Introduction
Head and neck cancer (HNC), the seventh most common
type of cancer worldwide, poses a challenge to public health, with
~860,000 new cases and 450,000 deaths annually, as reported by
GLOBOCAN 2020 (1). The key risk
factors for HNC include tobacco and alcohol use, betel quid/areca
nut consumption, human papillomavirus infection, and genetic
alterations (2). Although
certain HNC subgroups (such as human papillomavirus-associated HNC)
(2) exhibit better survival
prospects, the 5-year overall survival for HNC remains around 50%,
even with comprehensive multi-disciplinary treatment (2,3).
This situation underscores the need to identify novel therapeutic
targets to enhance treatment efficacy and improve outcomes in
patients with HNC.
Metabolic reprogramming, a hallmark of cancer,
involves changes in energy metabolism and the reconfiguration of
metabolic pathways. These adaptations, influenced by specific
metabolites and enzymes, such as enhanced lactate as a consequence
of aerobic glycolysis, create a tumor microenvironment conducive to
tumor growth and progression (4,5).
Despite the diverse and complex metabolic characteristics and
preferences of tumors, marked alterations in metabolism are key for
the initiation and progression of cancer (4,5).
Therefore, identifying biomarkers with enzyme signatures reflecting
altered metabolic pathways, particularly those associated with
patient survival, is crucial. These biomarkers are essential for
developing therapeutic strategies targeting tumor metabolism,
offering a refined and potentially more effective approach to
treating HNC.
Alcohol consumption increases the risk of numerous
types of cancers, including HNC. The pathogenesis of
alcohol-mediated cancer is influenced by various factors, notably
aldehydic products such as acetaldehyde and 4-hydroxy-2-nonenal
(4-HNE) (6,7). Acetaldehyde, the primary metabolite
of ethanol, induces direct DNA damage by forming DNA adducts and
compromises genomic stability by forming DNA-protein crosslinks and
acetaldehyde-histone adducts (6-8).
A previous study has highlighted the dose-dependent effect of
alcohol consumption on the prognosis of patients with HNC, with
this interplay influenced by aldehyde-detoxifying enzymes, such as
ALDH2 (9). Given the poor
prognosis of HNC and the pivotal role of metabolic reprogramming in
cancer progression, this study aimed to investigate the links
between metabolic pathway alterations and their contributions to
tumorigenesis and cancer progression in patients with HNC,
providing a foundation for potential therapeutic advancements.
Materials and methods
Metabolic pathway analysis
GSE6631 dataset, an mRNA microarray comparing 44 HNC
with paired normal tissue controls, was downloaded from the Gene
Expression Omnibus (GEO; ncbi.nlm.nih.gov/geo/). To identify genes with enzyme
annotations that were significantly differentially expressed,
fold-change was determined using GEO2R (ncbi.nlm.nih.gov/geo/info/geo2r.html#how_to_use).
Ingenuity Pathway Analysis (IPA) was used to identify the metabolic
pathways significantly altered in HNC tumor tissue (digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/).
In silico mRNA profiles and Kaplan-Meier
analysis
ENCORI database (starbase.sysu.edu.cn/index.php) was used to analyze
the correlation between ALDH2 and vascular endothelial
growth factor C (VEGFC) mRNA expression. VEGFC mRNA
levels in HNC tissue and its impact on overall survival of patients
with varied ALDH2 and VEGFC expression were
investigated using the OncoLnc platform (oncolnc.org/). Gene expression analyses were conducted
using GEPIA (gepia.cancer-pku.cn/), based on data from The Cancer
Genome Atlas (TCGA) (10).
Clinical specimens and tissue microarrays
for HNC
Tissue sample were fixed in 10% buffered formalin
for 24 h at room temperature and embedded in paraffin.
Paraffin-embedded sections of malignant tissue and their matched
adjacent non-cancerous tissue (distance, >1 cm) were obtained
from 106 patients diagnosed with HNC. The cohort included 91 male
patients and 15 female pateints (age range: 27 to 87). Specimens
were from patients without distant metastasis at the initial
presentation, with histologically confirmed squamous cell
carcinoma, and primary tumors located in the oral cavity,
oropharynx, hypopharynx, or larynx. All the patients underwent
surgical intervention at the Kaohsiung Veterans General Hospital,
Kaohsiung, Taiwan between 2010/01 and 2016/12. Relevant clinical
variables were recorded, including age, sex, American Joint
Committee on Cancer (AJCC) T and N classification 11, overall stage
(11), substance misuse,
treatment strategy and disease status. Each discrete position
within the initial tissue microarray (TMA-1) was configured to
accommodate two cancer specimens and one adjacent non-cancerous
specimen for quantification of protein levels. For additional
analyses, a second microarray (TMA-2) was created from the same
cohort. Due to sample loss during TMA-2 preparation, 84 samples
were available. Ethics approval, including a waiver of informed
consent, was granted by the Ethics Committee of Kaohsiung Veterans
General Hospital (approval no. KSVGH23-CT8-10).
Immunohistochemistry
The 4-µm slides of the TMA paraffin block were used
for IHC. TMA slides were deparaffinized in xylene, dehydrated using
graded ethanols at room temperature. Briflyantigen retrieval from
the TMA slides was performed using retrieval solution (Tris-EDTA
buffer, pH 9.0) at 95°C for 12 min and incuabated with peroxidase
Blocking buffer (3% hydrogen peroxide) at room temperature for 10
min. Slides were incubated with blocking reagent (ready to use,
BioTnA, TA00C2) for 30 min at room temperature. After blocking,
slides were incubated with a primary antibody against ALDH2 (1:200;
GTX101429, Genetex) and VEGFC (1:1,000, GTX113574, Genetex) for 1 h
at room temperature. Expression level was determined by employing
the HRP-conjugated secondary antibody (ready to use, TnAlink
Polymer Detection System, cat. no. TACH02D, BioTnA,) for 30 min at
room temperature, followed by 1 min of DAB (1:20) and 5 sec
hematoxylin counterstaining at room temperature (TnAlink Polymer
Detection System, TACH02D, BioTnA, Kaohsiung, Taiwan). The TMA
slides were scanned by MoticEasyScan Pro (light microscope, 400×).
A pathologist assessed the TMAs to exclude microarrays of
suboptimal quality. Finally, quantification of ALDH2 and VEGFC IHC
staining was performed using HistoQuest (TissueGnostics, version
7.1, Deep-learning nuclear segmentation program,). ALDH2 protein
expression in cancer tissues was categorized into the high
expression group, defined as >50% positive cells, and the low
expression group (≤50% positive cells).
Cell culture
HNC cell lines SAS (Japanese Collection of Research
Bioresources; cat. no. JCRB0260) and MTCQ1 (Bioresource Collection
and Research Center, cat. no. 60620) were maintained in DMEM
(HyClone; Cytiva; cat. no. SH30003.02) supplemented with 10% FBS
(Cytiva, SH30396.03) and 1% penicillin-streptomycin-amphotericin
(PSA; Biological Industries, 03-033-1B) at 37°C in 5%
CO2. DOK (oral dysplasia), TW1.5 and TW2.6 (buccal
mucosa) cell lines were maintained in DMEM/F12 (HyClone,
SH30004.04) supplemented with 10% FBS and 1% PSA. DOK, TW1.5
(12,13) and TW2.6 were kindly provided by
Dr Michael Hsiao (Academia Sinica, Taipei, Taiwan).
Lentivirus infection
Lentivir us vector control (pLKO-1-shLuc) and short
hairpin (sh)ALDH2 viral supernatants (TRCN0000026452 and
TRCN0000026486) were obtained from the National RNAi Core Facility
(Taipei, Taiwan). RNAi core based on 3rd Generation lentiviral
guide, shRNA lentiviruses were produced by co-transfecting with
hairpin-pLKO.1 vector (1 μg), VSV-G/pMDG2.G (0.1 μg),
and pΔ8.91 (0.9 μg) constructs into 293T (American Type
Culture Collection, CRL-3216™) cells using TransIT-LT1 transfection
reagent (Mirus Bio, LLC; cat. no. #MIR 2300) and DNA complex
incubated for 30 min at room temperature. After 40 h transfection,
the viral supernatants were then harvested. Target sequences are
listed in Table SI. Polybrene
(8 μg/ml) was used to infect DOK or TW2.6 cells
(1×105/well) using viral supernatants (5 multiplicity of
infection). Following 72 h infection, cells were selected and
maintained using 2 μg/ml puromycin (InvivoGen, ant-pr-5).
Using 3rd Generation lentiviral guide, overexpression lentiviruses
were produced by co-transfecting with cDNA-expressing vector (10
μg), pMDG (10 μg), and pΔ8.91 (1 μg)
constructs into 293T cells using calcium phosphate (DNA complex
incubated for 30 min at room temperature). After 48 h transfection,
the viral supernatants were then harvested and concentrated by the
Lentivirus Concentration kit (TopGen Biotechnology Co., Ltd.,
GM801-02). The lentiviral expression vector carrying human ALDH2
(NM_000690.4, pLVX-ALDH2-IRES-Neo) and control vector
(pLVX-IRES-Neo) were acquired from TopGen Biotechnology Co., Ltd.
Human ALDH2 cDNA was overexpressed in SAS and MTCQ1 cell lines. A
total of 8 μg/ml polybrene was used to infect SAS or MTCQ1
cells (1×105/well) using viral supernatants (10MOI).
After 72 h infection, the cells were selected and maintained using
400 μg/ml G418 Sulfate (GibcoTM, 10131035). After
48 h selected and cultured for 48 h for subsequent analyses.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from HNC cells using
TRIzol® (Thermo Fisher Scientific, Inc.; cat. no.
#15596018) following the manufacturer's instructions. cDNA was then
synthesized using a PrimeScript™ RT Reagent kit (cat. no. #RR037A;
Takara Biotechnology Co., Ltd.) according to the manufacturer's
protocol. qPCR was performed using a SYBR Green PCR Master Mix (PCR
Biosystems Ltd. qPCRBIO SyGreen Mix Lo-ROX) to evaluate target gene
expression. Thermocycling conditions were as follows: Initial
denaturation at 95°C for 3 min; followed by 40 cycles of 95°C for
10 sec and 60°C for 30 sec. The 2−ΔΔCq was used to
calculate the results, and ACTB was used for normalization
(14). The primers are listed in
Table SII.
Gene expression evaluation using
microarray analysis
Total RNA was isolated from TW2.6/shluc and
TW2.6/shALDH2 cells using TRIzol® (Thermo Fisher
Scientific, Inc.; cat. no. #15596018). Microarray analysis was
performed on isolated RNA using a Clariom S Array, human (Applied
Biosystems; Thermo Fisher Scientific, Inc.; cat. no. 902916) and
scanned using an Affymetrix GeneChip Scanner 3000. To analyze the
effects of ALDH2 knockdown, an interaction network was
generated using Qiagen GmbH IPA (QIAGEN 2000-2023) from
TW2.6/shALDH2 cells, with the threshold set at 2-fold change. The
microarray data were deposited in the GEO under accession no.
