si-Sirt1 + A/R,
prior to A/R injury, cells transfected with si-Sirt1 and pretreated
with 10 μM Cur at 37°C for 48 h and viii) Cur (10 μM)
+ API-2 + A/R, prior to A/R injury, cells pretreated with 10
μM Cur and 20 μM API-2 at 37°C for 48 h.
Cell counting kit-8 (CCK-8) and LDH
activity assay
Cellular viability was assessed using CCK-8 assay
(GLPBIO Technology LLC; cat. no. GK10001) according to the
manufacturer's instructions. Subsequent to A/R procedure, 100
μl serum-free DMEM containing 10 μl CCK-8 reaction
solution was introduced into a 96-well plate, and the cells were
incubated at 37°C for 30 min. Subsequently, the optical density
(OD) value was determined at 450 nm. Cell survival rate was
calculated as a percentage.
LDH activity was assessed using a detection kit
(cat. no. C0016; Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. In brief, the culture fluid or
cell lysate supernatant from each group was collected and LDH
activity was detected. The OD value was determined at 490 nm, and
the percentage of LDH activity was calculated.
Caspase 3 activity assay
Caspase 3 activity was quantified using an assay kit
(Beyotime Institute of Biotechnology; cat. no. C1116) according to
the manufacturer's instructions. First, supernatant was collected
from the H9c2 cell lysate, and protein concentration was measured
using the Bradford method. Subsequently, 40 μl activation
reagent, 50 μl cell homogenate and 10 μl caspase 3
substrates were incubated for 2 h at 37°C. Finally, the OD value
was measured at 405 nm.
Apoptosis assay
The ratio of apoptotic cells (early + late apoptotic
cells) was measured using an Annexin V-FITC/PI apoptosis assay kit
(BestBio, Inc.; cat. no. BB-4101) according to the manufacturer's
instructions. After harvesting treated H9c2 cells, 5 μl
Annexin V-FITC was added and incubated at 4°C for 15 min in the
dark, after which 5 μl PI was added and incubated for an
additional 5 min under the same conditions. Apoptotic cells were
detected using Cytomics FC500 flow cytometer at 488 and 578 nm,
respectively, and data were analyzed using NovoExpress (v.10.8;
Agilent Technologies, Inc.).
Measurement of total iron, superoxide
dismutase (SOD), malondialdehyde (MDA), glutathione disulfide
(GSSG) and glutathione (GSH) levels
As previously described (30), after collecting the cell lysis
supernatant from each group, the levels of total iron (Applygen;
cat. no. E1042), SOD (cat. no. S0131S), MDA (cat. no. S0053), GSSG
(cat. no. S0101S) and GSH (all Beyotime Institute of Biotechnology;
cat. no. S0101S) as well as the ratio of GSH/GSSG were assessed
according to the manufacturer's instructions.
Measurement of intracellular ROS
The intracellular level of ROS was detected using
DCFH-DA (Beyotime Institute of Biotechnology; cat. no. S0033S).
Treated cells were incubated with the cell culture DMEM containing
DCFH-DA (0.1%) for 15 min at 37°C in the dark. Subsequently, ROS
levels were measured using fluorescence microscopy (×200; Olympus
IX 73, Olympus Corporation). ImageJ (version 1.8.0; National
Institutes of Health) was employed for the analysis of alterations
in fluorescence intensity across the various experimental
groups.
Detection of lysosome level
Intracellular lysosomal generation was detected
using a LysoTracker Red detection kit (Beyotime Institute of
Biotechnology; cat. no. C1046). Treated cells were incubated with
the cell culture DMEM containing LysoTracker (0.1%) for 15 min at
37°C in the dark. Subsequently, lysosome levels were measured using
fluorescence microscopy (×200; Olympus IX 73; Olympus Corporation).
The ImageJ software (version 1.8.0; National Institutes of Health)
was employed for the analysis of alterations in fluorescence
intensity across the various experimental groups.
Detection of ferrous iron level
The level of ferrous ion was measured via an assay
kit (Dojindo Laboratories, Inc.; cat. no. F374). Threated cells
were incubated with the cell culture DMEM containing
FerroOrangeTracker (0.1%) for 15 min at 37°C in the dark.
Subsequently, intracellular ferrous iron levels were measured using
fluorescence microscopy (×200; Olympus IX 73; Olympus Corporation).
ImageJ software (version 1.8.0; National Institutes of Health) was
employed for the analysis of alterations in fluorescence intensity
across the various experimental groups.
Western blotting
Cytoplasmic and nuclear extracts were obtained using
commercially available kits (Beyotime Institute of Biotechnology;
cat. no. P0027), and total proteins were extracted from H9c2 cells
and cardiac tissue using RIPA buffer (Beijing Solarbio Science
& Technology; cat. no. R0010). A bicinchoninic acid kit (GLPBIO
Technology LLC; cat. no. GK10009) was used to measure the protein
concentration. The components of protein supernatants were
denatured by boiling for 5 min following the addition of a loading
buffer. Next, 20 μg/lane protein was separated using 8-12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis and
transferred onto polyvinylidene fluoride membranes. Protein bands
were incubated overnight at 4°C with the primary antibody (1:1,000)
following 1 h blocking using 5% skimmed milk or 5% bovine serum
albumin (Beijing Solarbio Science & Technology Co., Ltd.; cat.
no. A8020) at room temperature. Afterward, the membranes were
incubated with HRP-conjugated secondary antibodies (1:5,000;
ZEN-BIOSCIENCE Biotechnology Co., Ltd.; cat. no. 550076; 550047) at
room temperature for 2 h. The membranes were analyzed using an
ultra-high sensitivity enhanced chemiluminescence kit (GLPBIO
Technology LLC; cat. no. GK10008) and imaged with FluorChemFC3
(ProteinSimple). In this experiment, protein expression was
measured using ImageJ software (version 1.8.0; National Institutes
of Health). The relative expression of the target proteins was
obtained by comparing protein levels to β-actin levels.
RT-qPCR
As described previously (35), total RNA was extracted from H9c2
cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RT kit (Vazyme Biotech, cat. no. R412-01) was used to obtain
cDNA according to the manufacturer's protocol. The reaction
solution was prepared by mixing 12 SYBR Green Mix (Selleck
Chemicals, cat. no. B21202), 1 each forward and reverse primers (10
μM), 4 cDNA and 2 μl nuclease-free water. The
reaction solution was pre-denatured at 95°C for 30 sec, followed by
40 cycles at 95°C for 15 sec and 60°C for 1 min. Target gene
expression was detected using the BIO-RAD CFX detection system and
the relative expression of target genes in terms of RNA levels was
measured using the 2-ΔΔCq method (36). The primers were designed and
synthesized by Sangon Biotech Co., Ltd. (Table SII).
Statistical analysis
Data are expressed as the mean ± standard deviation
of three independent experimental repeats. GraphPad Prism software
(version 8.0.2; graphpad.com/) was used for
statistical analysis. One-way analysis of variance followed by
Tukey's post hoc multiple comparison test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Cur pretreatment attenuates A/R injury in
H9c2 cells
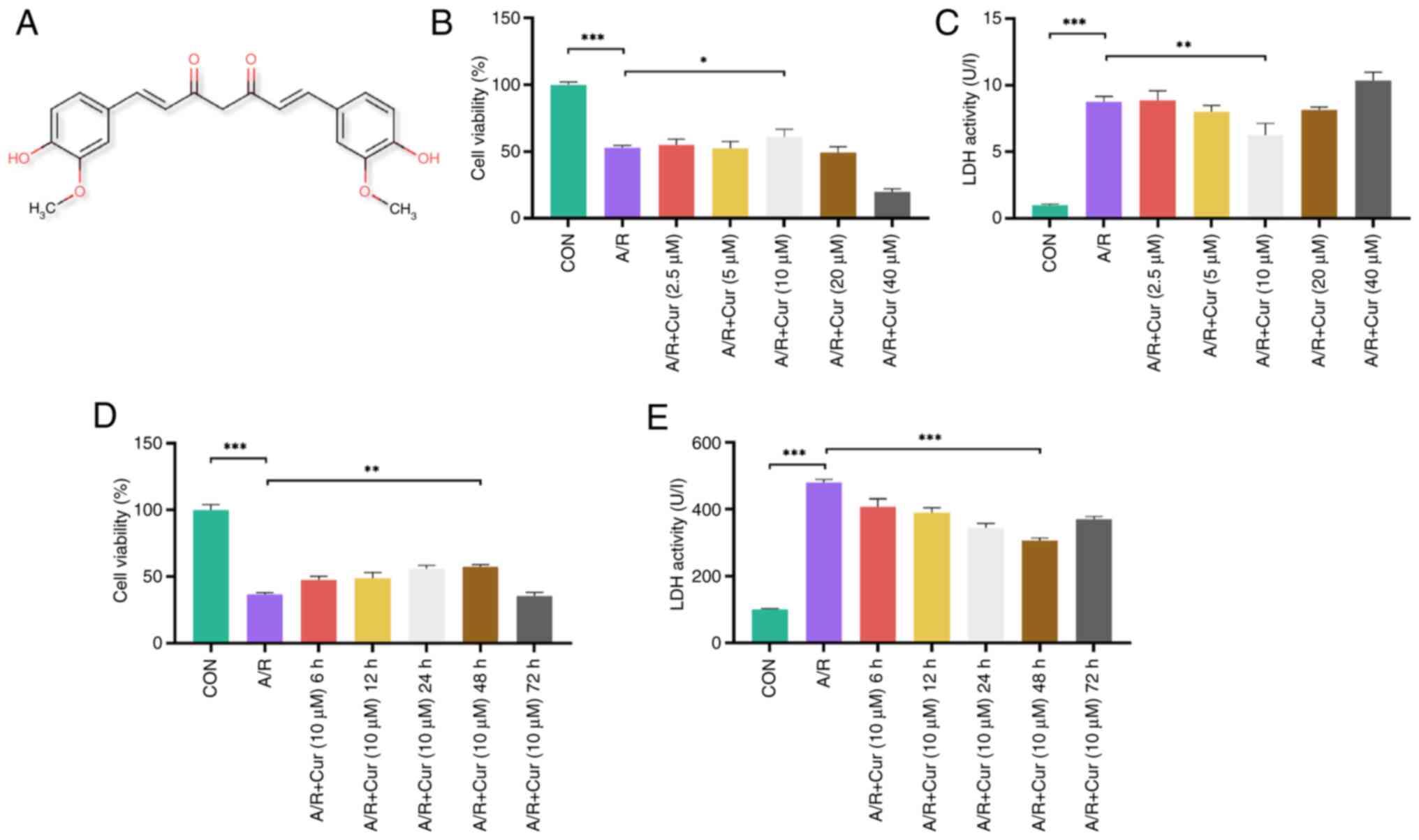
Fig. 1A
illustrates the chemical structure of Cur. CCK-8 and LDH assays
were used to identify the optimal drug treatment concentration.
