Introduction
Approximately 75% of breast cancer is categorized as
estrogen receptor α-positive, with estradiol-bound estrogen
receptor α as the key determinant in promoting breast cancer growth
(1). Blocking the action of
estrogen receptor α by selective estrogen receptor modulators, such
as tamoxifen, has been the most common treatment strategy for
breast cancer patients (2).
However, despite the significant benefits of tamoxifen treatment,
almost all patients with metastatic disease and as many as 40% of
the patients receiving adjuvant tamoxifen therapy do not respond,
or acquire resistance during treatment (3). Additionally, in estrogen receptor
α-negative breast cancer patients, therapeutic outcome for
anthracycline-based chemotherapy is unsatisfactory (4). Therefore, novel strategies are
required to improve therapeutic efficacy for these breast cancer
patients.
Bortezomib (BZ), a selective and potent inhibitor of
the 26S proteasome, has been approved for treating multiple myeloma
(5). Increasing evidence indicates
that inhibition of the 26S proteasome by BZ leads to the
accumulation of misfolded proteins in the endoplasmic reticulum
(ER), resulting in ER stress followed by a coordinated cellular
response known as unfolded protein response (UPR) (6–10).
Downstream effectors of UPR include activation of the chaperone
protein GRP78 (Bip) to maintain ER integrity, and the transcription
factor CHOP (the C/EBP homologous protein, also designated as
GADD153) to mediate cell death when ER stress is beyond the
tolerance of the cell adaptation (11–13).
Many in vitro studies have demonstrated that BZ potently
induces cell growth inhibition and apoptosis in breast cancer cell
lines via UPR (14–16). However, clinical trials using BZ as
a single agent for treating metastatic breast cancer were initially
disappointing (17). A new study
combining the pure anti-estrogen fulvestrant with BZ suggested that
the combination of anti-estrogen therapies with proteasome
inhibition might increase treatment efficacy in estrogen receptor
α-positive-breast cancer cell lines (18). It was also reported that BZ
inhibited breast cancer cell growth and reduced osteolysis by
down-regulating metastatic genes in xenograft mice (19).
Macroautophagy (hereafter, autophagy) is a highly
conserved cellular process in eukaryotes. Intracellular proteins
and organelles including ER are engulfed in a double-membrane
vesicle called an autophagosome and delivered to lysosomes for
degradation by lysosomal hydrolases (20,21).
Autophagy occurs in parallel with the ubiquitin-proteasome system
that is specific to short-lived protein turnover (22). Autophagy has been regarded as a
bulk non-selective degradation system for long-lived proteins and
organelles. However, recent reports focused on the selective
degradation pathway of ubiquitinated protein through autophagy via
p62 and the related protein NBR1, which are docking proteins having
both a microtubule-associated protein 1 light chain 3
(LC3)-interacting region and a ubiquitin-associated domain
(23). LC3 is essential for
autophagy and is associated with autophagosome membranes after
processing (24). Thus, by binding
ubiquitin via their C-terminal ubiquitin-associated domains,
p62-mediated degradation of ubiquitinated cargo occurs by selective
autophagy. The two major intra-cellular degradation systems are
thus directly linked (22,23).
We previously reported that combined treatment with
BZ and bafilomycin A1, resulted in synergistic
enhancement of the cytocidal effect along with the induction of ER
stress in myeloma cells (9).
Bafilomycin A1 is a macrolide antibiotic, a specific
inhibitor of vacuolar-ATPase, and is used as an autophagy inhibitor
in the late stage of this process (25). A recent report demonstrated that
clarithromycin (CAM), a semi-synthetic macrolide anti-biotic
derived from erythromycin, inhibited autophagy flux and exhibited
some cell growth inhibition in myeloma cells (26). High efficacy of the
chemotherapeutic regimen combining CAM with lenalidomide, a
derivative of thalidomide, in treating myeloma was recently
reported (27,28). Many lines of evidence indicate that
certain macrolide antibiotics exert some anti-tumor activities in
marginal zone B-cell lymphoma, leukemia, non-small lung cancer, and
melanoma (29–34). Although the underlying molecular
mechanism has not yet been clarified, this anti-tumor activity does
not appear to be mediated by eradication of Helicobacter
pylori, as with MALT lymphoma (35). A direct apoptosis inducing-effect
of CAM has been observed in lymphoma cells (36).
Based on previous data, we attempted to investigate
whether combined treatment with CAM and BZ for simultaneously
inhibit of two major intracellular protein degradation systems
enhances ER stress-mediated cell death in breast cancer cells.
Materials and methods
Reagents
BZ was purchased from Toronto Research Chemical Inc.
(North York, Ontario, Canada). BZ was dissolved in dimethyl
sulfoxide (DMSO) at a concentration of 1 mM as a stock solution.
CAM, purchased from Wako Pure Chemical Industries, Ltd. (Osaka,
Japan), was dissolved in ethanol at a concentration of 5 mg/ml as a
stock solution. E-64d and Pepstatin A, which are inhibitors of
lysosomal proteases, were purchased from Biomol International LP
(Plymouth Meeting, PA, USA).
Cell lines and culture conditions
For this study, we used the breast cancer cell lines
MDA-MB-231 and MCF7 (kind gifts of Dr Keiichi Iwaya, Department of
Basic Pathology, National Defense Medical College, Saitama, Japan),
and the MDA-MB-468 cells, obtained from the American Type Culture
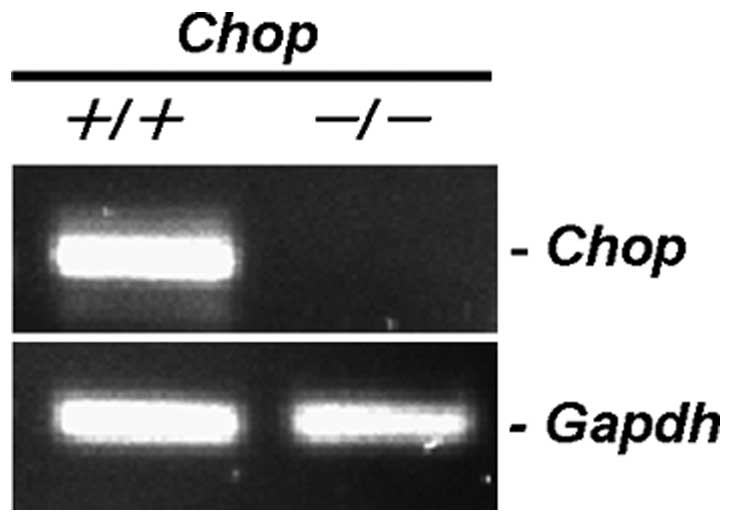
Collection (ATCC) (Manassas, VA). A CHOP−/− MEF cell
line (CHOP-KO-DR) established from a 13.5-day-old
CHOP−/− mouse embryo by SV-40 immortalization and a
CHOP+/+ MEF cell line (DR-wild-type) established by
SV-40 immortalization as a control cell line for CHOP-KO-DR were
obtained from ATCC (Rockville, MD). MDA-MB-231, MDA-MB-468, and
MCF7 cells were maintained in continuous culture in RPMI-1640
medium (Gibco, Grand Island, NY) supplemented with 10% FBS (PAA
Laboratories, Austria), 2 mM L-glutamine, penicillin (100 U/ml) and
streptomycin (100 μg/ml) (Wako Pure Chemical Industries, Ltd.).