GSE253622.
Boyden chamber assay
For invasion experiments, a polycarbonate membrane
was pre-coated with fibronectin (10 μg, Thermo Fisher,
33016015) on the lower side and Matrigel (Corning, Inc.; cat. no.
354234) on the upper side at room temperature, 3 min. Migration
assay used membranes without Matrigel pre-coating. The lower
chamber was filled with complete culture medium (containing DMEM,
10% FBS). The cells (3×105/ml) were suspended in a
serum-free DMEM, added to the 50 μl cells into upper chamber
of each well and incubated at 37°C in 5% CO2 for 15-18
h. NF-κB inhibitor (Bay11-7082) was included in cells to perform
the assay. To block VEGFC, we incubated TW2.6/shALDH2 cells with a
VEGFC-neutralizing antibody (10-20 μg, GTX52393, GeneTex)
for 30 min. After incubation, cells were trypsinized for migration
and invasion assays, and a VEGFC-neutralizing antibody was added to
the cells. After incubation, the cells were stained with crystal
violet for 30 min at room temperature and counted under a light
microscope (16×).
Cell viability assay
Cells (5,000 cells/well for DOK/shluc, DOK/shALDH2,
TW2.6/shluc, TW2.6/shALDH2, SAS/Vector, and SAS/ALDH2) were plated
in a 96 well plate with complete DMEM (10% FBS) mediaum for 24-72 h
at 37°C in 5% CO2.
Using MTT (Sigma-Aldrich, 10 μl) for 4 h, the
cell proliferation was assessed, and the signals were measured
using an ELIAS reader (Thermo Fisher Scientific; Multiskan FC).
Immunoblotting
After extracting protein from the cell lines
(DOK/shluc, DOK/shALDH2, TW2.6/shluc, TW2.6/shALDH2, SAS/Vector,
and SAS/ALDH2) using RIPA lysis buffer [50 mM Tris-HCl, pH 8.0, 150
mM NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% SDS, PMSF (0.1 mM),
Na3VO4 (2 mM, and NaF (1 mM)] and protein
concenteration were determined using Pierce™ BCA Protein Assay kits
(ThermoFisher, 23225). Protein (50 μg) were loaded onto 10%
SDS-PAGE for electrophoresis and subsequently transferred to a PVDF
membrane and blocked with 5% non-fat dry milk in 1xPBST for 1 h at
room temperature. The primary antibodies were incubated overnight
at 4°C. Table SIII lists the
primary antibodies used for immunoblotting. The membranes were
incubated with second antibodies [1:5,000, mouse (Jackson
ImmunoResearch, 115-035-003), 1:5,000, rabbit (Jackson
ImmunoResearch, 111-035-0031)] for 1 h at room temperature. The
signals were visualised using an ECL™ Western Blotting Reagents
(Cytiva, XR-IGE-RPN2106). ImageJ software (version 1.53t, National
Institutes of Health) was used to quantify the densitometry.
Small interfering (si)RNA
transfection
NF-κB p65 (RELA) siRNA, locus 11q13.1.; cat. no.
#sc-29410 and control siRNA-A (cat. no. #sc-37007) were purchased
from Santa Cruz Biotechnology, Inc. Next, 1×106 cells
with complete media were plated in 6-cm plates for 24 h at 37°C and
subsequently transfected with 100 nM NF-κB p65 or control siRNA
using TransIT-X2 (Mirus Bio, LLC; cat. no. #MIR 6000) and
incubacted room temperature 30 min. Subsequent experiments were
performed after 48 h.
Reactive oxygen species (ROS)
quantification
Briefly, 2.5×104 cells/well were plated
on a 96-well plate and incubated overnight at 37°C in 5%
CO2. DCFDA (Abcam; cat. no. ab113851) was added to the
cells and incubated at 37°C in 5% CO2 for 45 min. After
removing the DCFDA, cells were examined using a fluorescence plate
reader (FLUOstar Omega; BMG Labtech GmbH).
Reporter assay
TW2.6/shluc and TW2.6/shALDH2 cells were plated on 6
cm plates with complete DMEM/F12 (10% FBS) at a density of
7.5×105 cells/plate for 24 h at 37°C in 5%
CO2. Subsequently, 0.5 pCMV6-AV-GFP (OriGene; cat. no.
#PS100010) and 4.5 μg pGL4.32 (luc2P/NF-κB-RE/Hygro)
[Promega Corporation; E8491, NF-κ B response element (33-84),
5′-GGGAATTTCCGGGGACTTTCCGGGAATTTCCGGGGACTTTCCGGGAATTTCC-3′] plasmid
were co-transfected into cells using TransIT-X2 (Mirus Bio, LLC;
cat. no. #MIR 6000). Following 48 h incubation, 1×104
cells with DMEM/F12 (10% FBS) were plated in 96-well plates for 6 h
at 37°C in 5% CO2 and subsequent analyses. TW2.6/shALDH2
cells were treated with N-acetyl-L-cysteine (NAC, 10 mM; Table SIV) for 12 h at 37°C in 5%
CO2. A ONE-Glo Luciferase assay kit (Promega
Corporation; cat. no. #E6120) and a luminometer (FLUOstar Omega;
BMG Labtech GmbH) were used to measure the green fluorescence and
luciferase signals. pCMV6-AV-GFP was used as internal control.
H2O2 treatment
SAS/ALDH2 cells (5×105 cells/well) were
plated in a 6-well plate and incubated overnight at 37°C in 5%
CO2. H2O2 (80 μM; Table SIV) was added at 37°C in 5%
CO2 overnight. After 24 h, cells were examined using
RT-qPCR to examine VEGFC expression.
Colony formation assay
TW2.6/shluc, TW2.6/shALDH2, SAS/Vector, SAS/ALDH2,
MTCQ1/Vector, and MTCQ1/ALDH2 HNC cells (4,000 cells/well) were
plated in a 6-well plate. After being incubated for 7 days at 37°C
and 5% CO2, the resulting cell colonies were fixed with
1% formalin for 30 min at room tempature and stained with crystal
violet for 30 min at room tempature. ImageJ software (version
1.53t, National Institutes of Health) was used to count the number
of colonies (diameter >0.1 cm).
Statistical analysis
Data are shown as the mean ± standard deviation of
≥3 independent repeats. The data analyses used the Student's t- and
χ2 parametric tests, while Mann-Whitney U and Fisher's
exact test were employed for non-parametric tests. One-way ANOVA
followed by Tukey's post hoc test was used used for multiple group
comparisons. Correlation analysis was determined by Spearman's
correlation. Prognostic importance of covariates was evaluated
through Cox proportional hazards regression in both univariate and
multivariate models. Kaplan-Meier method was applied to examine
survival curves and the log-rank test was used to compare survival
rates between groups. All statistical tests were two-tailed.
P<0.05 was considered to indicate a statistically significant
difference.
Results
ALDH family genes are downregulated in
HNC tumor tissue
To identify the metabolic pathways involved in the
initiation and progression of HNC, genes with enzyme annotations
that showed differential expression in tumor compared with normal
tissues from patients with HNC in the GSE6631 dataset were analyzed
(Fig. 1A; Table SV). Based on 150 genes that
showed differential expression in HNC compared with normal tissue
(Table SV), pathways were
filtered based on the z-scores with z-score <-2.0 indicating
inhibition (Table SVI). IPA
revealed that three of the top 15 enriched pathways were 'ethanol
degradation II' and 'Ethanol Degradation IV' (Fig. 1B; Table SVI); the remaining 12 pathways
predominantly involved enzymes from the ALDH and ADH families.
Notably, all of these genes consistently showed significant
downregulation, with the commonly downregulated genes being
ADH1B, ALDH2, ALDH3A2 and ALDH9A1.
These enzymes were also found to be downregulated in tumor compared
with normal tissues in TCGA/HNC cohort (Fig. 1C). Survival analysis using
TCGA/HNC cohort identified ALDH2 as the only gene
significantly associated with overall survival (Fig. 1D).
