Introduction
Colon cancer is the second most prevalent cancer and
the third leading cause of cancer deaths worldwide, resulting in
almost half a million deaths every year (1,2).
Surgical resection remains the only curative treatment for
colorectal cancer, but the outcome is not always satisfactory. Only
70% of colorectal cancers are resectable, and 75% of the resectable
cancers are curable. Many patients require adjuvant chemotherapy
(3). Approximately 70–90% of colon
cancers seem to be associated with dietary habits; therefore, their
is interest in dietary factors that can exert cancer
chemopreventive/chemotherapeutic action against colon cancer cells
(4).
Resveratrol (Res;
3,4′,5-trihydroxy-trans-stilbene) is a natural polyphenolic
product and a phytoalexin produced by grapes, mulberries, and
peanuts, and it is widely present in red wine and other
constituents of the human diet. Res has various biological
activities, including anti-inflammatory, antifungal, antimutagenic
and anticancer effects (5). In
this respect, Res may be a potential anticancer agent in the humans
(6). Res is not overtly toxic to
animals when administered at doses high enough to achieve a
pharmacological effect (7), and is
gaining acceptance as a potential antitumor agent because of its
pleiotropic effects through many intracellular signaling pathways
(6,8). Res inhibits the growth of cancer cell
lines derived from various origins, and this effect was associated
with cell-cycle arrest and the induction of apoptosis (9–12).
Autophagy is an evolutionarily conserved catabolic
process of degrading damaged proteins and/or organelles and
recycling the materials to maintain the quality of cellular
components (13). During
autophagy, autophagosomes are formed by elongation of double
membrane-bound vesicles, and they sequester cytoplasmic
constituents. Subsequently, autophagosomes fuse with lysosomes to
form autolysosomes in which the incorporated organelles are
degraded. Autophagy also plays a role in human diseases including
cancer. Emerging evidence indicates that chemotherapeutic agents
induce autophagy in various types of cancer cells (14). On one hand, autophagy exerts a cell
protective role that allows cells to survive against cytotoxic
agents. On the other hand, autophagy results in cell death termed
autophagic cell death or type II cell death (15,16).
Recently, Res was shown to induce autophagy in several cancer cell
lines (17–20). However, the ability of Res to
induce autophagy and the role of autophagy in the production of a
cell death signal or a cell survival signal in human colon cancer
cells are largely unknown.
Several studies indicate that reactive oxygen
species (ROS) production may mediate apoptosis and/or autophagy
induction in several types of cancer cells (21–23).
The relationship between ROS, autophagy, and apoptosis induced by
Res in human colon cancer cells is still undefined. In the present
study, we demonstrated the Res-induced cytotoxic effect in human
colon cancer cells. A possible molecular mechanism involved is
Caspase-8/Caspase-3-dependent apoptosis via ROS-triggered
autophagy.
Materials and methods
Cell culture
A human colon cancer cell line, HT-29, was kindly
provided by Dr Y. Katakura (Faculty of Agriculture, Kyusyu
University, Fukuoka, Japan), and COLO 201 was obtained from the
Japanese Cancer Research Resource Bank. HT-29 and COLO 201 cells
were grown in DMEM and RPMI-1640, respectively, supplemented with
10% fetal bovine serum, and maintained at 37°C in a humidified
atmosphere containing 5% CO2.
Cell proliferation
Proliferation of HT-29 and COLO 201 cells was
determined by using the 3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyltetrazolium bromide (MTT, Sigma, St. Louis, MO, USA)
assay and the 2-(2-methoxy-4-nitophenyl)-3-(4-nitrophenyl)-5-(2,
4-disulfophenyl-2H-thetra-zolium, monosodium salt (WST-8, Wako
Chemical, Osaka, Japan) assay, respectively. Cells were seeded into
96-well plates at a concentration of 5×103 HT-29
cells/well and 2×103 COLO 201 cells/well. After 24 h of
incubation, the cells were treated with 5 different concentrations
(25, 50, 75, 100 and 150 μM) of Res (Sigma). The stock solution of
Res (200 mM) was prepared by using dimethyl sulfoxide (DMSO) as the
solvent. The absorbance was read at 540 nm for MTT (HT-29 cells)
and 450 nm for WST-8 (COLO 201) by using an iMark microplate reader
(Bio-Rad, Hercules, CA, USA).
Soft agar colony formation assay
A soft agar colony formation assay was performed as
described previously (24). First,
0.5% agarose in growth medium was added to six-well plates and
allowed to solidify. Then, 1×104 cells were plated
triplicate in 0.3% agarose and were added to each well. The cells
were incubated at 37°C in a 5% CO2 atmosphere for 13
days. Fresh growth medium (0.5 ml/well) was added after 1 week of
incubation. At the end of the incubation period, colonies were
stained with 0.005% crystal violet for 1 h and photographed.
Colonies were counted by using the Image J imaging software
developed at NIH.
Western blot analysis
Western blots were performed as described previously
(24). The following primary
antibodies were used: anti-poly(ADP-ribose) polymerase (PARP,
polyclonal) antibody, anti-Caspase-8 (1C12) antibody, anti-cleaved
Caspase-3 (Asp175) (5A1E) antibody, anti-Bcl-xL (54H6) antibody and
anti-Bax (polyclonal) antibody (Cell Signaling Technology, Beverly,
MA, USA); anti-microtubule-associated protein 1 light chain 3 (LC3,
polyclonal) antibody (Abgent, San Diego, CA, USA); and
HRP-conjugated anti-actin (polyclonal) antibody (Santa Cruz
Biotechnology, Santa Cruz, CA, USA).
Electron microscopy
The ultrastructure of HT-29 cells after 150 μM Res
treatment was determined by electron microscopy. After treatment
with DMSO or 150 μM Res for 24 h, the cells were collected by
centrifugation, washed with PBS, fixed in ice-cold Karnovsky’s
solution (4% paraformaldehyde, 5% glutaraldehyde and 30 mM
cacodylate buffer) at 4°C for 1 h, washed with 0.1 M cacodylate
buffer several times, and incubated at 4°C overnight. The cells
were then post-fixed in 2% osmium tetroxide at 4°C for 1 h,
resuspended in 1% sodium alginate, collected by centrifugation, and
gelated by adding 1 M calcium chloride. The gelated cells were
dehydrated through a graded series of ethanol (50–100%) and
propylene oxide and then processed for epon embedding. Semi-thin
sections were stained with toluidine blue, and representative areas
were chosen for ultrathin sectioning. Ultrathin sections were
stained with uranyl acetate and lead citrate and examined with a
JEM-1011 electron microscope (JEOL, Tokyo, Japan).