CHOP-KO-DR and DR-wild-type cells were maintained in Dulbecco’s
modified Eagle’s medium (Sigma, St. Louis, MO) supplemented with
10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). All
cell lines were cultured in a humidified incubator containing 5%
CO2 and 95% air at 37°C.
Assessment of the viable number of cells
among cultured cells
The number of viable cells was assessed by CellTiter
Blue, a cell viability assay kit (Promega Corp., Madison, WI), with
fluorescence measurements at 570 nm for excitation and 590 nm for
fluorescence emission.
Morphology assessment
After trypsinization, cell suspensions were
sedimented and fixed on slide glasses using a Shandon Cytospin II
(Shandon, Pittsburgh, PA). Preparations were then stained with
May-Grünwald-Giemsa, and examined using a digital microscope
BZ-9000 (Keyence Co., Osaka, Japan).
Immunoblotting
Immunoblotting was performed as previously described
(9). Cells were lysed with RIPA
lysis buffer (Nacalai Tesque Inc., Kyoto, Japan) containing 1 mM
PMSF, 0.15 U/ml aprotinin, 10 mM EDTA, 10 mg/ml sodium fluoride,
and 2 mM sodium orthovanadate. Cellular proteins were quantified
using a DC Protein Assay Kit of Bio-Rad (Richmond, CA). Equal
amounts of proteins were loaded onto the gels, separated by
SDS-PAGE, and transferred onto Immobilon-P membrane (Millipore
Corp., Bedford, MA). The membranes were probed with first
antibodies (Abs) such as anti-LC3B Ab (Novus Bioloicals, Inc.,
Littleton, CO), anti-p62 mAb (sequestsome-1) (Santa Cruz, CA),
anti-phospho-JNK Ab (Thr183/Tyr185) (Cell Signaling Technology,
Danvers, MA), anti-cleaved caspase-3 Ab (Asp175) (Cell Signaling
Technology), anti-phospho-eIF2α Ab (Ser51) (Cell Signaling
Technology), and anti-GAPDH mAb (Santa Cruz). Immunoreactive
proteins were detected with horseradish peroxidase-conjugated
second Abs and an enhanced chemiluminescence reagent (ECL)
(Millipore). Densitometry was performed using a Molecular Imager,
ChemiDoc XRS System (Bio-Rad).
Gene expression analysis
Total RNA was isolated from cell pellets using
Isogen (Nippon Gene, Tokyo, Japan) and genomic DNA was removed
using RQ1 RNase-Free DNase (Promega) at 37°C for 30 min, followed
by extraction with phenol chloroform and ethanol precipitation.
Reverse-transcription using a PrimeScript RT Master Mix (Takara Bio
Inc. Ohtsu, Japan) was performed according to the manufacturer’s
instructions. Real-time PCR was performed on 3 ng of cDNA using
validated SYBR Green gene expression assays for human GRP78,
CHOP, GADD34 and p62 in combination with SYBR Premix Ex
Taq II (Takara Bio Inc.). The primer for p62 was: forward
5′-AGCTGCCTTGTACCCACATC-3′. Reverse 5′-CAGAG AAGCCCATGGACAG-3′. The
sequences of primes for GRP78, CHOP, GADD34 and GAPDH
were as we previously described (9).
Quantitative real-time PCR was performed in
duplicates in a Thermal Cycler Dice Real Time System TP800 (Takara
Bio Inc.) with the following conditions: initial cDNA denaturation
at 95°C for 30 sec, followed by 45 cycles of the sequence of
denaturation at 95°C for 5 sec and simultaneous annealing and
extension at 60°C for 30 sec. The data were analyzed using Thermal
Cycler Dice Real Time System Software (Takara Bio Inc.), and the
comparative Ct method
(2−ΔΔCt) was used
for relative quantification of gene expression. The data of
real-time PCR products were standardized to GAPDH as an
internal control. To confirm the specific amplification of target
genes, each gene product after real-time PCR was further separated
by 1.5% agarose gel to detect a single band at the theoretical
product size, as well as analysis of the dissociation curve for
detecting a single peak.
Electron microscopy
Electron microscopy was performed as previously
described (37). Transfection
of CHOP siRNA. For the gene silence of CHOP in MDA-MB231
cells, CHOP siRNA and control scramble siRNA, whose sequences are
described below, were diluted to a final concentration of 20 nM in
Opti-MEM I (Invitrogen, Paisley, UK), and transfection was
performed with cells at 50% confluency using Oligofectamine
transfection reagent (Invitrogen) according to the manufacturer’s
instructions. CHOP sense: CUGAUUGACCGAAUGGUGATT. CHOP antisense:
UCACCAUUCGGUCAAUCAGTT. Control sense: GACUACUGGUCGUUGAACUTT.
Control antisense: AGU UCAACGACCAGUAGUCTT.
Statistical analyses
The data are given as the mean ± SD. Statistical
analysis was performed by using Mann-Whitney’s U test
(two-tailed).
Results
Cell growth inhibition and apoptosis
induction after treatment with BZ in breast cancer cell lines
MDA-MB-231 and MDA-MB-468 cells for estrogen
receptor α-negative-breast cancer cell lines and MCF7 cells for an
estrogen receptor α-positive breast cancer cell line were cultured
in the presence of BZ at various concentrations. Viable numbers of
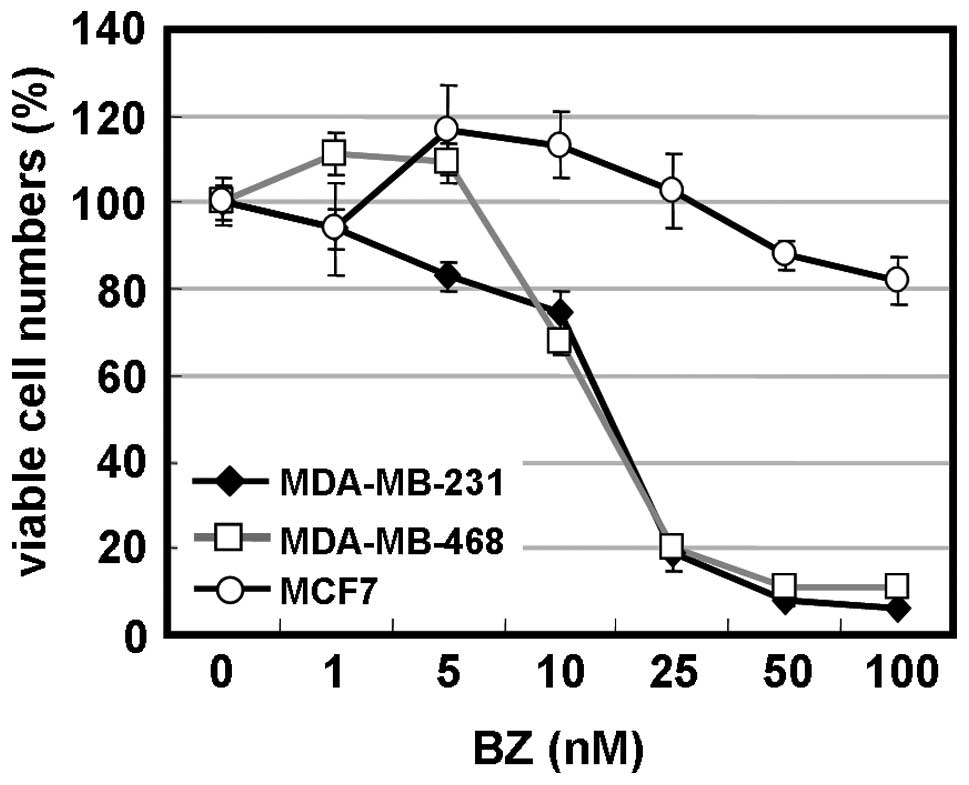
cells were assessed after 48-h treatment. As indicated in Fig. 1A, BZ at concentrations of >10 nM
exhibited potent cell growth inhibition of MDA-MB-231 and
MDA-MB-468 cells, but was less effective in MCF7 cells.