H9c2 cells were pretreated with Cur, followed by A/R injury, and
cell viability and LDH release levels were assessed. A/R injury
significantly decreased cell viability and increased LDH levels.
Treatment with 10 μM Cur resulted in a significant increase
in cell viability and a decrease in LDH levels compared with the
A/R group, whereas no significant difference was observed in the 20
with the 10 μM Cur group (Fig. 1B and C). Therefore, 10 μM
Cur was deemed to represent the threshold for a safe drug
concentration and used for subsequent experiments. Significantly
higher cell viability was observed after 48 h 10 μM Cur
treatment than in the A/R group, whereas significantly lower cell
viability was observed after 72 h compared with 48 h 10 μM
Cur treatment (Fig. 1D and E).
Thus, 10 μM Cur for 48 h was selected as the optimal drug
pretreatment.
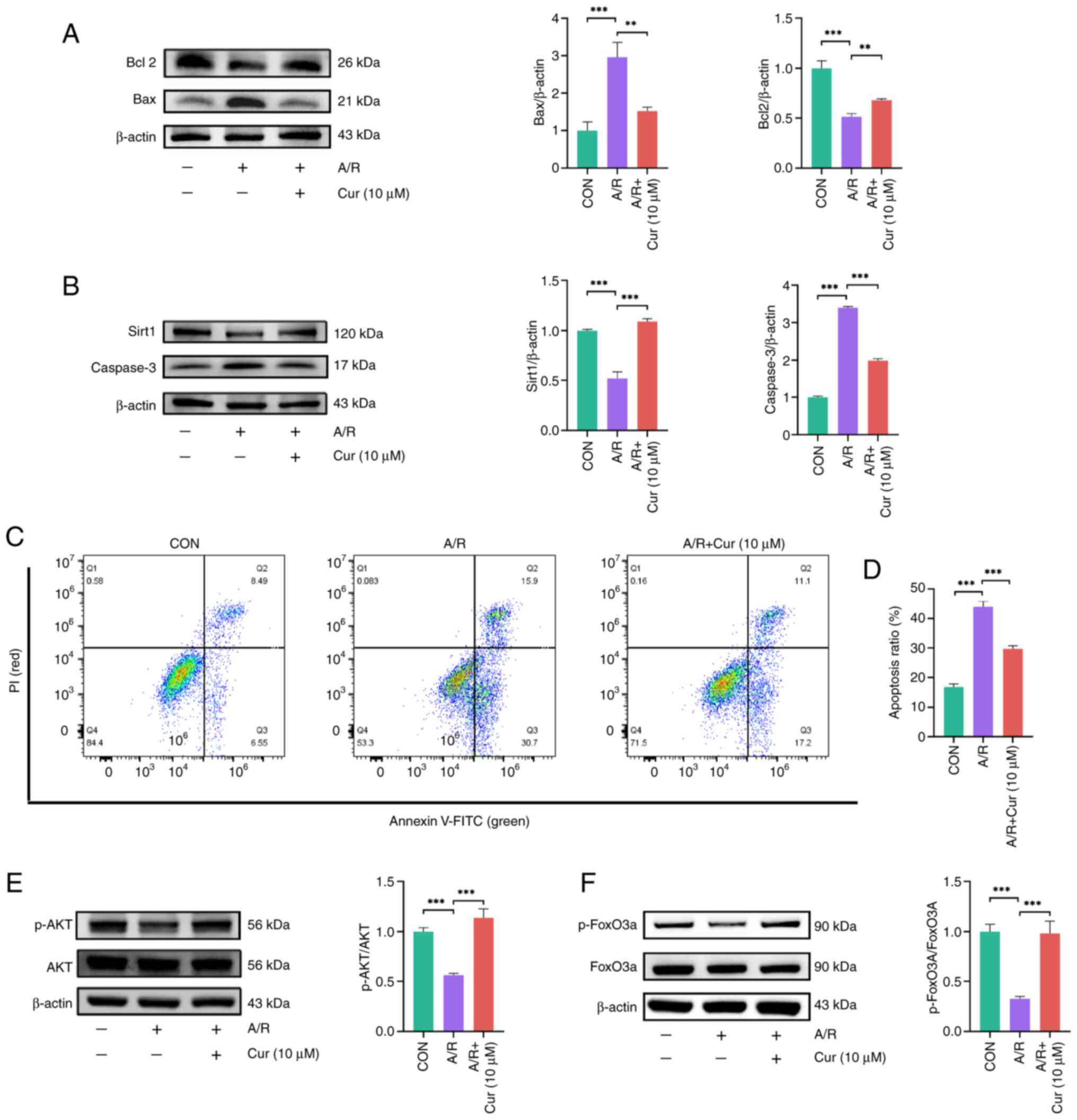
Cur attenuates A/R injury in H9c2 cells
via the Sirt1-based activation of Akt/FoxO3a
To assess the ability of Cur to attenuate A/R
injury, protein expression of Bcl-2, Bax and caspase 3, which are
apoptotic markers (37), and
that of Sirt1 were assessed in H9c2 cells. Caspase 3 and Bax
protein expression in the Cur group was lower than that in the A/R
group (Fig. 2A and B). By
contrast, Bcl-2 and Sirt1 protein expression was significantly
higher in Cur-pretreated than in A/R-treated H9c2 cells. These
results suggested that Cur attenuated A/R injury in H9c2 cells.
The detection of apoptosis in H9c2 cells was
performed via flow cytometry. The apoptosis rate of H9c2 cells in
the A/R group was significantly increased compared with the control
group. However, compared with A/R group, the apoptosis rate of H9c2
cells in the Cur group was significantly decreased (Fig. 2C and D). This finding indicated
that Cur pretreatment is an effective method for attenuating A/R
injury.
The AKT/FoxO3a signaling pathway is involved in the
regulation of autophagy and promotion of phagocytosis of abnormal
organelles and proteins (38).
Thus, the expression of AKT, p-AKT, FoxO3a and p-FoxO3a was
evaluated in cells. A/R injury decreased both p-AKT/AKT and
p-FoxO3a/FoxO3a ratios in H9c2 cells compared with the control
group. However, in the Cur groups, p-AKT/AKT and p-FoxO3a/FoxO3a
ratios were both increased (Fig.
2E-G). These results indicate that Cur decreased A/R injury in
H9c2 via the Sirt1/AKT/FoxO3a pathway.
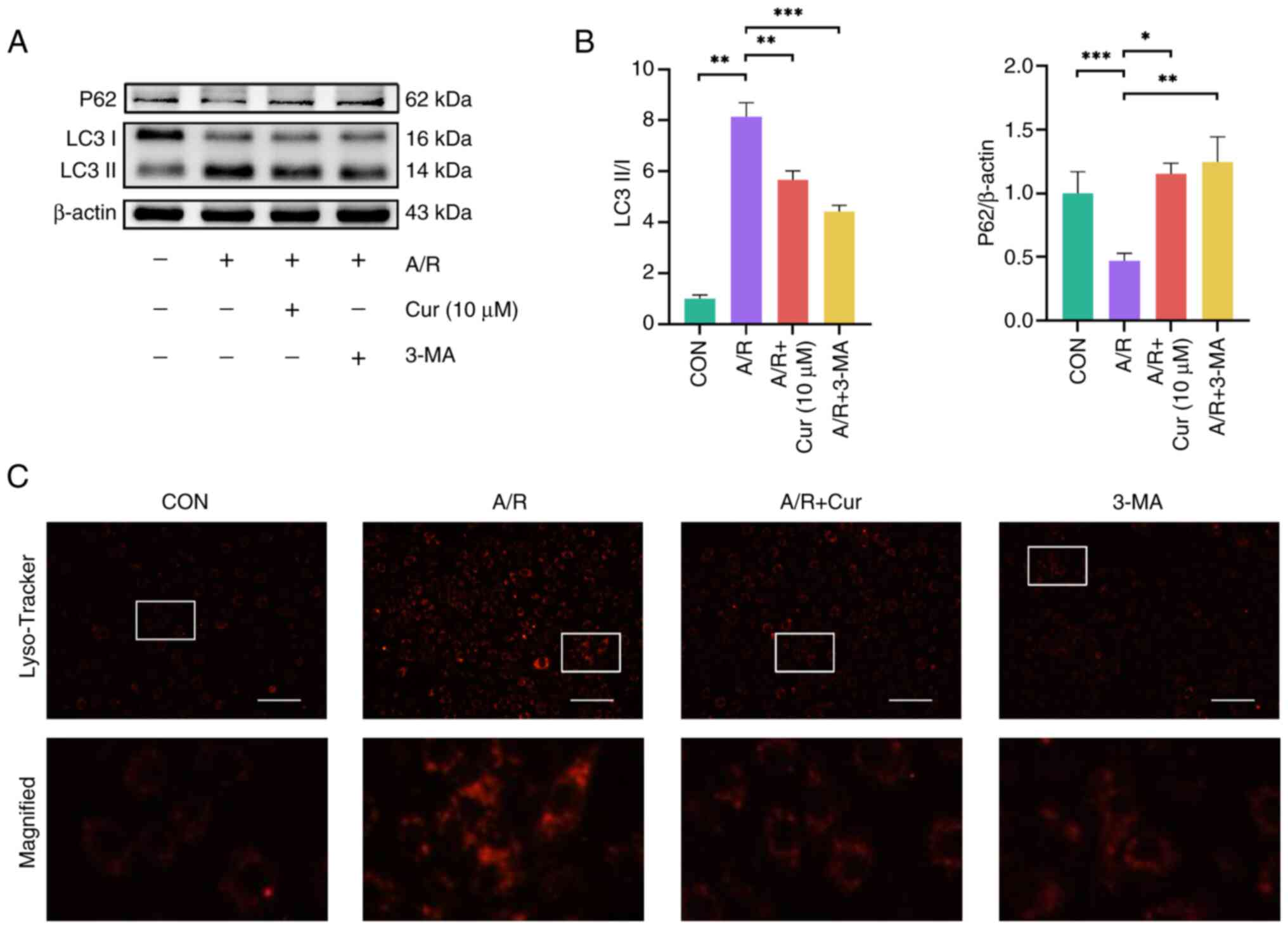
Cur inhibits excessive autophagy due to
A/R injury
A/R injury results in overactivation of
intracellular autophagy in H9c2 cells, which exacerbates myocardial
injury (9). Therefore, changes
in protein expression of autophagy markers LC3II and P62 were
analyzed (39). A/R injury
reduced P62 protein expression in H9c2 cells compared with the
control group, whereas Cur pretreatment of H9c2 cells reversed
these changes (Fig. 3A and B).
