Introduction
Myocardial ischemia (MI) is typically caused by
inadequate blood flow in the coronary arteries. A worldwide
epidemiological analysis indicated that the number of cases of MI
steadily increased from 1990-2019, reaching a total of 197 million
cases by 2019 (1). Myocardial
infarction, caused by prolonged and severe MI, is a prevalent
cardiovascular disease and a major cause of disability and
mortality worldwide (2,3). The accumulation of reactive oxygen
species (ROS) during MI creates conditions for the generation of
oxidative stress (4). Currently,
the primary treatment method for myocardial infarction is coronary
artery reperfusion (5). This
procedure is crucial for treating ischemic myocardial tissue damage
by removing harmful metabolites and restoring normal metabolism
(4). However, the sudden
reintroduction of high oxygen tension due to blood flow reperfusion
results in increased levels of oxygen free radicals, leading to a
surge in ROS levels. This causes more severe tissue damage than
that caused by ischemia (6-8),
a condition known as MI/reperfusion injury (MIRI). MIRI is a
complex pathological process related to the production of ROS and
mitochondrial dysfunction, involving reduced ATP production and
destruction of the mitochondrial ridge (9). Therefore, it is necessary to
clarify the molecular mechanisms underlying MIRI to develop
effective prevention strategies.
During reperfusion, uncontrolled ROS-mediated
oxidative stress has been considered a key triggering factor for
MIRI (10). ROS mainly originate
from the byproduct of the mitochondrial respiratory chain (11). Under physiological conditions,
ROS participate in signal transduction and regulate various
physiological activities of the heart (12). However, uncontrolled increases in
ROS levels cause damage to biological macromolecules, such as lipid
peroxidation and mitochondrial dysfunction, which may lead to
ferroptosis (13). Nuclear
factor erythroid 2-related factor 2 (Nrf2) is a transcription
factor that activates endogenous antioxidant enzymes in response to
oxidative stress and ferroptosis (14). It is widely expressed in tissues
of oxygen-consuming organs, such as the muscles, heart and blood
vessels (15). When oxidative
stress occurs, Nrf2 is activated and is transferred from the
cytoplasm to the nucleus to maintain cellular redox balance and
avoid the occurrence of lipid peroxidation (16).
The regulator of G-protein signaling (RGS) family
serves a crucial role in the regulatory processes of the
cardiovascular system (17). A
previous study reported a significant increase in the mRNA and
protein expression levels of RGS3 and RGS4 in failing heart tissues
(18). It has been shown that
the expression of RGS2 inhibits G-protein signaling in the
myocardium of adult mice, which is necessary to prevent the early
development of compensatory hypertrophy in response to pressure
overload (19). The deletion of
RGS6 exacerbates ischemic injury in the hearts of mice (20). RGS12 is currently the largest
known mammalian RGS protein (21). It has been reported that the
expression of RGS12 may contribute to pathological cardiac
hypertrophy (22). RGS12
contains a central RGS domain, a postsynaptic density
protein-95/discs-large/zona occludens homology domain, a
phosphotyrosine binding domain, tandem Ras binding domains and a
Gαi/o-Loco interaction motif (21), which suggests the versatility of
RGS12 and its association with multiple signaling pathways. RGS12
has been reported to regulate oxidative stress through Nrf2 in
various tissues, including neuronal cells (23) and osteoclasts (24); however, its role in the heart
remains unexplored. The deficiency of RGS12 activates Nrf2 and
impairs the production of ROS in osteoclast (24), but overexpression of RGS12
inhibited the activation of Nrf2 (24). Targeting RGS12 may thus alleviate
oxidative stress and inflammation by regulating the Nrf2 pathway in
the hippocampus of depressed rats (23). However, in MIRI, it is currently
unknown whether RGS12 affects the Nrf2 pathway.
Penehyclidine hydrochloride (PHC) is an
anticholinergic drug with antimuscarinic and antinicotinic activity
(25). Previous studies have
reported that preconditioning with PHC is effective in preventing
ischemia/reperfusion injury in multiple organs, which is achieved
by inhibiting apoptosis and relieving oxidative stress (26,27). Preconditioning with PHC
alleviates the mitochondrial dynamic imbalance and protects
myocardial cells against MIRI (28). This suggests a potential
protective role of PHC against MIRI; however, the underlying
molecular mechanisms have not yet been fully elucidated.
The present study aimed to establish a mouse model
of MIRI and a cell model subjected to hypoxia/reoxygenation (H/R)
to investigate the regulatory role and molecular mechanisms of
RGS12 in MIRI and the myocardium after PHC preconditioning.
Materials and methods
Adenovirus (Ad) construction
The short hairpin RNA (shRNA) sequence targeting
mouse RGS12 (shRGS12, 5′-GGACCTCAGCACTCGAGAAAG-3′), along with a
non-specific sequence (shNC, 5′-TTCTCCGAACGTGTCACGT-3′), were
synthesized and inserted into pShuttle-CMV plasmids (cat. no.
BR009; Hunan Fenghui Biotechnology Co., Ltd.) by General Biology,
Inc. A total of 25 μg shuttle plasmids were transfected into
293A cells at room temperature using Lipofectamine 3000 (cat. no.
L3000015; Thermo Fisher Scientific, Inc.) and P3000 (cat. no.
L3000015; Thermo Fisher Scientific, Inc.). The coding region of
RGS12 was cloned into pDC315 plasmids by YouBio (cat. no. VT1500)
to construct the RGS12 overexpression vector. The mass of nucleic
acid used was 9 μg. Empty vector without any exogenous
fragment was used as a negative control. A total of 27 μg
plasmids (shuttle plasmid: skeleton plasmid=1:2) were transfected
into the 293A cells at room temperature using Lipofectamine 3000
(41.8 μl) (cat. no. L3000015; Thermo Fisher Scientific,
Inc.) and P3000 (54 μl) (cat. no. L3000015; Thermo Fisher
Scientific, Inc.). After 14 days, the cell supernatant containing
Ad particles was collected.
Animal experiments
The present study utilized 210 10-week-old male
C57BL/6 mice (weighing 24.0±2.0 g) that underwent a 1-week
acclimatization period to the feeding regimen. During the
experiment, the mice were allowed to eat and drink freely. Mice
were subjected to a 12 h light/dark cycle under controlled
environmental conditions, including a temperature maintained at
22±1°C and humidity levels between 45-55%. The animal experiments
were approved by the Ethics Committee of Hebei North University
(approval no. HBNU202306022105; Zhangjiakou, China).
The mice were allocated into two groups (Sham and
MIRI) through a random process, and were anesthetized using an
intraperitoneal injection of 75 mg/kg ketamine and 10 mg/kg
xylazine. Mice in the MIRI group were subjected to left main
coronary artery ligation for a duration of 30 min to induce
ischemia, followed by the restoration of blood flow for 24 h to
establish the MIRI model (29).
The mice in the Sham group underwent a surgical procedure without
ligation as a control measure. After surgery, all the animals were
administered a subcutaneous injection of 5 mg/kg carprofen for pain
relief (30) and were placed in
a comfortable environment that included maintaining a constant
temperature and humidity, as well as providing a cage with adequate
space and non-toxic, non-irritating bedding material. Animal health
and behavior were monitored every 4-6 h. According to the National
Institutes of Health Guidelines for Endpoints in Animal Study
Proposals (31) the following
humane endpoints were used: Anorexia (lack or loss of appetite),
failure to drink, labored breathing, gasping, lethargy or
persistent recumbency and excessive hyperthermia or
hypothermia.
Next, the mice were randomly divided into two groups
[MIRI + Ad-RGS12 and MIRI + Ad-empty vector (EV)]. After the mice
were anesthetized, the plasmid vectors carrying the coding sequence
of RGS12 or EVs were packaged into Ads and injected into the left
ventricular free wall of the mice in both groups with a total
volume of 30 μl and a concentration of 1×1011
plaque forming units/ml, 72 h prior to the induction of MIRI
(32).
For further experiments, the mice were randomly
divided into four groups, denoted as the MIRI, MIRI + PHC, MIRI +
PHC + Ad-EV and MIRI + PHC + Ad-RGS12 groups. The mice in the
experimental MIRI + PHC, MIRI + PHC + Ad-EV and MIRI + PHC +
Ad-RGS12 groups received a tail vein injection of PHC (cat. no.
HY-137976; Merck KGaA) at a dose of 1 mg/kg body weight 1 h prior
to the induction of MIRI (33).
The mice in the MIRI + PHC + Ad-EV and MIRI + PHC + Ad-RGS12 groups
received Ad injections 72 h prior to undergoing MIRI, followed by
the administration of PHC 1 h prior to the induction of MIRI.
The left ventricular function of all the mice was
evaluated under anesthesia utilizing an echocardiographic imaging
system. Subsequently, the mice were sacrificed via exsanguination.
When the mice experienced cardiac arrest and their pupils were
dilated, they were considered deceased. Blood (~1 ml) from postcava
was collected. Serum and myocardial tissues were subsequently
obtained for further analysis.
A total of 210 animals were used in the present
study, of which 195 were euthanized (n=24/group), while 15 were
found deceased during the operation. Of the animals euthanized, 2
animals experienced persistent recumbency and were euthanized at
the humane endpoints and 1 animal was used to explore the
appropriate antibody concentrations and incubation times in a
preliminary experiment.
Cell culture
The mouse myocardial cell line HL-1 (cat. no.
iCell-m077; iCell) was cultured in Minimum Essential Medium (cat.
no. 41500; Beijing Solarbio Science & Technology Co., Ltd.)
containing 10% FBS and 1% penicillin streptomycin and incubated at
37°C and 5% CO2.
Cell induction
HL-1 cells were exposed to various concentrations of
PHC (1, 2.5 and 5 μg/ml) at 37°C for a duration of 2 h
(34), after which they were
subjected to H/R injury according to previously established
protocols (35). Briefly, the
cells were cultured in a medium devoid of FBS for a duration of 12
h. Following this, the cells were subjected to hypoxic conditions
consisting of 1% O2, 94% N2 and 5%
CO2 for a period of 8 h. Subsequently, the cells were
returned to normal culture conditions for 12 h to induce H/R
injury. Control cells were cultured under normal conditions. Cell
viability was assessed using the Cell Counting Kit-8 assay in
accordance with the manufacturer's protocol (cat. no. KGA317;
Nanjing KeyGen Biotech Co., Ltd.). The cells were incubated with
CCK-8 reagent at 37°C for 2 h.