IC50 was 17 nM for MDA-MB-231 cells and 16 nM for
MDA-MB-468 cells, which were almost equivalent to myeloma cell
lines (9). Extended BZ-exposure
time to 96 h indicated 50% cell growth inhibition at 25 nM in MCF7
cells (data not shown). Morphological studies confirmed chromatin
condensation and nuclear fragments in MDA-MB-231 and MDA-MB-468
cells in response to BZ. However, some cells exhibited increased
vacuoles in cytoplasm. Immunoblotting revealed the cleavage of
caspase-3 after treatment with BZ (Fig. 1B and C). These data indicate that
BZ induces apoptosis in MDA-MB-231 and MDA-MB-468 cells, as
previously reported for various cell lines including myeloma
(9).
BZ induces autophagy in breast cancer
cell lines
We previously reported that BZ induces autophagy,
along with ER stress, in myeloma cells (9). LC3B exists in two cellular forms,
LC3B-I and LC3B-II. LC3B-I is converted to LC3B-II by conjugation
to phosphatidyl ethanolamine during the formation of
autophagosomes. Therefore, the amount of LC3B-II is a good early
marker of the formation of autophagosomes (24). Treatment with BZ induced increased
ratios of LC3B-II/LC3B-I in a dose-dependent manner, whereas a
significant reduction of p62 was observed after 76 h with BZ
(Fig. 2A and B). Additionally,
combined treatment with BZ and lysosomal inhibitors such as E-64d
and pepstatin A, which were used to block the catabolic flux of the
autophagic process, resulted in further enhancement of the
LC3B-II/LC3B-I ratio, compared with treatment with BZ or lysosomal
inhibitors alone (Fig. 2C). These
data indicate that BZ induces autophagy, in agreement with previous
reports on myeloma cells (9,24).
However, it is noteworthy that an increased expression of p62 with
25 nM BZ for a 48-h treatment was observed (Fig. 2A and B).
Combination of clarithromycin and BZ
enhances cytotoxicity and autophagy
It has been reported that CAM attenuates autophagy
in myeloma cells (26). Although
the mechanism still must be clarified, it is suggested that CAM may
inhibit the latter part of autophagic flux, such as autolysosome
formation and lysosomal hydrolysis (26). This results in an accumulation of
autophagosomes in cytoplasm. Our recent study also demonstrated
that combined treatment with BZ and bafilomycin A1