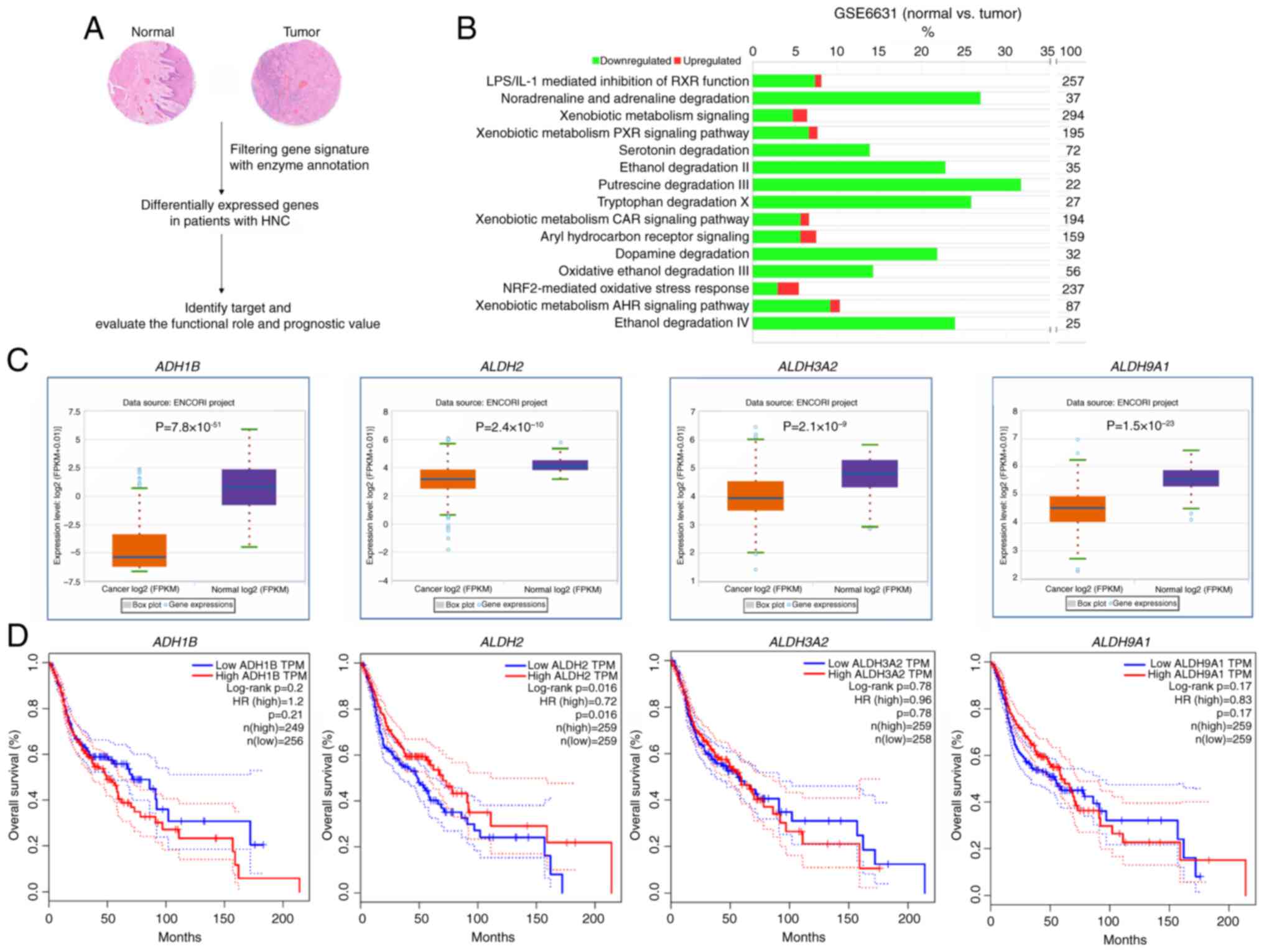 | Figure 1ALDH family of genes is
downregulated in patients with HNC. (A) Metabolic pathway screening
in normal and tumor tissue from patients with HNC. (B) Pathway
analysis of differentially expressed genes between normal and tumor
tissue in the GSE6631 dataset. (C) mRNA levels of ADH1B,
ALDH2, ALDH3A2 and ALDH9A1 in patients (n=502)
and healthy samples (n=44), based on data from TCGA database
(ENCORI). (D) Overall survival rates from TCGA/HNC cohort as
analyzed using GEPIA2 based on expression levels of ADH1B,
ALDH2, ALDH3A2 and ALDH9A1. ALDH, aldehyde
dehydrogenase; HNC, Head and Neck Cancer; TCGA, The Cancer Genome
Atlas; ADH, alcohol dehydrogenase; FPKM, Fragments Per Kilobase per
Million; HR, Hazard Ratio; TPM, Transcripts Per Million. |
ALDH2 downregulation is associated with
poor clinicopathological characteristics and outcomes in HNC
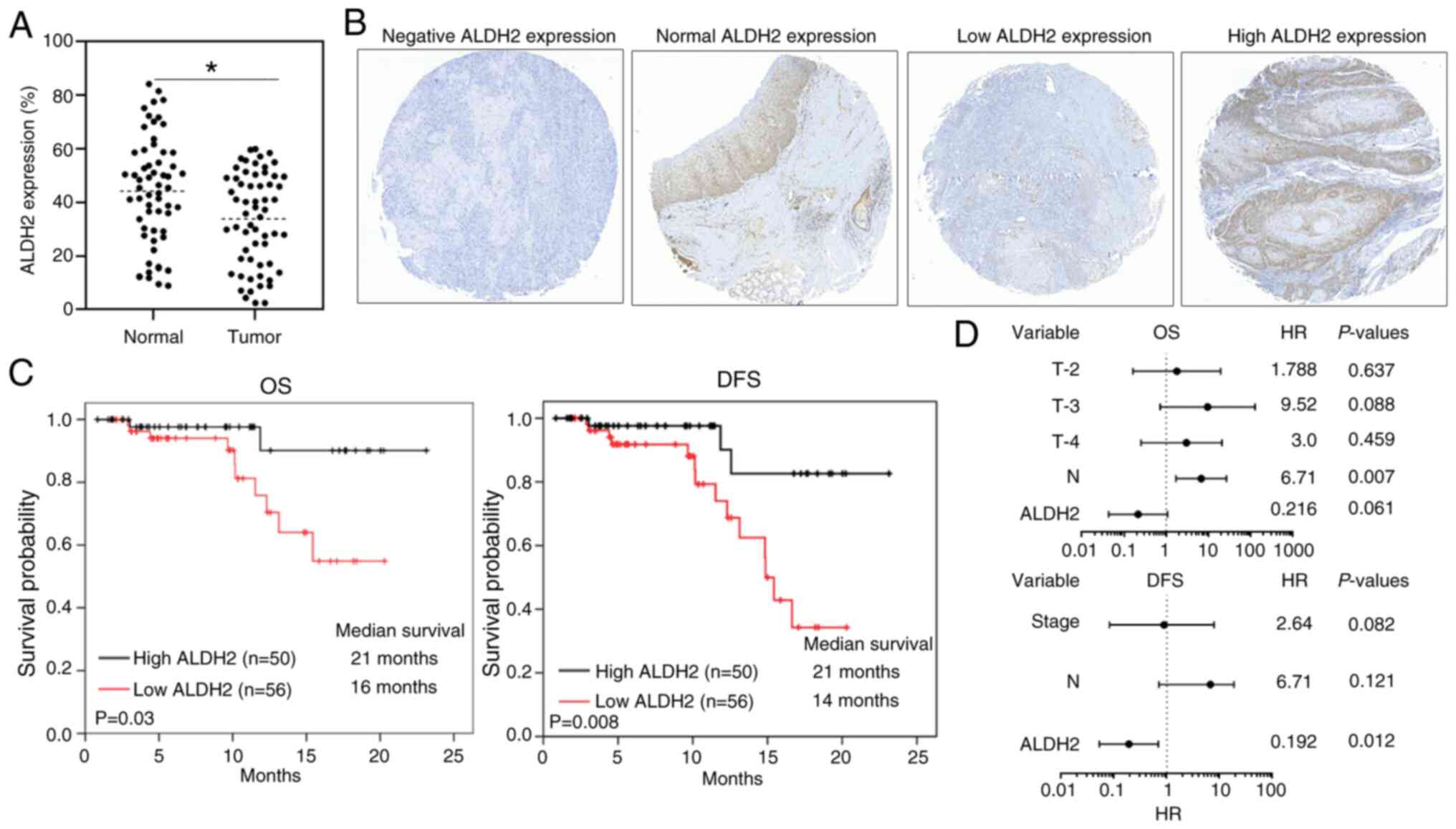
To investigate the differential protein expression
of ALDH2 in HNC, the present study analyzed 60 tumor specimens and
their paired adjacent normal tissues using IHC. ALDH2 was
significantly downregulated in tumor specimens compared with
adjacent normal tissue (Fig. 2A and
B), To assess the clinical role of ALDH2 expression in HNC,
TMA-1 from 106 patients was evaluated. Low ALDH2 expression in HNC
tissue was significantly correlated with higher AJCC T
classification, advanced overall stage and increased recurrence
rates (Table I). Survival
analysis using log-rank test revealed that patients with high ALDH2
expression (n=50) had significantly better overall survival (OS)
and disease-free survival (DFS) rates than those with low
expression (n=56) (Fig. 2C).
Univariate analyses identified low ALDH2 expression and high AJCC T
and N stage as poor prognostic factors for OS, whereas multivariate
analysis showed the no significance of ALDH2 after adjusting for
other variables (Fig. 2D;
Table SVII). High ALDH2
expression was an independent favorable factor for DFS (Fig. 2D; Table SVIII).
 | Table ICorrelation between ALDH2 expression
and clinicopathological characteristics in patients with head and
neck cancer. |
Table I
Correlation between ALDH2 expression
and clinicopathological characteristics in patients with head and
neck cancer.
| Characteristic | Total (n=106) | Low ALDH2
(n=56) | High ALDH2
(n=50) | P-value |
|---|
| Sex (%) | | | | |
| Female | 15 (14.2) | 5 (8.9) | 10 (20.0) | 0.162 |
| Male | 91 (85.8) | 51 (91.1) | 40 (80.0) | |
| Age, years (%) | | | | |
| <60 | 55 (51.9) | 27 (48.2) | 28 (56.0) | 0.443 |
| ≥60 | 51 (48.1) | 29 (51.8) | 22 (44.0) | |
| T stage (%) | | | | |
| 1 | 40 (37.7) | 17 (30.3) | 23 (46.0) | 0.013 |
| 2 | 29 (27.3) | 12 (21.4) | 17 (34.0) | |
| 3 | 6 (5.7) | 3 (5.4) | 3 (6.0) | |
| 4 | 31 (29.3) | 24 (42.9) | 7 (14.0) | |
| N stage (%) | | | | |
| 0 | 82 (77.4) | 42 (75.0) | 40 (80.0) | 0.644 |
| + | 24 (22.6) | 14 (25.0) | 10 (20.0) | |
| Stage (%) | | | | |
| I/II | 57 (53.8) | 24 (42.8) | 33 (66.0) | 0.020 |
| III/IV | 49 (46.2) | 32 (57.2) | 17 (34.0) | |
| Alcohol consumption
(%) | | | | |
| No | 25 (23.6) | 10 (17.9) | 15 (30.0) | 0.172 |
| Yes | 81 (76.4) | 46 (82.1) | 35 (70.0) | |
| Betel nut
consumption (%) | | | | |
| No | 37 (34.9) | 18 (32.1) | 19 (38.0) | 0.458 |
| Yes | 69 (65.1) | 38 (67.9) | 31 (62.0) | |
| Smoking status
(%) | | | | |
| No | 28 (22.6) | 11 (16.9) | 17 (28.8) | 0.135 |
| Yes | 96 (77.4) | 54 (83.1) | 42 (71.2) | |
| Radiotherapy
(%) | | | | |
| No | 64 (60.3) | 31 (55.4) | 33 (66.0) | 0.321 |
| Yes | 42 (39.6) | 25 (44.6) | 17 (34.0) | |
| Chemotherapy
(%) | | | | |
| No | 91 (85.8) | 49 (87.5) | 42 (84.0) | 0.781 |
| Yes | 15 (14.2) | 7 (12.5) | 8 (16.0) | |
| Recurrence (%) | | | | |
| No | 89 (84.0) | 43 (76.7) | 46 (92.0) | 0.037 |
| Yes | 17 (16.0) | 13 (23.3) | 4 (8.0) | |
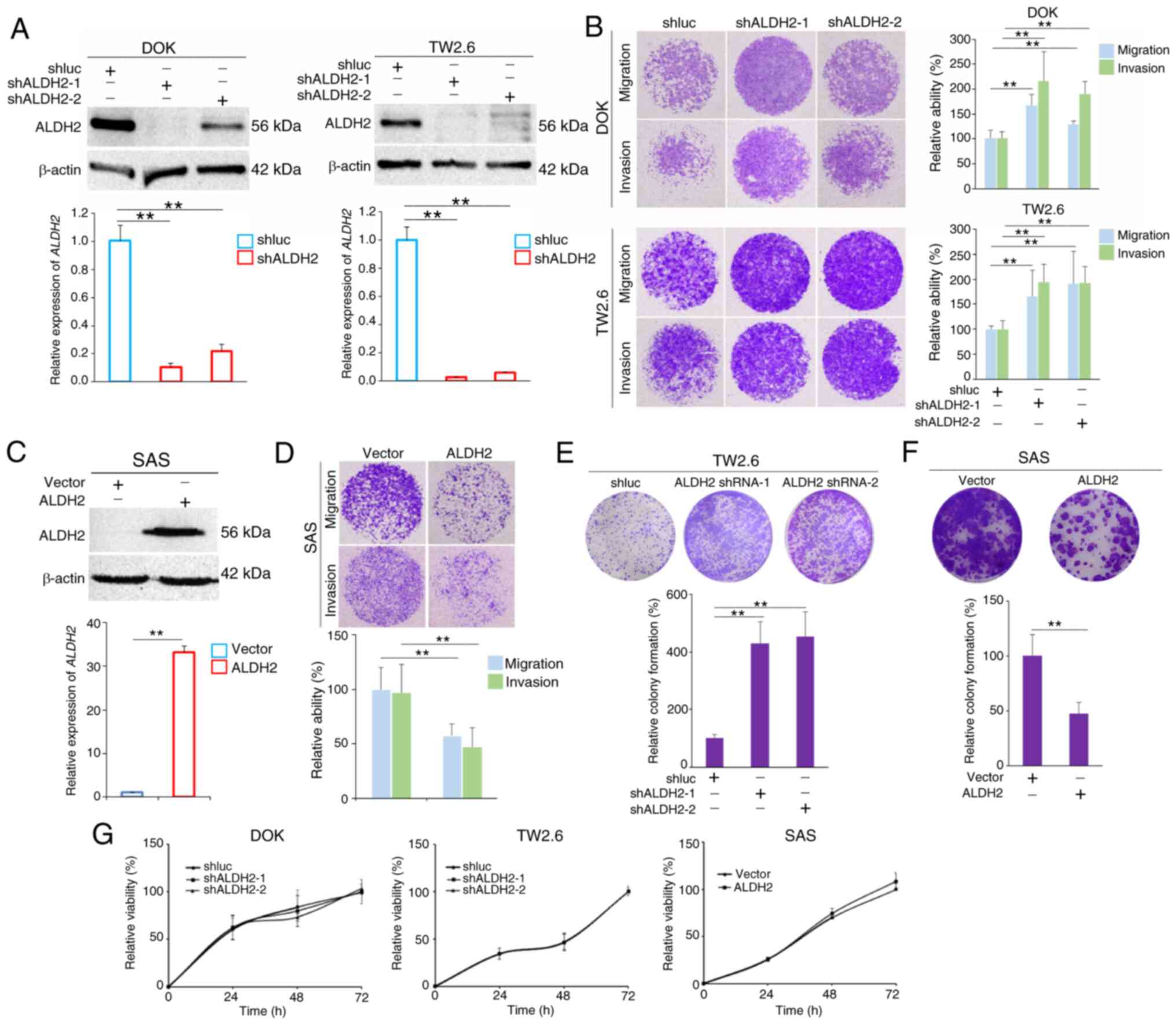
ALDH2 knockdown enhances the migration
and invasion abilities of HNC cells
Functional roles of ALDH2 in HNC cell proliferation,
migration and invasion were assessed using MTT and Boyden chamber
assay. ALDH2 was most highly expressed in DOK cells, an oral
dysplasia cell line (15) and
TW2.6 cells, with the lowest expression observed in SAS cells
(Fig. S1A). Based on these
expression levels, ALDH2 was knocked down in DOK and TW2.6 cells
and ALDH2 was overexpressed in SAS cells. As an oral dysplasia cell
line derived from a pre-malignant lesion, DOK cells exhibit higher
ALDH2 expression levels and lower invasive and migratory ability
compared with other HNC cell lines, making them an ideal model for