Infection
Ad was added to the culture medium. Following a 48-h
infection period, the HL-1 cells were exposed to PHC (5
μg/ml) at 37°C and H/R was induced in cells after 2 h. The
concentration of 5 μg/ml was selected on the basis that 5
μg/ml PHC treatment had the strongest inhibition of RGS12
expression and did not decrease cell viability.
Staining with 2,3,5-triphenyl tetrazolium
chloride (TTC)
Myocardial tissues cryopreserved at -2°C were
sectioned into 1 mm slices, followed by staining with a 0.4%
solution of TTC at 37°C for 15 min in the dark. The viable
myocardial tissues exhibited a red stain, while the infarcted
region displayed a white appearance.
Measurement of biochemical markers
The activity of LDH, CK and AST in serum were
measured. The content of MDA and the activity of SOD in ischemic
penumbra of myocardial tissues were detected. The content of MDA
and the activity of SOD and CAT in the cells were detected. Samples
were preprocessed according to the instructions provided by the
kits' manufacturer. The myocardial tissues were homogenized in
normal saline, then centrifuged at 4°C at 12,000 × g for 10 min and
the supernatant was collected for analysis. The cells were lysed
using ultrasound on ice (power, 200 W; 5 sec ultrasonic treatment;
15 sec interval, repeated 5 times) and then centrifuged at 4°C at
15,000 × g for 10 min to separate the supernatant for detection.
The serum was analyzed directly without any pretreatment. The
activities of lactate dehydrogenase (LDH; cat. no. A020), creatine
kinase (CK; cat. no. A032), aspartate aminotransferase (AST; cat.
no. C010), catalase (CAT; cat. no. A007), glutathione peroxidase
(GPX; cat. no. A005) and superoxide dismutase (SOD; cat. no.
A003-1), as well as the concentration of malondialdehyde (MDA; cat.
no. A001), were quantified utilizing test kits procured from
Nanjing Jiancheng Bioengineering Institute in accordance with the
manufacturer's protocols. The activity of all enzymes was measured
through enzymatic reaction at 37°C. MDA content was determined
based on the Thibabituric Acid assay at 95°C (cat. no. A001;
Nanjing Jiancheng Bioengineering Institute). Serum troponin T (TnT)
levels were assessed using an ELISA kit (cat. no. SEB820Mu; Wuhan
USCN Business Co., Ltd.).
Histopathological analysis
Myocardial tissues obtained from the left ventricle
were preserved in a 4% paraformaldehyde solution at room
temperature for 24 h, subsequently embedded in paraffin and
sectioned into 5 μm slices. The sections underwent a
dewaxing process and were stained with hematoxylin (5 min) and
eosin (3 min) at room temperature. Subsequently, the samples were
imaged using a light microscope (BX53; Olympus Corporation).
Immunofluorescence
The HL-1 cell and ischemic penumbra of myocardial
tissue were pretreated separately. The cells (5×104)
were seeded onto coverslips in a 24-well plate and subsequently
fixed using 4% paraformaldehyde at room temperature for 20 min.
Cells were permeabilized using 0.1% Triton X-100 (cat. no. ST795;
Beyotime Institute of Biotechnology) for 30 min at room
temperature. Tissues were embedded in paraffin and sectioned into
5-μm slices. The slices were rehydrated in descending
alcohol series (95, 85, and 75%), and boiled in an antigen
retrieval solution (1.8% 0.1 M citrate buffer and 8.2% 0.1 M sodium
citrate buffer) at 95°C for 10 min. The slices were washed in a PBS
buffer. Subsequently, the prepared tissue or cell specimens were
treated with 1% BSA (cat. no. A602440-0050; Sangon Biotech Co.,
Ltd.) and incubated for 15 min at room temperature. Samples were
incubated at 4°C overnight with the following primary antibodies:
RGS12 (cat. no. DF4415; 1:100; Affinity Biosciences) and Nrf2 (cat.
no. AF0639; 1:100; Affinity Biosciences). Following multiple washes
in PBS buffer, samples were incubated for 1 h at 37°C with Alexa
Fluor 555-conjugated goat anti-rabbit IgG secondary antibodies
(cat. no. 4413; 1:200; Cell Signaling Technology, Inc.) in the
dark. DAPI (cat. no. D106471-5mg; Shanghai Aladdin Biochemical
Technology Co., Ltd.) culture for 5 min at room temperature was
utilized as a nuclear counterstain. Subsequently, the stained
sections were imaged using a fluorescence microscope.
ROS and lipid ROS detection
The ischemic penumbra of myocardial tissue was
embedded in Optimal Cutting Temperature Compound freezing medium
(cat. no. 4583; Sakura Finetek USA, Inc.), frozen at −20°C and cut
into 10 μm sections for staining. Cells (1×105)
were seeded into a 12-well plate and cultured to a confluence of
70-80%, and subsequently prepared for staining procedures. DHE (4
μM; cat. no. D807594; Shanghai Macklin Biochemical Co.,
Ltd.) was added to the tissues and cells and incubated at 37°C for
30 min. The stained sections and cells were then examined using a
fluorescence microscope to determine ROS levels.
To detect lipid ROS levels, cells (5×105)
were seeded into a 6-well plate and incubated with C11-BODIPY
581/591 (cat. no. MX5211; Shanghai Maokang Biotechnology Co., Ltd.)
at 37°C for 30 min, and the alteration in fluorescence emission
peak wavelength from 590-510 nm was assessed using a flow cytometer
(NovoCyte; Agilent Technologies, Inc.).
Fe2+ detection
The presence of Fe2+ in tissues and cells
was identified using a Ferrous Iron Colorimetric Assay kit [cat.
no. E-BC-K773-M (for tissues); cat. no. E-BC-K881-M (for cells);
Wuhan Elabscience Biotechnology Co., Ltd.] according to the
manufacturer's instructions.
Reverse transcription-quantitative PCR
(RT-qPCR)
The HL-1 cells and ischemic penumbra of myocardial
tissue were homogenized in TRIpure lysis buffer (cat. no. RP1001;
BioTeke Corporation) and the total RNA concentration was assessed
using an ultraviolet spectrophotometer (NanoDrop 2000; Thermo
Fisher Scientific, Inc.). Subsequently, cDNA was synthesized using
the BeyoRT II M-MLV reverse transcriptase (cat. no. D7160L;
Beyotime Institute of Biotechnology) as per the manufacturer's
guidelines. The resulting cDNA was utilized as a template for qPCR.
The qPCR procedure was conducted with SYBR Green (cat. no. SY1020;
Beijing Solarbio Science & Technology Co., Ltd.) and 2×Taq PCR
MasterMix (cat. no. PC1150; Beijing Solarbio Science &
Technology Co., Ltd.) using a fluorescent qPCR instrument
(Exicycler 96; Bioneer Corporation). The following thermocycling
conditions were used: 95°C for 5 min, followed by 95°C for 10 sec,
60°C for 10 sec and 72°C for 15 s. The 2−ΔΔCq method was
used to analyze the target gene expression (36). The primers used were obtained
from General Biology, Inc. and the sequences were as follows: RGS12
forward (F), 5′-AAGCGGACTTTGTTTCGG-3′ and reverse (R),
5′-GGAGCACCTTTCTGTTTGT-3′; and β-actin F,
5′-CATCCGTAAAGACCTCTATGCC-3′ and R, 5′-ATGGAGCCACCGATCCACA-3′.
Western blotting
The HL-1 cells and ischemic penumbra of myocardial
tissue were used for western blotting. Total protein was extracted
using lysis buffer (cat. no. P0013; Beyotime Institute of
Biotechnology) containing phenylmethanesulfonyl fluoride (cat. no.
ST506; Beyotime Institute of Biotechnology) on ice for 5 min. The
resulting lysates were centrifuged at 10,000 x g for 5 min at 4°C
to obtain supernatants containing proteins. The cytoplasmic and
nuclear proteins were then separated using the Nuclear Protein and
Cytoplasmic Protein Extraction kit (cat. no. P0027; Beyotime
Institute of Biotechnology). The protein concentration was
determined using the BCA method (cat. no. P0011; Beyotime Institute
of Biotechnology). The 30 μg protein samples were then
separated using SDS-PAGE with varying concentrations of gel (8, 10
and 14%) and a 5% stacking gel, followed by transfer onto PVDF
membranes. The membranes were blocked using a blocking buffer (cat.
no. P023; Beyotime Institute of Biotechnology) at room temperature
for 1 h and subsequently incubated overnight at 4°C with primary
antibodies targeting RGS12 (cat. no. DF4415; 1:500; Affinity
Biosciences), GPX4 (cat. no. A1933; 1:1,000; ABclonal Biotech Co.,
Ltd.), solute carrier family 7 member 11 (SLC7A11; cat. no.
DF12509; 1:500; Affinity Biosciences) and Nrf2 (cat. no. AF0639;
1:1,000; Affinity Biosciences). After washing using a washing
reagent (cat. no. P023; Beyotime Institute of Biotechnology), the
blots were incubated for 45 min at 37°C with horseradish
peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG,
cat. no. A0208; goat anti-mouse IgG, cat. no. A0216; 1:5,000;
Beyotime Institute of Biotechnology). Histone H3 (cat. no. AF0009;
1:1,000; Beyotime Institute of Biotechnology) and β-actin (cat. no.
sc-47778; 1:1,000; Santa Cruz Biotechnology, Inc.) were used as
internal references for nuclear protein and total protein,
respectively. The blots were visualized using ECL reagent (cat. no.
P0018; Beyotime Institute of Biotechnology).
Transmission electron microscopy (TEM)
analysis
The cells were fixed using an Electron Microscope
Fixative (cat. no. G1102; Wuhan Servicebio Technology Co., Ltd.)
and incubated at 4°C for 2 h before being embedded in 1% agarose.
Cells were treated with 1% osmium tetroxide for at room temperature
2 h. Following dehydration in ascending alcohol series (30, 50, 70,
80, 95 and 100%) the cells were encased in 812 epoxy resin monomers
(cat. no. 02659-ABl; Structure Probe, Inc.) at 37°C overnight.
Embedded cells were sliced into 60-80 nm sections, stained with 2%
uranyl acetate (8 min) and 2.6% lead citrate (8 min) at room
temperature and subsequently examined using TEM (cat. no. H-7650;
Hitachi High-Technologies Corporation). The mitochondria were
manually counted.
Statistical analysis
Data were statistically analyzed using GraphPad
software (version 9.0; Dotmatics). The data are presented as the
mean ± SD. Statistical analysis was conducted using the unpaired
Student's t-test or one-way ANOVA with Tukey's post-hoc test to
identify significant differences between the experimental groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
RGS12 is highly expressed in a MIRI model
of mice and in cells subjected to H/R
The MIRI model was developed using a 30 min ischemia
and 24 h reperfusion (Fig. 1A).