Likewise, P62 protein levels increased significantly in the 3-MA
+A/R compared with the A/R injury group. Consequently, LC3II/LC3I
protein ratio was higher in H9c2 cells from the AR group than in
cells from the control group. By contrast, LC3II/LC3I protein ratio
was reduced in the Cur and 3-MA group. LysoTracker Red was used to
identify the number of lysosomes in a cell (9). A/R injury increased intracellular
lysosomal fluorescence intensity compared with the control group,
suggesting A/R injury increases intracellular lysosomes. Cur or
3-MA could effectively attenuate the intracellular lysosomal
fluorescence intensity (Fig.
3C). These findings indicated that Cur pretreatment can prevent
excessive autophagic injury induced by A/R and avoid further damage
to H9c2 cells.
Cur attenuates autophagy-dependent
ferroptosis induced by A/R injury in H9c2 cells
There may be crosstalk between autophagy and
ferroptosis pathways during MIRI (24). Therefore, whether autophagy
inhibition attenuates ferroptosis was explored by analyzing changes
in the protein levels of NCOA4 and FTH1, which are markers of
autophagy-dependent ferroptosis (21). A/R injury increased NCOA4
expression and decreased FTH1 expression in H9c2 cells, whereas Cur
and 3-MA reversed these changes (Fig. 4A and B). The features of
ferroptosis include Fe2+ accumulation, lipid
peroxidation and ROS overload (19). Thus, intracellular MDA, SOD, GSH,
GSSG, and total iron ion levels were measured. A/R injury increased
the total iron, GSSG and MDA levels in H9c2 cells, whereas Cur and
3-MA reduced these levels significantly (Fig. 4C-E). By contrast, A/R injury
decreased intracellular SOD and GSH levels as well as GSSG/GSH
ratio compared with the control group. Cur or 3-MA increased
intracellular SOD and GSH levels as well as GSSG/GSH ratio
(Fig. 4F-H). Of note, ROS and
Fe2+ levels were higher in H9c2 cells following A/R
injury than in the control group and Cur or 3-MA attenuated ROS and
Fe2+ accumulation in H9c2 cells (Fig. 4I and J). These results suggest
that autophagy-dependent ferroptosis is involved in A/R injury and
that Cur and 3-MA pretreatment can attenuate autophagy-dependent
ferroptosis injury in H9c2 cells.
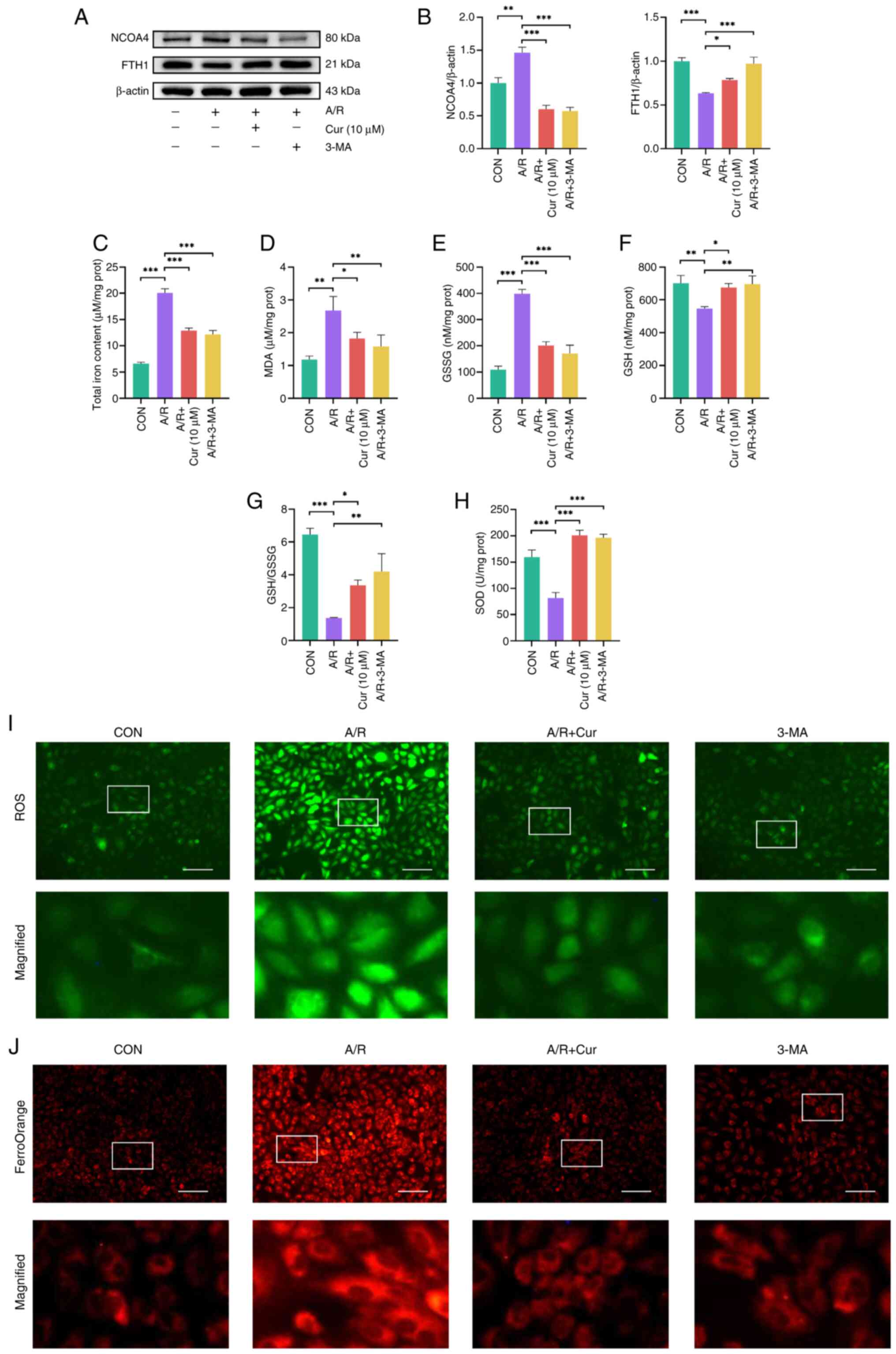 | Figure 4Cur attenuates autophagy-dependent
ferroptosis induced by A/R injury in H9c2 cells. (A) Expression of
autophagy-dependent ferroptosis-associated proteins (FTH1 and
NCOA4) was detected in A/R-injured H9c2 cells after Cur or 3-MA
treatment. (B) Relative protein expression of FTH1 and NCOA4.
Detection of (C) total iron ions, (D) MDA, (E) GSSG, (F) GSH, (G)
GSH/GSSG and (H) SOD in H9c2 cells with A/R injury by Cur or 3-MA
intervention. (I) ROS and (J) Fe2+ levels in H9c2 cells
following AR injury after Cur or 3-MA treatment (magnification,
×200; scale bar, 400 μm). *P<0.05,
**P<0.01, ***P<0.001. Cur, curcumin;
A/R, anoxia/reoxygenation; FTH1, ferritin heavy chain 1; NCOA4,
nuclear receptor coactivator 4; 3-MA, 3-Methyladenine; MDA,
malondialdehyde; GSSG, glutathione disulfide; GSH, glutathione;
SOD, superoxide dismutase; ROS, reactive oxygen species; CON,
control; prot, protein. |
Cur attenuates A/R injury via Sirt1
Sirt1 influences many biological processes,
including apoptosis, senescence and mitochondrial biogenesis in
cardiomyocytes (40). Sirt1
deficiency may exacerbate MIRI and there may be crosstalk between
Sirt1 and AKT/FoxO3a (41).
Consequently, it was hypothesized that Cur decreases
autophagy-dependent ferroptosis in MIRI by modulating the
AKT/FoxO3a pathway via Sirt1. siRNAs were used to silence Sirt1.
First, the infection efficiency of siRNAs was verified at both the
protein and RNA levels in cells (Fig. S1); all siRNAs reduced the
protein and mRNA expression of Sirt1, but si-Sirt1-002 reduced
Sirt1 expression most significantly. si-Sirt1-002 was selected for
subsequent experiments. Next, molecular regulation experiments were
performed to verify whether Cur affects AKT/FoxO3a signaling and
decreases apoptosis via Sirt1. The results showed that A/R
treatment caused apoptosis in H9c2 cells and pretreatment with Cur
reduced apoptosis caused by A/R (Fig. 5A-C). Of note, silencing of Sirt1
in H9c2 cells or API-2 counteracted the protective effect of Cur,
suggesting that Cur exerted its protective effect through Sirt1 and
AKT (Fig. 5D and E).