investigating the early stages of HNC progression and ALDH2
tumor-suppressive function (15,16). Following lentivirus-mediated
knockdown of ALDH2 via shRNA, western blot and RT-qPCR showed that
endogenous ALDH2 was downregulated in DOK and TW2.6 cells (Fig. 3A). ALDH2 knockdown in DOK and
TW2.6 cells significantly increased migration and invasion
abilities compared with shluc-infected control cells (Fig. 3B). Conversely, overexpression of
ALDH2 in SAS cell significantly reduced their ability to invade and
migrate (Fig. 3C and D).
Similarly, colony formation assays showed a significant increase in
colony-forming ability in ALDH2-knockdown HNC cells (Fig. 3E). Conversely, ALDH2
overexpression significantly reduced colony formation (Fig. 3F). However, neither
overexpression nor knockdown of ALDH2 significantly affected the
proliferation of HNC cells (Fig.
3G).
MTCQ1 cells (derived from 4-nitroquinoline
1-oxide-induced tongue carcinoma in C57BL/6 mice) were used to
validate the aforementioned results. MTCQ1 cells are characterized
by higher invasiveness and clonogenic potential but lower
proliferation rate than SAS cells (17). ALDH2 overexpression in
MTCQ1 cells significantly decreased migration, invasion and colony
formation without significantly affecting cell proliferation
(Fig. S1B-E). These results
indicate that ALDH2 influences migration, invasion and colony
formation in HNC cells, including DOK cell.
ALDH2 downregulation promotes
migration/invasion of HNC cells by activating NF-κB/VEGFC
signaling
To elucidate the molecular mechanism mediated by
ALDH2, gene expression microarray analysis of ALDH2-knockdown
(TW2.6/shALDH2) and control cells (TW2.6/shluc) was performed.
ALDH2 knockdown resulted in the upregulation of 980 and the
downregulation of 748 genes, with an absolute 2-fold change as the
threshold. IPA on these differentially expressed genes, identified
tumor necrosis factor (TNF) as the top upstream regulator (Fig. S2A; Table SIX). Pathway analysis showed
that 'NF-κB signaling' pathway was significantly upregulated
following ALDH2 knockdown, with a z-score of 2.324 (Table SX).
Effect of ALDH2 on NF-κB signaling was assessed in
ALDH2-knockdown cells. Knockdown of ALDH2 in TW2.6 cells resulted
in increased levels of phosphorylated (p)NF-κB (Fig. 4A). Conversely, ALDH2
overexpression in SAS cells led to a significant decrease in pNF-κB
expression (Fig. 4B). Silencing
RELA) using siRNA markedly inhibited the migration and
invasion activity of ALDH2-knockdown cells (Figs. 4B and S2B and C). These results suggested
that ALDH2 mediated cell migration and invasion through NF-κB.
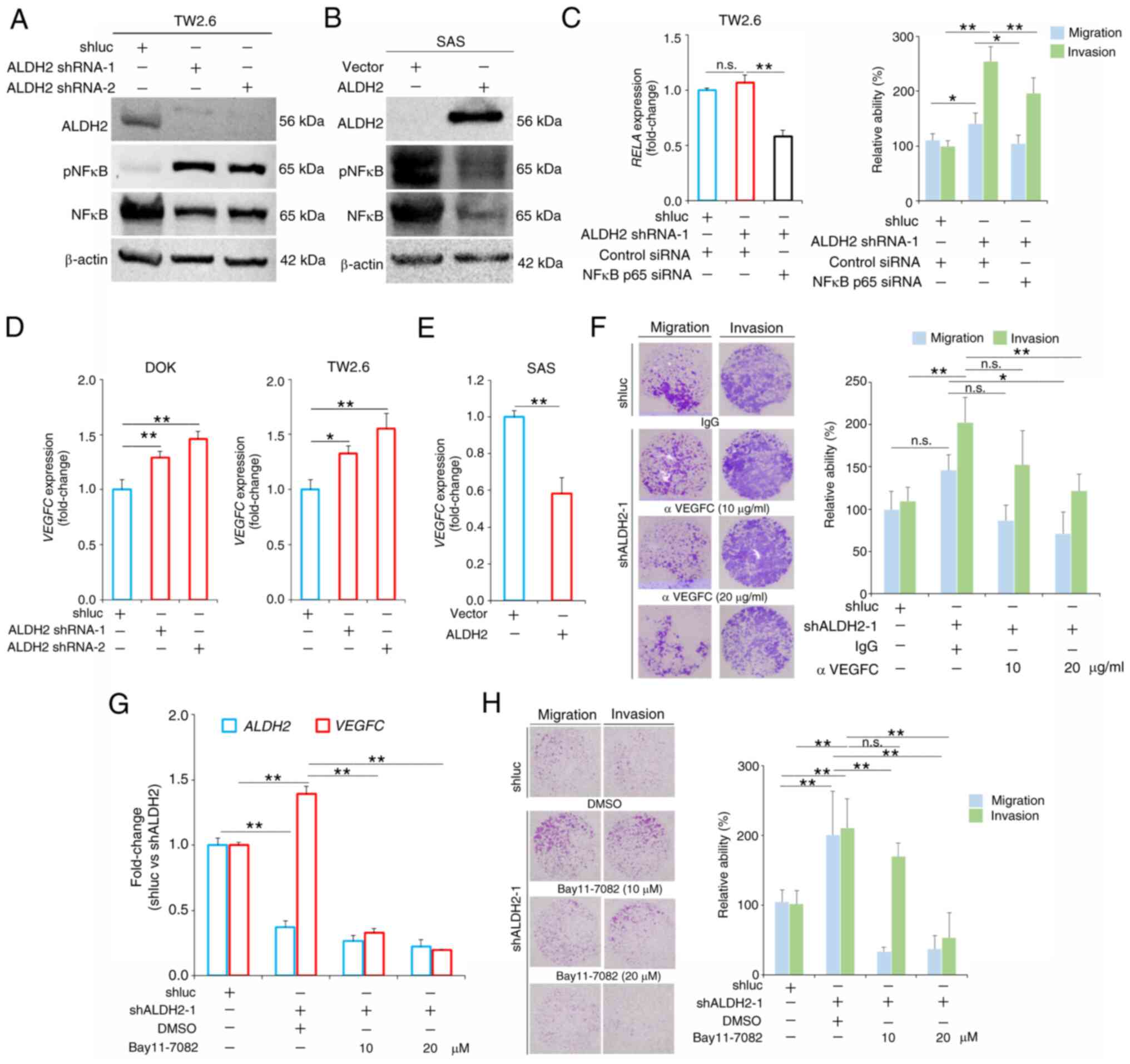 | Figure 4ALDH2 knockdown promotes head and
neck cancer cell migration/invasion via VEGFC. (A) Comparative
expression levels of ALDH2, pNFκB, and NF-κB in TW2.6 cells. (B)
Western blot analysis of ALDH2, pNF-κB, and NF-κB in SAS/ALDH2
cells. (C) RT-qPCR analysis of RELA expression and migration
and invasion of TW2.6/shALDH2 cells following RELA
silencing. RT-qPCR analysis of VEGFC expression in (D) DOK
and TW2.6 cells following ALDH2 knockdown and (E)
ALDH2-overexpressing SAS cells. (F) Migration and invasion
capabilities of TW2.6/shALDH2 cells following pretreatment with a
VEGFC-neutralizing antibody for 30 min. (G) RT-qPCR analysis of
ALDH2 and VEGFC expression and (H) migration and
invasion of TW2.6/shALDH2 cells after treatment with the NF-κB
inhibitor Bay11-7082 (16×). *P<0.05,
**P<0.01. n.s., not significant; ALDH, aldehyde
dehydrogenase; VEGFC, Vascular Endothelial Growth Factor C; p,
phosphorylated; RT-q, Reverse Transcription-quantitative; RELA,
NF-κB p65; sh, short hairpin; si, small interfering. |
Analysis of microarray data from ALDH2-knockdown
TW2.6 cells suggests that VEGFC is a key factor in cancer
progression and is regulated by NF-κB (18). RT-qPCR confirmed that
VEGFC was significantly upregulated in ALDH2-knockdown cells
(Fig. 4D), whereas its
expression was notably decreased in ALDH2-overexpressing cells
(Figs. 4E and S2D). ALDH2 knockdown enhanced cell
migration and invasion, which were significantly suppressed
following treatment with a VEGFC-neutralizing antibody (Fig. 4F). To determine whether NF-κB
mediates VEGFC expression in ALDH2-knockdown cells, NF-κB inhibitor
Bay11-7082 was used, which inhibits IκBα phosphorylation (19). The present results showed a
significant decrease in VEGFC levels and migration and
invasion of TW2.6/shALDH2 cells upon NF-κB inhibition (Fig. 4G and H). These findings confirm
that ALDH2 suppression activates the NF-κB/VEGFC axis, leading to
aggressiveness of HNC cells.