The present study assessed heart function and myocardial
damage-related markers in mice to confirm the successful
establishment of the MIRI model. The echocardiography results
indicated a significant reduction in the left ventricular ejection
fraction (LVEF) and left ventricular fractional shortening (LVFS)
in the mice with MIRI compared with the control group (Fig. 1B). The mice with MIRI exhibited
severe injury in the myocardial tissues, as indicated by an
increase in the size of the infarct (Fig. 1C) and significantly increased
activity levels of LDH and CK in comparison with the control mice
(Fig. 1D). The intensity of red
fluorescence in the ischemic penumbra of the mice with MIRI was
greater compared with that of the control mice, indicating an
increase in ROS levels caused by MIRI (Fig. 1E). The concentration of
Fe2+ in the myocardial tissues of mice with MIRI was
significantly higher compared with that in the control group
(Fig. 1F), suggesting the
occurrence of ferroptosis. These results indicated that MIRI was
successfully induced in the mice subjected to ischemia/reperfusion.
Subsequently, it was demonstrated that RGS12 was highly expressed
in the mice with MIRI compared with the control group, as evidenced
by the results of RT-qPCR, western blotting analysis and
immunofluorescence (Fig. 1G and
H). Moreover, a cell model of H/R-induced injury was developed
using 12 h serum starvation, 8 h hypoxia and 12 h reoxygenation
(Fig. 1I). The expression level
of RGS12 was assessed. Similar patterns of RGS12 expression levels
were also observed in HL-1 cells with H/R injury compared with
control cells (Fig. 1J and
K).
 | Figure 1RGS12 is highly expressed in MIRI
mice and H/R cells. (A) The MIRI model was developed using a 30 min
ischemia and 24 h reperfusion. (B) Echocardiogram, LVEF and LVFS of
mice. (C) Quantification of the infarct area in the myocardial
tissue and images of infarct tissue. (D) Serum LDH and CK activity.
(E) ROS levels (magnification, ×400) and (F) Fe2+
content in the myocardial tissue. RGS12 expression levels in the
myocardial tissue was analyzed using (G) reverse-transcription
quantitative PCR and western blotting and (H) immunofluorescence.
Magnification, ×400. n=6. (I) A cell model of H/R-induced injury
was developed using a 12 h serum starvation, 8 h hypoxia and 12 h
reoxygenation. RGS12 expression levels in H/R cells was analyzed
using (J) reverse transcription quantitative PCR and western
blotting and (K) immunofluorescence. Magnification, ×400. n=3. Data
were presented as the mean ± SD. MIRI, myocardial
ischemia/reperfusion injury; H/R, hypoxia/reoxygenation; RGS12,
regulator of G-protein signaling 12; LVEF, left ventricular
ejection fraction; LVFS, left ventricular fractional shortening;
LDH, lactate dehydrogenase; CK, creatine kinase; ROS, reactive
oxygen species. |
Downregulation of RGS12 relieves
oxidative stress in H/R cells
To investigate the involvement of RGS12 in MIRI, the
expression of RGS12 was knocked down in HL-1 cells (Fig. S1A and B). Increased protein
expression levels of RGS12 were observed in the cells subjected to
H/R injury, except for the cells in which RGS12 expression was
suppressed (Fig. 2A). The
viability of the cells subjected to H/R was significantly reduced
compared with the control cells; however, the silencing of RGS12
resulted in a significant increase in cell viability (Fig. 2B). Elevated levels of ROS and MDA
were detected in the cells subjected to H/R compared with those in
the controls (Fig. 2C and D).
Conversely, the silencing of RGS12 led to a reduction in ROS levels
and the inhibition of MDA production. The activities of SOD and CAT
significantly decreased following H/R-induced injury; however,
these significantly increased after the silencing of RGS12
(Fig. 2E and F).
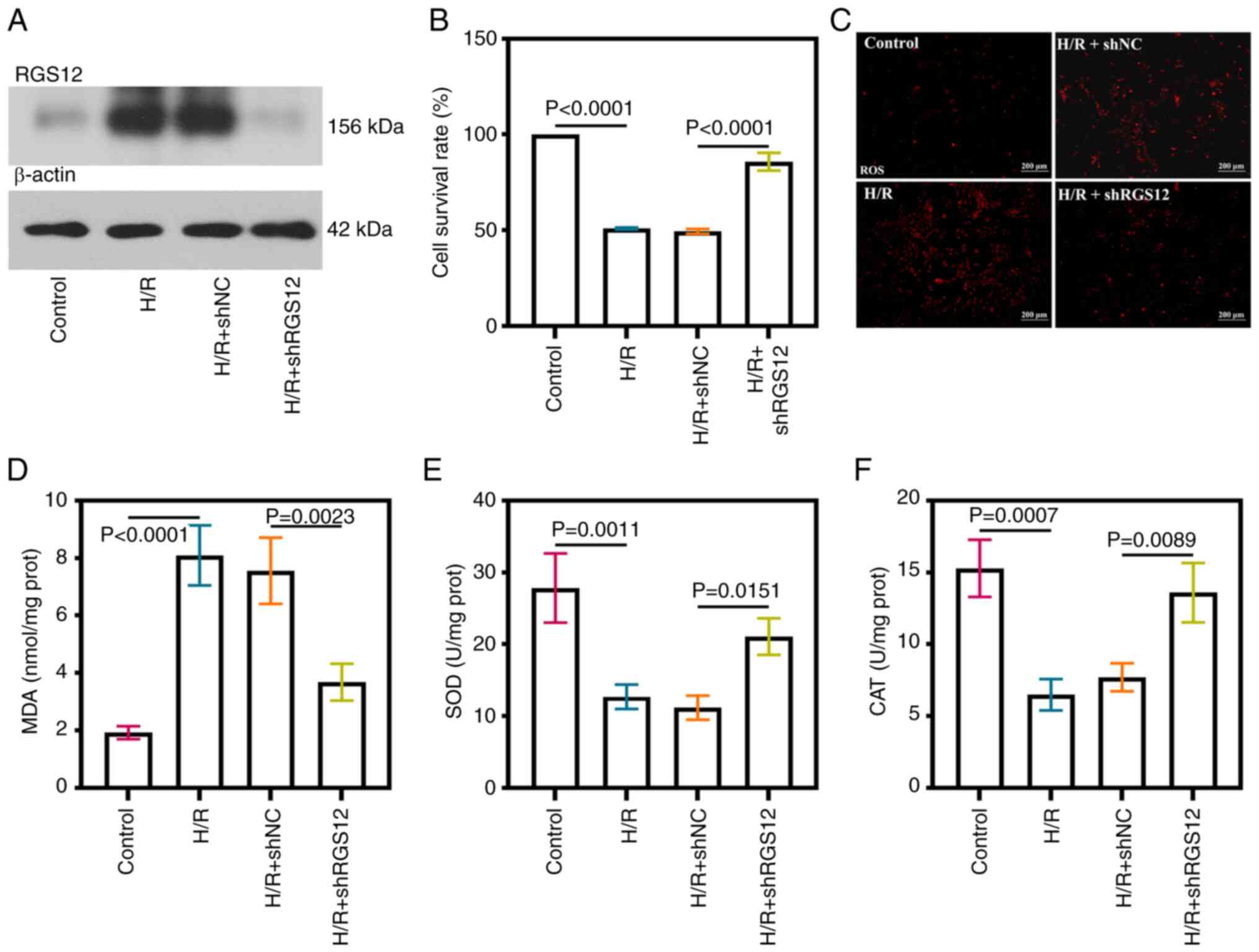 | Figure 2RGS12 silencing relieves oxidative
stress in H/R cells. (A) RGS12 protein expression levels in H/R
cells. (B) Cell survival rate. (C) ROS levels (magnification,
×100). (D) MDA content. (E) SOD and (F) CAT activity in H/R cells.
n=3. MDA, malondialdehyde; SOD, superoxide dismutase; CAT,
catalase; H/R, hypoxia/reoxygenation; RGS12, regulator of G-protein
signaling 12; sh, short hairpin RNA; NC, negative control; prot,
protein; ROS, reactive oxygen species. The data are presented as
the mean ± SD. |
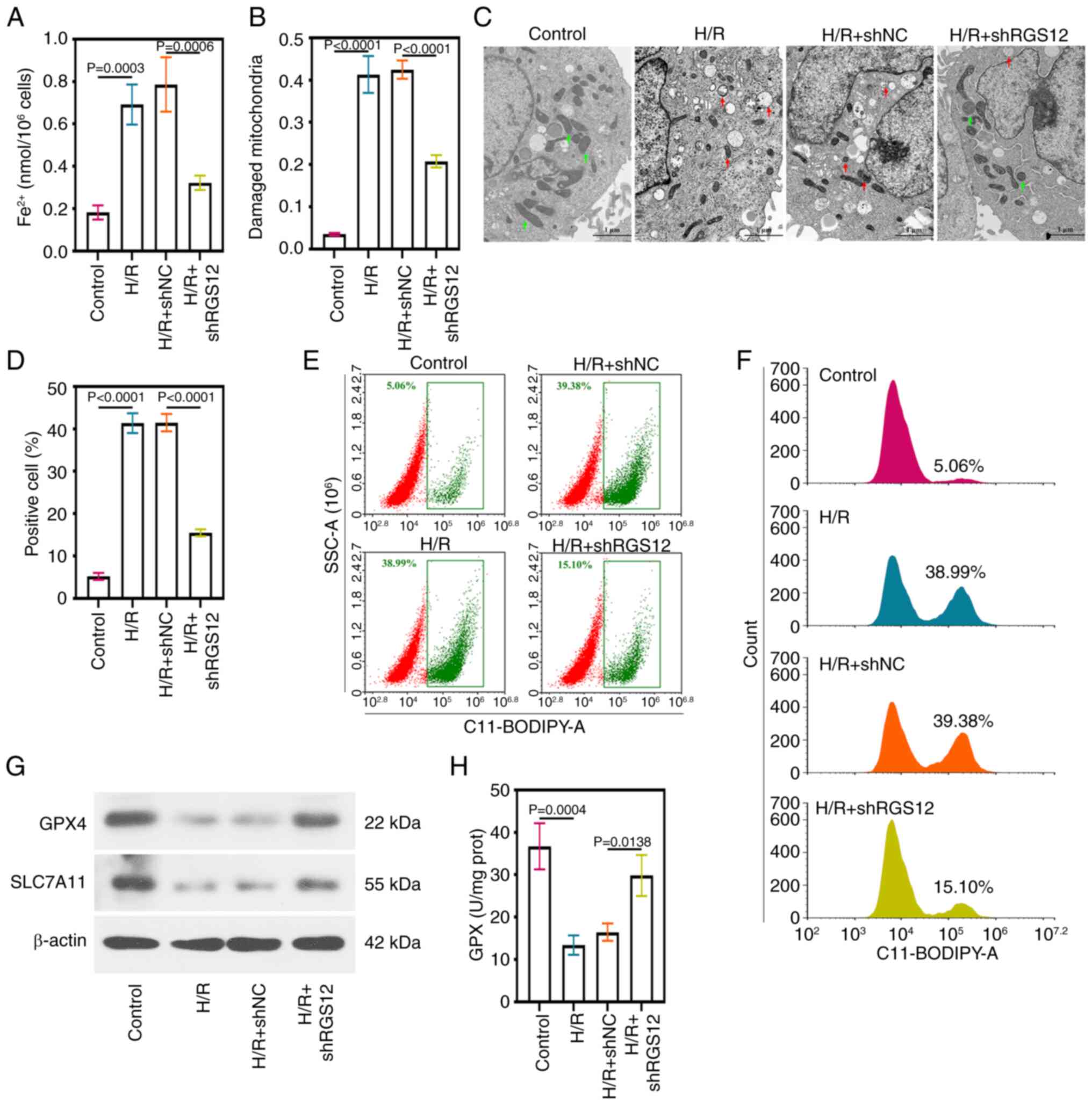
Downregulation of RGS12 inhibits
ferroptosis in H/R cells
Excess ROS and MDA levels often predict the
occurrence of ferroptosis (13).