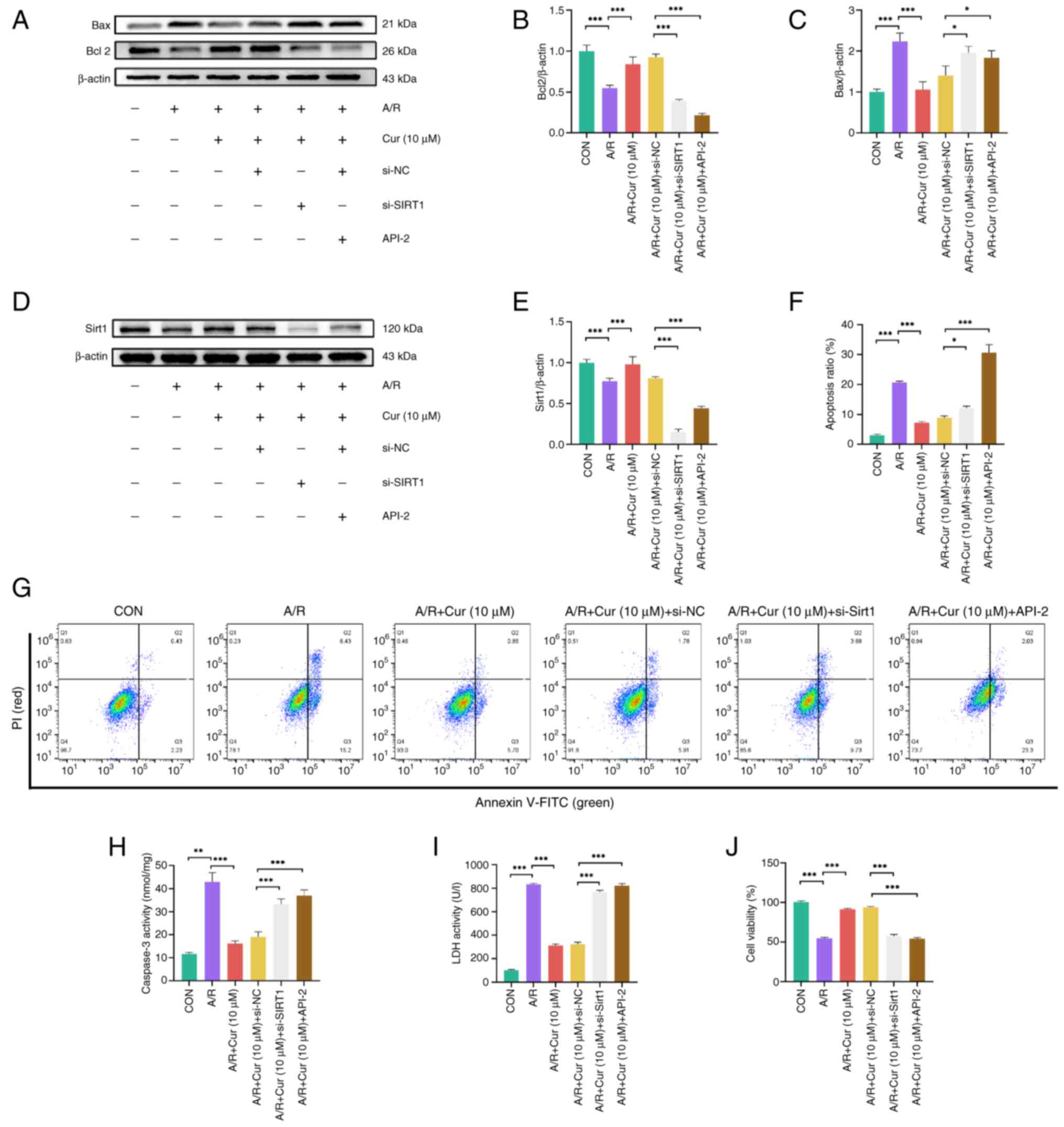 | Figure 5Cur decreases A/R injury in H9c2
cells via Sirt1. Expression of (A) apoptosis-associated proteins in
A/R-injured H9c2 cells following Cur pretreatment, Sirt1 silencing
and targeted inhibition of AKT activity. (B) Relative protein
expression of Bcl2. (C) represents the relative protein expression
of Bax. Expression of Sirt1 (D) in A/R-injured H9c2 cells following
Cur pretreatment, Sirt1 silencing and targeted inhibition of AKT
activity. (E) Relative protein expression of Sirt1. Apoptosis rate
of H9c2 cells was measured by flow cytometry (F and G) illustrates
the proportion of apoptotic cells as determined by flow cytometry.
(H) caspase 3, (I) LDH activity and (J) viability of A/R injured
H9c2 cells after Cur pretreatment, silencing of Sirt1 expression
and targeted inhibition of AKT activity. *P<0.05,
**P<0.01, ***P<0.001. Cur, curcumin;
A/R, anoxia/reoxygenation; Sirt, silent information regulator 1;
LDH, Lactate dehydrogenase; si, small interfering; NC,
non-targeting control; API-2, triciribine; CON, control. |
Flow cytometry demonstrated that A/R injury resulted
in elevated apoptosis rate in H9c2 cells and Cur effectively
reduced the apoptosis rate after A/R injury. si-Sirt1 increased the
apoptotic rate in H9c2 cells, thereby preventing the protective
effect of Cur (Fig. 5F and G).
A/R injury also increased caspase 3 activity in H9c2 cells, whereas
Cur effectively decreased caspase 3 activity. However, silencing of
Sirt1 protein expression or inhibition of AKT activity increased
caspase 3 activity in H9c2 cells (Fig. 5H). The same results were obtained
in CCK-8 and LDH activity assay; A/R resulted in release of LDH
from cells. The administration of Cur led to a reduction in LDH
levels. Furthermore, the silencing of Sirt1 protein expression or
the inhibition of Akt activity resulted in a stimulation of LDH
release (Fig. 5I); A/R injury
decreased H9c2 cell viability to ~50% of the control group.
However, the addition of Cur rescued cell viability, whereas
silencing of Sirt1 protein expression or inhibition of Akt activity
decreased viability (Fig. 5J).
These findings suggest that Cur attenuated A/R injury in H9c2 cells
through Sirt1.
Cur decreases autophagy-dependent
ferroptosis via Sirt1 in H9c2 cells following A/R injury
To validate the ability of Cur to attenuate
autophagy-dependent ferroptosis via Sirt1/AKT/FoxO3a in A/R-injured
H9c2 cells, changes in proteins associated with autophagy-dependent
ferroptosis injury were assessed. A/R injury led to an elevated
LC3II/I ratio as well as increased NCOA4 expression, whereas P62
and FTH1 protein expression decreased compared with the control
group. These changes were reversed in H9c2 cells pretreated with
Cur. Silencing of Sirt1 expression or cotreatment with API-2
effectively attenuated the protective effect of Cur. These results
indicated that Cur is effective in attenuating autophagy-dependent
ferroptosis in A/R. However, these effects were dependent on the
expression of Sirt1 and activity of AKT (Fig. 6A-D).
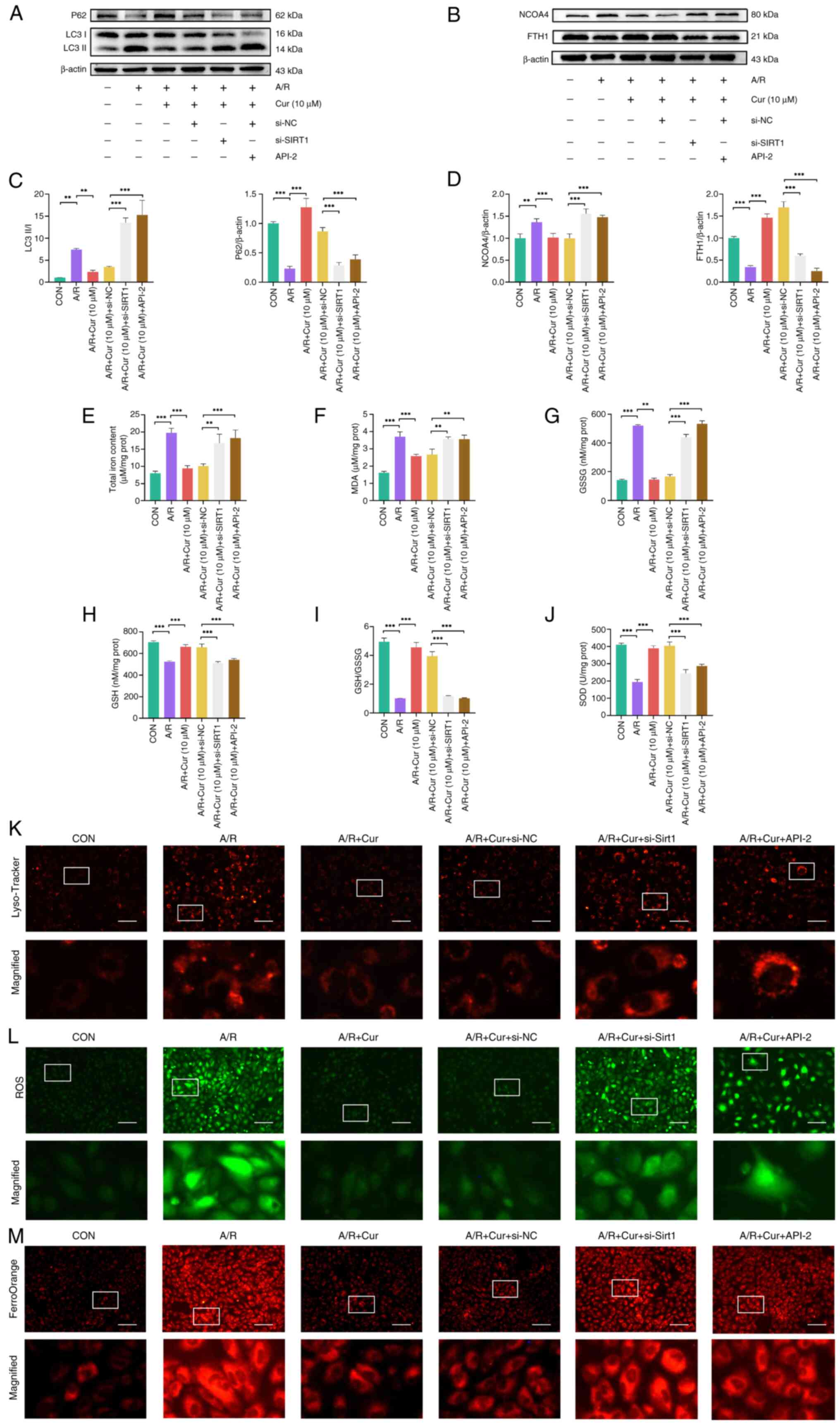 | Figure 6Cur reduces autophagy-dependent
ferroptosis in H9c2 cells associated with A/R injury via Sirt1.