(BAF), which is a macrolide antibiotic that is used as an autophagy
inhibitor by blocking autolysosome formation, synergistically
enhanced the cytocidal effect in myeloma cell lines, compared with
treatment with BZ or BAF alone (9). We therefore investigated whether the
combination of BZ and CAM enhances cytotoxicity in breast cancer
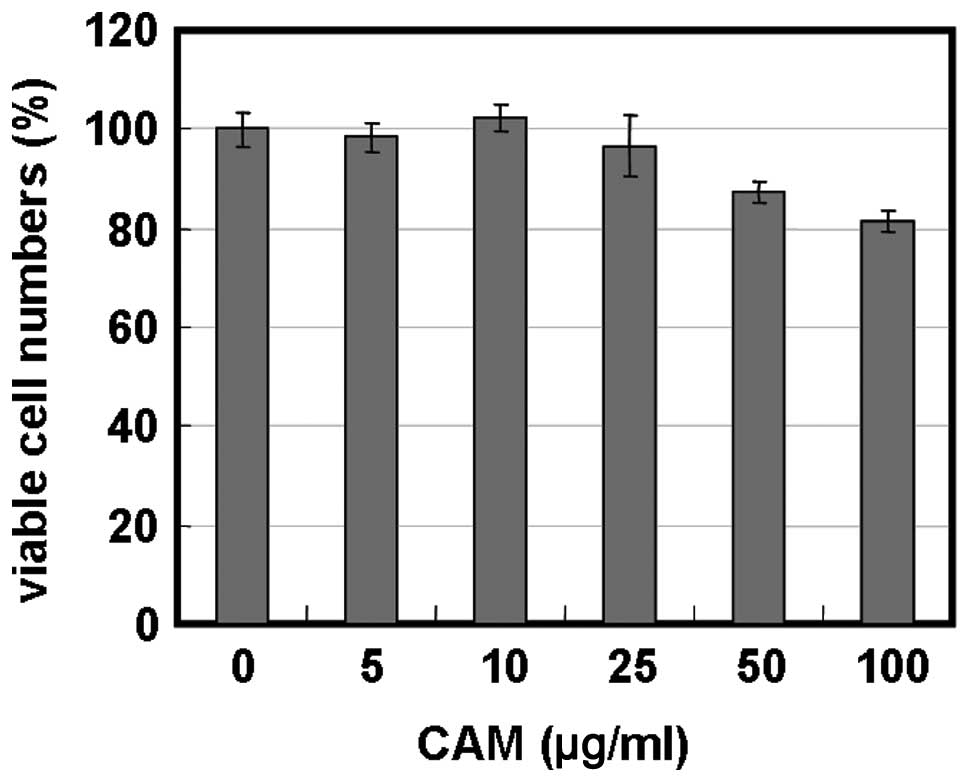
cells. Treatment of MDA-MB-231 cells with 5–100 μg/ml of CAM alone
resulted in little growth inhibition (Fig. 3A). However, treatment with BZ in
the presence of 25 and 50 μg/ml of CAM considerably enhanced cell
growth inhibition in MDA-MB-231 and MDA-MB-468 cells (Fig. 3A and B). Morphological studies with
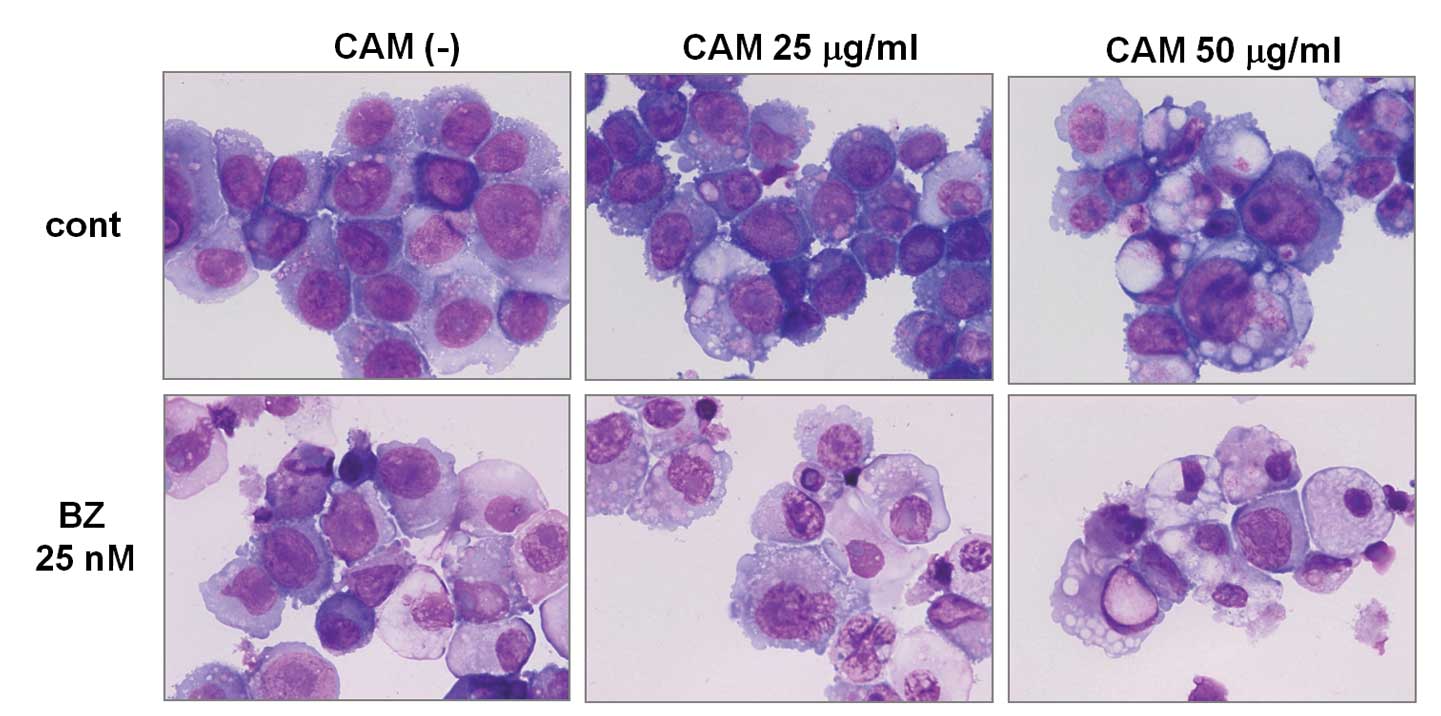
May-Grünwald-Giemsa staining revealed that CAM treatment resulted
in increased large vacuole formation in cytoplasm. Simultaneous
treatment with CAM and BZ further resulted in high levels of
vacuolization in cytoplasm, as well as nuclear chromatin
condensation in the majority of cells (Fig. 4A). Electron microscopy also
demonstrated that treatment with either BZ or CAM alone increased
the number of autophagosomes, and combined treatment with BZ and
CAM further increased the number of autophagosomes and
autolysosomes (Fig. 4B).
First, we examined whether the increased number of
auto-phagosomes in response to CAM was due to the blocking of the
catabolic process of autophagosomes or the inducement of autophagy
(26). CAM treatment increased the
ratio of LC3B-II to LC3B-I (Fig.
5A). Treatment with lysosomal inhibitors also increased this
ratio by blocking the catabolic flux of autophagy. However,
combined treatment with CAM and lysosomal inhibitors did not
further increase the LC3B-I/LC3B-II ratio, compared with treatment
with either CAM or lysosomal inhibitors alone (Fig. 5B). This result indicated that CAM
blocks autophagy flux, but does not induce autophagy (24).
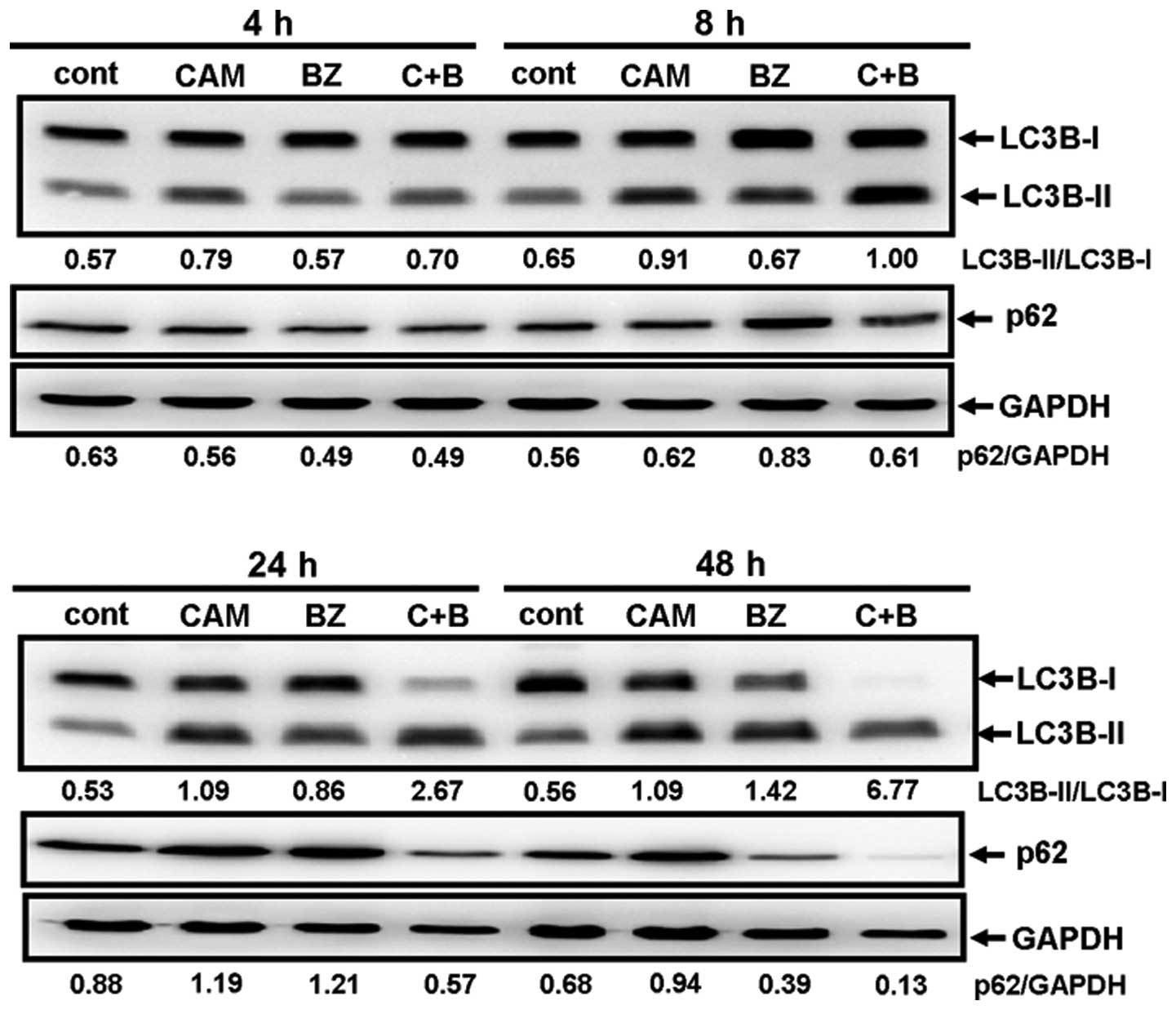
Next, MDA-MB-231 cells were cultured with/without
CAM in the presence/absence of BZ for various lengths of times and
the ratios of LC3B-I/LC3B-II, as well as the expression levels of
p62, were monitored. Although either CAM or BZ alone increased the
LC3B-I/LC3B-II ratios, combined treatment with BZ and CAM for 24–48
h further increased the ratios (Fig.