The levels of Fe2+ in the cells subjected to H/R
significantly increased, while these levels were significantly
reduced by the silencing of RGS12 (Fig. 3A). Mitochondrial morphology was
assessed using TEM and the control cells displayed intact
mitochondria (Fig. 3B).
Conversely, H/R-induced damage was evident in the mitochondrial
membrane and cristae, with protective effects observed after the
silencing of RGS12. The proportion of damaged mitochondria was
calculated by evaluating the ratio of the number of damaged
mitochondria to the total number of mitochondria in the visual
field (Fig. 3B), which was
consistent with the results shown in the representative images
(Fig. 3C). Lipid ROS detection
using C11-BODIPY 581/591 staining, combined with flow cytometric
analysis, indicated that RGS12 silencing reduced lipid ROS
production (Fig. 3D).
Additionally, the proportion of C11-BODIPY 581/591-positive cells
in the samples were measured (Fig.
3E and F). Cells undergoing ferroptosis exhibited significantly
elevated lipid ROS levels, which were significantly suppressed by
the silencing of RGS12. In addition, the protein expression levels
of GPX4 and SLC7A11 were significantly reduced in the cells
subjected to H/R compared with those in the control cells. However,
the silencing of RGS12 expression significantly increased the
protein expression levels of GPX4 and SLC7A11.
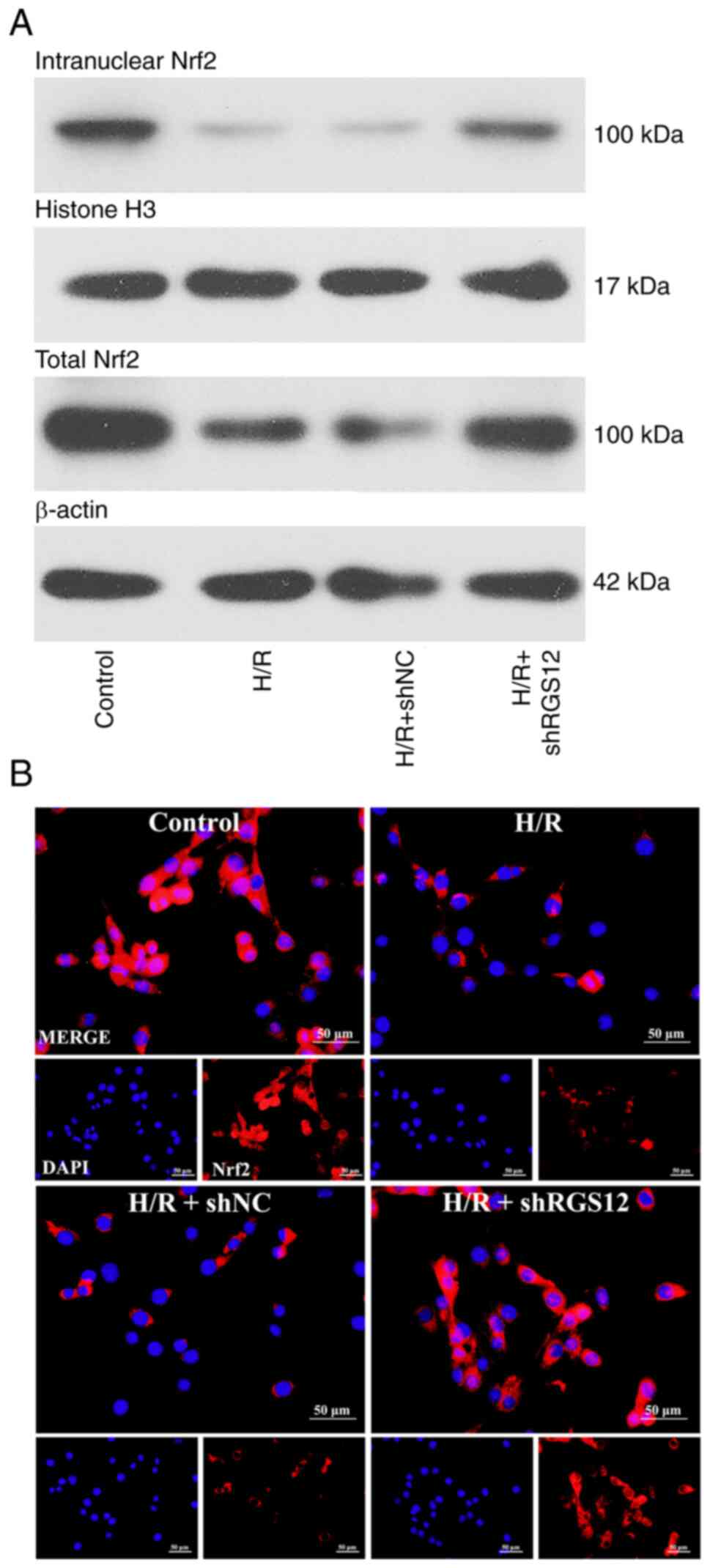
Downregulation of RGS12 activates the
Nrf2 pathway
Nrf2 serves a crucial role in the regulation of
ferroptosis and oxidative stress (37). Therefore, the present study
investigated the effects of RGS12 on Nrf2 expression. Upon the
induction of H/R, the protein expression levels of both nuclear and
total Nrf2 decreased compared with those in the control cells
(Fig. 4A). Conversely, the
silencing of RGS12 led to a significant increase in Nrf2 protein
expression levels. Immunofluorescence analysis demonstrated a
similar trend, indicating that the low expression level of RGS12
induced the expression of Nrf2 in cells subjected to H/R (Fig. 4B).
Overexpression of RGS12 exacerbates MIRI
in mice
In the aforementioned experiments, it was
demonstrated that the silencing of RGS12 mitigated H/R-injury in
HL-1 cells. Subsequently, overexpression of RGS12 was induced in
mice to further elucidate its potential role in MIRI. The LVEF and
LVFS significantly decreased in the mice with RGS12 overexpression,
compared with the control group (Fig. 5A). In addition, compared with the
MIRI + Ad-EV group, the increase in TnT serum levels of the MIRI +
Ad-RGS12 group also indicated the damage inflicted on cardiac
function (Fig. S2). In the mice
with RGS12 overexpression, a significant increase in the infarct
area of the myocardial tissue (Fig.
5B), accompanied by pronounced leukocyte infiltration in the
myocardial tissue (Fig. 5C) and
the significantly increased activity of myocardial enzymes, such as
LDH, CK and AST (Fig. 5D-F),
were observed compared with the control group. The expression of
RGS12 was examined in the myocardial tissue and it was demonstrated
that the protein expression level of RGS12 was successfully
increased in the RGS12-overexpressing mice compared with that in
the control group (Fig. 5G).
Furthermore, oxidative stress levels were significantly increased
in the mice in which RGS12 was overexpressed (Fig. 5H), resulting in significantly
increased MDA levels and decreased SOD activity compared with the
control group (Fig. 5I and J).
The overexpression of RGS12 in mice exacerbated ferroptosis, as
evidenced by the significantly elevated levels of Fe2+
and the reduced protein expression level of GPX4 (Fig. 5K and L). Additionally, the
upregulation of RGS12 inhibited the activation of the Nrf2 pathway
(Fig. 5L).