Expression of autophagy-(A) and ferroptosis-related proteins (B) in
A/R-injured H9c2 cells following Cur pretreatment, Sirt1 silencing
and targeted inhibition of AKT activity. (C) Relative protein
expression of P62 and the ratio of LC3II/I. (D) Relative protein
expression of NCOA4 and FTH1. Detection of (E) total iron ions, (F)
MDA, (G) GSSG, (H) GSH, (I) GSH/GSSG and (J) SOD in A/R injured
H9c2 cells following Cur pretreatment, Sirt1 silencing and targeted
inhibition of AKT activity. Fluorescence intensity of (K)
lysosomes, (L) ROS and (M) Fe2+ in A/R-injured H9c2
cells following Cur pretreatment, Sirt1 silencing and targeted
inhibition of AKT activity (magnification, ×200; scale bar, 400
μm). **P<0.05, ***P<0.01. Cur,
curcumin; A/R, anoxia/reoxygenation; Sirt, silent information
regulator 1; MDA, malondialdehyde; GSSG, glutathione disulfide;
GSH, glutathione; SOD, superoxide dismutase; ROS, reactive oxygen
species; NCOA4, nuclear receptor coactivator 4; FTH1, ferritin
heavy chain 1; CON, control; si, small interfering; prot, protein;
API, triciribine; LC3II, microtubule-associated protein 1 light
chain 3 β; NC, non-targeting control. |
To validate whether Cur can attenuate
autophagy-dependent ferroptosis via Sirt1/AKT/FoxO3a in A/R-injured
H9c2 cells, ferroptosis- and autophagy-related biochemical indices
were compared (Fig. 6E-J). The
results revealed that A/R injury increased total iron ion, MDA and
GSSG levels and decreased GSH and SOD levels as well as the
GSH/GSSG ratio relative to the control group. These changes were
reversed in H9c2 cells pretreated with Cur. By contrast, silencing
of Sirt1 expression or API-2 cotreatment led to a weakened
protective effect of Cur, increased total iron ion, MDA, and GSSG
levels and decreased GSH and SOD levels as well as the GSH/GSSG
ratio in H9c2 cells. LysoTracker Red, DCFH-DA and
FerroOrangeTracker staining (Fig.
6K-M) showed that autophagic lysosomes, ROS, and
Fe2+ in H9c2 cells were increased following A/R injury.
However, Cur pretreatment decreased levels of autophagic lysosomes,
ROS and Fe2+ relative to the A/R injury group.
Fluorescence intensity of ROS and Fe2+ was significantly
enhanced following the addition of si-Sirt1 or API-2. These results
indicate that Cur attenuates autophagy-dependent ferroptosis
induced by A/R.
Cur mediates nuclear localization of
FoxO3a via Sirt1/Akt
FoxO3a is retained in the cytoplasm following
phosphorylation by Akt, which inhibits the transcription of the
target genes of FoxO3a (42).
Previous studies have shown that FoxO3a may regulate autophagy
levels by regulating genes such as LC3II, γ-aminobutyric acid
receptor-associated protein-like 1 (Gabarapl1) and autophagy
related 12 homolog (ATG12) (27,43). To demonstrate that FoxO3a is a
key gene for adaptive autophagy in A/R injury, the phosphorylation
of AKT and FoxO3a and the distribution of FoxO3a proteins were
analyzed. Western blotting showed that A/R injury decreased the
phosphorylation of AKT and FoxO3a compared with the control group
and expression of FoxO3a was significantly increased in nuclear
extracts of H9c2 cells but decreased in the cytoplasmic extracts.
Cur pretreatment increased phosphorylation levels of AKT and FoxO3a
compared with the A/R group. Expression of FoxO3a was significantly
decreased in nuclear extract of H9c2 cells and significantly
increased in the cytoplasmic extract in the Cur group compared with
the A/R group. si-Sirt1 or API-2 with Cur pretreatment restored
phosphorylation levels of AKT and FoxO3a to those of the A/R group.
Expression of FoxO3a in the nuclear extracts of H9c2 cells in the
si-Sirt1 and API-2 groups also increased to that of the A/R group.
By contrast, there was a decrease in the expression of FoxO3a in
the cytoplasm of H9c2 cells (Fig.
7A-D). These results indicated that Cur regulated the
phosphorylation of AKT through Sirt1, which subsequently regulated
phosphorylation of FoxO3a and affected the localization of FoxO3a
in H9c2 cells.
 | Figure 7Cur mediates nuclear localization of
FoxO3a via Sirt1/AKT. (A) Western blot analysis of (B)
phosphorylation of AKT and FoxO3a in A/R-injured H9c2 cells after
Cur pretreatment, Sirt1 silencing and targeted inhibition of AKT
activity. (C) Western blot analysis of (D) distribution of FoxO3a
in the cytoplasm and nucleus of A/R-injured H9c2 cells following
Cur pretreatment, Sirt1 silencing and targeted inhibition of AKT
activity. ***P<0.01. Cur, curcumin; PCNA,
proliferating cell nuclear antigen; Sirt, silent information
regulator 1; A/R, anoxia/reoxygenation; p-, phosphorylated; si,
small interfering; NC, non-targeting control; API, Triciribine;
CON, control. |
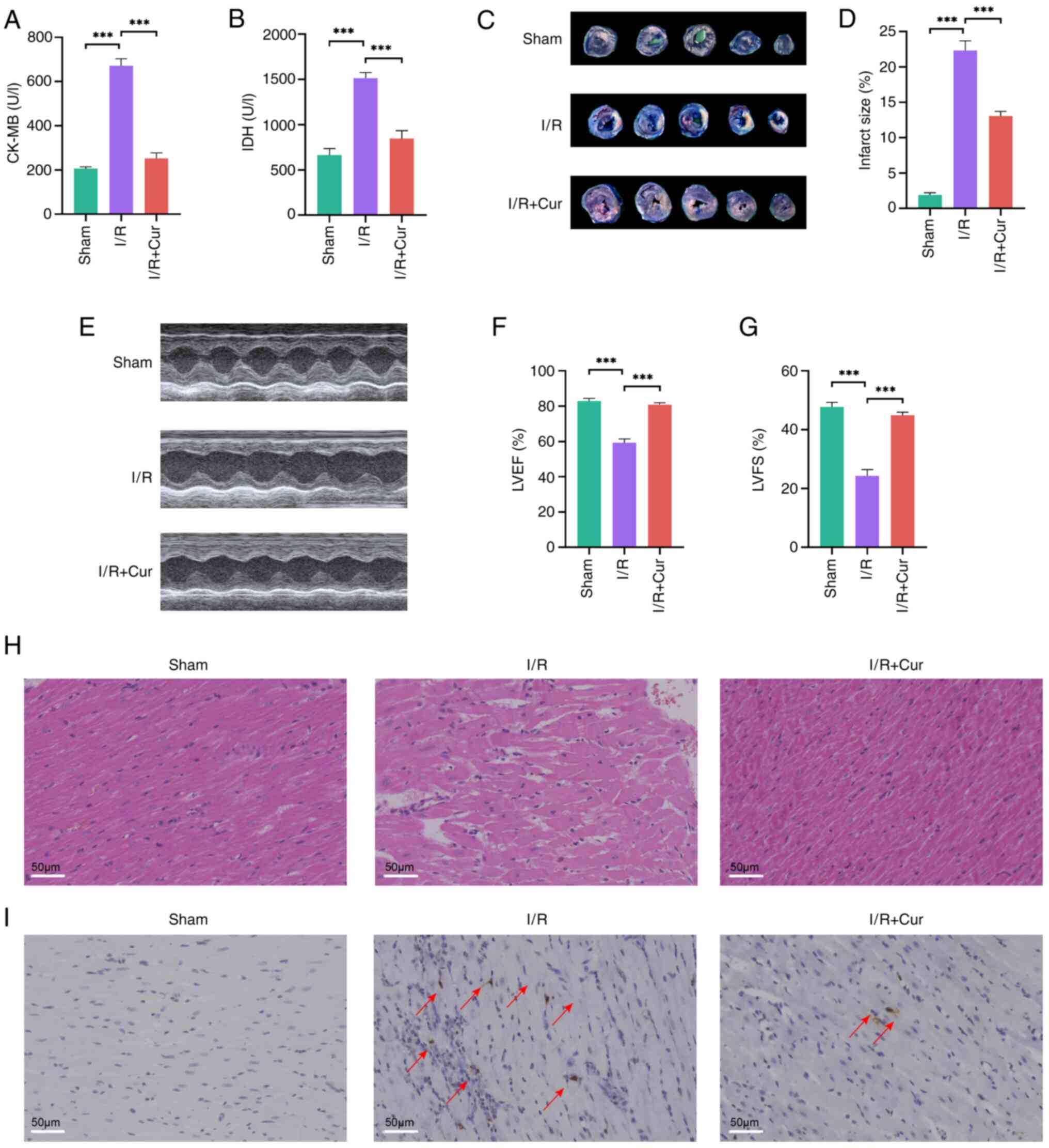
Cur protects cardiomyocytes from MIRI via
Sirt1/AKT/FoxO3a signaling
I/R model in the rat heart was established to
confirm the protective effect of Cur against MIRI. CK-MB and LDH
(myocardial injury marker enzymes) (13) levels were significantly elevated
in the serum samples of I/R-injured rats and Cur effectively
restored these abnormal enzymatic indices (Fig. 8A and B). Evans blue and TTC
staining revealed MI area was significantly increased in
I/R-injured rats compared with that in the sham group, whereas Cur
pretreatment significantly decreased the infarct area after MIRI
(Fig. 8C and D).
Echocardiography assessment of left ventricle function in rats
showed that MIRI impaired cardiac function by decreasing LVEF and
LVFS. Cur was effective in restoring these abnormal functions
(Fig. 8E-G). HE staining showed
that the myocardium exhibited myofiber separation, cardiomyocyte
swelling and interstitial cell hypertrophy and TUNEL staining
revealed an increase in TUNEL-positive cardiomyocytes following
MIRI. However, Cur pretreatment eliminated these MIRI-induced
morphological changes in myocardial tissue (Fig. 8H and I).
The protein expression of Sirt1, LC3II, P62, NCOA4
and FTH1 and the ratios of p-AKT/AKT and p-FoxO3a/FoxO3a in rat
myocardium were assessed to validate the molecular mechanism of Cur
(Fig. 9A-H). I/R decreased the
expression of Sirt1 in cardiomyocytes, whereas Cur-pretreated rats
did not exhibit a decrease in expression of Sirt1 after I/R, which
suggested that Cur can activate Sirt1 in cardiomyocytes to
attenuate MIRI (Fig. 9A and B).
I/R increased the ratio of LC3II/LC3Ⅰ and expression of NCOA4 in
rat cardiomyocytes, with a corresponding decrease in expression of
P62 and FTH1, which indicated that I/R induced autophagy-dependent
ferroptosis in rat cardiomyocytes and Cur pretreatment eliminated
these effects (Fig. 9C-F). p-AKT
and p-FoxO3a levels showed a decreasing trend after I/R, whereas
Cur increased p-AKT and p-FoxO3a levels in the rat myocardium
(Fig. 9G and H). These results
indicated that Cur attenuated MIRI by regulating
autophagy-dependent ferroptosis via Sirt1/AKT/FoxO3a signaling
(Fig. 9I).