6). This result agrees well with the light microscopic and the
electron microscopic findings (Fig.
4). The pronounced clearance of p62 was also detected by
combined treatment with CAM and BZ for 48 h (Fig. 6). These data indicate that CAM plus
BZ enhances autophagy, compared with BZ alone.
Involvement of CHOP induction for
enhanced cytotoxicity by combined treatment with CAM and BZ
To clarify the underlying molecular mechanism for
cytotoxic enhancement by CAM plus BZ, we further investigated
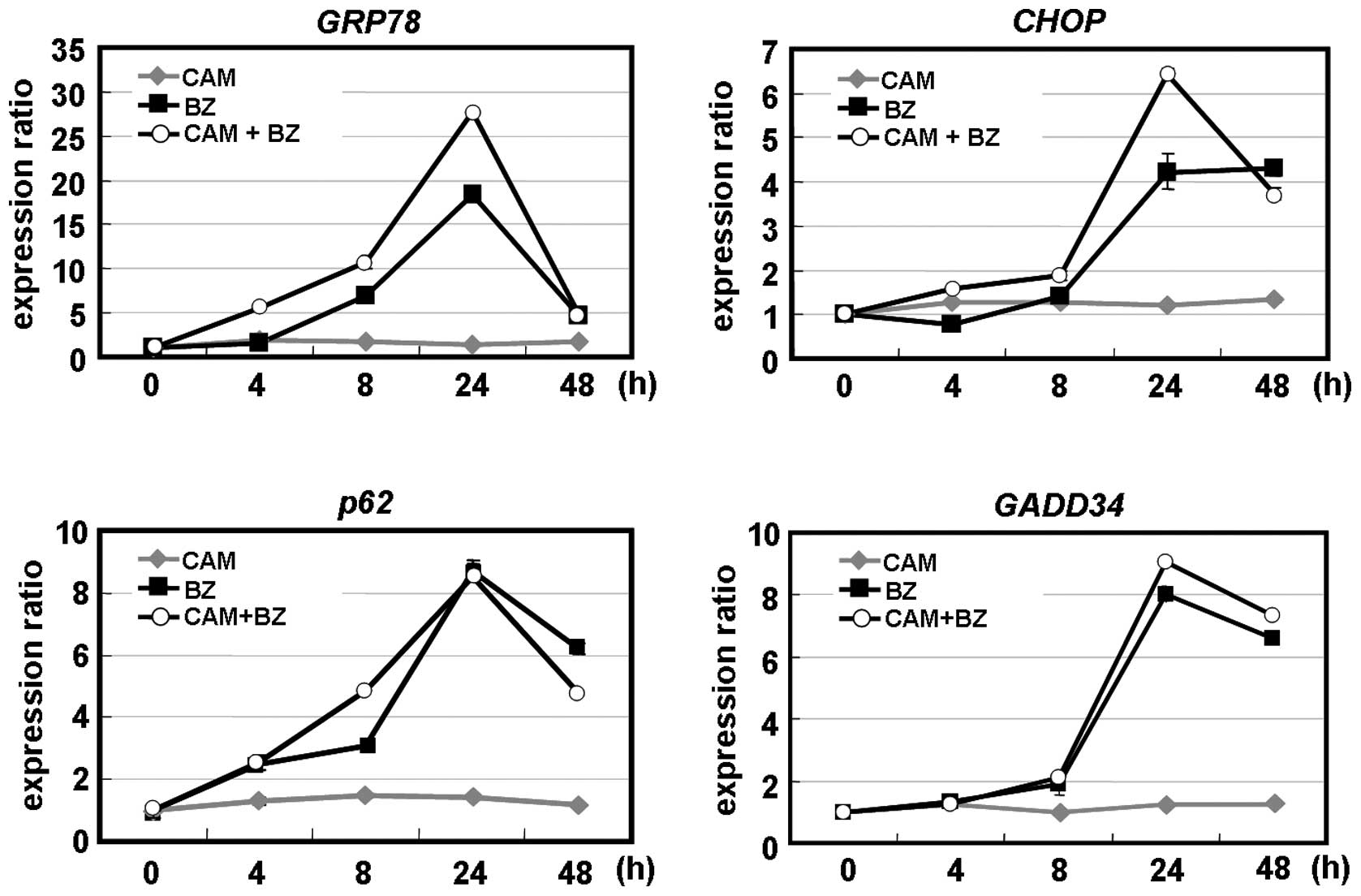
ER-stress induction. Real-time PCR indicated that the chaperone
protein GRP78 and the pro-apoptotic transcription factor CHOP. CAM
alone did not induce these ER-stress-mediated genes. However, the
combination of CAM and BZ enhanced GRP78 and CHOP after 24-h
treatment. An increased expression of GADD34, one of the genes
transcriptionally regulated by CHOP, was observed with the
combination of CAM and BZ (13).
Notably, BZ, but not CAM, induced p62 mRNA. BZ plus CAM did not
further increase p62 gene expression, compared with BZ alone
(Fig. 7A). This result appeared to
reflect the transient increase of p62 protein expression after 48-h
treatment with BZ (Fig. 2A and B),
because intracellular protein expression is determined by dynamic
equilibrium between synthesis and degradation. Immunoblottings
detected phosphorylation of JNK, as well as cleaved caspase-3,
after treatment with BZ alone and BZ plus CAM (Fig. 7B).
It has been suggested that ER-stress-mediated CHOP
induction exerts cytotoxicity of BZ in myeloma cells (7–10).
Transient knockdown for CHOP by siRNA in MDA-MB-231 cells appeared
less sensitive to BZ than control siRNA-treated cells (Fig. 8). Furthermore, CHOP−/−
MEF cells were more resistant to BZ than wild-type MEF cells
(Fig. 9). Combined treatment with
CAM and BZ resulted in pronounced cytotoxicity in wild-type MEF
cells, whereas this enhancement was completely cancelled in
CHOP−/− MEF cells. All these data suggest that
ER-stress-mediated CHOP induction was involved in the cytotoxic
effect of BZ in breast cancer cell lines. Additionally, enhanced
cytotoxicity by combining two reagents appeared to be due to
increased CHOP induction via UPR.
Discussion
In the present study, we demonstrated that combined
treatment with BZ and CAM enhanced cytotoxicity in breast cancer
cells (Fig. 3A and B). Treatment
with CAM alone had little effect on cell growth inhibition and UPR;
however, simultaneous treatment with BZ plus CAM enhanced UPR,
leading to induction of the transcription factor CHOP and the
chaperone protein GRP78 in MDA-MB-231 cells (Fig. 7). Notably, this enhancement was
completely cancelled in CHOP−/− MEF cells, whereas
wild-type MEF cells clearly exhibited pronounced cytotoxicity with
BZ plus CAM, as observed in MDA-MB-231 cells (Fig. 9). Therefore, it is strongly
suggested that CHOP is involved in enhanced cytotoxicity. It has
been reported that CHOP not only transcriptionally induces
pro-apoptotic proteins such as Bim, BAX, and DR5 but also
down-regulates the anti-apoptotic protein Bcl-2 (13,39).