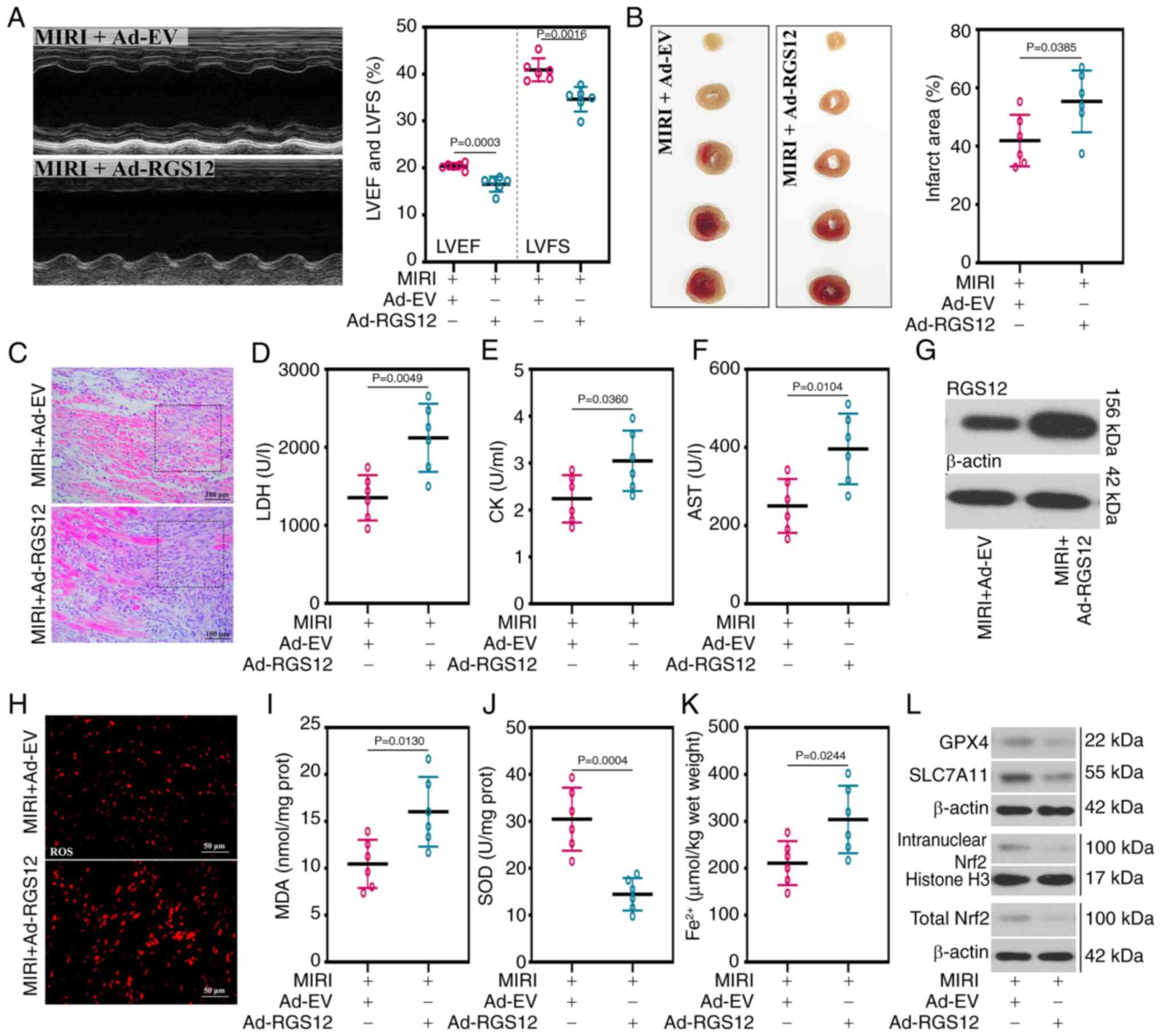 | Figure 5RGS12 upregulation exacerbates MIRI
in mice. (A) Echocardiogram, LVEF and LVFS of mice with MIRI. (B)
Representative images of 2,3,5-triphenyl tetrazolium chloride
staining and quantification of the infarct area in myocardial
tissues. (C) Representative images of hematoxylin and eosin
staining of myocardial tissues (magnification, ×200). The activity
of (D) LDH, (E) CK and (F) AST in serum. (G) RGS12 protein
expression levels in myocardial tissues. (H) ROS level
(magnification, ×400), (I) MDA content, (J) SOD activity and (K)
Fe2+ content. (L) Protein expression levels of GPX4,
SLC7A11, intranuclear Nrf2 and total Nrf2 in myocardial tissues.
Data are presented as the mean ± SD (n=6). Ad, adenovirus; Ad-EV,
Ad-empty vectors; AST, aspartate aminotransferase; GPX4,
glutathione peroxidase 4; SLC7A11, solute carrier family 7 member
11; MIRI, myocardial ischemia/reperfusion injury; LVEF, left
ventricular ejection fraction; LVFS, left ventricular fractional
shortening; LDH, lactate dehydrogenase; CK, creatine kinase; MDA,
malondialdehyde; SOD, superoxide dismutase; RGS12, regulator of
G-protein signaling 12; ROS, reactive oxygen species; nrf2Nrf2,
nuclear factor erythroid 2-related factor 2. |
RGS12 attenuates the protective effect of
PHC on oxidative stress and ferroptosis in cells subjected to
H/R
As aforementioned, in a preliminary experiment, the
present study identified the effect of RGS12 on MIRI. Subsequently,
the current study aimed to explore whether RGS12 mediates the
protective effects of PHC on the heart. PHC was shown to exert a
positive effect on the survival of HL-1 cells under normal
conditions, with cell viability significantly increasing as the
concentration of PHC treatment increased to 2.5 and 5 μg/ml,
compared with the viability of control cells and those treated with
1 μg/ml PHC (Fig. 6A).
Upon the induction of H/R, the beneficial effects of PHC on cell
survival became more pronounced, with cell viability significantly
increasing as the PHC concentration increased, peaking at 5
μg/ml (Fig. 6B).
Conversely, the expression level of RGS12 decreased as the PHC
concentration increased (Fig.
6C). A concentration of 5 μg/ml PHC was selected to be
used in subsequent experiments, resulting in a significant
downregulation of RGS12 expression in HL-1 cells. To explore the
role of RGS12 in the effect of PHC against H/R damage, RGS12 was
overexpressed in HL-1 cells (Fig.
S3A). The elevated levels of RGS12 led to a significantly
decreased cell survival rate and negated the beneficial effects of
PHC treatment in H/R-induced HL-1 cells (Fig. 6D). Furthermore, PHC treatment
reduced ROS production (Fig.
6E), MDA level (Fig. 6F) and
SOD activity (Fig. 6G) in
H/R-induced cells. PHC treatment significantly reduced the
production of lipid ROS (Fig.
6H). Additionally, PHC treatment reduced the number of cell
points within the gate (Fig. 6I)
and a reduction in the number of cells with high fluorescence
intensity (Fig. 6J). PHC
treatment increased the protein expression levels of GPX4 and
SLC7A11 in cells subjected to H/R (Fig. 6K) and promoted the activation of
the Nrf2 signaling pathway (Fig.
6L). The overexpression of RGS12 reversed the protective
effects of PHC. In addition, in cells overexpressing RGS12,
increased concentrations of PHC treatment caused increased cell
viability (Fig. S3B), decreased
MDA levels (Fig. S3C) and
increased SOD activity (Fig.
S3D). This suggested that PHC prevented MIRI through inhibiting
RGS12 in a concentration-dependent manner.
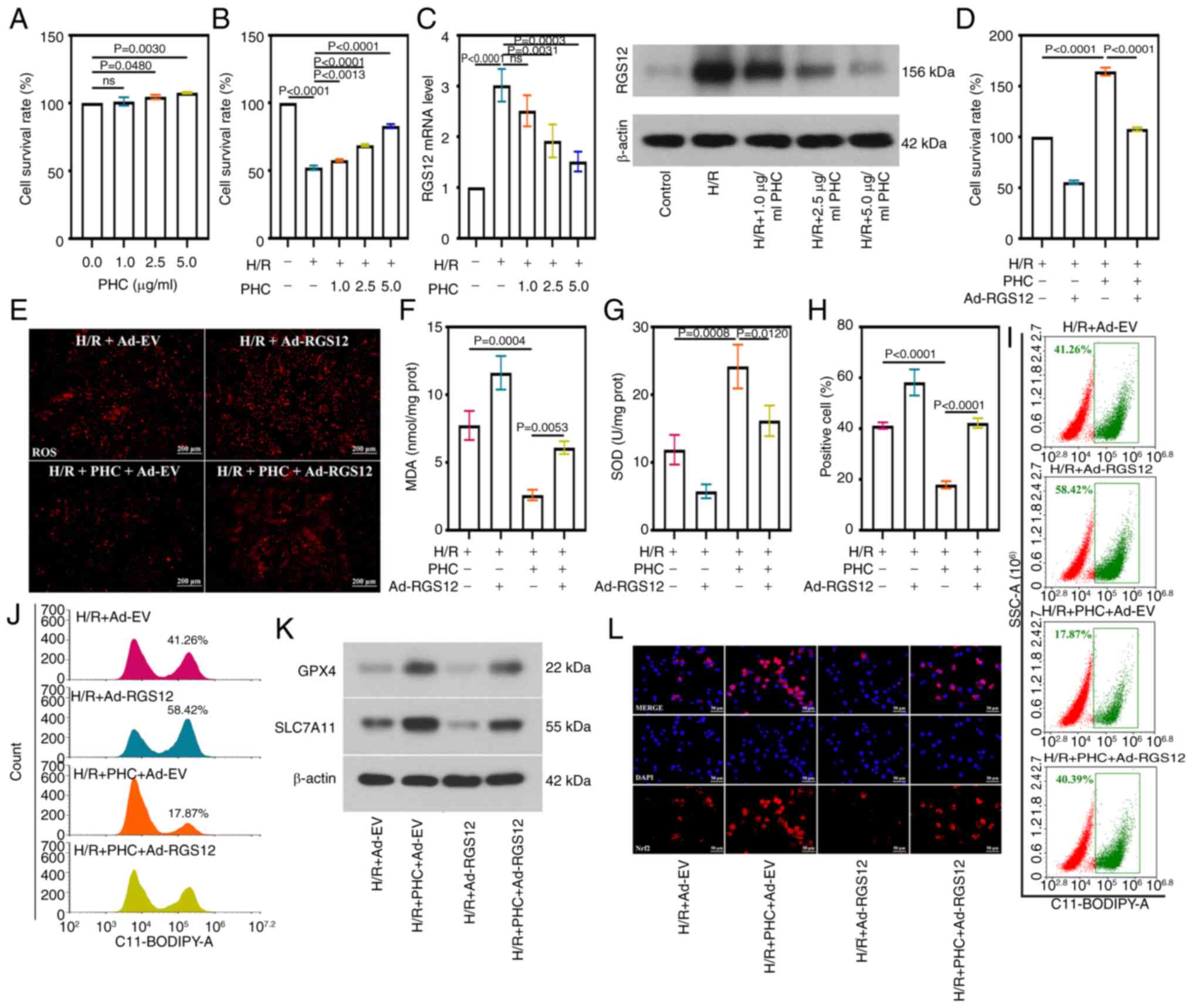 | Figure 6RGS12 attenuates the protective
effect of PHC on oxidative stress and ferroptosis in H/R cells. (A)
Survival rate of HL-1 cells treated with PHC. (B) Survival rate of
HL-1 cells treated with H/R and PHC. (C) RGS12 expression levels in
H/R cells induced with PHC. (D) Survival rate of HL-1 cells. (E)
ROS levels (magnification, ×100), (F) MDA content, and (G) SOD
activity in H/R cells. Lipid reactive oxygen species levels were
analyzed using flow cytometry (H), and the result was shown by
scatter diagram (I) and histogram. (J,K) Protein expression levels
of GPX4 and SLC7A11 and (L) the cellular location of Nrf2 in H/R
cells (magnification, ×400). Data are presented as the mean ± SD
(n=3). Ad, adenovirus; Ad-EV, Ad-empty vectors; PHC, penehyclidine
hydrochloride; RGS12, regulator of G-protein signaling 12; ROS,
reactive oxygen species; H/R, hypoxia/reoxygenation; MDA,
malondialdehyde; SOD, superoxide dismutase; GPX4, glutathione
peroxidase 4; SLC7A11, solution carrier family 7 member 11; Nrf2,
nuclear factor erythroid 2-related factor 2; prot, protein. |
RGS12 attenuates the protective effect of
PHC on MIRI
Administration of PHC resulted in the remission of
MIRI in mice in vivo. PHC treatment led to significant
improvements in LVEF and LVFS in mice with MIRI (Fig. 7A). Compared with MIRI + Ad-RGS12
group, the TnT level of MIRI + PHC + Ad-RGS12 group was reduced,
indicating that PHC administration improved cardiac function
(Fig. S2). Treatment with PHC
relieved the alleviated MIRI and ameliorated the damaged myocardial
tissue morphology (Fig. 7B and
7C), as myocardial fiber tissue
structure was clear and inflammatory cell infiltration was
decreased. Furthermore, PHC treatment significantly decreased the
activity of myocardial enzymes compared with those in the control
group, including LDH (Fig. 7D),
CK (Fig. 7E) and AST (Fig. 7F). Notably, the RGS12 protein
expression levels were lower in the myocardial tissue of all
PHC-treated mice compared with the mice with MIRI, apart from those
in the mice in which RGS12 was overexpressed (Fig. 7G). PHC administration reduced ROS
production (Fig. 7H) and
inhibited oxidative stress-induced injury, including increased MDA
level (Fig. 7I) and decreased
SOD activity (Fig. 7J).