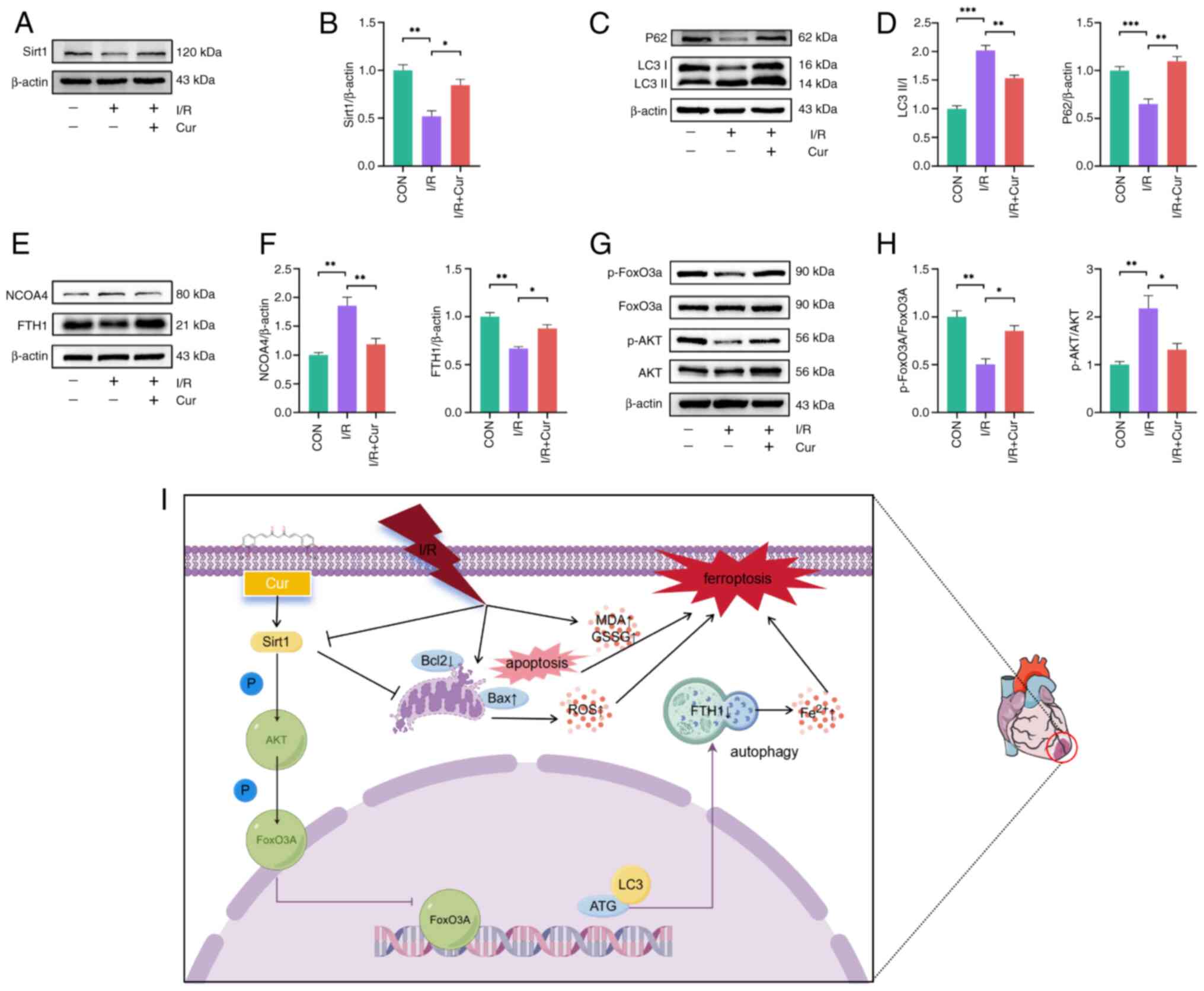 | Figure 9Potential mechanism of action of Cur
in MIRI. (A) Western blot was performed to detect protein
expression of Sirt1 in MIRI heart tissues after Cur treatment, (B)
Relative protein expression of Sirt1. Western blot was performed to
detect protein expression of LC3II and P62 (C) in MIRI heart
tissues after Cur treatment, (D represents the relative protein
expression of P62 and the ratio of LC3II/I. Western blot was
performed to detect protein expression of NCOA4 and FTH1 (E) in
MIRI heart tissues after Cur treatment, (F) represents the relative
protein expression of NCOA4 and FTH1. Western blot was performed to
detect the ratios of p-AKT/AKT and p-FoxO3a/FoxO3a (G) in MIRI
heart tissues after Cur treatment, (H) showed statistical figures.
(I) Cur inhibits MIRI-induced autophagy-dependent ferroptosis and
apoptosis in cardiomyocytes via Sirt1/AKT/FoxO3a, which increases
cardiomyocyte survival and maintains cardiac function. Part of the
figure was created using Figdraw (https://www.figdraw.com). *P<0.05,
**P<0.01, ***P<0.001. Cur, curcumin;
MIRI, myocardial ischemia/reperfusion injury; Sirt, silent
information regulator 1; LC3II, microtubule-associated protein 1
light chain 3 beta; NCOA4, nuclear receptor coactivator 4; FTH1,
ferritin heavy chain 1; p-, phosphorylation; I/R,
ischemia/reperfusion; ROS, reactive oxygen species; SOD, superoxide
dismutase; MDA, malondialdehyde; GSH, glutathione; GSSG,
glutathione disulfide; A/R, anoxia/reoxygenation; ATG, autophagy
related gene. |
Discussion
MIRI, a notable complication with poor prognosis
among patients with cardiac disorder, causes irreversible damage to
the heart (44). Therefore, it
is key to understand molecular mechanisms underlying MIRI and
investigate the potential of effective therapeutic agents in
mitigating MIRI. Ferroptosis process is iron-dependent and involves
enhanced accumulation of ROS and impairment of the GSH-dependent
antioxidant system, which are the primary causes of IRI (19). Cur exhibits potent modulatory
activity on multiple signaling pathways associated with
inflammation and oxidative stress (45). Consequently, Cur may be a
promising candidate compound for MIRI attenuation (46). The present study provided
insights into the molecular mechanisms underlying the protective
role of Cur in MIRI. MIRI induced autophagy-dependent ferroptosis
and apoptosis. In addition, Cur modulated Sirt1 to attenuate
autophagy-dependent ferroptosis and apoptosis in cardiomyocytes.
Modulation of Sirt1 demonstrated that Sirt1 regulated the
phosphorylation level of AKT and FoxO3a, which localized FoxO3a in
the cytoplasm and blocked its entry into the nucleus to prevent
initiation of transcription. In turn, reduced translation decreased
expression of autophagy and ferroptosis biomarkers, thereby
protecting cardiomyocyte function. These findings elucidated the
molecular mechanisms by which Cur attenuates MIRI, offering novel
insights and avenues for future therapeutic strategies for
MIRI.
In addition to traditional treatment options for
cardiovascular disease, a growing body of research has suggested
that functional food compounds treat cardiovascular disease via
their effect on the epigenome (13,47-49). Cur is a bioactive component of
turmeric that exerts multiple protective effects on the
cardiovascular system, and its pleiotropic effects in
cardiovascular disease have been studied extensively to establish
it as a potential candidate for the treatment of MI (17,46). Cur attenuates MIRI by modulating
redox dysregulation; however, Cur also modulates downstream protein
activity and expression to attenuate MIRI (50,51). Duan et al (17) showed that Cur activates the
JAK2/STAT3 pathway and decreases oxidative damage, which in turn
inhibits MIRI. In addition, Kim et al (52) suggested that Cur modulates the
toll-like receptor 2/NF-κB pathway to attenuate MIRI. The present
study revealed that Cur effectively reduced serum LDH and CK-MB
levels after MIRI and changes in histopathology and myocardial
apoptosis after MIRI were notably suppressed. Of note, both in
vivo and in vitro experiments showed that IRI or A/R
injury decreased Sirt1 protein expression in cardiomyocytes,
whereas Cur effectively increased Sirt1 protein expression,
improved cardiomyocyte viability and decreased apoptosis. Further
studies should determine whether Sirt1 activates the downstream
pathway proteins to regulate cardiomyocyte phenotype.
Ferroptosis is an iron ion-dependent mode of cell
death in which stress conditions decrease the antioxidant capacity
of the cell, causing ROS accumulation, peroxidation and ultimately
cell death (26). Autophagy is a
conserved cell survival mechanism that maintains cell function and
morphology by removing damaged organelles or proteins; however,
uncontrolled autophagy can cause cell death (9,53). Autophagy is an upstream mechanism
of ferroptosis that promotes iron oxidation by regulating
intracellular iron metabolism and ROS production (54-56). Ferroptosis occurs predominantly
during the reperfusion phase in the myocardium, as intracellular
iron overload occurs only when myocardial blood flow is restored
(57). This leads to
accumulation of intracellular Fe2+ and ROS, resulting in
cardiomyocyte dysfunction (19).
Excessive autophagy also occurs in cardiomyocytes at this stage
(9). Consequently, it was
hypothesized that autophagy-dependent ferroptosis is involved in
MIRI. In the present study, 3-MA (autophagy inhibitor) was used to
inhibit excessive autophagy in cardiomyocytes, leading to an
increase in FTH (a protein that stores iron and serves a key role
in maintaining intracellular iron homeostasis) (55) and decrease in NCOA4 (a specific
mediator of ferritin phagocytosis that selectively degrades
ferritin) (58). This indicates
that autophagy-dependent ferroptosis occurs during MIRI. Cur
pretreatment effectively inhibited ferroptosis induced by excessive
autophagy during MIRI, although the molecular mechanisms were not
fully investigated. Based on the present results, it is reasonable
to speculate that Cur further attenuates MIRI-induced
autophagy-dependent ferroptosis via Sirt1-based activation of
downstream pathways.