Therefore, the profile of all downstream gene expressions in
response to CHOP appears to direct the cells to undergo apoptosis
(13,39).
Treatment of MDA-MB-231 cells with CAM increased
LC3B-II/LC3B-I ratios. In the presence of lysosomal inhibitors,
these ratios did not increase any further with the accumulation of
p62 (Fig. 4). These data suggest
that the increased number of autophagosomes in cytoplasm in
response to CAM, as observed in light and electron microscopy, is
caused by the accumulation of autophagosomes by blocking autophagy
flux, but not by the induction of autophagy. It has been reported
that treatment with CAM attenuates autophagy by blocking the late
phase of the autophagic process, probably after the fusion of
autophagosomes with lysosomes in myeloma cells (26). It has also been reported that, in
addition to proteasome-mediated degradation, the ubiquitinated
proteins are selectively degraded by autophagy via the docking
protein p62, which has both an LC3-interacting region and a
ubiquitin-binding domain (23).
Therefore, it makes sense that blocking two major proteolytic
pathways, such as the ubiquitin-proteasome system by BZ and the
autophagy-lysosome system by CAM, results in the accumulation of
unfolded proteins in ER and subsequently the induction of CHOP. We
have reported on a similar phenomenon in myeloma cells using BZ and
the autophagy inhibitor bafilomycin A1 (9). However, previous reports demonstrated
that inhibiting formation of autophagosome by either
3-methyladenine or siRNA for LC3 or knockout of atg5gene,
somewhat attenuated BZ-induced cytotoxicity in myeloma cells
(9,40). We cannot clearly explain this
discrepancy. All these treatments inhibit autophagosome formation,
whereas treatment with either CAM or bafilomycin A1
inhibits the late stage of autophagy, such as inhibiting
autolysosome formation or lysosomal hydrolysis (9,26).
Further studies will be required to assess the intracellular
loading volume for ER-stress by blocking autophagosome formation,
and by blocking autolysosome formation or lysosomal hydrolysis.
Recent reports demonstrated that the non-canonical autophagic
pathway does not require the entire set of autophagy-related (Atg)
proteins (41,42). Therefore, an alternative pathway to
form autolysosome may provide a bypass to avoid loading ER-stress,
even though the canonical autophagy pathway is blocked.
Some contradiction still exists regarding autophagy
induction in response to BZ. It has been reported that BZ blocks
the catabolic process of autophagy via a cathepsin-dependent
mechanism in estrogen receptor-positive breast cancer cells
(16), whereas other reports
demonstrated that BZ induces autophagy in myeloma cells, prostatic
cancer cells, endothelial cells, and breast cancer cells (9,15,40,43,44).
BZ has been reported to induce autophagy via proteasomal
stabilization of activating transcription factor 4 (ATF4) and
up-regulation of LC3B by ATF4, thus preventing BZ-induced cell
death in MCF7 breast cancer cells (15). ATF4 is a transcription factor
reported to be induced under severe hypoxia and a component of the
PERK pathway involved in the UPR (45). ATF4 facilitates autophagy through
direct binding to a cyclic AMP response element-binding site in the
LC3B promoter, resulting in LC3B up-regulation (45,46).
It has also been reported that ER-stress evokes up-regulation of
the transcriptional co-activator p8 and its target, the
pseudo-kinase tribbles homolog 3 (TRB3), and subsequently induced
autophagy via inhibition of the Akt/mTORC1 axis in human glioma
cells (47). These data indicate a
direct signaling pathway from ER-stress to autophagy induction.
Therefore, autophagy induction in response to BZ may occur via
ER-stress-mediated AFT4 induction. In addition, enhanced autophagy
induction by combined treatment with CAM and BZ might be explained
by ATF4 mediate autophagy during ER-stress loading. A similar
phenomenon was previously reported regarding enhanced cytotoxicity
by the combination of BZ and an inhibitor for the
autophagy-lysosome pathway in myeloma cells (48). The microtubule-organizing center
(MTOC) transports unfolded proteins to lysosomes and are degraded
through the autophagy-lysosome pathway. Histone deacetylase 6
(HDAC6) deacetylates α-tubulin, which is thought to be a component
of the MTOC. HDAC6 knockdown results in decreased LC3B protein and
reduces autophagy (49). Tubacin,
a small molecule inhibitor of HDAC6, prevents deacetylation of
α-tubulin and produces accumulation of polyubiquitinated proteins
and apoptosis, and further acts synergistically with BZ to induce
cytotoxicity in multiple myeloma (48). Based on our results presented here,
these data also can be explained by enhanced loading on ER-stress
by simultaneously targeting the autophagy-lysosome pathway by
tubacin and the ubiquitin-protease pathway by BZ.
One may ask whether the enhanced cytotoxicity
archived by combining BZ and CAM is simply due to the
pharmacointeraction between CYP3A4 and CAM. CAM is known to inhibit
CYP3A4, the isoenzyme responsible for metabolizing BZ (50,51).
Therefore, concomitant administration of BZ with CAM may lead to
intracellular elevation of BZ concentrations. We cannot completely
exclude this possibility. However, it is unlikely because combined
treatment with CAM and either docetaxel or paclitaxel, both of
which are chemotherapeutic reagents for breast cancer and are
metabolized by CYP3A4 as well as BZ, did not result in any enhanced
cytotoxicity in MDA-MB-231 cells, compared with treatment with each
reagent alone (data not shown).
As indicated in Figs.
2 and 7A, p62 was transiently
increased in response to BZ after 24–48-h treatment. P62 is a
stress response protein that is strongly induced at the mRNA and
protein levels by exposure to oxidants, sodium arsenite, cadmium,
ionophores, and proteasome inhibitors (52). Interestingly, p62 is over-abundant
in malignant breast tissue, compared with normal breast tissue
(53). The proteasome inhibitor
PSI has been reported to increase p62 mRNA and protein as well as
BZ does (52). Since the
intracellular protein level is determined by dynamic equilibrium
between protein synthesis and degradation, a transient increase in
response to BZ appears to reflect the predominance of p62 synthesis
at that time-point. However, abundance of intracellular p62 in
breast cancer cells, which may be caused by some intrinsic stress
response, may determine sensitivity to BZ (53). We are now in the process of
clarifying the molecular mechanism in order to explain why MCF7
cells are less sensitive to BZ than MDA-MB-231 and MDA-MB-468 cells
(Fig. 1). This will identify the
determinant factor for indicating BZ-therapy in breast cancer
patients.
Considering all the data, ER-stress-mediated CHOP
appears to be involved in the cytotoxicity of BZ in breast cancer
cells. Combining BZ and CAM is suggested to be a promising option
for improving the therapeutic outcome of breast cancer therapy.
Additionally, simultaneously targeting the ubiquitin-proteasome
pathway and the autophagy-lysosome pathway appears to be a rational
strategy to enhance chemo-sensitivity and overcome chemo-resistance
in cancer therapy.