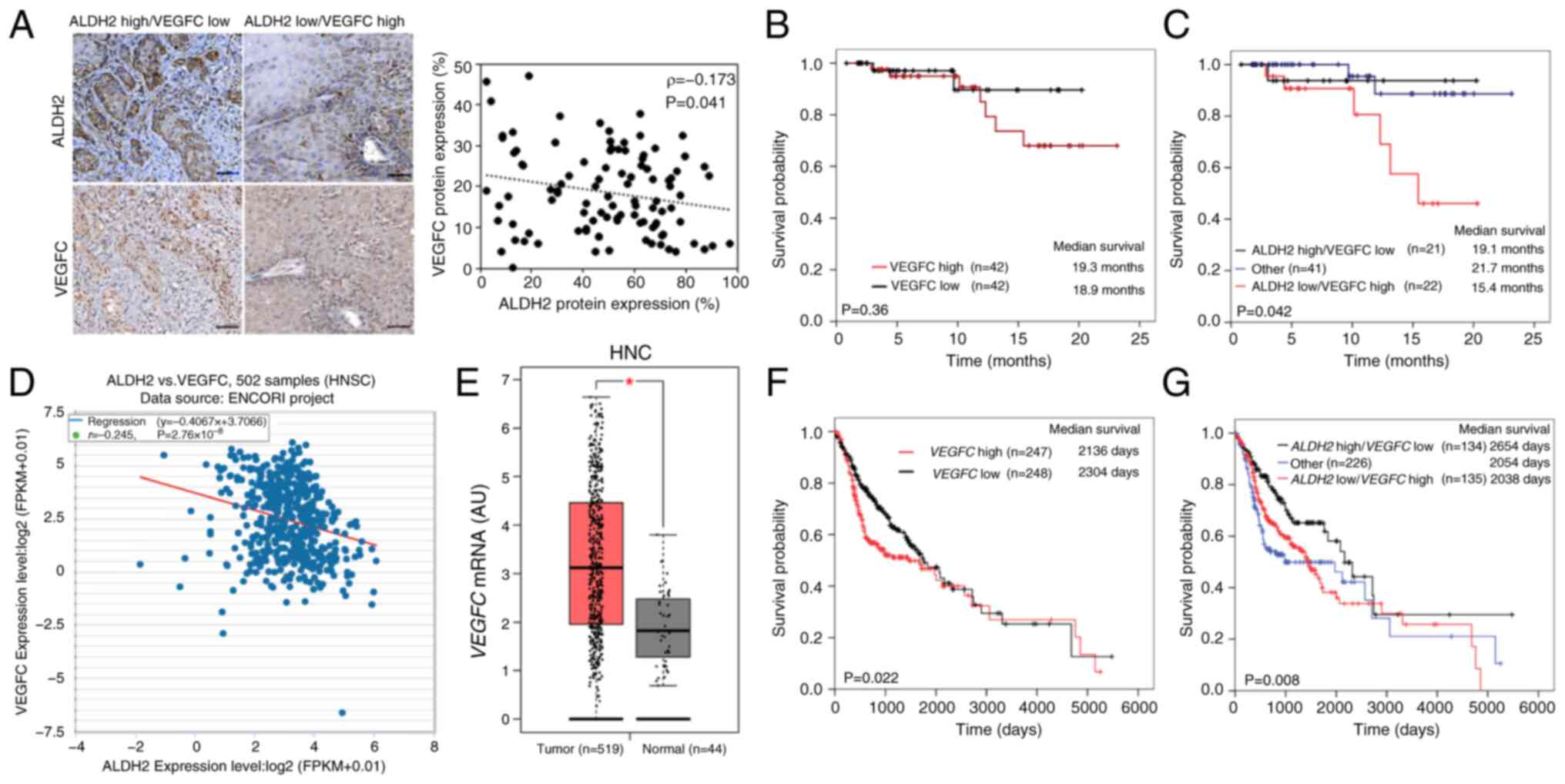
VEGFC levels are negatively correlated
with ALDH2 and positively correlated with poor survival in HNC
ALDH2 and VEGFC expression in HNC tissue was
examined via immunohistochemistry in 84 HNC samples from TMA-2.
This revealed a negative correlation (Spearman correlation
coefficient=-0.173; Fig. 5A),
suggesting a potential suppressive effect of ALDH2 on VEGFC.
Although VEGFC expression alone did not significantly influence OS
(Fig. 5B), the combination of
high ALDH2 and low VEGFC levels was associated with greater
survival probability compared with the low ALDH2 and high VEGFC
(Fig. 5C). In transcriptomic
data of 502 patients from TCGA/HNC cohort, there was a consistent
inverse relationship between ALDH2 and VEGFC
(r=-0.245, Fig. 5D), supporting
the protein-level findings. Additionally, GEPIA2 platform showed
significantly higher VEGFC levels in tumor tissue (n=519)
compared with normal (n=44; Fig.
5E). Survival analysis further demonstrated that patients with
low VEGFC levels (VEGFClow, n=248) had
improved OS (Fig. 5F). Further
analysis of the combined expression levels of ALDH2 and
VEGFC revealed that
ALDH2high/VEGFClow (n=134) had the
most favorable survival outcome, whereas patients with
ALDH2low/VEGFChigh (n=135)
exhibited the poorest prognosis (Fig. 5G).
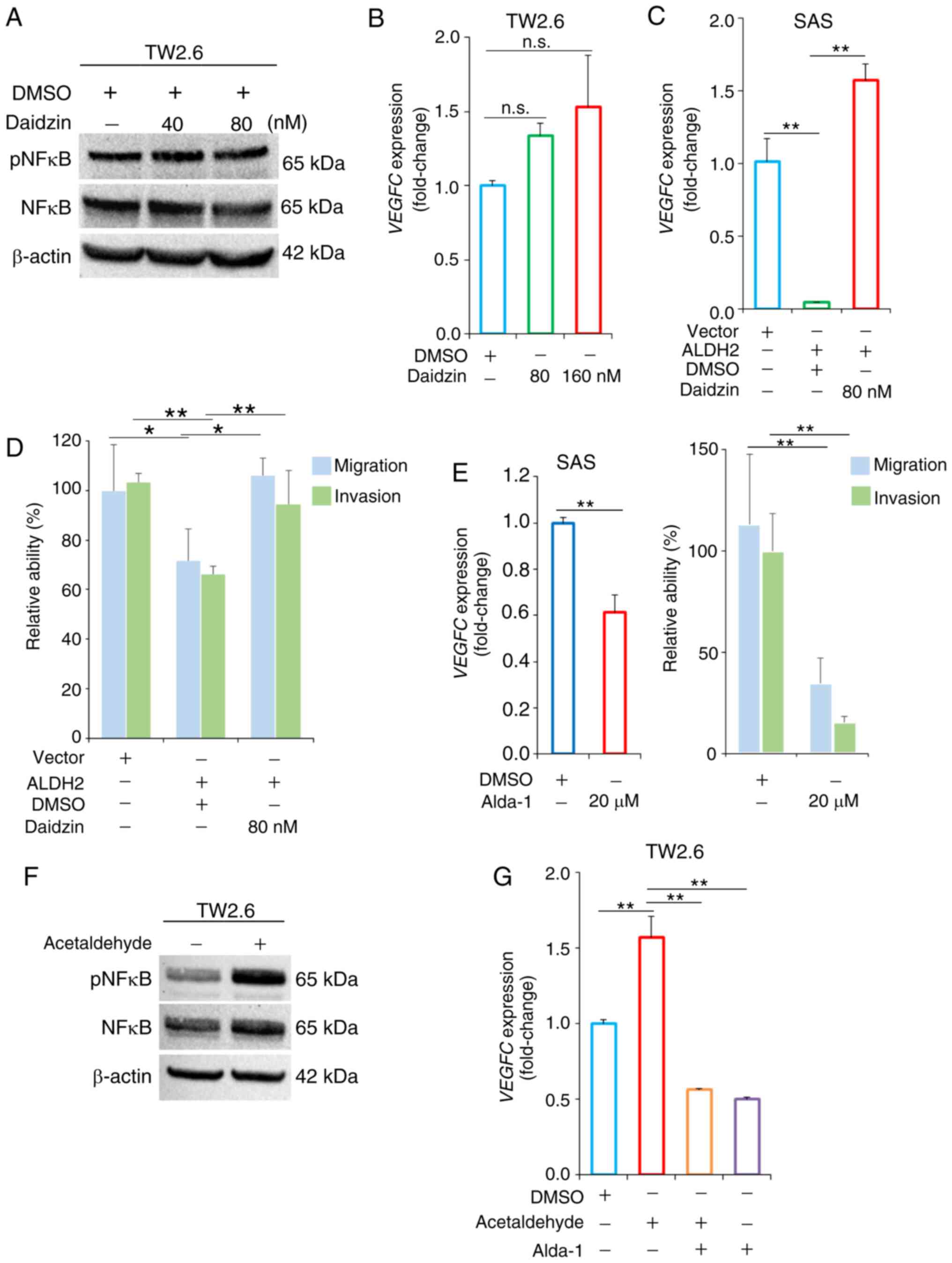
ALDH2 agonist Alda-1 mitigates
acetaldehyde-induced VEGFC expression of HNC cells
Based on enhancement of malignant traits in HNC
cells following ALDH2 knockdown, the present study aimed to
determine the effect of modulating the enzyme activity of ALDH2
using an antagonist and agonist, particularly focusing on its
effects mediated by NF-κB/VEGFC signaling pathway. Daidzin
(isoflavone glycoside), a specific ALDH2 antagonist, was used to
treat TW2.6 cells. Daidzin resulted in marked increase in pNF-κB
expression in TW2.6 cells (Fig.