Immunofluorescence
Immunofluorescence detection for LC3 was performed
as described previously (25).
HT-29 cells were grown on a 35-mm glass-bottom dish (Matsunami
Glass, Osaka, Japan), treated with DMSO or 150 μM Res for 24 h,
washed with PBS, and fixed in 10% neutral buffered formalin for 30
min at room temperature. The cells were washed in Tris-buffer (TBS)
and blocked in TBS containing 5% bovine serum albumin at room
temperature for 30 min. Cells were subsequently incubated overnight
with anti-LC3 antibody (Cell Signaling), washed several times and
then incubated with secondary antibody (Alexa Fluor 488 anti-rabbit
IgG; Molecular Probes, Eugene, OR, USA) for 1 h. The cells were
then counterstained with 4′6-diamidino-2-phenylindole (Dojindo
Laboratories, Kumamoto, Japan) for 5 min. A laser scanning
microscope (LSM510-META, Zeiss, Jena, Germany) was used to collect
images of the cells.
Apoptosis analysis
Apoptosis was analyzed by flow cytometry with
Annexin V (Becton Dickinson, Franklin Lakes, NJ, USA) and propidium
iodide (PI). A total of 4×105 cells were seeded in each
50-mm culture dish. Twenty-four hours later, 150 μM of Res with the
autophagy inhibitor 3-methyladenine (3-MA, Sigma) or the
pan-caspase inhibitor Z-VAD (OMe)-FMK (Z-VAD, Enzo Life Sciences,
Plymouth Meeting, PA, USA) were added. The cells were trypsinized,
washed in cold PBS, and resuspended in 1X binding buffer at a
concentration of 1×106 cells/ml. Annexin V and PI
solution were added to the cell preparations and incubated for 15
min in the dark at room temperature. Binding buffer was then added
to each tube, and the samples were analyzed by using a FACSCalibur
flow cytometer (Becton Dickinson) and CellQuest software (Becton
Dickinson). For each sample, 10,000 cells were analysed.
Flow cytometric methods for determination
of total ROS
For determination of intracellular ROS levels, cells
were grown in 6-well plates and treated with 150 μM Res for 24 to
72 h with or without ROS scavenger N-acetyl cysteine (NAC, Sigma).
Cells were incubated with 5 μm
5-(and-6)-chloromethyl-2′,7′-dichlorodihydro-fluorescein diacetate,
acetyl ester (CM-DCHF-DA, Invitrogen, Carlsbad, CA, USA), which is
cleaved by intracellular esterases and transformed into a
fluorescent dye when oxidized at 37°C for 30 min. The samples were
analyzed by using a FACSCalibur flow cytometer and CellQuest
software. For each sample, 10,000 cells were analyzed.
Statistical analysis
All discrete values, expressed as mean ± SD, were
analyzed with the Student’s t-test. P-values less than 0.05 and
0.01 were considered to be significant and highly significant,
respectively.
Results
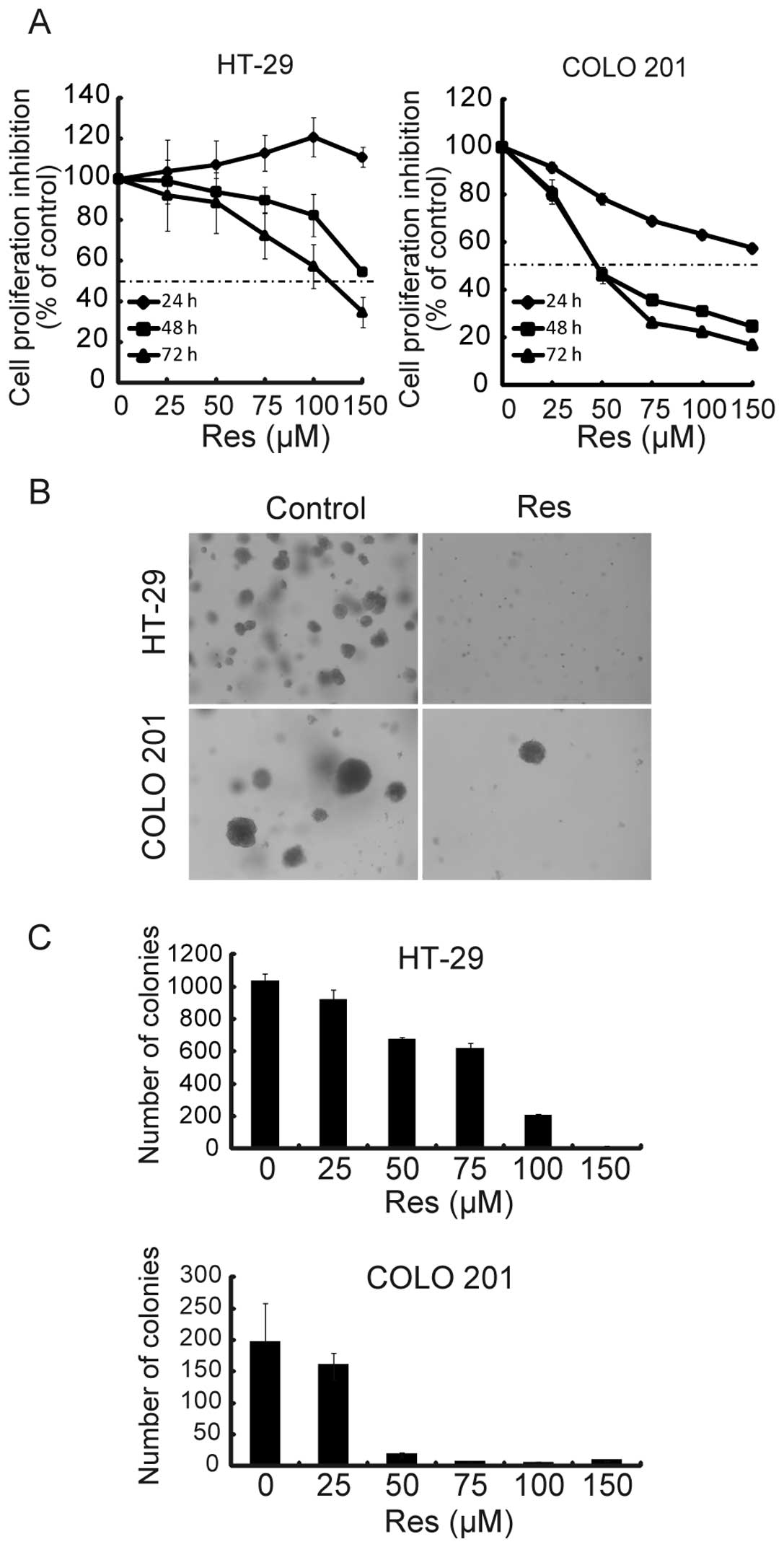
Res inhibits anchorage-dependent and
-independent growth of HT-29 and COLO 201 cells
To evaluate the effect of Res on cell proliferation,
HT-29 and COLO 201 cells were treated with 5 different
concentrations (25, 50, 75, 100 and 150 μM) of Res for up to 72 h.