Ferroptosis in mice with MIRI was also alleviated by PHC
administration, which was evidenced by reduced Fe2+
(Fig. 7K) and enhanced
expression of GPX4 (Fig. 7L).
Moreover, treatment with PHC induced the activation of the Nrf2
pathway in the model of MIRI (Fig.
7L). This was also observed in the MIRI + PHC + Ad-EV group.
However, the overexpression of RGS12 abrogated the effects of PHC
treatment. These findings suggested that RGS12 attenuated the
protective effects of PHC in the myocardial tissue of mice with
MIRI.
 | Figure 7RGS12 attenuates the protective
effect of PHC on a MIRI mouse model. (A) Echocardiogram, LVEF and
LVFS of mice with MIRI. (B) Representative images of
2,3,5-triphenyl tetrazolium chloride staining and quantification of
the infarct area in myocardial tissues. (C) Representative images
of hematoxylin and eosin staining in myocardial tissues
(magnification, ×200). Black boxes represent areas with significant
pathological changes. The activity of (D) LDH, (E) CK and (F) AST
in serum. (G) RGS12 protein expression levels in myocardial
tissues. (H) ROS levels (magnification, ×400), (I) MDA content, (J)
SOD activity and (K) Fe2+ content in myocardial tissues.
(L) Protein expression levels of GPX4 and intranuclear Nrf2. The
data are presented as the mean ± SD (n=6). RGS12, regulator of
G-protein signaling 12; PHC, penehyclidine hydrochloride; MIRI,
myocardial ischemia/reperfusion injury; LVEF, left ventricular
ejection fraction; LVFS, left ventricular fractional shortening;
LDH, lactate dehydrogenase; CK, creatine kinase; AST, aspartate
aminotransferase; ROS, reactive oxygen species; MDA,
malondialdehyde; SOD, superoxide dismutase; GPX4, glutathione
peroxidase 4; prot, protein; Nrf2, nuclear factor erythroid
2-related factor 2. |
Discussion
The present study observed a high expression level
of RGS12 in a mouse model of MIRI. The mechanism of action of RGS12
in PHC pretreatment was investigated through cell experiments and
verified in a mouse model. These results suggested that the
silencing of RGS12 alleviated H/R-induced damage to myocardial
cells, including reducing oxidative stress and lipid peroxidation,
thereby preventing ferroptosis, which may involve the activation of
the Nrf2 pathway. The overexpression of RGS12 led to the opposite
results in vivo and in vitro. Preconditioning with
PHC relied on the RGS12-mediated regulatory mechanism to protect
the heart from MIRI in a concentration-dependent manner.
Oxidative stress is a key physiological process
involved in MIRI (38). The
present study demonstrated that RGS12 promoted oxidative stress in
the MIRI process by inhibiting the Nrf2 pathway, a typical
antioxidant pathway. The regulation of the Nrf2 pathway by RGS12
may involve a direct effect. For instance, it has been demonstrated
that RGS12 promotes the degradation of the Nrf2 protein in RAW
264.7 cells (24). In addition,
RGS12 may indirectly regulate the Nrf2 pathway as, for example, it
has been reported that targeting RGS12 leads to a decrease in the
expression of Kelch-like ECH-associated protein 1, an upstream
regulatory molecule of Nrf2, thereby activating the Nrf2 pathway
(23,39). Therefore, based on the
aforementioned research findings, the effects of RGS12 on the Nrf2
pathway in MIRI may be either direct or indirect, both of which
support the conclusions reported herein. In addition to the Nrf2
pathway, RGS12 may be involved in the regulation of oxidative
stress by affecting other pathways. RGS12 activates the classical
NF-κB inflammatory pathway and contributes to the onset of
inflammatory arthritis (40).
Inflammation and oxidative stress are interrelated processes that
mutually promote one another, potentially due to crosstalk between
the NF-κB and Nrf2 pathways (41). Thus, RGS12 may indirectly promote
oxidative stress by activating the NF-κB pathway.
Ferroptosis is an iron-dependent mechanism of cell
death closely related to oxidative stress. It is characterized by
the accumulation of Fe2+ and lipid peroxidation
(42). The regulatory effects of
RGS12 on ferroptosis remain unclear; however, targeting of the Nrf2
pathway by RGS12 may be a key mechanism involved in the promotion
of ferroptosis. Nrf2 is a crucial transcriptional regulator of
anti-ferroptosis genes and its target genes participate in the
ferroptosis cascade reaction (14). The activation of the Nrf2 pathway
mediates the inhibition of ferroptosis and exerts protective
effects on the heart, which supports the conclusion obtained in the
present study (43).
Furthermore, the regulation of ferroptosis by RGS12 may involve
other pathways, such as the MAPK pathway. It has been reported that
RGS12 activates the MAPK pathway in pathological cardiac
hypertrophy (22). The
activation of the MAPK pathway may mediate ferroptosis in rat
myocardial cells (44).
Therefore, RGS12 may enhance ferroptosis by activating the MAPK
pathway, indicating the complex mechanism by which RGS12 regulates
ferroptosis.
The present study demonstrated that RGS12 promoted
oxidative stress and aggravated MIRI by inhibiting the Nrf2 pathway
in myocardial cells. The Nrf2 signaling pathway is a classical
pathway associated with oxidative stress and ferroptosis. Previous
studies have demonstrated the therapeutic potential of the Nrf2
pathway in MIRI (15,43,45). The present study showed the
regulatory effects of RGS12 on this pathway, which further
highlighted the influence of RGS12 on the MIRI process. However,
RGS12 may affect oxidative stress and ferroptosis through various
factors. It has been reported that RGS12 is an activator of the
NF-κB and MAPK pathways (22,40), which are well-established
inflammatory signaling pathways. The activation of these pathways
stimulates the production of inflammatory factors and amplifies the
inflammatory response, while simultaneously decreasing the
expression levels of certain anti-ferroptosis-related proteins,
such as heme oxygenase 1, ultimately leading to ferroptosis
(46). Targeting the NF-κB/MAPK
pathway effectively inhibits ferroptosis in osteoarthritis
(47). In addition, the
activation of the NF-κB and MAPK pathways is a marker of oxidative
stress in peripheral blood mononuclear cell and endothelial cells
(48,49) and inhibiting the activation of
the NF-κB and MAPK pathways alleviates oxidative stress in
bronchial epithelial cells and nerve cells (50,51). Therefore, the promotion of
oxidative stress and ferroptosis by RGS12 may involve inflammatory
processes, such as the activation of the NF-κB and MAPK
pathways.
The present study primarily investigated the
regulatory mechanisms involving Nrf2-mediated ferroptosis in
myocardial cells, which are the predominant cell type in the heart
and are essential for its contractile function (52). Myocardial cell death ultimately
results in the structural and functional impairment of the heart
tissue. Preventing MIRI-induced myocardial cell damage effectively
improves cardiac function (53).
Oxidative stress leads to the ferroptosis and apoptosis of
myocardial cells, which is a typical manifestation of MIRI at the
cellular level (54). Therefore,
the present study was conducted using the mouse myocardial cell
line, HL-1. However, the regulatory pattern of RGS12 may differ
between different types of cells. Macrophages are key immune cells
in MIRI, and M1 and M2 macrophage polarization mediates the
enhancement and decrease of MIRI-induced inflammation, respectively
(55). RGS12 has been reported
to induce M1 macrophage polarization (56). IL-6, IL-1β and TNF-α secreted by
M1 macrophages enhance the occurrence of ferroptosis (57), and the expression of RGS12 is
increased and the NF-κB pathway is activated in macrophages under
inflammatory conditions (58,59). This suggests that RGS12-induced
M1 macrophage polarization may mediate ferroptosis and the
activation of the NF-κB pathway.
The therapeutic effects of PHC on
ischemia/reperfusion injury have been previously well-established
in animal models and this effect on MIRI is dose-dependent
(33,60). According to previous reports
(33,60), the present study adopted an
optimal dose of 1 mg/kg PHC, which was shown to be effective in
alleviating MIRI in the mouse model. Moreover, it was demonstrated
that PHC prevented MIRI through the inhibition of RGS12 in a
dose-dependent manner using rescue experiments. The present study
demonstrated the molecular mechanism by which PHC attenuates MIRI;
therefore, it could be suggested that targeting RGS12 may be a
promising approach for preventing MIRI by alleviating oxidative
stress and ferroptosis. The present study also provided insights
into the pharmacological effects of the treatment of MIRI using PHC
and may lead to the development of novel strategies for the gene
therapy of MIRI in the future.