Sirt1 is a member of the NAD-dependent histone
deacetylases (59). Sirt1 exerts
a protective effect in various models of cardiovascular oxidative
stress and mitigate doxorubicin-induced cardiotoxicity through the
regulation of H2A histone family member X (60,61). In addition, there is evidence
suggesting its involvement in other processes, such as
cardiomyocyte apoptosis, autophagy, oxidative stress, cellular
senescence and metabolic regulation (62). These findings are consistent with
the present results, which demonstrated that increased expression
of Sirt1 by Cur was accompanied by decreased cell death, autophagy
and ferroptosis. However, silencing of Sirt1 expression resulted in
a decreased protective effect of Cur, indicating that the
protective effect of Cur was dependent on Sirt1. Sirt1 has been
shown to mediate the activity of several key cellular proteins
(AMPK, Bcl2, and Nrf2) associated with cardioprotection in addition
to deacetylating lysine residues in histones for epigenetic
regulation (63-65). AKT regulates the activity of
several targets, including the pro-apoptotic protein Bad, caspase 9
and members of the FoxO transcription factor/protein family (FoxO1,
FoxO3a and FoxO4) (42). The
present study results indicated that Cur is an effective activator
of the phosphorylation of AKT and FoxO3a. However, phosphorylation
level of AKT and FoxO3a was reduced following the intervention of
si-Sirt1. Cur failed to induce FoxO3a phosphorylation after AKT
phosphorylation was blocked, which indicated that Cur activated AKT
phosphorylation via Sirt1, followed by the subsequent activation of
FoxO3a phosphorylation by AKT. Previous studies have shown that the
AKT-induced phosphorylation of FoxO proteins leads to their nuclear
export, localizing FoxO in the cytoplasm (42,66). This is in line with the present
results. Nucleoplasm separation experiments showed that A/R injury
retained FoxO3a in the nucleus and Cur impaired this effect.
However, after inhibiting Sirt1 protein expression, FoxO3a was
translocated from cytoplasm to the nucleus; the blockade of AKT
activity also promoted FoxO3a translocation to the nucleus. FoxO3a
entry into the nucleus during MIRI stimulates translation of
autophagy-associated proteins (LC3, Gabarapl1 and ATG12), which
impairs mitochondrial metabolism, promotes apoptosis and
exacerbates ischemic injury in cardiomyocytes (27). The present study revealed that
autophagy and ferroptosis levels were increased and viability
decreased in H9c2 cells after the A/R-promoted FoxO3a entry into
the nucleus. By contrast, apoptosis rate, number of autophagic
lysosomes and accumulation of intracellular ROS and Fe2+
decreased in H9c2 cells after Cur retained FoxO3a in the cytoplasm.
These results indicated that Cur attenuates both apoptosis and
autophagy-dependent ferroptosis induced by A/R injury in H9c2 cells
via the Sirt1/AKT/FoxO3a pathway. As post-translational
modification alters subcellular localization of FoxO3a and its
functional release (40), it is
unclear whether phosphorylation or acetylation exerts a greater
effect on nuclear translocation of FoxO3a. It is unclear whether
target genes of FoxO3a directly affect ferroptosis.
Taken together, the present study showed that
MIRI-induced excessive autophagy caused intracellular iron ion
accumulation, which promoted ferroptosis and impaired cardiac
function. By contrast, Cur inhibited MIRI-induced
autophagy-dependent ferroptosis and apoptosis in cardiomyocytes via
Sirt1/AKT/FoxO3a, thereby increasing cardiomyocyte survival and
maintaining cardiac function. These findings provide novel
perspectives to understand autophagy-dependent ferroptosis during
MIRI.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
STZ, ZCQ and ZQX performed experiments, analyzed
data and wrote the manuscript. LFZ and HZP performed experiments
and analyzed data. RBQ and EDT analyzed and interpreted data. SQL
and LW confirm the authenticity of all the raw data. SQL and LW
designed the experiments. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
The present research protocol was reviewed and
approved by the Ethics Committee of the First Affiliated Hospital
of Nanchang University (approval no. CDYFY-IACUC-202211QR010).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Nature Science
Foundation of China (grant nos. 82460057 and 82160073) and Jiangxi
Provincial Natural Science Foundation (grant nos. 20212ACB206011,
20224ACB206002 and 20232BAB206009).
References
|
1
|
Abdollahi E, Momtazi AA, Johnston TP and
Sahebkar A: Therapeutic effects of curcumin in inflammatory and
immune-mediated diseases: A nature-made jack-of-all-trades? J Cell
Physiol. 233:830–848. 2018. View Article : Google Scholar
|
|
2
|
Xie W, Gan J, Zhou X, Tian H, Pan X, Liu
W, Li X, Du J, Xu A, Zheng M, et al: Myocardial infarction
accelerates the progression of MASH by triggering
immunoinflammatory response and induction of periosti. Cell Metab.
36:1269–1286.e9. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen H, Yao Z, Zhao W, Zhang Y, Yao C and
Tong C: miR-21 enhances the protective effect of loperamide on rat
cardiomyocytes against hypoxia/reoxygenation, reactive oxygen
species production and apoptosis via regulating Akap8 and Bard1
expression. Exp Ther Med. 17:1312–1320. 2019.PubMed/NCBI
|
|
4
|
Wang Z, Yao M, Jiang L, Wang L, Yang Y,
Wang Q, Qian X, Zhao Y and Qian J: Dexmedetomidine attenuates
myocardial ischemia/reperfusion-induced ferroptosis via
AMPK/GSK-3β/Nrf2 axis. Biomed Pharmacother. 154:1135722022.
View Article : Google Scholar
|
|
5
|
Hausenloy DJ, Garcia-Dorado D, Bøtker HE,
Davidson SM, Downey J, Engel FB, Jennings R, Lecour S, Leor J,
Madonna R, et al: Novel targets and future strategies for acute
cardioprotection: Position paper of the european society of
cardiology working group on cellular biology of the heart.
Cardiovasc Res. 113:564–585. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Davidson SM, Ferdinandy P, Andreadou I,
Bøtker HE, Heusch G, Ibáñez B, Ovize M, Schulz R, Yellon DM,
Hausenloy DJ, et al: Multitarget strategies to reduce myocardial
ischemia/reperfusion injury: JACC review topic of the week. J Am
Coll Cardiol. 73:89–99. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao GL, Yu LM, Gao WL, Duan WX, Jiang B,
Liu XD, Zhang B, Liu ZH, Zhai ME, Jin ZX, et al: Berberine protects
rat heart from ischemia/reperfusion injury via activating
JAK2/STAT3 signaling and attenuating endoplasmic reticulum stress.
Acta Pharmacol Sin. 37:354–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yao M, Wang Z, Jiang L, Wang L, Yang Y,
Wang Q, Qian X, Zeng W, Yang W, Liang R and Qian J: Oxytocin
ameliorates high glucose- and ischemia/reperfusion-induced
myocardial injury by suppressing pyroptosis via AMPK signaling
pathway. Biomed Pharmacother. 153:1134982022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wen L, Cheng X, Fan Q, Chen Z, Luo Z, Xu
T, He M and He H: TanshinoneIIA inhibits excessive autophagy and
protects myocardium against ischemia/reperfusion injury via
14-3-3η/Akt/Beclin1 pathway. Eur J Pharmacol. 954:1758652023.
View Article : Google Scholar
|
|
10
|
Anand P, Thomas SG, Kunnumakkara AB,
Sundaram C, Harikumar KB, Sung B, Tharakan ST, Misra K,
Priyadarsini IK, Rajasekharan KN and Aggarwal BB: Biological
activities of curcumin and its analogues (Congeners) made by man
and mother nature. Biochem Pharmacol. 76:1590–1611. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aggarwal BB, Sundaram C, Malani N and
Ichikawa H: Curcumin: The Indian solid gold. Adv Exp Med Biol.
595:1–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kumar G, Mittal S, Sak K and Tuli HS:
Molecular mechanisms underlying chemopreventive potential of
curcumin: Current challenges and future perspectives. Life Sci.
148:313–328. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He H, Luo Y, Qiao Y, Zhang Z, Yin D, Yao
J, You J and He M: Curcumin attenuates doxorubicin-induced
cardiotoxicity via suppressing oxidative stress and preventing
mitochondrial dysfunction mediated by 14-3-3γ. Food Funct.
9:4404–4418. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang C, Shi Q, Yang J, Ren H, Zhang L,
Chen S, Si J, Liu Y, Sha D, Xu B and Ni J: Ceria nanozyme
coordination with curcumin for treatment of sepsis-induced cardiac
injury by inhibiting ferroptosis and inflammation. J Adv Res.
63:159–170. 2024. View Article : Google Scholar :
|
|
15
|
El-Far AH, Elewa YHA, Abdelfattah EZA,
Alsenosy AWA, Atta MS, Abou-Zeid KM, Al Jaouni SK, Mousa SA and
Noreldin AE: Thymoquinone and curcumin defeat aging-associated
oxidative alterations induced by D-galactose in rats' brain and
heart. Int J Mol Sci. 22:68392021. View Article : Google Scholar
|
|
16
|
Hong D, Zeng X, Xu W, Ma J, Tong Y and
Chen Y: Altered profiles of gene expression in curcumin-treated
rats with experimentally induced myocardial infarction. Pharmacol
Res. 61:142–148. 2010. View Article : Google Scholar
|
|
17
|
Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou
J, Zhang J, Jin Z and Yi D: The effects of curcumin post-treatment
against myocardial ischemia and reperfusion by activation of the
JAK2/STAT3 signaling pathway. Basic Res Cardiol. 107:2632012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang JY, Kim H, Mun D, Yun N and Joung B:
Co-delivery of curcumin and miRNA-144-3p using heart-targeted
extracellular vesicles enhances the therapeutic efficacy for
myocardial infarction. J Control Release. 331:62–73. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang B, Xu Y, Yu J, Wang Q, Fan Q, Zhao X,
Qiao Y, Zhang Z, Zhou Q, Yin D, et al: Salidroside pretreatment
alleviates ferroptosis induced by myocardial ischemia/reperfusion
through mitochondrial superoxide-dependent AMPKα2 activation.
Phytomedicine. 128:1553652024. View Article : Google Scholar
|
|
21
|
Fang X, Ardehali H, Min J and Wang F: The
molecular and metabolic landscape of iron and ferroptosis in
cardiovascular disease. Nat Rev Cardiol. 20:7–23. 2023. View Article : Google Scholar
|
|
22
|
Chen YR and Zweier JL: Cardiac
mitochondria and reactive oxygen species generation. Circ Res.
114:524–537. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pasricha SR, Tye-Din J, Muckenthaler MU
and Swinkels DW: Iron deficiency. Lancet. 397:233–248. 2021.
View Article : Google Scholar
|
|
24
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kremastinos DT and Farmakis D: Iron
overload cardiomyopathy in clinical practice. Circulation.