Res induced growth inhibition in a dose- and time-dependent manner
(Fig. 1A). The half maximal
inhibitory concentration (IC50) against HT-29 and COLO
201 cells after a 72-h treatment was 115.9 and 47.3 μM,
respectively. To ascertain whether Res affected
anchorage-independent growth, we assessed the ability of
Res-treated cells to form colonies in soft agar. As shown in
Fig. 1B and C, Res significantly
reduced the number of colonies compared with the untreated cells in
a dose-dependent manner. For the following studies, Res doses
greater than the IC50 for 72 h were chosen (HT-29, 150
μM; COLO 201, 75 μM).
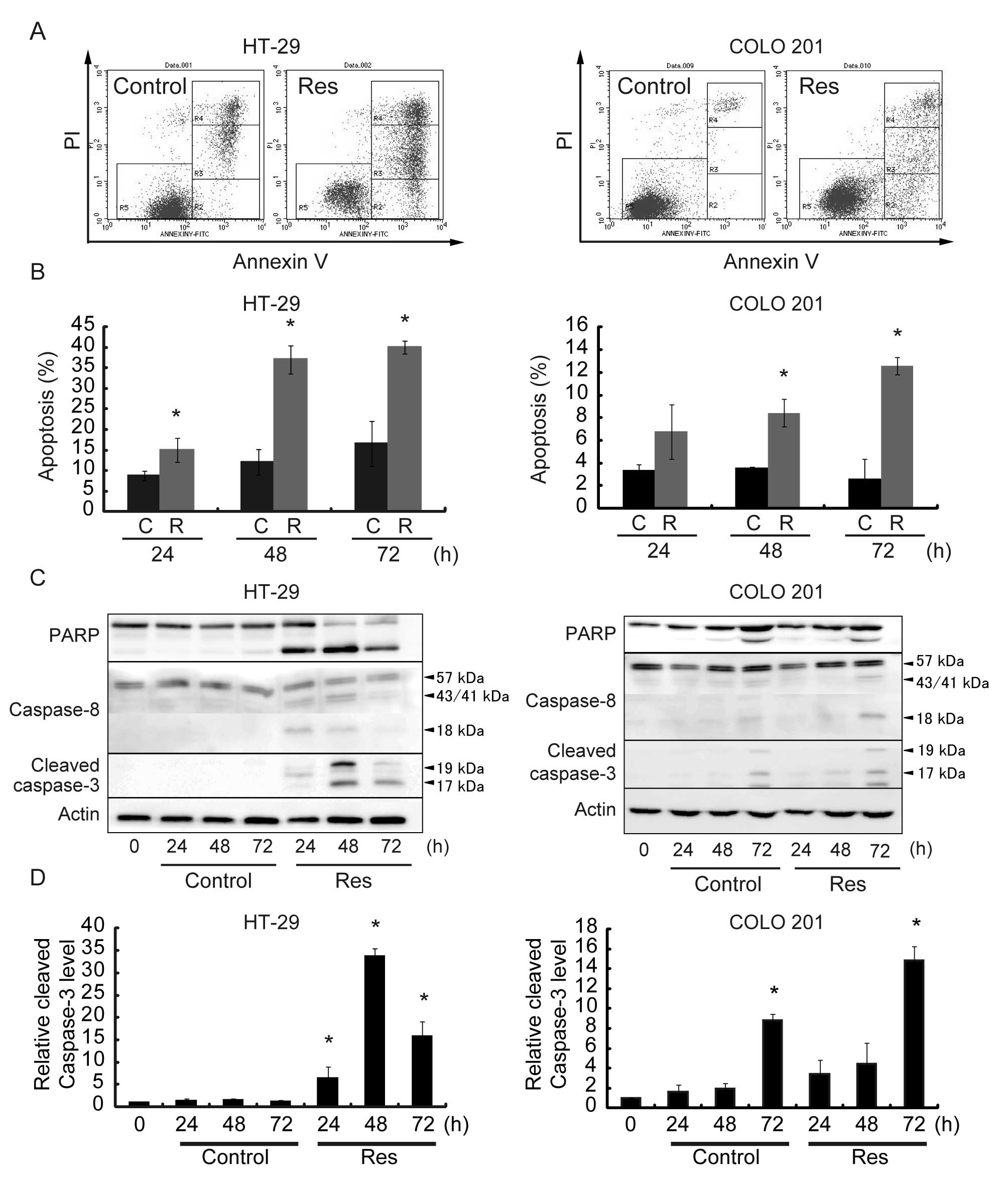
Res induces apoptosis in HT-29 and COLO
201 cells
An Annexin V assay showed that Res induced apoptotic
cell death in a time-dependent manner. Representative results are
shown in Fig. 2A, and quantitative
data from 3 different experiments are summarized in Fig. 2B. The percentage of apoptotic cells
was ~40% in HT-29 cells and ~12% in COLO 201 cells after a 72-h Res
treatment. We then examined changes in the protein levels of
apoptosis-related molecules. Representative results are shown in
Fig. 2C, and quantitative data
from 3 different experiments are summarized in Fig. 2D. Cells treated with Res exhibited
an increase in the levels of PARP, Caspase-8 and cleaved Caspase-3.
The levels of PARP, Caspase-8 and cleaved Caspase-3 peaked at 48 h
after Res treatment in HT-29 cells and 72 h after Res treatment in
COLO 201 cells. Res did not affect the levels of Bax or Bcl-xL in
HT-29 and COLO 201 cells (data not shown). Thus, Res-induced
apoptosis may be mediated through Caspase-8/Caspase-3
activation.