6A), accompanied by enhanced VEGFC levels, however this
was not significant (Fig. 6B).
In ALDH2-overexpressing SAS cells, daidzin treatment not only
increased VEGFC levels but also restored the migration and
invasion capacities of SAS/ALDH2 cells (Figs. 6C and D and S3A). To investigate the potential of
ALDH2 agonist Alda-1 for decreasing cancer aggressiveness, its
effect on viability in SAS cells was assessed at various
concentrations. Alda-1 treatment showed minimal cytotoxicity in SAS
cells, with <25 μM showing no significant decrease in
cell viability across the tested range.Hovever, 50 μM of
Alda-1 significantly reduced cell viability in SAS cells (Fig. S3B). Furthermore, a previous
study showed that treatment with 20 μM Alda-1 increased
ALDH2-mediated VISTA (V-domain Ig suppressor of T-cell activation)
expression in breast cancer cells (20). Based on this evidence, we used a
concentration of 20 μM of Alda-1 for subsequent experiments.
At this concentration, Alda-1 significantly reduced VEGFC
levels and markedly decreased the migratory and invasive ability of
SAS cells (Figs. 6E and S3C).
Since pharmacological modulation of ALDH2 activity
regulated VEGFC-mediated HNC migration and invasion
similarly to ALDH2 expression modulation, the present study
investigated whether these effects were associated with alcohol
metabolism. pNF-κB and VEGFC expression were significantly
upregulated in TW2.6 cells following treatment with acetaldehyde,
the primary metabolite of ethanol (21) (Fig. 6F and G). Furthermore,
VEGFC expression levels in the groups treated with both
acetaldehyde and Alda-1 were comparable with those treated with
Alda-1 alone (Fig. 6G).
ALDH2 knockdown increases NF-κB activity
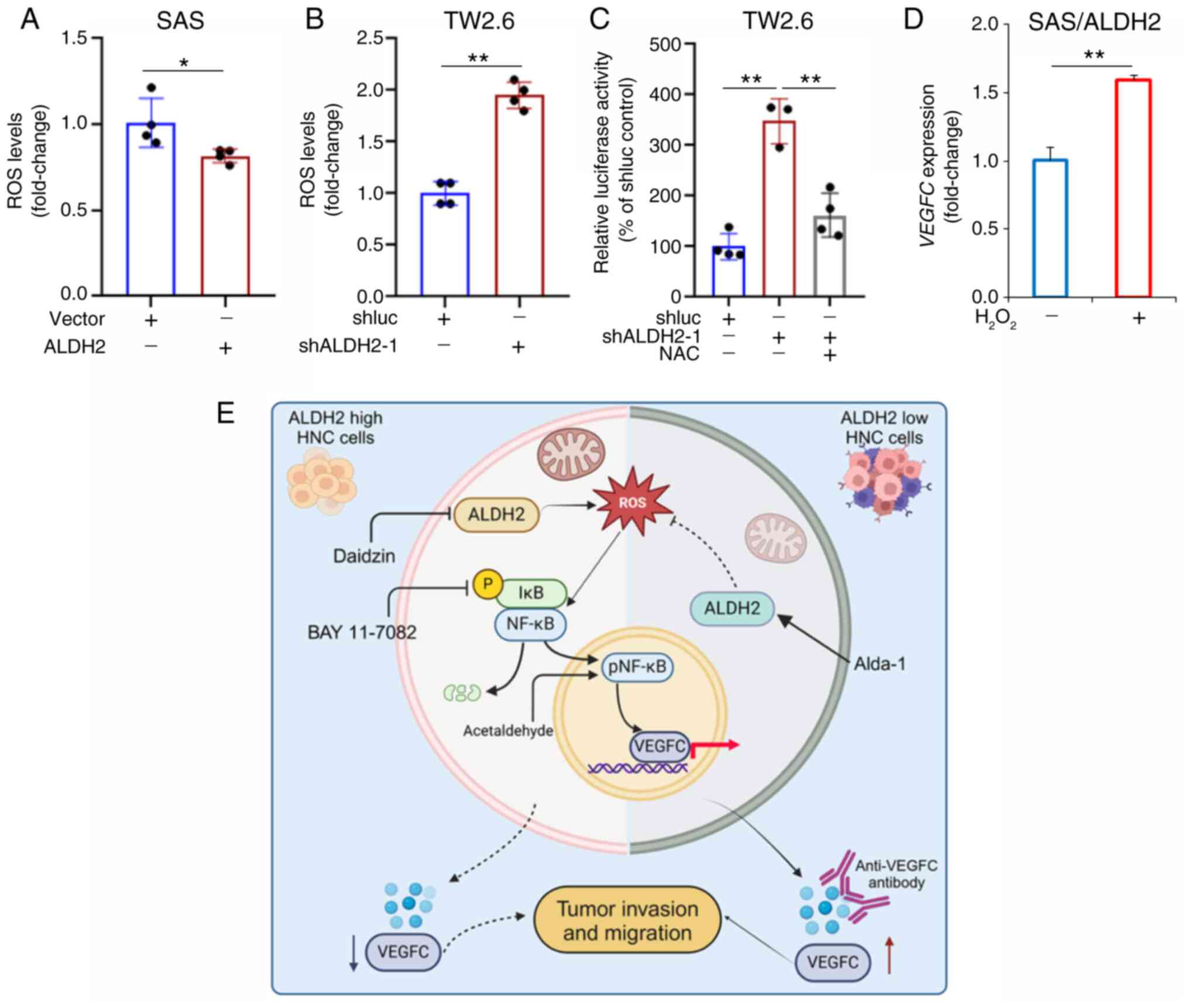
via ROS production in HNC cells
Acetaldehyde may promote ROS production to enhance
oxidative stress (22). NF-κB is
a redox-sensitive transcription factor that can be either activated
or deactivated by ROS (23). The
present study investigated whether ALDH2 regulates NF-κB through
ROS modulation. ALDH2 overexpression significantly reduced ROS
levels, while ALDH2 knockdown increased ROS levels in HNC cells
(Fig. 7A and B). NAC), a free
radical scavenger, in ALDH2-knockdown TW2.6 cells significantly
suppressed NF-κB activity (Fig.
7C). H2O2 led to a marked increase in
VEGFC levels in ALDH2-overexpressing SAS cells compared with
those without H2O2 treatment (Fig. 7D). These results collectively
suggested that ALDH2 modulates the NF-κB/VEGFC axis by regulating
ROS production in HNC cells (Fig.
7E).
Discussion
The present study demonstrated that ALDH2, a common
gene with an enzyme signature within the top-ranked altered
metabolic pathways in tumor tissue, influences the migration,
invasion and colony formation capacities of HNC cells, but not
their proliferation. Silencing NF-κB p65 significantly inhibited
migration and invasion in ALDH2-knockdown cells, with the
regulatory mechanism between ALDH2 and NF-κB mediated through its
control of ROS production. Treatment of ALDH2-knockdown cells with
NF-κB inhibitors or VEGFC-neutralizing antibodies mitigated these
enhanced activities by decreasing VEGFC expression,
confirming that ALDH2-driven migration and invasion are dependent
on modulation of the NF-κB/VEGFC axis. ALDH2-overexpressing
MTCQ1 cells exhibited similar changes in migration, invasion,
colony formation and VEGFC expression, supporting the
present in vitro findings. However, the present study lacked
in vivo validation; animal models are required to determine
the effect of ALDH2 on the NF-κB/VEGFC pathway within a complex
biological environment. Moreover, the present results suggest that
modulating the enzymatic function of ALDH2 induces phenotypical
changes similar to those observed when altering ALDH2 expression
levels, through established signaling pathways, even in HNC cells
overexpressing ALDH2. Modulating ALDH2 activity with Alda-1
mitigated the acetaldehyde-induced enhancement of the NF-κB/VEGFC
axis, consistent with the present IPA findings on altered alcohol
metabolism. The inverse association between ALDH2 and VEGFC and the
detrimental impact of their combined expression on patient
outcomes, positions ALDH2 as a potential metabolic target for HNC
treatment, offering novel avenues for therapeutic intervention.
Additionally, previous data revealed lower ALDH2 expression in
cancer tissues and showed that ALDH2 knockdown increased the
half-maximal inhibitory concentration of 5-fluorouracil in HNC
cells (24), further emphasizing
the role of the metabolic gene ALDH2 in the characteristic
behavior of HNC.
ALDH2 is a key mitochondrial enzyme that forms
tetramers and serves a pivotal role in detoxification of
acetaldehyde and endogenous aldehydes. Defective ALDH2 enzymes lead
to the accumulation of acetaldehyde, inducing DNA damage through
formation of DNA adducts and causing genetic and epigenetic
instability by forming DNA-protein crosslinks and histone adducts
(6-8). Exposure to ethanol-derived
acetaldehyde causes chromosomal rearrangements and genomic
instability in hematopoietic stem cells, potentially initiating
malignancy despite recombination repair activation and p53 deletion
(25). Additionally,
acetaldehyde-induced DNA damage, as indicated by γH2AX levels, is
decreased in ALDH2-overexpressing A549 cells after
acetaldehyde exposure, suggesting that ALDH2 suppression leads to
acetaldehyde accumulation, which increases DNA damage and enhances
the migratory ability of these cells (26). Acetaldehyde further amplifies
oxidative stress by producing ROS, similar to the effects of
endogenous aldehydes such as 4-HNE, which are generated during
redox stress-induced lipid peroxidation (6,7).
The cycle of mutual amplification between toxic reactive aldehydes
and ROS may exacerbate lipid peroxidation (6,7),
highlighting the key role of ALDH2 in maintaining redox
homeostasis. Therefore, when cellular antioxidant defenses and
energetic adaptability are insufficient to mitigate damage caused
by either oxidative stress or aldehydic products, cells may incur
mutations in oncogenes or tumor suppressor genes that induce
carcinogenesis (27). Western
blot analysis revealed background bands in the 45-60 kDa range in
ALDH2 knockdown HNC cells. Similar background bands have also been
reported in a previous study on lung cancer (26), which may be attributed to the
presence of two ALDH2 isoforms at the protein level, with molecular
weights of 56 and 46 kDa (28).