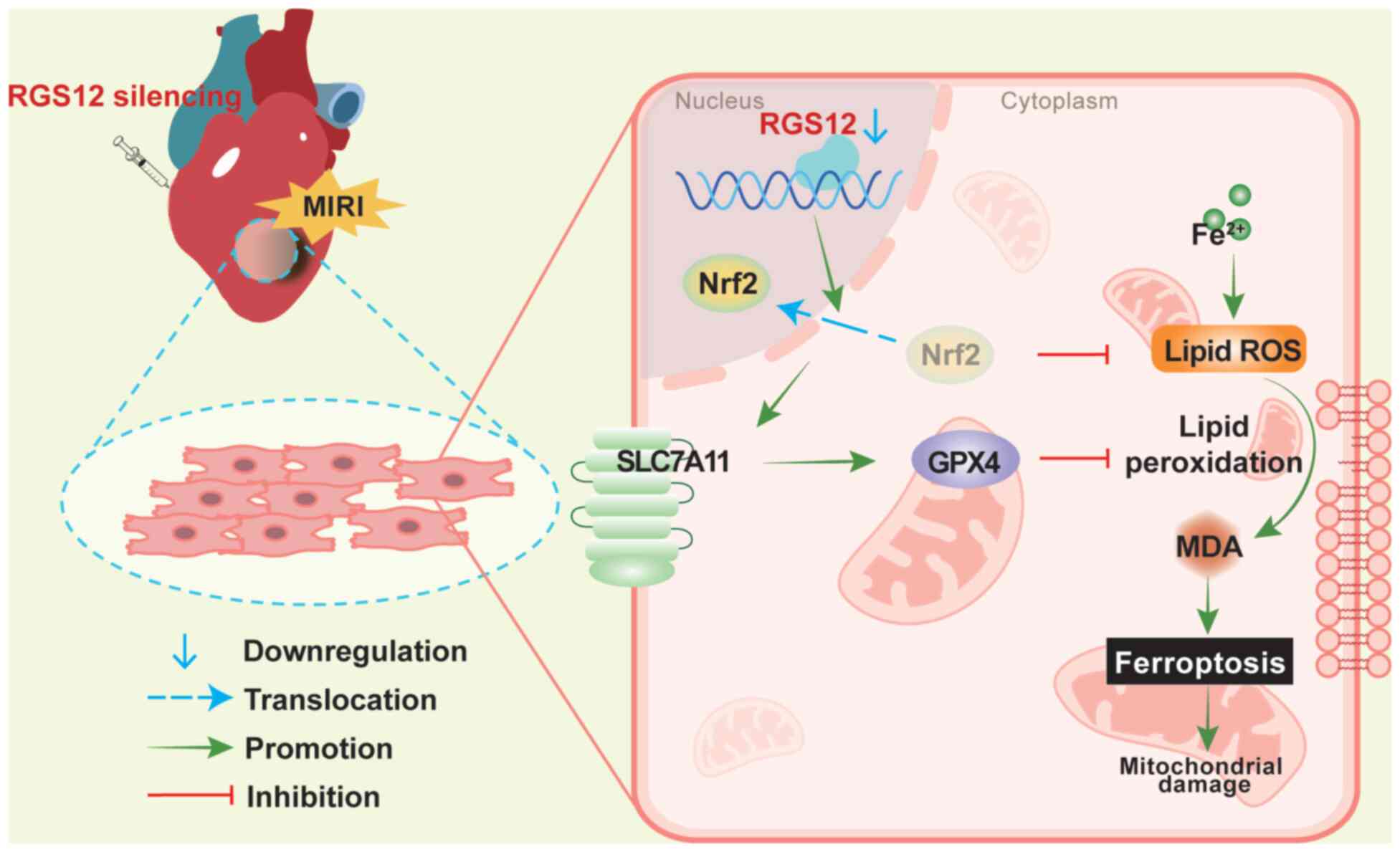
In conclusion, the present study demonstrated that
the silencing of RGS12 mediated the protective effects of PHC by
activating the Nrf2 pathway in MIRI, which contributed to the
reduction of oxidative stress and the inhibition of ferroptosis
(Fig. 8). This mechanism could
potentially provide new insights into the prevention and treatment
of MIRI.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
CZ was responsible for the conceptualization,
methodology conducting the experiment, data analysis, and drafting
of the manuscript. YW was responsible for conducting the
experiments, data analysis and writing, reviewing and editing of
the manuscript. YZ was responsible for conducting the experiment,
data analysis, data visualization and obtaining resources. JF was
responsible for the use of software. YW and JF confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The present study was carried out in accordance with
the requirements in the Guide for the Care and Use of Laboratory
Animals of the Ministry of Science and Technology of China. All
animal experiments were approved by the Ethics Committee of Hebei
North University (approval no. HBNU202306022105; Zhangjiakou,
China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
RGS12
|
regulator of G-protein signaling
12
|
|
MI
|
myocardial ischemia
|
|
MIRI
|
MI/reperfusion injury
|
|
H/R
|
hypoxia/reoxygenation
|
|
PHC
|
penehyclidine hydrochloride
|
|
ROS
|
reactive oxygen species
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
Ad
|
adenovirus
|
|
DHE
|
dihydroethidium
|
|
TTC
|
2,3,5-triphenyl tetrazolium
chloride
|
|
LDH
|
lactate dehydrogenase
|
|
CK
|
creatine kinase
|
|
AST
|
aspartate aminotransferase
|
|
CAT
|
catalase
|
|
GPX
|
glutathione peroxidase
|
|
MDA
|
malondialdehyde
|
|
SOD
|
superoxide dismutase
|
|
TnT
|
troponin T
|
|
TEM
|
transmission electron microscopy
|
|
LVEF
|
left ventricular ejection
fraction
|
|
LVFS
|
left ventricular fractional
shortening
|
Acknowledgements
Not applicable.
Funding
Funding for the present study was provided by the Hebei Province
Natural Science Foundation Project (grant no. H2022405031).
References
|
1
|
Roth GA, Mensah GA, Johnson CO, Addolorato
G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ,
Benziger CP, et al: Global burden of cardiovascular diseases and
risk factors, 1990-2019: Update from the GBD 2019 study. J Am Coll
Cardiol. 76:2982–3021. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang J, Liu Y, Liu Y, Huang H, Roy S, Song
Z and Guo B: Recent advances in nanomedicines for imaging and
therapy of myocardial ischemia-reperfusion injury. J Control
Release. 353:563–590. 2023. View Article : Google Scholar
|
|
3
|
Galeone A, Grano M and Brunetti G: Tumor
necrosis factor family members and myocardial ischemia-reperfusion
injury: State of the art and therapeutic implications. Int J Mol
Sci. 24:46062023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen QM and Maltagliati AJ: Nrf2 at the
heart of oxidative stress and cardiac protection. Physiol Genomics.
50:77–97. 2018. View Article : Google Scholar :
|
|
5
|
Bangalore S, Pursnani S, Kumar S and Bagos
PG: Percutaneous coronary intervention versus optimal medical
therapy for prevention of spontaneous myocardial infarction in
subjects with stable ischemic heart disease. Circulation.
127:769–781. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiang M, Lu Y, Xin L, Gao J, Shang C,
Jiang Z, Lin H, Fang X, Qu Y, Wang Y, et al: Role of oxidative
stress in reperfusion following myocardial ischemia and its
treatments. Oxid Med Cell Longev. 2021:66140092021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Algoet M, Janssens S, Himmelreich U, Gsell
W, Pusovnik M, Van den Eynde J and Oosterlinck W: Myocardial
ischemia-reperfusion injury and the influence of inflammation.
Trends Cardiovasc Med. 33:357–366. 2023. View Article : Google Scholar
|
|
8
|
Deng F, Zhang LQ, Wu H, Chen Y, Yu WQ, Han
RH, Han Y, Zhang XQ, Sun QS, Lin ZB, et al: Propionate alleviates
myocardial ischemia-reperfusion injury aggravated by Angiotensin II
dependent on caveolin-1/ACE2 axis through GPR41. Int J Biol Sci.
18:858–872. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen M, Zhong G, Liu M, He H, Zhou J, Chen
J, Zhang M, Liu Q, Tong G, Luan J and Zhou H: Integrating network
analysis and experimental validation to reveal the
mitophagy-associated mechanism of Yiqi Huoxue (YQHX) prescription
in the treatment of myocardial ischemia/reperfusion injury.
Pharmacol Res. 189:1066822023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Peoples JN, Saraf A, Ghazal N, Pham TT and
Kwong JQ: Mitochondrial dysfunction and oxidative stress in heart
disease. Exp Mol Med. 51:1–13. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bleier L and Dröse S: Superoxide
generation by complex III: From mechanistic rationales to
functional consequences. Biochim Biophys Acta. 1827:1320–1331.
2013. View Article : Google Scholar
|
|
12
|
Burgoyne JR, Mongue-Din H, Eaton P and
Shah AM: Redox signaling in cardiac physiology and pathology. Circ
Res. 111:1091–1106. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang B, Wang Y, Zhang J, Hu C, Jiang J, Li
Y and Peng Z: ROS-induced lipid peroxidation modulates cell death
outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis.
Arch Toxicol. 97:1439–1451. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dodson M, Castro-Portuguez R and Zhang DD:
NRF2 plays a critical role in mitigating lipid peroxidation and
ferroptosis. Redox Biol. 23:1011072019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen Y, Liu X, Shi J and Wu X: Involvement
of Nrf2 in myocardial ischemia and reperfusion injury. Int J Biol
Macromol. 125:496–502. 2019. View Article : Google Scholar
|
|
16
|
Hybertson BM, Gao B, Bose SK and McCord
JM: Oxidative stress in health and disease: The therapeutic
potential of Nrf2 activation. Mol Aspects Med. 32:234–246. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McNeill SM and Zhao P: The roles of RGS
proteins in cardiometabolic disease. Br J Pharmacol. 181:2319–2337.
2024. View Article : Google Scholar
|
|
18
|
Owen VJ, Burton PB, Mullen AJ, Birks EJ,
Barton P and Yacoub MH: Expression of RGS3, RGS4 and Gi alpha 2 in
acutely failing donor hearts and end-stage heart failure. Eur Heart
J. 22:1015–1020. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takimoto E, Koitabashi N, Hsu S, Ketner
EA, Zhang M, Nagayama T, Bedja D, Gabrielson KL, Blanton R,
Siderovski DP, et al: Regulator of G protein signaling 2 mediates
cardiac compensation to pressure overload and antihypertrophic
effects of PDE5 inhibition in mice. J Clin Invest. 119:408–420.