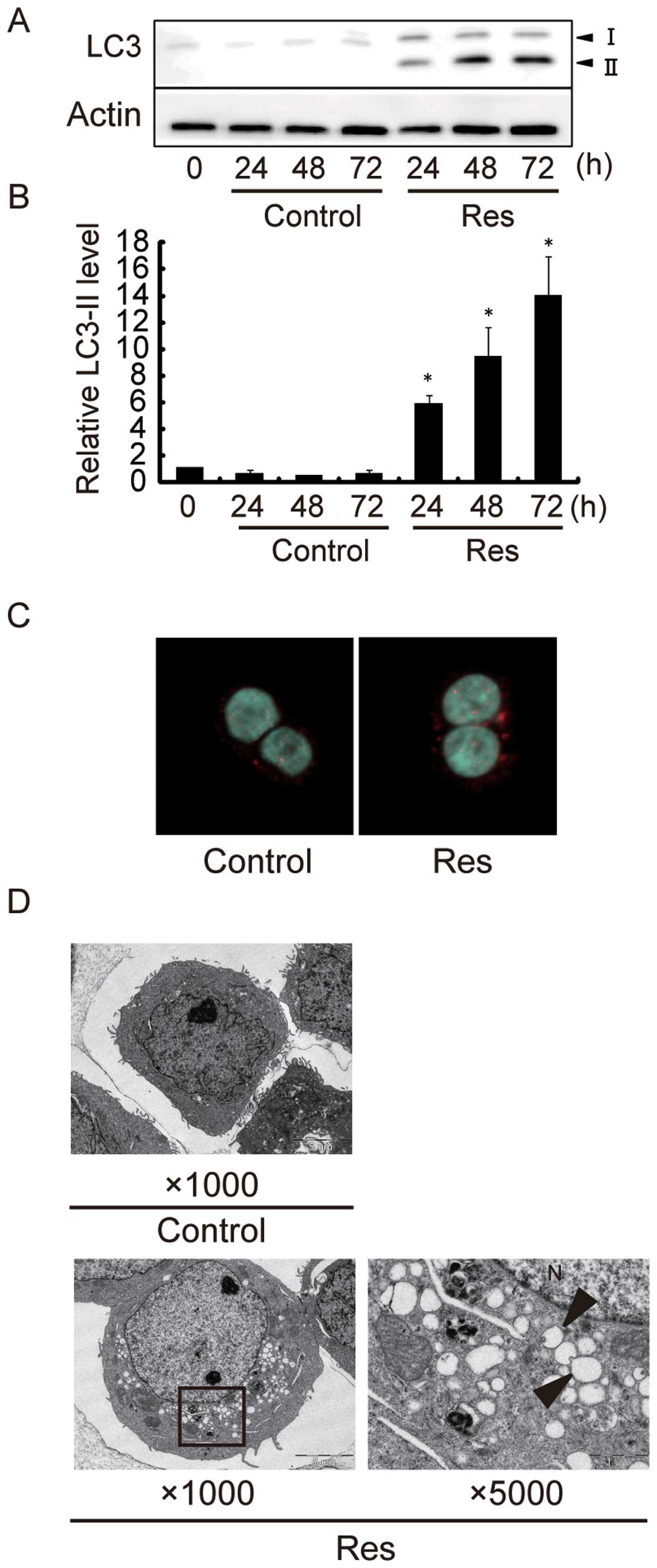
Resveratrol induces autophagy in HT-29
and COLO 201 cells
Next, we investigated the effect of Res on autophagy
induction. Representative results are shown in Fig. 3A, and quantitative data from 3
different experiments are summarized in Fig. 3B. Western blot analysis revealed
that the protein level of LC3-II increased by a maximum of ~15-fold
in HT-29 cells after 72-h Res treatment as compared to control
cells. The protein level of LC3-II increased by a maximum of
~3-fold in COLO 201 cells after 24-h Res treatment (data not shown)
as compared to control cells. These data indicate that Res induced
autophagy in HT-29 and COLO 201 cells. Although Res effectively
suppressed the growth of both human colon cancer cell lines, HT-29
cells were chosen for the following detailed studies because the
apoptotic as well as autophagic magnitude was much larger in HT-29
cells than in COLO 201 cells. We examined the localization of LC3
in HT-29 cells by immunofluorescence, and found punctuate patterns
of LC3 fluorescence signals in Res-treated cells (Fig. 3C). By electron microscopy, numerous
membranous vacuoles, autophagosomes containing residual materials,
appeared in the cytoplasm of Res-treated cells, while there were
relatively few such structures in the cytoplasm of control cells
(Fig. 3D).