Acknowledgments
This study was supported in part by funds from the
Private University Strategic Research-Based Support Project
(Molecular Information-based Intractable Disease Research Project)
from the Ministry of Education, Culture, Sports, Science and
Technology of Japan to A.T. and K.M. (2008–2012), and a
Grant-in-Aid for Scientific Research (C) from the Ministry of
Education, Culture, Sports, Science and Technology of Japan to K.M.
(#22591050).
References
|
1
|
Burstein HJ: Novel agents and future
directions for refractory breast cancer. Semin Oncol. 38(Suppl 2):
S17–S24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Higgins MJ and Stearns V: Pharmacogenetics
of endocrine therapy for breast cancer. Annu Rev Med. 62:281–293.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al Saleh S, Sharaf LH and Luqmani YA:
Signalling pathways involved in endocrine resistance in breast
cancer and associations with epithelial to mesenchymal transition.
Int J Oncol. 38:1197–1217. 2011.PubMed/NCBI
|
|
4
|
Goldhirsch A, Wood WC, Gelber RD, Coates
AS, Thürlimann B and Senn HJ; 10th St. Gallen conference. Progress
and promise: highlights of the international expert consensus on
the primary therapy of early breast cancer 2007. Ann Oncol.
18:1133–1144. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laubach J, Richardson P and Anderson K:
Multiple myeloma. Annu Rev Med. 62:249–264. 2011. View Article : Google Scholar
|
|
6
|
Obeng EA, Carlson LM, Gutman DM,
Harrington WJ Jr, Lee KP and Boise LH: Proteasome inhibitors induce
a terminal unfolded protein response in multiple myeloma cells.
Blood. 107:4907–4916. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meister S, Schubert U, Neubert K, Herrmann
K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann
M, Jäck HM and Voll RE: Extensive immunoglobulin production
sensitizes myeloma cells for proteasome inhibition. Cancer Res.
67:1783–1792. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fels DR, Ye J, Segan AT, Kridel SJ,
Spiotto M, Olson M, Koong AC and Koumenis C: Preferential
cytotoxicity of bortezomib toward hypoxic tumor cells via
overactivation of endoplasmic reticulum stress pathways. Cancer
Res. 68:9323–9330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kawaguchi T, Miyazawa K, Moriya S, Ohtomo
T, Che XF, Naito M, Itoh M and Tomoda A: Combined treatment with
bortezomib plus bafilomycin A1 enhances the cytocidal effect and
induces endoplasmic reticulum stress in U266 myeloma cells:
crosstalk among proteasome, autophagy-lysosome and ER stress. Int J
Oncol. 38:643–654. 2011.
|
|
10
|
Ri M, Iida S, Nakashima T, Miyazaki H,
Mori F, Ito A, Inagaki A, Kusumoto S, Ishida T, Komatsu H, Shiotsu
Y and Ueda R: Bortezomib-resistant myeloma cell lines: a role for
mutated PSMB5 in preventing the accumulation of unfolded proteins
and fatal ER stress. Leukemia. 24:1506–1512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Herr I and Debatin K-M: Cellular stress
response and apoptosis in cancer therapy. Blood. 98:2603–2613.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verfaillie T, Salazar M, Velasco G and
Agostinis P: Linking ER stress to autophagy: potential implications
for cancer therapy. Int J Cell Biol. 2010:1–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cardoso F, Durbecq V, Laes JF, Badran B,
Lagneaux L, Bex F, Desmedt C, Willard-Gallo K, Ross JS, Burny A,
Piccart M and Sotiriou C: Bortezomib (PS-341, Velcade) increases
the efficacy of trastuzumab (Herceptin) in HER-2-positive breast
cancer cells in a synergistic manner. Mol Cancer Ther. 5:3042–3051.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Milani M, Rzymski T, Mellor HR, Pike L,
Bottini A, Generali D and Harris AL: The role of ATF4 stabilization
and autophagy in resistance of breast cancer cells treated with
Bortezomib. Cancer Res. 69:4415–4423. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Periyasamy-Thandavan S, Jackson WH,
Samaddar JS, Erickson B, Barrett JR, Raney L, Gopal E, Ganapathy V,
Hill WD, Bhalla KN and Schoenlein PV: Bortezomib blocks the
catabolic process of autophagy via a cathepsin-dependent mechanism,
affects endoplasmic reticulum stress and induces caspase-dependent
cell death in antiestrogen-sensitive and resistant ER+
breast cancer cells. Autophagy. 6:19–35. 2010. View Article : Google Scholar
|
|
17
|
Engel RH, Brown JA, Von Roenn JH, O’Regan
RM, Bergan R, Badve S, Rademaker A and Gradishar WJ: A phase II
study of single agent bortezomib in patients with metastatic breast
cancer: a single institution experience. Cancer Invest. 25:733–737.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ishii Y, Papa L, Bahadur U, Yue Z,
Aguirre-Ghiso J, Shioda T, Waxman S and Germain D: Bortezomib
enhances the efficacy of fulvestrant by amplifying the aggregation
of the estrogen receptor, which leads to a proapoptotic unfolded
protein response. Clin Cancer Res. 17:2292–2300. 2011. View Article : Google Scholar
|
|
19
|
Jones MD, Liu JC, Barthel TK, Hussain S,
Lovria E, Cheng D, Schoonmaker JA, Mulay S, Ayers DC, Bouxsein ML,
Stein GS, Mukherjee S and Lian JB: A proteasome inhibitor,
bortezomib, inhibits breast cancer growth and reduces osteolysis by
down-regulating metastatic genes. Clin Cancer Res. 16:4978–4989.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Janku F, McConkey DJ, Hong DS and Kurzrock
R: Autophagy as a target for anticancer therapy. Nat Rev Clin
Oncol. 8:528–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Korolchuk VI, Menzies FM and Rubinsztein
DC: Mechanisms of cross-talk between the ubiquitin-proteasome and
autophagy-lysosome systems. FEBS Lett. 584:1393–1398. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kirkin V, McEwan DG, Novak I and Dikic I:
A role for ubiquitin in selective autophagy. Mol Cell. 34:259–269.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mizushima N and Yoshimori T: How to
interpret LC3 immuno-blotting. Autophagy. 3:542–545. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yamamoto A, Tagawa Y, Yoshimori T,
Moriyama Y, Masaki R and Tashiro Y: Bafilomycin A1 prevents
maturation of autophagic vacuoles by inhibiting fusion between
autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E
cells. Cell Struct Funct. 123:33–42. 1998. View Article : Google Scholar
|
|
26
|
Nakamura M, Kikukawa Y, Takeya M, Mitsuya
H and Hata H: Clarithromycin attenuates autophagy in myeloma cells.