2009.PubMed/NCBI
|
|
20
|
Rorabaugh BR, Chakravarti B, Mabe NW,
Seeley SL, Bui AD, Yang J, Watts SW, Neubig RR and Fisher RA:
Regulator of G protein signaling 6 protects the heart from ischemic
injury. J Pharmacol Exp Ther. 360:409–416. 2017. View Article : Google Scholar :
|
|
21
|
Siderovski DP, Diversé-Pierluissi M and De
Vries L: The GoLoco motif: A Galphai/o binding motif and potential
guanine-nucleotide exchange factor. Trends Biochem Sci. 24:340–341.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang J, Chen L, Yao Y, Tang C, Ding J, Fu
C, Li H and Ma G: Pivotal role of regulator of G-protein signaling
12 in pathological cardiac hypertrophy. Hypertension. 67:1228–1236.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lan T, Li Y, Fan C, Wang L, Wang W, Chen S
and Yu SY: MicroRNA-204-5p reduction in rat hippocampus contributes
to stress-induced pathology via targeting RGS12 signaling pathway.
J Neuroinflammation. 18:2432021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ng AYH, Li Z, Jones MM and Yang S, Li C,
Fu C, Tu C, Oursler MJ, Qu J and Yang S: Regulator of G protein
signaling 12 enhances osteoclastogenesis by suppressing
Nrf2-dependent antioxidant proteins to promote the generation of
reactive oxygen species. Elife. 8:e429512019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang YA, Zhou WX, Li JX, Liu YQ, Yue YJ,
Zheng JQ, Liu KL and Ruan JX: Anticonvulsant effects of
phencynonate hydrochloride and other anticholinergic drugs in soman
poisoning: Neurochemical mechanisms. Life Sci. 78:210–223. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang YP, Li G, Ma LL, Zheng Y, Zhang SD,
Zhang HX, Qiu M and Ma X: Penehyclidine hydrochloride ameliorates
renal ischemia-reperfusion injury in rats. J Surg Res. 186:390–397.
2014. View Article : Google Scholar
|
|
27
|
Yu C and Wang J: Neuroprotective effect of
penehyclidine hydrochloride on focal cerebral ischemia-reperfusion
injury. Neural Regen Res. 8:622–632. 2013.PubMed/NCBI
|
|
28
|
Yang Y, Zhao L and Ma J: Penehyclidine
hydrochloride preconditioning provides cardiac protection in a rat
model of myocardial ischemia/reperfusion injury via the mechanism
of mitochondrial dynamics mechanism. Eur J Pharmacol. 813:130–139.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jin JK, Blackwood EA, Azizi K, Thuerauf
DJ, Fahem AG, Hofmann C, Kaufman RJ, Doroudgar S and Glembotski CC:
ATF6 decreases myocardial ischemia/reperfusion damage and links ER
stress and oxidative stress signaling pathways in the heart. Circ
Res. 120:862–875. 2017. View Article : Google Scholar :
|
|
30
|
Wei Z, Fei Y, Wang Q, Hou J, Cai X, Yang
Y, Chen T, Xu Q, Wang Y and Li YG: Loss of Camk2n1 aggravates
cardiac remodeling and malignant ventricular arrhythmia after
myocardial infarction in mice via NLRP3 inflammasome activation.
Free Radic Biol Med. 167:243–257. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Guidelines for Endpoints in Animal Study
Proposals. Animal Research Advisory Committee NIH:
|
|
32
|
Chen L, Luo G, Liu Y, Lin H, Zheng C, Xie
D, Zhu Y, Chen L, Huang X, Hu D, et al: Growth differentiation
factor 11 attenuates cardiac ischemia reperfusion injury via
enhancing mitochondrial biogenesis and telomerase activity. Cell
Death Dis. 12:6652021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Q, Li L, Liu Z, Li C, Yu L and Chang
Y: Penehyclidine hydrochloride ameliorates renal ischemia
reperfusion-stimulated lung injury in mice by activating Nrf2
signaling. Bioimpacts. 12:211–218. 2022.PubMed/NCBI
|
|
34
|
Liu Z, Li Y, Yu L, Chang Y and Yu J:
Penehyclidine hydrochloride inhibits renal
ischemia/reperfusion-induced acute lung injury by activating the
Nrf2 pathway. Aging (Albany NY). 12:13400–13421. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang
JJ, Luo XJ and Peng J: Ubiquitin-specific protease 7 promotes
ferroptosis via activation of the p53/TfR1 pathway in the rat
hearts after ischemia/reperfusion. Free Radic Biol Med.
162:339–352. 2021. View Article : Google Scholar
|
|
36
|
Liu Y, Wang T, Zhang M, Chen P and Yu Y:
Down-regulation of myocardial infarction associated transcript 1
improves myocardial ischemia-reperfusion injury in aged diabetic
rats by inhibition of activation of NF-κB signaling pathway. Chem
Biol Interact. 300:111–122. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen GH, Song CC, Pantopoulos K, Wei XL,
Zheng H and Luo Z: Mitochondrial oxidative stress mediated
Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol
Med. 180:95–107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gumpper-Fedus K, Park KH, Ma H, Zhou X,
Bian Z, Krishnamurthy K, Sermersheim M, Zhou J, Tan T, Li L, et al:
MG53 preserves mitochondrial integrity of cardiomyocytes during
ischemia reperfusion-induced oxidative stress. Redox Biol.
54:1023572022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Dong J, Liu M, Bian Y, Zhang W, Yuan C,
Wang D, Zhou Z, Li Y and Shi Y: MicroRNA-204-5p ameliorates renal
injury via regulating Keap1/Nrf2 pathway in diabetic kidney
disease. Diabetes Metab Syndr Obes. 17:75–92. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yuan G and Yang S, Ng A, Fu C, Oursler MJ,
Xing L and Yang S: RGS12 Is a novel critical NF-κB activator in
inflammatory arthritis. iScience. 23:1011722020. View Article : Google Scholar
|
|
41
|
McGarry T, Biniecka M, Veale DJ and Fearon
U: Hypoxia, oxidative stress and inflammation. Free Radic Biol Med.
125:15–24. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li D, Pi W, Sun Z, Liu X and Jiang J:
Ferroptosis and its role in cardiomyopathy. Biomed Pharmacother.
153:1132792022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yan J, Li Z, Liang Y, Yang C, Ou W, Mo H,
Tang M, Chen D, Zhong C, Que D, et al: Fucoxanthin alleviated
myocardial ischemia and reperfusion injury through inhibition of
ferroptosis via the NRF2 signaling pathway. Food Funct.
14:10052–10068. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chen W, Zhang Y, Wang Z, Tan M, Lin J,
Qian X, Li H and Jiang T: Dapagliflozin alleviates myocardial
ischemia/reperfusion injury by reducing ferroptosis via MAPK
signaling inhibition. Front Pharmacol. 14:10782052023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang T, Liu H, Yang C, Mo H, Wang X, Song
X, Jiang L, Deng P, Chen R, Wu P, et al: Galangin attenuates
myocardial ischemic reperfusion-induced ferroptosis by targeting
Nrf2/Gpx4 signaling pathway. Drug Des Devel Ther. 17:2495–2511.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen Y, Fang ZM, Yi X, Wei X and Jiang DS:
The interaction between ferroptosis and inflammatory signaling
pathways. Cell Death Dis. 14:2052023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhao C, Sun G, Li Y, Kong K, Li X, Kan T,
Yang F, Wang L and Wang X: Forkhead box O3 attenuates
osteoarthritis by suppressing ferroptosis through inactivation of
NF-κB/MAPK signaling. J Orthop Translat. 39:147–162. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
van den Berg R, Haenen GR, van den Berg H
and Bast A: Transcription factor NF-kappaB as a potential biomarker
for oxidative stress. Br J Nutr. 86(Suppl 1): S121–S127. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Son Y, Cheong YK, Kim NH, Chung HT, Kang
DG and Pae HO: Mitogen-activated protein kinases and reactive
oxygen species: How can ROS activate MAPK pathways? J Signal
Transduct. 2011:7926392011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dang X, He B, Ning Q, Liu Y, Guo J, Niu G
and Chen M: Alantolactone suppresses inflammation, apoptosis and
oxidative stress in cigarette smoke-induced human bronchial
epithelial cells through activation of Nrf2/HO-1 and inhibition of
the NF-κB pathways. Respir Res. 21:952020. View Article : Google Scholar
|
|
51
|
Tang Y, Gu W and Cheng L: Evodiamine
attenuates oxidative stress and ferroptosis by inhibiting the MAPK
signaling to improve bortezomib-induced peripheral neurotoxicity.
Environ Toxicol. 39:1556–1566. 2024. View Article : Google Scholar
|
|
52
|
Litviňuková M, Talavera-López C, Maatz H,
Reichart D, Worth CL, Lindberg EL, Kanda M, Polanski K, Heinig M,
Lee M, et al: Cells of the adult human heart. Nature. 588:466–472.
2020. View Article : Google Scholar
|
|
53
|
Zhang CX, Cheng Y, Liu DZ, Liu M, Cui H,
Zhang BL, Mei QB and Zhou SY: Mitochondria-targeted cyclosporin A
delivery system to treat myocardial ischemia reperfusion injury of
rats. J Nanobiotechnology. 17:182019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang S, Yan F, Luan F, Chai Y, Li N, Wang
YW, Chen ZL, Xu DQ and Tang YP: The pathological mechanisms and
potential therapeutic drugs for myocardial ischemia reperfusion
injury. Phytomedicine. 129:1556492024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhuang Q, Li M, Hu D and Li J: Recent
advances in potential targets for myocardial ischemia reperfusion
injury: Role of macrophages. Mol Immunol. 169:1–9. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yuan G, Huang Y, Yang ST, Ng A and Yang S:
RGS12 inhibits the progression and metastasis of multiple myeloma
by driving M1 macrophage polarization and activation in the bone
marrow microenvironment. Cancer Commun (Lond). 42:60–64. 2022.
View Article : Google Scholar
|
|
57
|
Yang Y, Wang Y, Guo L, Gao W, Tang TL and
Yan M: Interaction between macrophages and ferroptosis. Cell Death
Dis. 13:3552022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yuan G and Yang S and Yang S: RGS12
represses oral squamous cell carcinoma by driving M1 polarization
of tumor-associated macrophages via controlling ciliary
MYCBP2/KIF2A signaling. Int J Oral Sci. 15:112023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yuan G and Yang S and Yang S: Macrophage
RGS12 contributes to osteoarthritis pathogenesis through enhancing
the ubiquitination. Genes Dis. 9:1357–1367. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lin D, Ma J, Xue Y and Wang Z:
Penehyclidine hydrochloride preconditioning provides
cardioprotection in a rat model of myocardial ischemia/reperfusion
injury. PLoS One. 10:e01380512015. View Article : Google Scholar : PubMed/NCBI
|