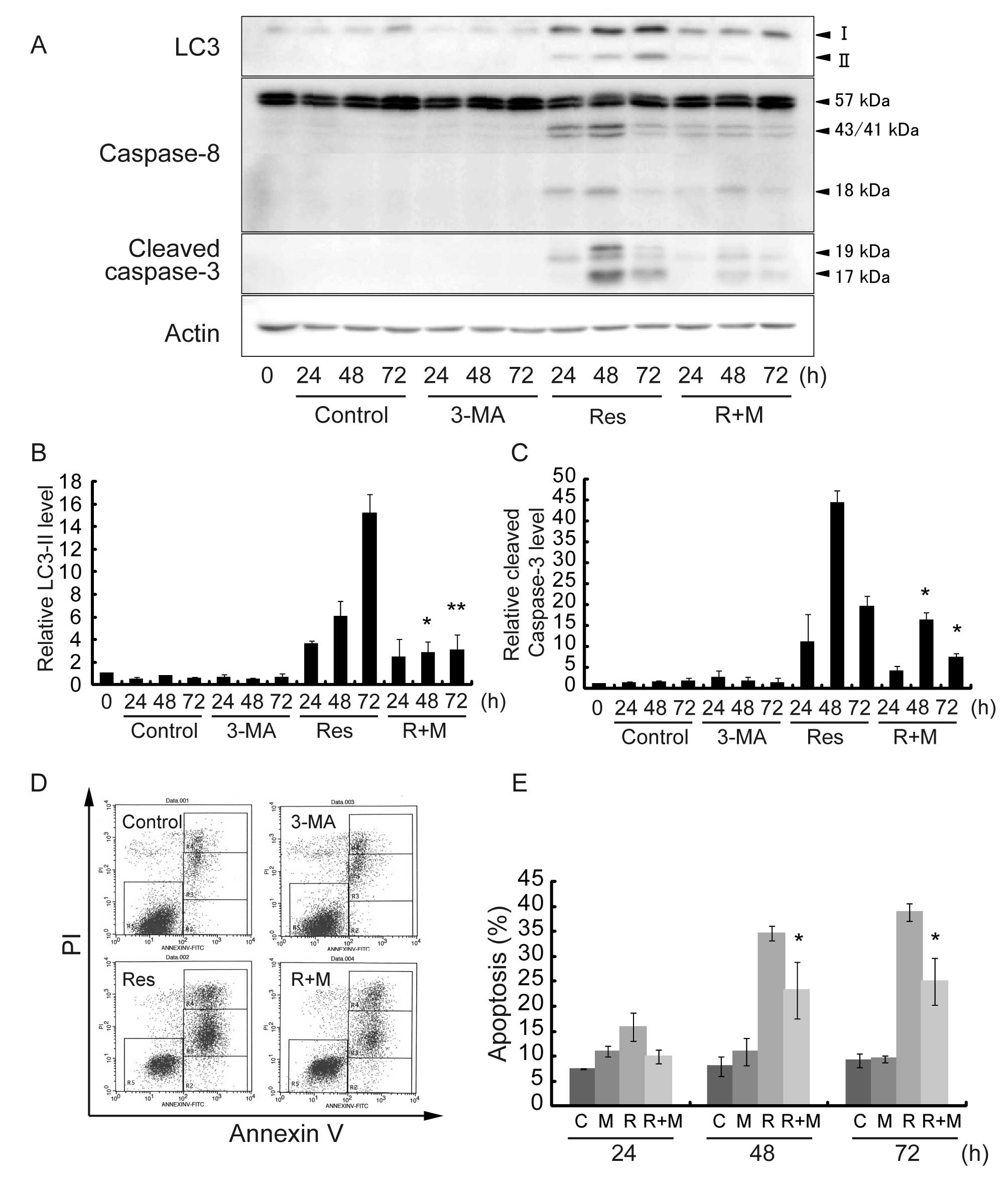
Resveratrol induces autophagic cell death
in HT-29 cells
To investigate the role of Res-induced autophagy in
HT-29 cells, autophagy specific inhibitor 3-MA was used.
Representative results are shown in Fig. 4A, and quantitative data from 3
different experiments are summarized in Fig. 4B and C. After 48- and 72-h Res
treatment in the presence of 3-MA, the LC3-II level was
significantly decreased as compared to Res treatment alone
(Fig. 4A and B). The effect of
3-MA on cell viability and the induction of apoptosis was
determined by Annexin V assay (Fig.
4D). Res treatment in the presence of 3-MA significantly
reduced the percentage of apoptotic cells from 35 to 23% for a 48-h
treatment and from 40 to 25% for a 72-h treatment as compared to
Res treatment alone (Fig. 4E). We
then examined the alterations in the protein levels of Caspase-8
and cleaved Caspase-3 after Res treatment and in the presence of
3-MA (Fig. 4A). The cleavage of
Caspase-8 and Caspase-3 levels (Fig.
4C) were significantly deceased after Res treatment in the
presence of 3-MA. These results indicate that Res induced
autophagic cell death via the Caspase-8/Caspase-3 pathway in HT-29
cells.
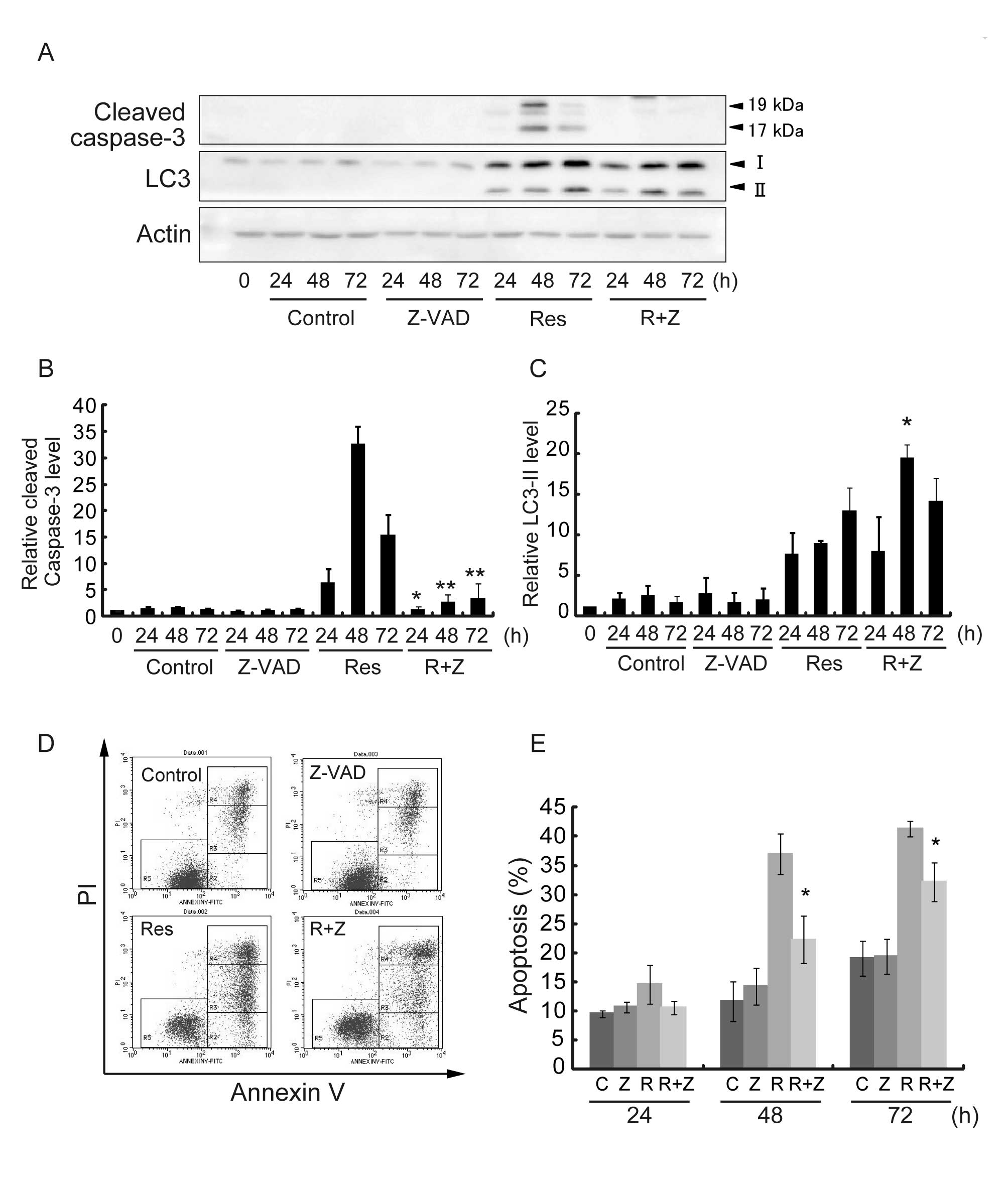
Z-VAD inhibits Res-induced cell death but
not autophagy
To investigate the relationship between apoptosis
and autophagy induced by Res in HT-29 cells, pan-caspase inhibitor