Int J Oncol. 37:815–820. 2010.PubMed/NCBI
|
|
27
|
Gay F, Rajkumar SV, Coleman M, Kumar S,
Mark T, Dispenzieri A, Pearse R, Gertz MA, Leonard J, Lacy MQ,
Chen-Kiang S, Roy V, Jayabalan DS, Lust JA, Witzig TE, Fonseca R,
Kyle RA, Greipp PR, Stewart AK and Niesvizky R: Clarithromycin
(Biaxin)-lenalidomide-low-dose dexamethasone (BiRd) versus
lenalidomide-low-dose dexamethasone (Rd) for newly diagnosed
myeloma. Am J Hematol. 85:664–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Niesvizky R, Jayabalan DS, Christos PJ,
Furst JR, Naib T, Ely S, Jalbrzikowski J, Pearse RN, Zafar F, Pekle
K, Larow A, Lent R, Mark T, Cho HJ, Shore T, Tepler J, Harpel J,
Schuster MW, Mathew S, Leonard JP, Mazumdar M, Chen-Kiang S and
Coleman M: BiRD (Biaxin [clarithromycin]/Revlimid
[lenalidomide]/dexamethasone) combination therapy results in high
complete- and overall-response rates in treatment-naive symptomatic
multiple myeloma. Blood. 111:1101–1109. 2008.
|
|
29
|
Govi S, Dognini GP, Licata G, Crocchiolo
R, Resti AG, Ponzoni M and Ferreri AJ: Six-month oral
clarithromycin regimen is safe and active in extranodal marginal
zone B-cell lymphomas: final results of a single-centre phase II
trial. Br J Haematol. 50:226–229. 2010.PubMed/NCBI
|
|
30
|
Ohe M and Hashino S: Successful treatment
with clarithromycin for mixed phenotype acute leukemia, T/myeloid,
NOS (in Japanese). Rinsho Ketsueki. 51:297–299. 2010.PubMed/NCBI
|
|
31
|
Mikasa K, Sawaki M, Kita E, Hamada K,
Teramoto S, Sakamoto M, Maeda K, Konishi M and Narita N:
Significant survival benefit to patients with advanced
non-small-cell lung cancer from treatment with clarithromycin.
Chemotherapy. 43:288–296. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wada T, Sata M, Sato J, Tokairin Y,
Machiya J, Hirama N, Arao T, Inoue S, Takabatake N, Shibata Y and
Kubota I: Clarithromycin suppresses invasiveness of human lung
adenocarcinoma cells. Chemotherapy. 53:77–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yatsunami J, Fukuno Y, Nagata M, Tsuruta
N, Aoki S, Tominaga M, Kawashima M, Taniguchi S and Hayashi S:
Roxithromycin and clarithromycin, 14-membered ring macrolides,
potentiate the antitumor activity of cytotoxic agents against mouse
B16 melanoma cells. Cancer Lett. 147:17–24. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hamada K, Kita E, Sawaki M, Mikasa K and
Narita N: Antitumor effect of erythromycin in mice. Chemotherapy.
41:59–69. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Malfertheiner P, Megraud F, O’Morain C,
Bazzoli F, El-Omar E, Graham D, Hunt R, Rokkas T, Vakil N and
Kuipers EJ: Current concepts in the management of Helicobacter
pylori infection: the Maastricht III Consensus Report. Gut.
56:772–781. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ohara T, Morishita T, Suzuki H, Masaoka T,
Ishii H and Hibi T: Antibiotics directly induce apoptosis in B cell
lymphoma cells derived from BALB/c mice. Anticancer Res.
24:3723–3730. 2004.PubMed/NCBI
|
|
37
|
Ohtomo T, Miyazawa K, Naito M, Moriya S,
Kuroda M, Itoh M and Tomoda A: Cytoprotective effect of imatinib
mesylate in non-BCR-ABL-expressing cells along with autophagosome
formation. Biochem Biophys Res Commun. 391:310–315. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moriya S, Miyazawa K, Kawaguchi T, Che XF
and Tomoda A: Involvement of endoplasmic reticulum stress-mediated
CHOP (GADD153) induction in the cytotoxicity of
2-aminophenoxazine-3-one in cancer cells. Int J Oncol. 39:981–988.
2011.PubMed/NCBI
|
|
39
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hoang B, Benavides A, Shi Y, Frost P and
Lichtenstein A: Effect of autophagy on multiple myeloma cell
viability. Mol Cancer Ther. 8:1974–1984. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nishida Y, Arakawa S, Fujitani K,
Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y
and Shimizu S: Discovery of Atg5/Atg7-independent alternative
macroautophagy. Nature. 461:654–658. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shimizu S, Arakawa S and Nishida Y:
Autophagy takes an alternative pathway. Autophagy. 6:290–291. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Belloni D, Veschini L, Foglieni C,
Dell’Antonio G, Caligaris-Cappio F, Ferrarini M and Ferrero E:
Bortezomib induces autophagic death in proliferating human
endothelial cells. Exp Cell Res. 316:1010–1018. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhu K, Dunner K Jr and McConkey DJ:
Proteasome inhibitors activate autophagy as a cytoprotective
response in human prostate cancer cells. Oncogene. 29:451–462.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rzymski T, Milani M, Singleton DC and
Harris AL: Role of ATF4 in regulation of autophagy and resistance
to drugs and hypoxia. Cell Cycle. 8:3838–3847. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rzymski T, Milani M, Pike L, Buffa F,
Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I and Harris
AL: Regulation of autophagy by ATF4 in response to severe hypoxia.
Oncogene. 29:4424–4435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Salazar M, Carracedo A, Salanueva IJ,
Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C,
Torres S, García S, Nowak J, Fimia GM, Piacentini M, Cecconi F,
Pandolfi PP, González-Feria L, Iovanna JL, Guzmán M, Boya P and
Velasco G: Cannabinoid action induces autophagy-mediated cell death
through stimulation of ER stress in human glioma cells. J Clin
Invest. 119:1359–1372. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Hideshima T, Bradner JE, Wong J, Chauhan
D, Richardson P, Schreiber SL and Anderson KC: Small-molecule
inhibition of proteasome and aggresome function induces synergistic
antitumor activity in multiple myeloma. Proc Natl Acad Sci USA.
102:8567–8572. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lee JY, Koga H, Kawaguchi Y, Tang W, Wong
E, Gao YS, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo
AM and Yao TP: HDAC6 controls autophagosome maturation essential
for ubiquitin-selective quality-control autophagy. EMBO J.
29:969–980. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Iwamoto T, Ishibashi M, Fujieda A, Masuya
M, Katayama N and Okuda M: Drug interaction between itraconazole
and bortezomib: exacerbation of peripheral neuropathy and
thrombocytopenia induced by bortezomib. Pharmacotherapy.
30:661–665. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Suzuki A, Iida I, Hirota M, Akimoto M,
Higuchi S, Suwa T, Tani M, Ishizaki T and Chiba K: CYP isoforms
involved in the metabolism of clarithromycin in vitro: comparison
between the identification from disappearance rate and that from
formation rate of metabolites. Drug Metab Pharmacokinet.
18:104–113. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jain A, Lamark T, Sjøttem E, Larsen KB,
Awuh JA, Øvervatn A, McMahon M, Hayes JD and Johansen T: p62/SQSTM1
is a target gene for transcription factor NRF2 and creates a
positive feedback loop by inducing antioxidant response
element-driven gene transcription. J Biol Chem. 285:22576–22591.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Thompson HG, Harris JW, Wold BJ, Lin F and
Brody JP: p62 overexpression in breast tumors and regulation by
prostate-derived Ets factor in breast cancer cells. Oncogene.
22:2322–2333. 2003. View Article : Google Scholar : PubMed/NCBI
|