However, specific roles of ALDH2 isoforms in HNC remain unclear and
warrant further investigation.
As TNF modulates NF-κB signaling (29,30), knockdown of ALDH2 activated TNF
signal, these findings highlight its potential involvement in
ALDH2-mediated pathways. The impact of aldehydic products on NF-κB
has been established (31,32). Acetaldehyde activates NF-κB in
HepG2 cells via IκBα degradation and protein kinase C signaling
(31). By contrast, 4-HNE may
inhibit the NF-κB pathway and inactivate Bcl-2 via IKK-mediated
phosphorylation, suggesting NF-κB functions as an anti-apoptotic
element (32). ROS can either
activate or suppress NF-κB, depending on cellular context. This
dual role of ROS is complex, as ROS can activate NF-κB by promoting
alternative phosphorylation of IκBα or enhancing IKK activity via
NF-κB essential modulator (NEMO) dimerization (23). Conversely, ROS may inhibit NF-κB
by oxidizing key cysteine residues in its subunits, which decreases
the ability of NF-κB to bind to DNA (23). A previous study demonstrated that
elevated ROS levels from increased mitochondrial fission may
activate NF-κB via NF-κB inhibitor alpha (NFKBIA) and IKK,
promoting liver cancer cell survival (33). The present findings demonstrated
that, in HNC, ROS enhanced NF-κB activity, which served a key role
in activating VEGFC. This regulation underscores the involvement of
ROS in promoting HNC progression. However, further mechanistic
studies are warrated to elucidate redox-sensitive sites within the
NF-κB that respond to ROS changes induced by ALDH2.
The present study identified ALDH2 as a tumor
suppressor in HNC, similar to its roles in lung and liver cancer
(26,34,35). The present data highlight the
metabolic role of ALDH2 in modulating cancer cell phenotypes, as
Alda-1 administration reversed acetaldehyde-induced VEGFC
expression. This confirms that the pharmacological modulation of
ALDH2 significantly impacts HNC cell behavior regardless of ALDH2
expression levels. Alda-1, a selective activator of ALDH2, enhances
ALDH2 activity by functioning as a structural chaperone (36) and protects ALDH2 from
aldehyde-induced inactivation by preventing access to key cysteine
residues (37). Preclinical
in vivo studies have shown promising results regarding the
efficacy and safety of Alda-1 in oxidative stress-related disorders
affecting the brain, heart, lung, liver and retina (6,7,38). The underlying mechanism primarily
involves reducing ROS and aldehyde-mediated signaling pathways
(6,7,38). In esophageal cancer, previous
in vivo findings have demonstrated that Alda-1 reduces
alcohol-induced esophageal DNA damage in genetically modified mice
with ALDH2 polymorphisms (39) and inhibits the expansion of
CD44-high cancer stem cells, which are associated with tumor
initiation and chemoresistance (40). The present investigation
confirmed that ALDH2 is a prognostic factor with significant
correlations to T classification and overall stage in human HNC
tissue. The activation of ALDH2 by Alda-1 inhibited the malignant
features of HNC cells through the NF-κB/VEGFC axis. Overall, these
findings suggested that Alda-1 is a viable therapeutic strategy for
HNC. However, further studies are warranted to clarify the specific
dose-effect association, potential side effects and clinical
feasibility of Alda-1 to support its application as a therapeutic
agent for HNC.
VEGFC, a member of the VEGF/platelet-derived growth
factor family, contains conserved NF-κB binding sites in its
promoter, indicating regulation via the NF-κB pathway (41). Once processed into its mature
form, VEGFC binds to VEGFR-3 and neuropilin-2 (NRP2) on lymphatic
endothelial cells to facilitate lymphatic metastasis (42,43). VEGFC signaling is crucial for
establishing premetastatic niches in sentinel lymph nodes (44,45), facilitating spread, colonization
and maintenance of disseminated tumor cells (46-48). In HNC, elevated VEGFC expression
is associated with larger tumor size, higher recurrence rate and
poorer survival outcome (49-51) than low expression, highlighting
its significance as a therapeutic target. Current VEGF-targeted
therapies that use tyrosine kinases against VEGFR have shown
promise in clinical trials for metastatic HNC (52-54). For example, a phase II trial of
axitinib reported a disease control rate of 76.7% and median OS of
10.9 months (55). However,
evidence supporting the effectiveness of anti-VEGFC monoclonal
antibodies in this context is lacking. While studiy in clear cell
renal cell carcinoma suggests benefits in targeting VEGFC (53), especially in cells overexpressing
VEGFR-3 and NRP2, resistance to anti-angiogenic therapy can develop
through the upregulation of hypoxia-inducible factor-1α,
angiopoietin-2 and basic fibroblast growth factor (56). Thus, addressing resistance to
anti-VEGFC treatment may require targeting upstream regulatory
molecules simultaneously. The present findings indicate that ALDH2
regulated VEGFC by modulating NF-κB activity, further highlighting
the potential of pharmacologically enhancing ALDH2 function to
combat metastatic HNC.
Taken together, the present findings confirmed that
ALDH2 served a critical role in regulating alcohol metabolism in
HNC, with its downregulation linked to poor survival outcome.
Alda-1 restored ALDH2 activity, effectively inhibiting
acetaldehyde-induced upregulation of NF-κB/VEGFC axis and
inhibiting migration and invasion in HNC. While in vivo
validation is warranted to confirm these effects, the present
results highlight the therapeutic potential of targeting alcohol
metabolism via ALDH2 modulation to improve treatment outcome in
HNC.
Supplementary Data
Availability of data and materials
The data generated in the present study may be
found in the Gene Expression Omnibus under accession number
(GSE253622) or at the following URL: ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE253622.
Authors' contributions
YHL and YFY interpreted data and wrote the
manuscript. YHL and JBL performed histopathology experiments. YCL,
PLY and CYC performed experiments. YHL and YFY confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The Kaohsiung Veterans General Hospital ethics
committee approved the study (approval no. KSVGH23-CT8-10) and
waived the requirement for informed consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgements
The authors would like to thank Professor Michael
Hsiao from Academia Sinica, Taipei, Taiwan for kindly providing HNC
cell lines.
Funding
The present study was supported by Kaohsiung Veterans General
Hospital, Taiwan (grant nos. KSVGH111-098, KSVGH112-094 and
KSVGH-113-062), Yen Tjing Ling Medical Foundation (grant no.
CI-112-5) and National Science and Technology Council, Taiwan
(grant nos. MOST-110-2314-B-075B-014, NSTC-113-2314-B-075B-001,
MOST-110-2314-B-075B-009-MY3 and NSTC-113-2314-B-075B-002).
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnson DE, Burtness B, Leemans CR, Lui
VWY, Bauman JE and Grandis JR: Head and neck squamous cell
carcinoma. Nat Rev Dis Primers. 6:922020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cancer Genome Atlas Network: Comprehensive
genomic characterization of head and neck squamous cell carcinomas.
Nature. 517:576–582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Faubert B, Solmonson A and DeBerardinis
RJ: Metabolic reprogramming and cancer progression. Science.
368:eaaw54732020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N,
Yi P, Tang L, Pan Q, Rao S, et al: The cancer metabolic
reprogramming and immune response. Mol Cancer. 20:282021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen CH, Ferreira JC, Gross ER and
Mochly-Rosen D: Targeting aldehyde dehydrogenase 2: New therapeutic
opportunities. Physiol Rev. 94:1–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gao J, Hao Y, Piao X and Gu X: Aldehyde
dehydrogenase 2 as a therapeutic target in oxidative stress-related
diseases: Post-translational modifications deserve more attention.
Int J Mol Sci. 23:26822022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen D, Fang L, Li H and Jin C: The
effects of acetaldehyde exposure on histone modifications and
chromatin structure in human lung bronchial epithelial cells.
Environ Mol Mutagen. 59:375–385. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee WT, Hsiao JR, Ou CY, Huang CC, Chang
CC, Tsai ST, Chen KC, Huang JS, Wong TY, Lai YH, et al: The
influence of prediagnosis alcohol consumption and the polymorphisms
of ethanol-metabolizing genes on the survival of head and neck
cancer patients. Cancer Epidemiol Biomarkers Prev. 28:248–257.
2019. View Article : Google Scholar
|
|
10
|
Tang Z, Li C, Kang B, Gao G, Li C and
Zhang Z: GEPIA: A web server for cancer and normal gene expression
profiling and interactive analyses. Nucleic Acids Res. 45(W1):
W98–W102. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Edge SB and Compton CC: The American joint
committee on cancer: The 7th edition of the AJCC cancer staging
manual and the future of TNM. Ann Surg Oncol. 17:1471–1474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tseng HH, Tseng YK, You JJ, Kang BH, Wang
TH, Yang CM, Chen HC, Liou HH, Liu PF, Ger LP and Tsai KW:
Next-generation sequencing for microRNA profiling: MicroRNA-21-3p
promotes oral cancer metastasis. Anticancer Res. 37:1059–1066.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu CW, Hua KT, Li KC, Kao HF, Hong RL, Ko
JY, Hsiao M, Kuo ML and Tan CT: Histone Methyltransferase G9a
drives chemotherapy resistance by regulating the glutamate-cysteine
ligase catalytic subunit in head and neck squamous cell carcinoma.
Mol Cancer Ther. 16:1421–1434. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Chang SE, Foster S, Betts D and Marnock
WE: DOK, a cell line established from human dysplastic oral mucosa,
shows a partially transformed non-malignant phenotype. Int J
Cancer. 52:896–902. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bs A, P A, As SG, A P and J VP: Analysis
of differentially expressed genes in dysplastic oral keratinocyte
cell line and their role in the development of HNSCC. J Stomatol
Oral Maxillofac Surg. 125:1019282024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen YF, Chang KW, Yang IT, Tu HF and Lin
SC: Establishment of syngeneic murine model for oral cancer
therapy. Oral Oncol. 95:194–201. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lin C, Song L, Gong H, Liu A, Lin X, Wu J,
Li M and Li J: Editor's Note: Nkx2-8 downregulation promotes
angiogenesis and activates NF-κB in esophageal cancer. Cancer Res.
82:16702022. View Article : Google Scholar
|
|
19
|
Strickson S, Campbell DG, Emmerich CH,
Knebel A, Plater L, Ritorto MS, Shpiro N and Cohen P: The
anti-inflammatory drug BAY 11-7082 suppresses the MyD88-dependent
signalling network by targeting the ubiquitin system. Biochem J.