Z-VAD was tested. Representative results are shown in Fig. 5A, and quantitative data from 3
different experiments are summarized in Fig. 5B and C. The cleaved Caspase-3 level
after Res treatment in the presence of Z-VAD was significantly
decreased at 24, 48 and 72 h after treatment, as compared with Res
treatment alone (Fig. 5B). The
effect of Z-VAD on cell viability and the induction of apoptosis
were determined by Annexin V assay (Fig. 5D). Res treatment in the presence of
Z-VAD reduced apoptotic cells from 35 to 23% for 48 h of treatment
and from 40 to 32% for 72 h of treatment as compared to Res
treatment alone at respective treatment periods (Fig. 5E). We then examined the alterations
in the protein levels of LC3-II after Res treatment in the presence
of Z-VAD. The protein level of LC3-II relative to the control cells
significantly increased after Res treatment in the presence Z-VAD
for 48 h after the treatment (10- vs. 20-fold) when Res-induced
apoptosis peaked in HT-29 cells (Fig.
5C). These results indicate that Z-VAD inhibited Res-induced
apoptosis but not autophagy and suggest that Res-induced autophagy
may be located upstream of apoptosis.
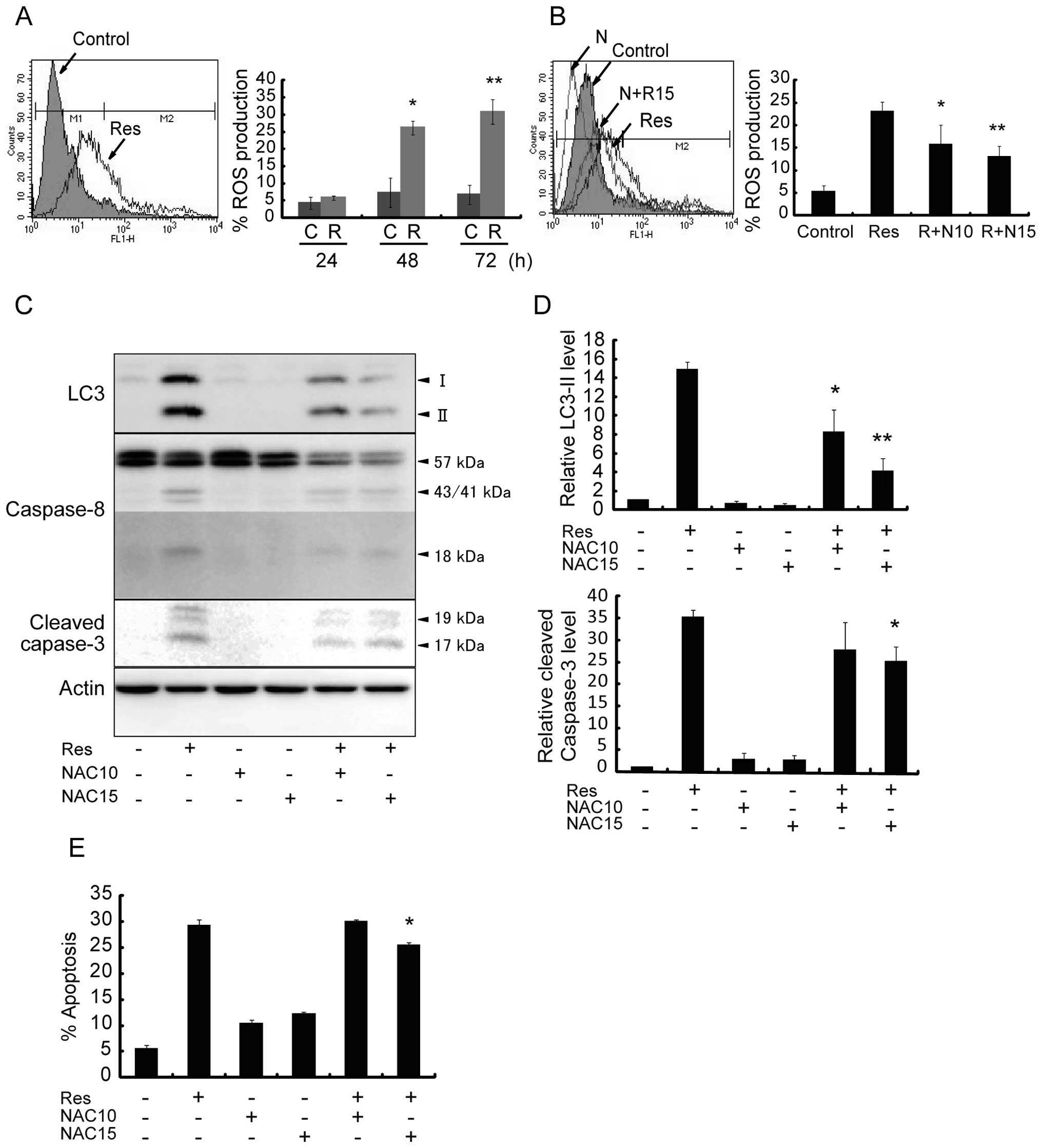
ROS mediates Res-induced autophagy and
apoptosis
We evaluated the effect of Res on the production of
ROS and its involvement with apoptosis and/or autophagy. Res
significantly increased intracellular ROS levels in a
time-dependent manner (Fig. 6A).