124:2253–2263. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim CH and Leitch HA: Iron
overload-induced oxidative stress in myelodysplastic syndromes and
its cellular sequelae. Crit Rev Oncol Hematol. 163:1033672021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang C, Xu W, Zhang Y, Zhang F and Huang
K: PARP1 promote autophagy in cardiomyocytes via modulating FoxO3a
transcription. Cell Death Dis. 9:10472018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
D'Onofrio N, Servillo L and Balestrieri
ML: SIRT1 and SIRT6 signaling pathways in cardiovascular disease
protection. Antioxid Redox Signal. 28:711–732. 2018. View Article : Google Scholar :
|
|
29
|
Karbasforooshan H and Karimi G: The role
of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother.
90:386–392. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Verdin E: The many faces of sirtuins:
Coupling of NAD metabolism, sirtuins and lifespan. Nat Med.
20:25–27. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li W, Du D, Wang H, Liu Y, Lai X, Jiang F,
Chen D, Zhang Y, Zong J and Li Y: Silent information regulator 1
(SIRT1) promotes the migration and proliferation of endothelial
progenitor cells through the PI3K/Akt/eNOS signaling pathway. Int J
Clin Exp Pathol. 8:2274–2287. 2015.PubMed/NCBI
|
|
32
|
National Research Council Committee for
the Update of the Guide for the C and Use of Laboratory A: The
National Academies Collection: Reports funded by National
Institutes of Health. Guide for the Care and Use of Laboratory
Animals. National Academies Press; US: Copyright © 2011, National
Academy of Sciences, Washington (DC). 2011
|
|
33
|
Nolen RS: New AVMA guidelines aim to limit
animal suffering in emergencies. J Am Vet Med Assoc. 254:1242–1243.
2019.
|
|
34
|
He H, Wang L, Qiao Y, Yang B, Yin D and He
M: Epigallocatechin-3-gallate pretreatment alleviates
doxorubicin-induced ferroptosis and cardiotoxicity by upregulating
AMPKα2 and activating adaptive autophagy. Redox Biol.
48:1021852021. View Article : Google Scholar
|
|
35
|
Zhao ST, Qiu ZC, Zeng RY, Zou HX, Qiu RB,
Peng HZ, Zhou LF, Xu ZQ, Lai SQ and Wan L: Exploring the molecular
biology of ischemic cardiomyopathy based on ferroptosis-related
genes. Exp Ther Med. 27:2212024. View Article : Google Scholar
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
37
|
Teringova E and Tousek P: Apoptosis in
ischemic heart disease. J Transl Med. 15:872017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, An M, Fu X, Meng X, Ma Y, Liu H, Li
Q, Xu H and Chen J: Bushen Wenyang Huayu Decoction inhibits
autophagy by regulating the SIRT1-FoXO-1 pathway in endometriosis
rats. J Ethnopharmacol. 308:1162772023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dong Y, Chen H, Gao J, Liu Y, Li J and
Wang J: Molecular machinery and interplay of apoptosis and
autophagy in coronary heart disease. J Mol Cell Cardiol. 136:27–41.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen L, Li S, Zhu J, You A, Huang X, Yi X
and Xue M: Mangiferin prevents myocardial infarction-induced
apoptosis and heart failure in mice by activating the Sirt1/FoxO3a
pathway. J Cell Mol Med. 25:2944–2955. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang X, Jiang T, Wang Y and Guo L: The
role and mechanism of SIRT1 in resveratrol-regulated osteoblast
autophagy in osteoporosis rats. Sci Rep. 9:184242019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tang L, Zeng Z, Zhou Y, Wang B, Zou P,
Wang Q, Ying J, Wang F, Li X, Xu S, et al: Bacillus
amyloliquefaciens SC06 Induced AKT-FOXO signaling pathway-mediated
autophagy to alleviate oxidative stress in IPEC-J2 cells.
Antioxidants (Basel). 10:15452021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sengupta A, Molkentin JD and Yutzey KE:
FoxO transcription factors promote autophagy in cardiomyocytes. J
Biol Chem. 284:28319–28331. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tsao CW, Aday AW, Almarzooq ZI, Anderson
CAM, Arora P, Avery CL, Baker-Smith CM, Beaton AZ, Boehme AK,
Buxton AE, et al: Heart disease and stroke statistics-2023 update:
A report from the american heart association. Circulation.
147:e93–e621. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li H, Sureda A, Devkota HP, Pittalà V,
Barreca D, Silva AS, Tewari D, Xu S and Nabavi SM: Curcumin, the
golden spice in treating cardiovascular diseases. Biotechnol Adv.
38:1073432020. View Article : Google Scholar
|
|
46
|
Mokhtari-Zaer A, Marefati N, Atkin SL,
Butler AE and Sahebkar A: The protective role of curcumin in
myocardial ischemia-reperfusion injury. J Cell Physiol.
234:214–222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jabczyk M, Nowak J, Hudzik B and
Zubelewicz-Szkodzińska B: Curcumin in metabolic health and disease.
Nutrients. 13:44402021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li M, Wu H, Yuan Y, Hu B and Gu N: Recent
fabrications and applications of cardiac patch in myocardial
infarction treatment. VIEW. 3:202001532022. View Article : Google Scholar
|
|
49
|
Lin L, Liu S, Chen Z, Xia Y, Xie J, Fu M,
Lu D, Wu Y, Shen H, Yang P and Qian J: Anatomically resolved
transcriptome and proteome landscapes reveal disease-relevant
molecular signatures and systematic changes in heart function of
end-stage dilated cardiomyopathy. VIEW. 4:202200402023. View Article : Google Scholar
|
|
50
|
Guo S, Meng XW, Yang XS, Liu XF, Ou-Yang
CH and Liu C: Curcumin administration suppresses collagen synthesis
in the hearts of rats with experimental diabetes. Acta Pharmacol
Sin. 39:195–204. 2018. View Article : Google Scholar :
|
|
51
|
Ramachandran C, Rodriguez S, Ramachandran
R, Raveendran Nair PK, Fonseca H, Khatib Z, Escalon E and Melnick
SJ: Expression profiles of apoptotic genes induced by curcumin in
human breast cancer and mammary epithelial cell lines. Anticancer
Res. 25:3293–3302. 2005.PubMed/NCBI
|
|
52
|
Kim YS, Kwon JS, Cho YK, Jeong MH, Cho JG,
Park JC, Kang JC and Ahn Y: Curcumin reduces the cardiac
ischemia-reperfusion injury: Involvement of the toll-like receptor
2 in cardiomyocytes. J Nutr Biochem. 23:1514–1523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hu F, Hu T, Qiao Y, Huang H, Zhang Z,
Huang W, Liu J and Lai S: Berberine inhibits excessive autophagy
and protects myocardium against ischemia/reperfusion injury via the
RhoE/AMPK pathway. Int J Mol Med. 53:492024. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Chen HY, Xiao ZZ, Ling X, Xu RN, Zhu P and
Zheng SY: ELAVL1 is transcriptionally activated by FOXC1 and
promotes ferroptosis in myocardial ischemia/reperfusion injury by
regulating autophagy. Mol Med. 27:142021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhou B, Liu J, Kang R, Klionsky DJ,
Kroemer G and Tang D: Ferroptosis is a type of autophagy-dependent
cell death. Semin Cancer Biol. 66:89–100. 2020. View Article : Google Scholar
|
|
56
|
Wu D, Zhang K and Hu P: The role of
autophagy in acute myocardial infarction. Front Pharmacol.
10:5512019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cai W, Liu L, Shi X, Liu Y, Wang J, Fang
X, Chen Z, Ai D, Zhu Y and Zhang X: Alox15/15-HpETE aggravates
myocardial ischemia-reperfusion injury by promoting cardiomyocyte
ferroptosis. Circulation. 147:1444–1460. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mancias JD, Wang X, Gygi SP, Harper JW and
Kimmelman AC: Quantitative proteomics identifies NCOA4 as the cargo
receptor mediating ferritinophagy. Nature. 509:105–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo Z, Liao Z, Huang L, Liu D, Yin D and
He M: Kaempferol protects cardiomyocytes against
anoxia/reoxygenation injury via mitochondrial pathway mediated by
SIRT1. Eur J Pharmacol. 761:245–253. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Huang L, He H, Liu Z, Liu D, Yin D and He
M: Protective effects of isorhamnetin on cardiomyocytes against
anoxia/reoxygenation-induced injury is mediated by SIRT1. J
Cardiovasc Pharmacol. 67:526–537. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kuno A, Hosoda R, Tsukamoto M, Sato T,
Sakuragi H, Ajima N, Saga Y, Tada K, Taniguchi Y, Iwahara N and
Horio Y: SIRT1 in the cardiomyocyte counteracts doxorubicin-induced
cardiotoxicity via regulating histone H2AX. Cardiovasc Res.
118:3360–3373. 2023. View Article : Google Scholar
|
|
62
|
Ding X, Zhu C, Wang W, Li M, Ma C and Gao
B: SIRT1 is a regulator of autophagy: Implications for the
progression and treatment of myocardial ischemia-reperfusion.
Pharmacol Res. 199:1069572024. View Article : Google Scholar
|
|
63
|
Wei YJ, Wang JF, Cheng F, Xu HJ, Chen JJ,
Xiong J and Wang J: miR-124-3p targeted SIRT1 to regulate cell
apoptosis, inflammatory response, and oxidative stress in acute
myocardial infarction in rats via modulation of the
FGF21/CREB/PGC1α pathway. J Physiol Biochem. 77:577–587. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu S, Qian H, Tian D, Yang M, Li D, Xu H,
Chen J, Yang J, Hao X, Liu Z, et al: Linggui Zhugan Decoction
activates the SIRT1-AMPK-PGC1α signaling pathway to improve
mitochondrial and oxidative damage in rats with chronic heart
failure caused by myocardial infarction. Front Pharmacol.
14:10748372023. View Article : Google Scholar
|
|
65
|
Zhang W, Wang X, Tang Y and Huang C:
Melatonin alleviates doxorubicin-induced cardiotoxicity via
inhibiting oxidative stress pyroptosis and apoptosis by activating
Sirt1/Nrf2 pathway. Biomed Pharmacother. 162:1145912023. View Article : Google Scholar
|
|
66
|
Xie W, Zhu T, Zhang S and Sun X:
Protective effects of Gypenoside XVII against cerebral
ischemia/reperfusion injury via SIRT1-FOXO3A- and
Hif1a-BNIP3-mediated mitochondrial autophagy. J Transl Med.
20:6222022. View Article : Google Scholar : PubMed/NCBI
|