451:427–437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Y, Sun J, Liu J, Wei Y, Wang X, Fang
H, Du H, Huang J, Li Q, Ren G, et al: Aldehyde dehydrogenase
2-mediated aldehyde metabolism promotes tumor immune evasion by
regulating the NOD/VISTA axis. J Immunother Cancer. 11:e0074872023.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lachenmeier DW and Sohnius EM: The role of
acetaldehyde outside ethanol metabolism in the carcinogenicity of
alcoholic beverages: Evidence from a large chemical survey. Food
Chem Toxicol. 46:2903–2911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li SY, Gomelsky M, Duan J, Zhang Z,
Gomelsky L, Zhang X, Epstein PN and Ren J: Overexpression of
aldehyde dehydrogenase-2 (ALDH2) transgene prevents
acetaldehyde-induced cell injury in human umbilical vein
endothelial cells: Role of ERK and p38 mitogen-activated protein
kinase. J Biol Chem. 279:11244–11252. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morgan MJ and Liu ZG: Crosstalk of
reactive oxygen species and NF-kappaB signaling. Cell Res.
21:103–115. 2011. View Article : Google Scholar
|
|
24
|
Lin YH, Yang YF, Liao JB, Chang TS,
Janesha UGS and Shiue YL: Analysis of aldehyde dehydrogenase 2 as a
prognostic marker associated with immune cell infiltration and
chemotherapy efficacy in head and neck squamous cell carcinoma. J
Cancer. 14:1689–1706. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Garaycoechea JI, Crossan GP, Langevin F,
Mulderrig L, Louzada S, Yang F, Guilbaud G, Park N, Roerink S,
Nik-Zainal S, et al: Alcohol and endogenous aldehydes damage
chromosomes and mutate stem cells. Nature. 553:171–177. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li K, Guo W, Li Z, Wang Y, Sun B, Xu D,
Ling J, Song H, Liao Y, Wang T, et al: ALDH2 repression promotes
lung tumor progression via accumulated acetaldehyde and DNA damage.
Neoplasia. 21:602–614. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dalleau S, Baradat M, Guéraud F and Huc L:
Cell death and diseases related to oxidative stress:
4-Hydroxynonenal (HNE) in the balance. Cell Death Differ.
20:1615–1630. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zahn-Zabal M, Michel PA, Gateau A, Nikitin
F, Schaeffer M, Audot E, Gaudet P, Duek PD, Teixeira D, Rech de
Laval V, et al: The neXtProt knowledgebase in 2020: Data, tools and
usability improvements. Nucleic Acids Res. 48(D1): D328–D334.
2020.
|
|
29
|
Legler DF, Micheau O, Doucey MA, Tschopp J
and Bron C: Recruitment of TNF receptor 1 to lipid rafts is
essential for TNFalpha-mediated NF-kappaB activation. Immunity.
18:655–664. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang YF, Jan YH, Liu YP, Yang CJ, Su CY,
Chang YC, Lai TC, Chiou J, Tsai HY, Lu J, et al: Squalene synthase
induces tumor necrosis factor receptor 1 enrichment in lipid rafts
to promote lung cancer metastasis. Am J Respir Crit Care Med.
190:675–687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Román J, Giménez A, Lluis JM, Gassó M,
Rubio M, Caballeria J, Parés A, Rodés J and Fernández-Checa JC:
Enhanced DNA binding and activation of transcription factors
NF-kappa B and AP-1 by acetaldehyde in HEPG2 cells. J Biol Chem.
275:14684–14690. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Timucin AC and Basaga H: Pro-apoptotic
effects of lipid oxidation products: HNE at the crossroads of NF-κB
pathway and anti-apoptotic Bcl-2. Free Radic Biol Med. 111:209–218.
2017. View Article : Google Scholar
|
|
33
|
Huang Q, Zhan L, Cao H, Li J, Lyu Y, Guo
X, Zhang J, Ji L, Ren T, An J, et al: Increased mitochondrial
fission promotes autophagy and hepatocellular carcinoma cell
survival through the ROS-modulated coordinated regulation of the
NFKB and TP53 pathways. Autophagy. 12:999–1014. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen X, Legrand AJ, Cunniffe S, Hume S,
Poletto M, Vaz B, Ramadan K, Yao D and Dianov GL: Interplay between
base excision repair protein XRCC1 and ALDH2 predicts overall
survival in lung and liver cancer patients. Cell Oncol (Dordr).
41:527–539. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Seo W, Gao Y, He Y, Sun J, Xu H, Feng D,
Park SH, Cho YE, Guillot A, Ren T, et al: ALDH2 deficiency promotes
alcohol-associated liver cancer by activating oncogenic pathways
via oxidized DNA-enriched extracellular vesicles. J Hepatol.
71:1000–1011. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Perez-Miller S, Younus H, Vanam R, Chen
CH, Mochly-Rosen D and Hurley TD: Alda-1 is an agonist and chemical
chaperone for the common human aldehyde dehydrogenase 2 variant.
Nat Struct Mol Biol. 17:159–164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen CH, Budas GR, Churchill EN, Disatnik
MH, Hurley TD and Mochly-Rosen D: Activation of aldehyde
dehydrogenase-2 reduces ischemic damage to the heart. Science.
321:1493–1495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kimura M, Yokoyama A and Higuchi S:
Aldehyde dehydrogenase-2 as a therapeutic target. Expert Opin Ther
Targets. 23:955–966. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hirohashi K, Ohashi S, Amanuma Y, Nakai Y,
Ida T, Baba K, Mitani Y, Mizumoto A, Yamamoto Y, Kikuchi O, et al:
Protective effects of Alda-1, an ALDH2 activator, on
alcohol-derived DNA damage in the esophagus of human ALDH2*2
(Glu504Lys) knock-in mice. Carcinogenesis. 41:194–202. 2020.
View Article : Google Scholar :
|
|
40
|
Flashner S, Shimonosono M, Tomita Y,
Matsuura N, Ohashi S, Muto M, Klein-Szanto AJ, Alan Diehl J, Chen
CH, Mochly-Rosen D, et al: ALDH2 dysfunction and alcohol cooperate
in cancer stem cell enrichment. Carcinogenesis. 45:95–106. 2024.
View Article : Google Scholar :
|
|
41
|
Chilov D, Kukk E, Taira S, Jeltsch M,
Kaukonen J, Palotie A, Joukov V and Alitalo K: Genomic organization
of human and mouse genes for vascular endothelial growth factor C.
J Biol Chem. 272:25176–25183. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang J, Huang Y, Zhang J, Wei Y, Mahoud S,
Bakheet AM, Wang L, Zhou S and Tang J: Pathway-related molecules of
VEGFC/D-VEGFR3/NRP2 axis in tumor lymphangiogenesis and lymphatic
metastasis. Clin Chim Acta. 461:165–171. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang J, Huang Y, Zhang J, Xing B, Xuan W,
Wang H, Huang H, Yang J and Tang J: NRP-2 in tumor
lymphangiogenesis and lymphatic metastasis. Cancer Lett.
418:176–184. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liersch R, Hirakawa S, Berdel WE, Mesters
RM and Detmar M: Induced lymphatic sinus hyperplasia in sentinel
lymph nodes by VEGF-C as the earliest premetastatic indicator. Int
J Oncol. 41:2073–2078. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hirakawa S, Brown LF, Kodama S, Paavonen
K, Alitalo K and Detmar M: VEGF-C-induced lymphangiogenesis in
sentinel lymph nodes promotes tumor metastasis to distant sites.
Blood. 109:1010–1017. 2007. View Article : Google Scholar
|
|
46
|
Karaman S and Detmar M: Mechanisms of
lymphatic metastasis. J Clin Invest. 124:922–928. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kong D, Zhou H, Neelakantan D, Hughes CJ,
Hsu JY, Srinivasan RR, Lewis MT and Ford HL: VEGF-C mediates tumor
growth and metastasis through promoting EMT-epithelial breast
cancer cell crosstalk. Oncogene. 40:964–979. 2021. View Article : Google Scholar
|
|
48
|
Banerjee K, Kerzel T, Bekkhus T, de Souza
Ferreira S, Wallmann T, Wallerius M, Landwehr LS, Agardy DA,
Schauer N, Malmerfeldt A, et al: VEGF-C-expressing TAMs rewire the
metastatic fate of breast cancer cells. Cell Rep. 42:1135072023.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Neuchrist C, Erovic BM, Handisurya A,
Fischer MB, Steiner GE, Hollemann D, Gedlicka C, Saaristo A and
Burian M: Vascular endothelial growth factor C and vascular
endothelial growth factor receptor 3 expression in squamous cell
carcinomas of the head and neck. Head Neck. 25:464–474. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fei J, Hong A, Dobbins TA, Jones D, Lee
CS, Loo C, Al-Ghamdi M, Harnett GB, Clark J, O'Brien CJ and Rose B:
Prognostic significance of vascular endothelial growth factor in
squamous cell carcinomas of the tonsil in relation to human
papillomavirus status and epidermal growth factor receptor. Ann
Surg Oncol. 16:2908–2917. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Siemert J, Wald T, Kolb M, Pettinella I,
Böhm U, Pirlich M, Wiegand S, Dietz A and Wichmann G:
Pre-therapeutic VEGF level in plasma is a prognostic bio-marker in
head and neck squamous cell carcinoma (HNSCC). Cancers (Basel).
13:37812021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Limaye S, Riley S, Zhao S, O'Neill A,
Posner M, Adkins D, Jaffa Z, Clark J and Haddad R: A randomized
phase II study of docetaxel with or without vandetanib in recurrent
or metastatic squamous cell carcinoma of head and neck (SCCHN).
Oral Oncol. 49:835–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Swiecicki PL, Zhao L, Belile E, Sacco AG,
Chepeha DB, Dobrosotskaya I, Spector M, Shuman A, Malloy K, Moyer
J, et al: A phase II study evaluating axitinib in patients with
unresectable, recurrent or metastatic head and neck cancer. Invest
New Drugs. 33:1248–1256. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Adkins D, Mehan P, Ley J, Siegel MJ,
Siegel BA, Dehdashti F, Jiang X, Salama NN, Trinkaus K and Oppelt
P: Pazopanib plus cetuximab in recurrent or metastatic head and
neck squamous cell carcinoma: An open-label, phase 1b and expansion
study. Lancet Oncol. 19:1082–1093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dumond A, Montemagno C, Vial V, Grépin R
and Pagès G: Anti-vascular endothelial growth factor C antibodies
efficiently inhibit the growth of experimental clear cell renal
cell carcinomas. Cells. 10:12222021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ansari MJ, Bokov D, Markov A, Jalil AT,
Shalaby MN, Suksatan W, Chupradit S, Al-Ghamdi HS, Shomali N,
Zamani A, et al: Cancer combination therapies by angiogenesis
inhibitors; a comprehensive review. Cell Commun Signal. 20:492022.
View Article : Google Scholar : PubMed/NCBI
|