The percentage of ROS production was ~30% after Res treatment for
72 h, while incubation of cells with Res together with ROS
scavenger NAC at a dose of 10 or 15 mM for 48 h significantly
blocked the increase in ROS level from 23 to 15% and to 12%,
respectively (Fig. 6B). At the
same time, NAC decreased the Res-induced protein level of LC3-II
relative to the control cells from 15-fold to ~8-fold and 4-fold,
respectively (Fig. 6C and D). The
protein levels of Caspase-8 and cleaved Caspase-3 were decreased,
and the cleaved Caspase-3 level dropped from 35-fold to ~25-fold
after cells were treated with Res in the presence of 15 mM NAC for
48 h (Fig. 6C and D), which was
associated with a decrease in apoptosis (Fig. 6E). These data may indicate
intracellular ROS as the upstream stimulus that controls both
autophagy and apoptosis in Res-treated HT-29 cells.
Discussion
We demonstrated that Res inhibited human colon
cancer cell growth and found that Res induced
Caspase-8/Caspase-3-dependent apoptosis through autophagy via ROS
production. A concentration of 40 μM is relevant in terms of the
possible biological effects of Res consumed from grape beverages
(26). Thus, a Res dose of 150 μM
for HT-29 cells and 75 μM for COLO 201 cells was relatively high,
compared with physiological doses. However, here we demonstrated
that exposure of HT-29 and COLO 201 cells to Res reduced cell
proliferation rates in a dose- and time-dependent manner (Fig. 1). Induction of growth arrest and
apoptosis is the central mechanism by which Res exerts antitumor
effects against various types of cancers (9–12).
When HT-29 and COLO 201 cells were exposed to 150 and 75 μM of Res,
respectively, apoptosis was induced in a time-dependent manner, and
the activity of apoptosis executor Caspase-3 was increased
(Fig. 2). Then, the apoptosis
cascade was examined. On one hand, Res was reported to induce
apoptosis through the mitochondria pathway (27,28).
On the other hand, the effect of Res involves Caspase-8/Caspase-3
signaling and induction of apoptosis via the death receptor pathway
in several cancer cell lines (29–31).
Our study showed that Res did not affect the Bax and Bcl-xL levels
(data not shown), whereas the increased activity of
Caspase-8/Caspase-3 (Fig. 2)
indicates that Res-induced apoptosis may be mediated through the
death-receptor pathway.
Res was found to induce autophagy in several cancer
cells from different origin (17–20).
In the present study, we found that Res induced autophagy in HT-29
and COLO 201 cells and that the magnitude of Res-induced apoptosis
and autophagy was different in the different colon cancer cell
lines (Figs. 2 and 3). ‘Autophagic cell death’ can be
verified by experiments with an autophagy inhibitor, and it can be
defined by characteristic cell morphology (32–34).
We demonstrated that inhibition of autophagy by 3-MA significantly
lowered Res-induced cytotoxicity by decreasing Caspase-8 and
Capase-3 levels; thus, autophagy functioned as the cell death
mechanism (Fig. 4). To determine
whether autophagy and apoptosis may precede each other or co-occur,
inhibition studies were performed. The inhibition of autophagy by
3-MA suppressed apoptosis. In contrast, inhibition of apoptosis by
Z-VAD accelerated autophagy (LC3-II accumulation) (Fig. 5). Blocking autophagy significantly
decreased the Caspase-8/Caspase-3 levels, whereas blocking
apoptosis increased the LC3-II levels, suggesting that autophagy
initiates apoptosis. There is evidence that LC3 mediates apoptosis
via the Caspas-8/Caspase-3 pathway (35).
Not only apoptosis but also autophagy are mediated
via ROS production (22,23). Res is an antioxidant (36), and antioxidants exert different
biological activities in cancer cells and in non-transformed cells.
Antioxidants including Res effectively induced apoptosis in HT-29
cells via increased ROS production (37–39).
In the present study, Res treatment time-dependently increased ROS
production, and the quenching of ROS by NAC abolished Res-induced
autophagy (reduced LC3-II levels) and apoptosis (decreased
Caspase-8/Caspase-3 levels). Therefore, Res caused apoptosis and
autophagy via the production of ROS (Fig. 6).
In conclusion, Res effectively suppressed the growth
of HT-29 and COLO 201 human colon cancer cells. The possible
molecular mechanisms involved are Caspase-8/Caspase-3-dependent
apoptosis via ROS-triggered autophagy. Res in combination with ROS-
and autophagy-inducers may be a possible therapy for colon cancer
control.
Acknowledgements
We thank Dr Y. Katakura (Faculty of Agriculture,
Kyushu University, Fukuoka, Japan) for providing the HT-29 human
colon cancer cell line and Dr Akitsugu Yamamoto (Nagahama Institute
of Bio-Science and Technology, Nagahama, Shiga, Japan) for
providing expertise on the judgment of autophagic vacuoles on
electron microscopy photographs. We thank Mr. H. Gonda and Ms. T.
Akamatsu for technical assistance and Ms. A. Shudo for manuscript
preparation.
References
|
1
|
Xu R, Zhou B, Fung PC and Li X: Recent
advances in the treatment of colon cancer. Histol Histopathol.
21:867–872. 2006.PubMed/NCBI
|
|
2
|
Chau I and Cunningham D: Adjuvant therapy
in colon cancer - what, when and how? Ann Oncol. 17:1347–1359.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huerta S, Goulet EJ and Livingston EH:
Colon cancer and apoptosis. Am J Surg. 191:517–526. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schatzkin A and Kelloff G: Chemo- and
dietary prevention of colorectal cancer. Eur J Cancer.
31A:1198–1204. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jang M, Cai L, Udeani GO, et al: Cancer
chemopreventive activity of resveratrol, a natural product derived
from grapes. Science. 275:218–220. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Athar M, Back JH, Tang X, et al:
Resveratrol: a review of preclinical studies for human cancer
prevention. Toxicol Appl Pharmacol. 224:274–283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Johnson WD, Morrissey RL, Usborne AL, et
al: Subchronic oral toxicity and cardiovascular safety pharmacology
studies of resveratrol, a naturally occurring polyphenol with
cancer preventive activity. Food Chem Toxicol. Sept 10–2011.(Epub
ahead of print).
|
|
8
|
Fulda S and Debatin KM: Resveratrol
modulation of signal transduction in apoptosis and cell survival: a
mini-review. Cancer Detect Prev. 30:217–223. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakagawa H, Kiyozuka Y, Uemura Y, et al:
Resveratrol inhibits human breast cancer cell growth and may
mitigate the effect of linoleic acid, a potent breast cancer cell
stimulator. J Cancer Res Clin Oncol. 127:258–264. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shih A, Davis FB, Lin HY and Davis PJ:
Resveratrol induces apoptosis in thyroid cancer cell lines via a
MAPK- and p53-dependent mechanism. J Clin Endocrinol Metab.
87:1223–1232. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liang YC, Tsai SH, Chen L, Lin-Shiau SY
and Lin JK: Resveratrol-induced G2 arrest through the inhibition of
CDK7 and p34CDC2 kinases in colon carcinoma HT29 cells. Biochem
Pharmacol. 65:1053–1060. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao PC, Ng LT, Lin LT, Richardson CD,
Wang GH and Lin CC: Resveratrol arrests cell cycle and induces
apoptosis in human hepatocellular carcinoma Huh-7 cells. J Med
Food. 13:1415–1423. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kondo Y and Kondo S: Autophagy and cancer
therapy. Autophagy. 2:85–90. 2006. View Article : Google Scholar
|
|
16
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Opipari AW Jr, Tan L, Boitano AE, Sorenson
DR, Aurora A and Liu JR: Resveratrol-induced autophagocytosis in
ovarian cancer cells. Cancer Res. 64:696–703. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Trincheri NF, Follo C, Nicotra G,
Peracchio C, Castino R and Isidoro C: Resveratrol-induced apoptosis
depends on the lipid kinase activity of Vps34 and on the formation
of autophagolysosomes. Carcinogenesis. 29:381–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Scarlatti F, Maffei R, Beau I, Codogno P
and Ghidoni R: Role of non-canonical Beclin 1-independent autophagy
in cell death induced by resveratrol in human breast cancer cells.
Cell Death Differ. 15:1318–1329. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Puissant ARG, Fenouille N, Luciano F,
Cassuto JP, Raynaud S and Auberger P: Resveratrol promotes
autophagic cell death in chronic myelogenous leukemia cells via
JNK-mediated p62/SQSTM1 expression and AMPK activation. Cancer Res.
70:1042–1052. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fawcett H, Mader JS, Robichaud M,
Giacomantonio C and Hoskin DW: Contribution of reactive oxygen
species and caspase-3 to apoptosis and attenuated ICAM-1 expression
by paclitaxel-treated MDA-MB-435 breast carcinoma cells. Int J
Oncol. 27:1717–1726. 2005.PubMed/NCBI
|
|
22
|
Wang Q, Liang B, Shirwany NA and Zou MH:
2-Deoxy-D-glucose treatment of endothelial cells induces autophagy
by reactive oxygen species-mediated activation of the AMP-activated
protein kinase. PLoS One. 6:e172342011. View Article : Google Scholar
|
|
23
|
Wong CH, Iskandar KB, Yadav SK, Hirpara
JL, Loh T and Pervaiz S: Simultaneous induction of non-canonical
autophagy and apoptosis in cancer cells by ROS-dependent ERK and
JNK activation. PLoS One. 5:e99962010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Uehara N, Matsuoka Y and Tsubura A:
Mesothelin promotes anchorage-independent growth and prevents
anoikis via extracellular signal-regulated kinase signaling pathway
in human breast cancer cells. Mol Cancer Res. 6:186–193. 2008.
View Article : Google Scholar
|
|
25
|
Kanematsu S, Uehara N, Miki H, et al:
Autophagy inhibition enhances sulforaphane-induced apoptosis in
human breast cancer cells. Anticancer Res. 30:3381–3390.
2010.PubMed/NCBI
|
|
26
|
Huang C, Ma WY, Goranson A and Dong Z:
Resveratrol suppresses cell transformation and induces apoptosis
through a p53-dependent pathway. Carcinogenesis. 20:237–242. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ganapathy S, Chen Q, Singh KP, Shankar S
and Srivastava RK: Resveratrol enhances antitumor activity of TRAIL
in prostate cancer xenografts through activation of FOXO
transcription factor. PLoS One. 5:e156272010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Z, Jin K, Gao L, et al: Anti-tumor
effects of bakuchiol, an analogue of resveratrol, on human lung
adenocarcinoma A549 cell line. Eur J Pharmacol. 643:170–179. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shankar S, Siddiqui I and Srivastava RK:
Molecular mechanisms of resveratrol
(3,4,5-trihydroxy-trans-stilbene) and its interaction with
TNF-related apoptosis inducing ligand (TRAIL) in
androgen-insensitive prostate cancer cells. Mol Cell Biochem.
304:273–285. 2007. View Article : Google Scholar
|
|
30
|
Kim MY, Trudel LJ and Wogan GN: Apoptosis
induced by capsaicin and resveratrol in colon carcinoma cells
requires nitric oxide production and caspase activation. Anticancer
Res. 29:3733–3740. 2009.PubMed/NCBI
|
|
31
|
Reis-Sobreiro M, Gajate C and Mollinedo F:
Involvement of mitochondria and recruitment of Fas/CD95 signaling
in lipid rafts in resveratrol-mediated antimyeloma and antileukemia
actions. Oncogene. 28:3221–3234. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Levine B and Yuan J: Autophagy in cell
death: an innocent convict? J Clin Invest. 115:2679–2688. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gozuacik D and Kimchi A: Autophagy and
cell death. Curr Top Dev Biol. 78:217–245. 2007. View Article : Google Scholar
|
|
35
|
Chen ZH, Lam HC, Jin Y, et al: Autophagy
protein microtubule-associated protein 1 light chain-3B (LC3B)
activates extrinsic apoptosis during cigarette smoke-induced
emphysema. Proc Natl Acad Sci USA. 107:18880–18885. 2010.
View Article : Google Scholar
|
|
36
|
Roig R, Cascon E, Arola L, Blade C and
Salvado MJ: Moderate red wine consumption protects the rat against
oxidation in vivo. Life Sci. 64:1517–1524. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Juan ME, Wenzel U, Daniel H and Planas JM:
Resveratrol induces apoptosis through ROS-dependent mitochondria
pathway in HT-29 human colorectal carcinoma cells. J Agric Food
Chem. 56:4813–4818. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wenzel U, Nickel A and Daniel H:
alpha-Lipoic acid induces apoptosis in human colon cancer cells by
increasing mitochondrial respiration with a concomitant
O2-*-generation. Apoptosis. 10:359–368. 2005. View Article : Google Scholar
|
|
39
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|